")
Back to Journals » OncoTargets and Therapy » Volume 13
Inhibition of Serine Metabolism Promotes Resistance to Cisplatin in Gastric Cancer
Authors Zhao X, Fu J , Tang W, Yu L, Xu W
Received 17 January 2020
Accepted for publication 11 May 2020
Published 29 May 2020 Volume 2020:13 Pages 4833—4842
DOI https://doi.org/10.2147/OTT.S246430
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjay Singh
Xiaoya Zhao,1,2,* Jianfei Fu,3,* Wanfen Tang,3 Liangliang Yu,4 Wenxia Xu1
1Central Laboratory, Jinhua Hospital of Zhejiang University, Jinhua 321000, Zhejiang Province, People’s Republic of China; 2Department of Medical Oncology, Sir Run Run Shaw Hospital, Medical School of Zhejiang University, Hangzhou 310000, Zhejiang Province, People’s Republic of China; 3Department of Medical Oncology, Jinhua Hospital of Zhejiang University, Jinhua 321000, Zhejiang Province, People’s Republic of China; 4Department of Gastroenterology, Sir Run Run Shaw Hospital, Medical School of Zhejiang University, Hangzhou 310000, Zhejiang Province, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Liangliang Yu
Department of Gastroenterology, Sir Run Run Shaw Hospital, Medical School of Zhejiang University, No. 3 Qingchun Road, Jianggan District, Hangzhou 310000, Zhejiang Province, People’s Republic of China
Tel +86 571-86006646
Email [email protected]
Wenxia Xu
Central Laboratory, Jinhua Hospital of Zhejiang University, No. 351 Mingyue Street, Wucheng District, Jinhua 321000, Zhejiang Province, People’s Republic of China
Tel +86 579 82553851
Email [email protected]
Background: Serine provides important precursors of protein, lipid, and nucleotide synthesis needed for tumor cell growth. Phosphoglycerate dehydrogenase (PHGDH), a key rate-limiting enzyme in the serine de novo synthesis pathway, is highly expressed in many tumor types (including gastric cancer) and negatively correlated with overall survival. Cisplatin is a chemotherapeutic drug commonly used in the treatment of gastric cancer. In this study, we mainly investigated the relationship between serine metabolism and resistance to cisplatin in gastric cancer cells, as well as the regulatory mechanism involved in this process.
Materials and Methods: We determined the effect of different concentrations of serine or a PHGDH inhibitor combined with cisplatin or oxaliplatin on the viability and apoptosis of SGC7901, BGC823, and MGC803 cells via the Cell Counting Kit-8 and Hoechst 33258 staining, respectively. Western blotting was utilized to detect the relative protein expression. Furthermore, we investigated DNA damage through the micrococcal nuclease sensitivity assay detected using agarose gels.
Results: We found that reduced concentrations of serine or inhibition of PHGDH hindered the toxicity and pro-apoptotic effects of cisplatin on gastric cancer cells. Moreover, the addition of serine could reverse the sensitivity of gastric cancer cells to cisplatin. Moreover, we found that DNA damage was reduced by treatment with PHGDH inhibitor NCT-503 or CBR-5884. Inhibition of serine metabolism induced a decrease in H3K4 tri-methylation, which was reversed by JIB-04 (inhibitor of H3K4 demethylase). The tolerance of gastric cancer cells to cisplatin was relieved by JIB-04. Through micrococcal nuclease experiments, we further found that inhibiting the activity of PHGDH strengthened chromatin tightness.
Conclusion: Inhibition of serine metabolism reduced H3K4 tri-methylation and increased the density of chromatin, which leads to decreased toxicity and pro-apoptotic effect of platinum chemotherapeutic drugs on gastric cancer cells.
Keywords: serine metabolism, cisplatin resistance, PHGDH, DNA damage, gastric cancer
Introduction
Gastric cancer is one of the most common malignant tumors, with the second-highest incidence and third highest mortality in China.1 Patients with gastric cancer show “three high and three low characteristics”, including high incidence, metastasis, and mortality rates, as well as low early diagnosis, radical cure, and 5-year survival rates.2 At present, gastric cancer is mainly treated with surgery, radiotherapy, and chemotherapy. However, the mechanism of occurrence and development of gastric cancer remains unclear. The high rates of metastasis and recurrence, along with drug tolerance have become the main obstacles to improve the long-term survival rate of patients.3,4 Therefore, the identification of new molecular prognostic markers and therapeutic targets is necessary to improve the clinical prognosis of patients with gastric cancer.
Cisplatin is one of the most commonly used chemotherapeutic agents for the treatment of numerous types of cancer (eg, gastric, non-small-cell lung, bladder, testicular, ovarian, head and neck, and other malignancies). However, the use of cisplatin in clinical treatment is limited by two major problems. Cisplatin exerts many toxic and side effects (eg, kidney damage, deafness, peripheral neuropathy, etc.), leading to a decrease in the overall efficacy of the drug. Furthermore, tumors that initially respond to treatment with cisplatin often develop resistance to further treatment with platinum after tumor recurrence. The overall mechanisms of resistance to platinum include increased DNA repair, altered drug accumulation in the cell, and increased intracytoplasmic inactivation of drugs.5
Metabolic reprogramming is one of the 10 characteristics of tumors, among which amino acid metabolic reprogramming has attracted increasing attention.6 In addition to glutamine metabolism, serine/glycine metabolism is also necessary for tumor cells. Serine is a non-essential amino acid in the human body; however, it is an essential amino acid in specific circumstances for tumors. It is obtained from the external environment through transporters and also synthesized by the serine synthesis pathway, as necessary. In 1955, the endogenous de novo synthesis pathway of serine was first observed in tumor cells.7 Phosphoglycerate dehydrogenase (PHGDH) converts approximately 10% of the 3-phosphoglyceric acid produced during glycolysis into 3-phosphatedehydropyruvate, a precursor of serine, and is subsequently catalyzed by phosphoserine aminotransferase 1 and phosphoserine phosphatase to produce serine. The reduction of exogenous serine concentration or targeted inhibition of PHGDH can effectively play an anti-tumor role.8–10 However, the effects of reducing the exogenous serine concentration or inhibiting PHGDH combined with chemotherapy remain unclear.
In this study, we discovered the mechanism of inhibiting serine metabolism leads to cisplatin resistance in gastric cancer. Which laid a foundation for further investigation of the relation between nutrient metabolism and tumor chemotherapy.
Materials and Methods
Reagents
Human gastric cancer cell lines SGC7901, BGC823, and MGC803 were purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). RPMI-1640 medium (31800–105) was obtained from Gibco (Grand Island, New York, USA). RPMI-1640 w/o Amino, sodium phosphate (powder) was purchased from US Biological (Salem, MA, USA). Fetal bovine serum (11011–8611) was obtained from Every Green (Hangzhou, China). Penicillin-streptomycin solution (GNM15140) and trypsin (GNM25200) were purchased from Genome (Hangzhou, Zhejiang, China). Arginine, asparagine, aspartic acid, cystine dihydrochloride, glutamic acid, glycine, histidine, hydroxyproline oxyproline, isoleucine, leucine, lysine hydrochloride, methionine, phenylalanine, proline, serine, threonine, tryptophan, tyrosine, valine, glutamine were obtained from Sigma (Louis, MO, USA). Hoechst 33258 (C1017), horseradish peroxidase-conjugated goat anti-Mouse, and anti-Rabbit IgG were purchased from Beyotime Biotechnology (Shanghai, China). Micrococcal nuclease (MNase; 2910A) was obtained from Takara (Osaka, Japan). Radioimmunoprecipitation assay lysis buffer (FD009), BCA Protein Assay Kit (FD2001), and enhanced chemiluminescence kit (FD800) were purchased from Fdbio Science (Hangzhou, Zhejiang, China). CBR-5884 (HY-100012) and NCT-503 (HY-101966) were purchased from MedChemExpression (Shanghai, China). JIB-04 (S7281) was obtained from Selleck (Houston, Texas, USA). Rabbit anti-poly-ADP ribose polymerase (anti-PARP; 13371-1-AP) and rabbit anti-Caspase3 (66470-2-Ig) were purchased from Proteintech (IL, USA). Rabbit anti-Tubulin (AF0001) and mouse anti-Actin (AA128) were obtained from Beyotime Biotechnology. Rabbit anti-γH2AX (ab81299) was purchased from Abcam (Cambridge, UK); rabbit anti-H3 (#4499) and rabbit anti-H3k4 me3 (#9751) were purchased from Cell Signaling Technology (Danvers, MA, USA). TRIzol (15596026) was purchased from Ambion (Carlsbad CA, USA) and dimethyl sulfoxide (D2650) was obtained from Sigma.
Cell Culture
Human gastric cancer cell lines SGC7901, BGC823, and MGC803 were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 mg/mL streptomycin. All cells were incubated at 37°C in a 5% CO2 humidified incubator. Cells were grown to 70–90% confluence.
Preparation of Different Concentrations of Serine
RPMI-1640 w/o Amino, Sodium Phosphate (powder), sodium bicarbonate, and 19 types of amino acids (excluding serine), were dissolved in double-distilled water to generate serine-free 1640 medium, termed 0×Ser. We defined the concentration of serine in the 1640 complete medium (30 μg/mL) as 1×Ser. The 100×Ser (3,000 μg/mL) solution was prepared in advance, and subsequently added to serine-free 1640 medium to produce 1/4×Ser (7.5 μg/mL), 1×Ser (30 μg/mL), 4×Ser (120 μg/mL), and 8×Ser (240 μg/mL), which were used in the following experiments.
Cell Viability
Cells were plated onto 96-well plates at a density of 1×104 cells per well, and cultured in 100 μL complete medium for 24 h. Subsequently, a complete medium was replaced with medium containing 0.6 μg/mL, 0.8 μg/mL, and 3 μg/mL cisplatin under different concentrations of serine or the inhibitor of PHGDH for 24 h. This was followed by the addition of 100 μL medium containing 10 μL Cell Counting Kit-8 (Beyotime Biotechnology) agent to each well for 1 h after discarding the supernatant. The optical density values at 450 nm were determined using a Microplate Reader (Synergy HT ZX-22; Bio-Tek Instruments, USA).
Western Blotting Analysis
Briefly, the whole-cell protein was obtained using radio-immunoprecipitation assay lysis buffer (RIPA) containing 1 mM phenylmethanesulfonyl fluoride. Equal loading of lysate protein was determined by standardization using the BCA Protein Assay Kit and a Microplate Reader (Synergy HT ZX-22; Bio-Tek Instruments). Proteins were separated through electrophoresis using sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels and transferred to polyvinylidene difluoride membranes (BioRad, Hercules, California, USA). The membranes were blocked at room temperature for 1 h in tris-buffered saline containing 5% fat-free powdered milk and incubated overnight with primary antibodies at 4°C. Subsequently, the membranes were incubated with secondary antibody for 2 h at room temperature after washing with tris-buffered saline containing 0.1% Tween 20. Proteins were detected using the FDbio-Pico enhanced chemiluminescence kit and quantified with the ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Hoechst 33258 Staining
Cells were plated onto 24-well plates at a density of 1×105 cells per well, and cultured in 1,000 μL complete medium for 24 h. Cells were cultured for 24 h after replacing the supernatant with the medium including 0.6 μg/mL cisplatin and extra serine. The cells were fixed with methanol for 10 min and washed thrice with phosphate-buffered saline. Three hundred μL of Hoechst 33258 staining solution was added for 3–5 min at room temperature. The staining solution was subsequently removed and the cells were washed thrice with phosphate-buffered saline, each time for 3–5 min. Cells were observed under a fluorescence microscope and quantified with the ImageJ software (National Institutes of Health).
MNase Sensitivity Assay
SGC7901 cells were treated with NCT-503, JIB-04 and cisplatin for 24 h. Cells were treated with lysis buffer (10 mM Tris-hydrochloride [pH 8.0], 10 Mm magnesium dichloride, 1 mM dithiothreitol and 0.5% NP-40), incubated on ice for 15 min, centrifuged at 13,000 g for 4 min at 4°C. The supernatant was removed, nuclei were pelleted and digested with MNase at a concentration of 4 U/200 μL digestion buffer (15 mM Tris-hydrochloride [pH 7.4], 60 mM potassium chloride, 15 mM sodium chloride, 0.25 M sucrose, 1Mm calcium chloride and 0.5 mM dithiothreitol) for 10 min at 37°C. Genomic DNA was extracted using a genomic DNA mini preparation kit (Beyotime Biotechnology), purified and separated through electrophoresis using 1.5% agarose gels.
Statistical Analysis
Data are presented as the mean ± standard deviation. Statistical analyses were performed with the unpaired, two-tailed Student’s t-test using GraphPad Prism 6.02 for Mac (GraphPad Prism Software Inc., San Diego, CA, USA). A p<0.05 denoted a statistically significant difference.
Results
Serine Concentration Was Positively Correlated with the Sensitivity of Gastric Cancer Cells to Cisplatin
We examined the activity of gastric cancer cells treated with cisplatin under different concentrations of serine to investigate the effect of serine concentration on sensitivity to this drug. The concentration of serine in the 1640 complete medium was 30 μg/mL, which was defined as 1×Ser. We treated gastric cancer cells SGC7901, BGC823 and MGC803 with 0×Ser, 1/4×Ser, 1×Ser, 4×Ser and 8×Ser. The lower (0×Ser, 1/4×Ser) and higher (4×Ser, 8×Ser) concentrations of serine had no effect on cell toxicity and apoptosis (Figures 1A–E and S1A). Cisplatin had a significantly higher toxic effect on gastric cancer cells in the 4×Ser and 8×Ser groups than the 1×Ser group (Figure 1A–C). However, the toxicity of cisplatin in the 0×Ser and 1/4×Ser groups was significantly lower than that observed in the 1×Ser group (Figure 1A–C). Typical nucleus condensation, as the morphological marker of apoptosis, was observed using Hoechst 33258 staining. The experiment showed that higher concentrations of serine (4×Ser, 8×Ser) significantly promoted the apoptosis of cells induced by cisplatin (Figure 1D and E). These results indicate that the concentration of serine was positively related to the sensitivity of gastric cancer cells to cisplatin.
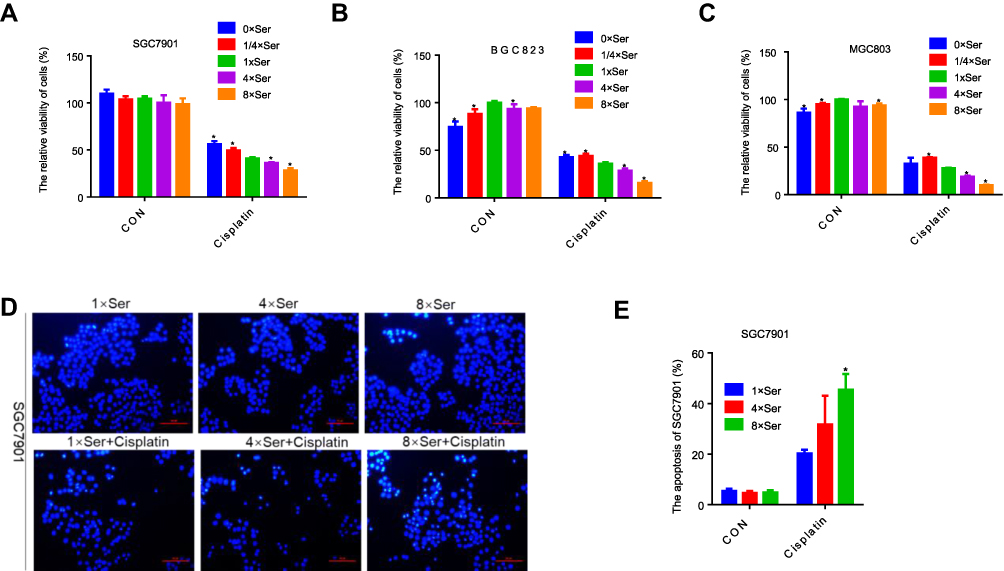 |
Figure 1 Serine concentration was positively correlated with the sensitivity of gastric cancer cells to cisplatin. (A–C) CCK-8 analysis of cell viability in SGC7901, BGC823 and MGC803 cells with 0.6 μg/mL, 0.8 μg/mL, 3 μg/mL cisplatin, respectively, under different concentrations of serine. The concentrations of 1/4×Ser, 1×Ser, 4×Ser and 8×Ser are 7.5 μg/mL, 30 μg/mL, 120 μg/mL, 240 μg/mL, respectively. Error bars represent SD (n = 3). (D and E) Hoechst 33,258 staining analysis of apoptosis in SGC7901 with 0.6 μg/mL cisplatin under different concentrations of serine and the apoptosis index analyzed by ImageJ software. *p<0.05. Compared to 1×Ser. |
Inhibition of PHGDH Leads to Resistance to Cisplatin in Gastric Cancer Cells
Serine can be obtained from the external environment, and also synthesized through the serine synthesis pathway from the glycolysis flux (Figure 2A). PHGDH is highly expressed in numerous types of human tumors and positively correlated with the progression of breast cancer, colorectal cancer and poor prognosis.11–15 We found that high PHGDH expression was significantly associated with decreased overall survival, progression-free survival and post-progression survival in patients with gastric cancer (Figures 2B, S1B, and S1C). We inhibited PHGDH by NCT-503 or CBR-5884 (the specific inhibitors of PHGDH) and found that the proliferation of BGC823 cells was suppressed (Figure S1G), while the apoptosis was not observed (Figures 2E, F, S1I and S1J). Moreover, the knockdown of PHGDH cannot induce apoptosis of cells (Figure S1H). Then, we treated gastric cancer cells with NCT-503 or CBR-5884 in combination with platinum-based drugs (cisplatin and oxaliplatin), to investigate the relationship between PHGDH and the sensitivity to these agents. Cisplatin or oxaliplatin combined with PHGDH inhibitors led to a significant increase in cell survival rate versus monotherapy with cisplatin or oxaliplatin (Figures 2C, D and S1D–F). Moreover, the expression levels of cleaved PARP and Caspase3 (two apoptotic marker proteins) were significantly reduced in the combined treatment group versus the monotherapy group (Figures 2E, F, S1I and S1J). We found lower survival and higher apoptosis rates in cells treated with NCT-503, cisplatin and extra serine relative to those recorded in cells treated with cisplatin and NCT-503 (Figure 2G and H). These results suggest that inhibition of PHGDH induced resistance to cisplatin in gastric cancer cells, whereas supplementation with serine restored the sensitivity of gastric cancer cells to this chemotherapeutic agent.
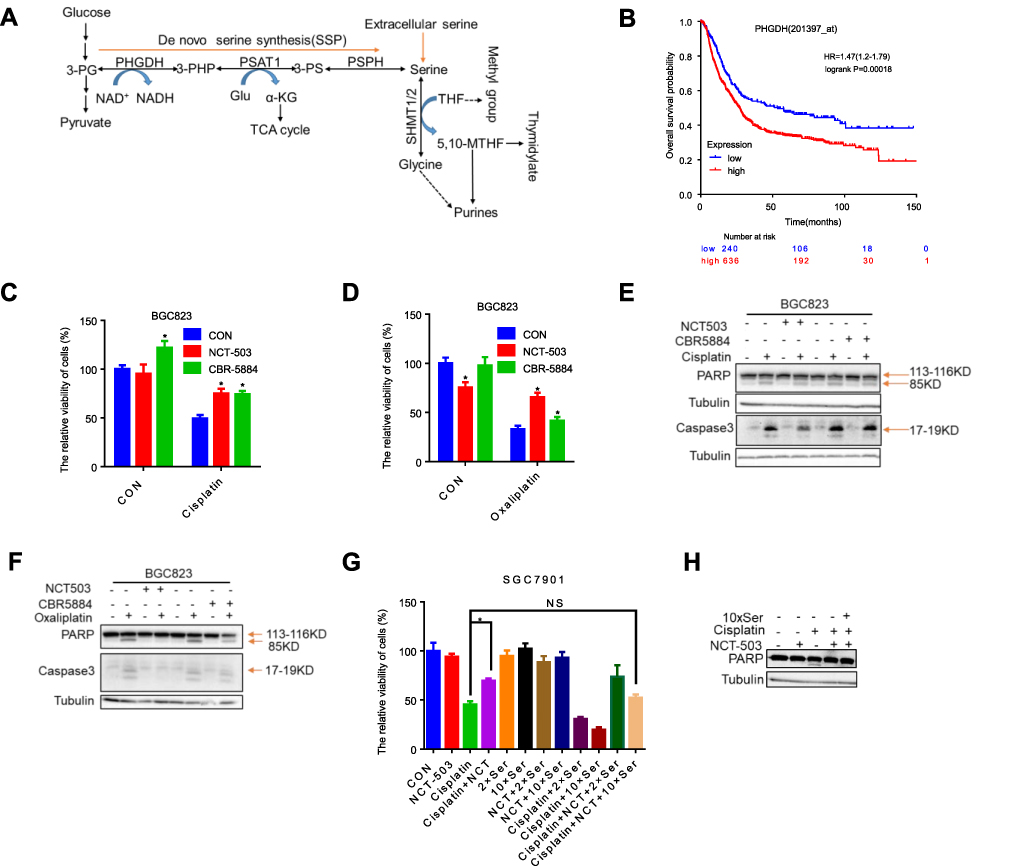 |
Figure 2 Inhibition of PHGDH leads to resistance to cisplatin in gastric cancer cells. (A) L-Serine synthesis pathway. PHGDH first catalyzes the oxidation of 3-phosphoglycerate (3-PG) to 3-phosphohydroxypyruvate (3-PHP) and coinstantaneous reduction of the cofactor NAD+ to NADH. The subsequent transamination reaction is catalyzed by phosphoserine aminotransferase (PSAT), which uses glutamate (Glu) as a nitrogen donor and thereby produces 3-phosphoserine (3-PS) and α-ketoglutarate (α-KG) into tricarboxylic acid (TCA) cycle. Dephosphorylation of phosphoserine by phosphoserine phosphatase (PSPH) gives rise to serine; then, hydroxymethyltransferase (SHMT) converts serine into glycine and 5,10-Methyltetrahydrofolate (5,10-MTHF) via tetrahydrofolate (THF) supplying methyl. (B) Kaplan–Meier overall survival for PHGDH expression in gastric tumors (GEO, EGA, and TCGA data set). (C and D) CCK-8 analysis of cell viability in BGC823 cells with 0.8 μg/mL cisplatin or 8 μg/mL oxaliplatin under treatment of 50 μΜ NCT-503 and 10 μΜ CBR-5884. Error bars represent SD (n = 3). See also Figures S1D-F. (E and F) Western blotting of PARP and Caspase3 in BGC823 with 0.8 μg/mL cisplatin or 8 μg/mL oxaliplatin under treatment of 10 μΜ CBR-5884. 85 KD and 17–19 KD were cleaved from PARP and Caspase3, respectively. The cleaved PARP and Caspase3 levels were quantified against tubulin. See also Figures S1I and S1J. (G) CCK-8 analysis of cell viability in SGC7901 cells with 0.6 μg/mL cisplatin under treatment of 50 μΜ NCT-503 and different concentrations of serine. Error bars represent SD (n = 3). (H) Western blotting of PARP in BGC823 cells with 0.6 μg/mL cisplatin, 50 μΜ NCT-503 and 10×Ser. *p<0.05. |
Inhibition of PHGDH Decreases Cisplatin-Induced DNA Damage in Gastric Cancer Cells
Cisplatin kills cancer cells mainly by destroying DNA in the form of Pt-d (GpG), leading to DNA double-strand breaks.16,17 It has been reported that the levels of γH2AX are positively correlated with the severity of DNA damage.18 We treated gastric cancer cells with cisplatin for 2 h, and replaced the medium with fresh complete medium with or without NCT-503 and CBR5884 for 0, 4, 8, 12 and 24 h to examine the cisplatin-induced DNA damage in gastric cancer cells following the inhibition of PHGDH. The levels of γH2AX protein in the NCT-503 and CBR5884 treatment groups were significantly lower than those reported in the control group at all time points (Figure 3A–C). The results of the gray-scale quantitative analysis showed that the growth rate of γH2AX was significantly lower than that observed in the control group (Figure 3D–F). These results illustrate that the inhibition of PHGDH reduced the DNA damage induced by cisplatin in gastric cancer cells.
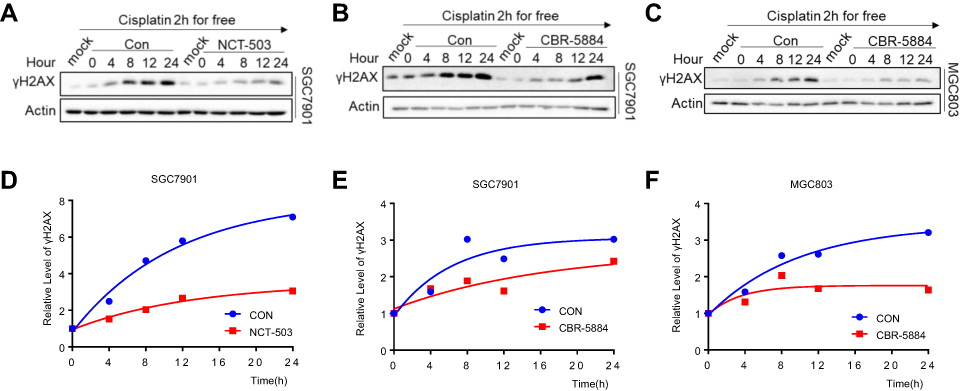 |
Figure 3 Inhibition of PHGDH decreases the cisplatin-induced DNA damage in gastric cancer cells. (A–C) Western blotting of γH2AX in SCC7901 with 50 μΜ NCT-503 or 10 μΜ CBR-5884 after 0.6 μg/mL cisplatin treatment for 2 h and in MGC803 with 10 μΜ CBR-5884 after 3 μg/mL cisplatin treatment for 2 h. γH2AX levels were quantified against actin. (D–F) The gray analysis of γH2AX levels quantified against actin of A-C by ImageJ software. The γH2AX levels of 4, 8, 12 and 24 h were normalized against the γH2AX level of 0 h. |
Inhibition of Serine Metabolism Leads to Histone Demethylation
We also examined the methylation of H3K4 to obtain a deep insight into the blockage of DNA damage on account of inhibition of serine metabolism. The results of our previous study showed lower H3K4 tri-methylation in drug-resistant gastric cancer cells.19 Moreover, decreased H3K4 tri-methylation was observed after serine deprivation or inhibition of PHGDH (Figure 4A). JIB-04 is a broad-spectrum selective inhibitor of Jumonji histone demethylase. We treated cells with cisplatin and NCT-503 combined with JIB-04. This treatment significantly decreased cell activity versus cisplatin co-administered with NCT-503 only (Figure 4B). The levels of H3K4 tri-methylation were promoted by treatment with JIB-04 (Figure 4C). Furthermore, we examined the DNA damage in cells treated with JIB-04 to extend these findings. Strikingly, the expression of γH2AX was markedly increased (Figure 4D and E). Studies have shown that low coagulation DNA encased in chromatin is more severely damaged than tightly packed chromatin under ionizing radiation,20,21 while the spatial structure of chromatin is regulated by histone post-translational modifications (eg, acetylation and methylation).22–26 Based on this evidence, we used an indirect method of MNase digestion to analyze chromatin condensation and investigate the mechanism through which NCT-503 reduces the cisplatin-induced DNA damage in gastric cancer cells. We found that the abundance of mononucleosomes and binucleosomes in the cisplatin and NCT-503 combination treatment group was lower than that noted in the cisplatin monotherapy group (Figure 4F). In contrast, the abundance of mononucleosomes and binucleosomes could be significantly increased by treatment with JIB-04 combined with cisplatin and NCT-503 (Figure 4F). Thus, a low concentration of serine enhances chromatin compactness by inhibiting H3K4 tri-methylation, resulting in decreased cisplatin-induced DNA damage.
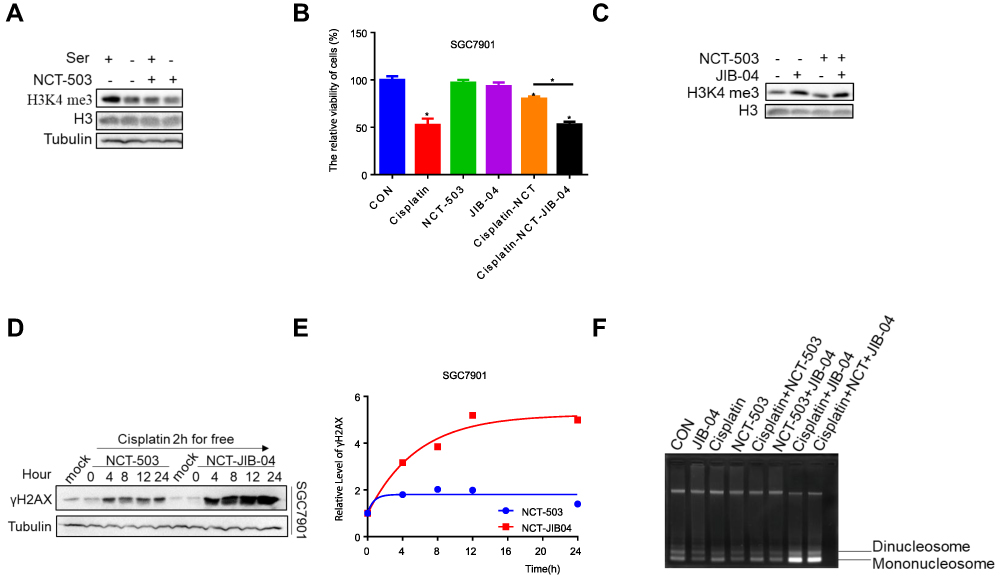 |
Figure 4 Inhibition of serine metabolism leads to histone demethylation. (A) Western blotting of H3K4 me3 in SGC7901 under the treatment of 50 μΜ NCT-503 or serine. H3K4 me3 levels were quantified against H3. (B) CCK-8 analysis of cell viability in SGC7901with 0.6 μg/mL cisplatin, 50 μΜ NCT-503, 25 μΜ JIB-04, joint use of 0.6 μg/mL cisplatin as well as 50 μΜ NCT-503 and joint use of 0.6 μg/mL cisplatin, 50 μΜ NCT-503 and 25 μΜ JIB-04. Error bars represent SD (n = 3). (C) Western blotting of H3K4 me3 in SCC7901 with 50 μΜ NCT-503 and 25 μΜ JIB-04, H3K4 me3 levels were quantified against H3. (D) Western blotting of γH2AX in SCC7901 with 25 μΜ JIB-04 combined with 50 μΜ NCT-503 after 0.6 μg/mL cisplatin treatment for 2 h. γH2AX levels were quantified against actin. (E) The gray analysis of γH2AX levels of 4, 8, 12 and 24 h was normalized against the γH2AX level of 0 h by ImageJ software. (F) SGC7901 cells were treated with MNase (4 U/mL) and chromatin was isolated and run on 1.5% agarose gel. Positions of mononucleosomes and dinucleosomes were indicated for comparing their abundances. *p<0.05. |
Discussion
Tumors require a variety of nutrients for the synthesis of biological macromolecules to maintain their growth and proliferation. Serine provides the necessary precursor for the synthesis of proteins, nucleic acids, and lipids essential for the growth of cancer cells.27 The association between cancer and serine biosynthesis was first demonstrated in rat liver tissue.28 We observed the activity and apoptosis of cells induced by cisplatin under different concentrations of serine to investigate the link between the sensitivity of gastric cancer cells to cisplatin or oxaliplatin and serine metabolism. Unexpectedly, lower serine concentration alleviated the sensitivity of gastric cancer cells to cisplatin and oxaliplatin. Serine can be obtained through the de novo serine synthesis pathway catalyzed by PHGDH, a rate-limiting enzyme that initiates this pathway. Numerous studies have linked the expression levels of PHGDH with the activity of the serine synthesis pathway. Thus far, PHGDH has been found to be highly expressed in various types of cancer (eg, breast,29 bladder,30 glioma,31 cervical,32 melanoma,33 non-small-cell lung,34 colorectal11 and gastric35), dysregulating serine metabolism and contributing to tumor progression. Inhibition of PHGDH activity by si-RNA or inhibitors can disrupt the serine synthesis pathway and restrain the growth and proliferation of tumor cells.9,36 Recent studies have shown that the combination of PHGDH inhibitors and chemotherapeutic drugs in breast cancer,13 multiple myeloma,37 melanoma,38 lung adenocarcinoma,39 renal cell carcinoma40 and hepatocellular carcinoma41 can increase the sensitivity of cells to chemotherapy. It is meaningful to clarify the mechanism of the combination of PHGDH inhibitors with chemotherapy in different types of tumors for the development of targeted drugs. However, thus far, studies have not investigated this mechanism in gastric cancer cells. In this study, we found that the combination of cisplatin with PHGDH inhibitors decreased the efficacy of cisplatin in gastric cancer. Notably, the addition of extra serine reversed the resistance of gastric cancer cells to cisplatin. The mechanism may involve the suppression of serine synthesis by inhibiting the metabolic enzyme activity of PHGDH, reducing the concentration of serine in cells. The relationship between resistance to cisplatin and serine metabolism in vivo, and whether controlling the concentration of serine can guide chemotherapy in clinical practice remains to be investigated.
The continuous use of cisplatin will activate the protective signaling regulatory network resulting in the development of drug resistance.42,43 It has been reported that the mechanism of DNA damage and repair can be affected by cell metabolism through three main links.44 First, covalent modifications of DNA and histones are regulated by different metabolic pathways (eg, methylation, hydroxylation, acetylation, O-GlcNAcylation and hydroxylation), which affect the folding and remodeling of chromatin.45,46 Second, glutamine, aspartate, serine and other nutrients are necessary for the de novo synthesis of nucleotides as well as the content of nucleotides affects the repair and replication of DNA. Thirdly, the regulation of reactive oxygen species through different metabolic pathways can increase the oxidative damage to DNA, thus increasing the load of the DNA repair mechanism. Although tumor metabolism has attracted considerable attention in the past decade, the link between metabolism and DNA damage, as well as repair in cancer remains to be explored. Therefore, we examined the degree of DNA damage in gastric cancer cells after treatment with PHGDH inhibitor, discovering that inhibition of PHGDH reduced DNA damage caused by cisplatin in gastric cancer cells.
Serine can be catalyzed to glycine by serine hydroxymethyl transferase. Subsequently, the dragged hydroxyl group is added to tetrahydrofolate to form N5, N10-methyl-tetrahydrofolate, which is incorporated into the folate cycle to provide a carbon unit for nucleotide synthesis and biological methyl groups, such as histone and DNA methyltransferase coenzyme, S-adenosine methionine.47 Histone methylation mainly occurs at sites such as H3K4, H3K9, H3K27, H3K36, H3K79 and H4K20.24,25,48 Previous studies provided evidence that the consequence of DNA damage and genome instability because of the demethylation of tri- or di-methylation of H3K9 caused by overexpression of lysine demethylase 4B (KDM4B) facilitated the failure of cytotoxic anti-cancer treatment.49,50 Furthermore, the depletion of PHD finger protein 2 (PHF2) increased the global levels of K3K9 me3 in progenitor cells, which could delay or impair DNA repair.51 Pippa et al demonstrated that following DNA damage, local H3K4 demethylation induced by lysine demethylase 5B (KDM5B) is vital for the recruitment of Ku70/80 and BReast-CAncer susceptibility gene 1 (BRCA1) in DNA repair.52 In addition, lower expression of H3K4 tri-methylation was found in drug-resistant gastric cancer cells.19 We found that the levels of H3K4 tri-methylation were decreased following the removal of exogenous serine and endogenous serine synthesis was blocked by treatment with NCT-503. We used JIB-04 to improve the methylation modification state of H3K4 and found that the sensitivity of cells to cisplatin was reversed and DNA damage was increased. These results indicate that a low concentration of serine disrupted the damage and repair mechanism of DNA by reducing H3K4 tri-methylation, which promotes the tolerance of gastric cancer cells to cisplatin.
The spatial structure of chromatin plays an important role in regulating the response to DNA damage. It is mainly regulated by post-translational modifications, such as acetylation and methylation on histone lysine residues.26,53 Demethylated H3K4 can adjust the spatial structure of chromatin to fully expose the damaged site after the occurrence of DNA damage, thus facilitating the accurate repair by DNA repair molecules.19 Condensation of chromatin structure is mainly controlled by inter-nucleosomal and/or linker DNA–core interaction.54 We used the indirect method of MNase digestion to investigate the mechanism through which the expression of H3K4 tri-methylation regulates the structure of chromatin after treatment with NCT-503. We found that NCT-503 enhanced the spatial density of chromatin, while JIB-04 relieved the condensation of chromatin caused by NCT-503. According to the above results, we hypothesized that low concentrations of serine promote resistance of gastric cancer cells to cisplatin by decreasing H3K4 tri-methylation and strengthening the spatial density of chromatin to avoid the exposure of DNA to chemotherapeutic drugs. Further detailed investigation of the regulatory mechanism of H3K4 tri-methylation for the condensation of chromatin and the concentration of cisplatin binding to DNA will be performed in future studies.
Conclusion
The results of the present study revealed that the demethylation of H3K4, generated by inhibition of serine metabolism, prevented DNA damage and gave rise to resistance to cisplatin in gastric cancer cells. These findings provide an opportunity to combine serine supplement with chemotherapy for the treatment of patients with gastric cancer in the clinical setting.
Abbreviations
MNase, micrococcal nuclease; PHGDH, phosphoglycerate dehydrogenase; PSAT1, phosphoserine aminotransferase 1; PHF2, PHD finger protein 2; KDM4B, lysine demethylase 4B; KDM5B, lysine demethylase 5B; BRCA1, BReast-CAncer susceptibility gene 1.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Chen WQ, Li H, Sun KX, et al. Report of cancer incidence and mortality in China, 2014. Zhonghua Zhong Liu Za Zhi [Chin J Oncol]. 2018;40(1):5–13. doi:10.3760/cma.j.issn.0253-3766.2018.01.002
2. Wu H, Wang W, Tong S, Wu C. Nucleostemin regulates proliferation and migration of gastric cancer and correlates with its malignancy. Int J Clin Exp Med. 2015;8(10):17634–17643.
3. Fukuda Y, Yamamoto K, Hirao M, et al. Sarcopenia is associated with severe postoperative complications in elderly gastric cancer patients undergoing gastrectomy. Gastric Cancer. 2016;19(3):986–993. doi:10.1007/s10120-015-0546-4
4. Russi S, Verma HK, Laurino S, et al. Adapting and surviving: intra and extra-cellular remodeling in drug-resistant gastric cancer cells. Int J Mol Sci. 2019;20(15):3736. doi:10.3390/ijms20153736
5. Amable L. Cisplatin resistance and opportunities for precision medicine. Pharmacol Res. 2016;106:27–36. doi:10.1016/j.phrs.2016.01.001
6. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi:10.1016/j.cell.2011.02.013
7. Kit S. The biosynthesis of free glycine and serine by tumors. Cancer Res. 1955;15(11):715–718.
8. Guo J, Gu X, Zheng M, Zhang Y, Chen L, Li H. Azacoccone E inhibits cancer cell growth by targeting 3-phosphoglycerate dehydrogenase. Bioorg Chem. 2019;87:16–22. doi:10.1016/j.bioorg.2019.02.037
9. Mullarky E, Lucki NC, Beheshti Zavareh R, et al. Identification of a small molecule inhibitor of 3-phosphoglycerate dehydrogenase to target serine biosynthesis in cancers. Proc Natl Acad Sci U S A. 2016;113(7):1778–1783. doi:10.1073/pnas.1521548113
10. Zheng M, Guo J, Xu J, et al. Ixocarpalactone A from dietary tomatillo inhibits pancreatic cancer growth by targeting PHGDH. Food Funct. 2019;10(6):3386–3395. doi:10.1039/C9FO00394K
11. Jia XQ, Zhang S, Zhu HJ, et al. Increased expression of PHGDH and prognostic significance in colorectal cancer. Transl Oncol. 2016;9(3):191–196. doi:10.1016/j.tranon.2016.03.006
12. Yoon S, Kim JG, Seo AN, et al. Clinical implication of serine metabolism-associated enzymes in colon cancer. Oncology. 2015;89(6):351–359. doi:10.1159/000439571
13. Zhang X, Bai W. Repression of phosphoglycerate dehydrogenase sensitizes triple-negative breast cancer to doxorubicin. Cancer Chemother Pharmacol. 2016;78(3):655–659. doi:10.1007/s00280-016-3117-4
14. Gromova I, Gromov P, Honma N, et al. High level PHGDH expression in breast is predominantly associated with keratin 5-positive cell lineage independently of malignancy. Mol Oncol. 2015;9(8):1636–1654. doi:10.1016/j.molonc.2015.05.003
15. Zhao X, Fu J, Du J, Xu W. The role of D-3-phosphoglycerate dehydrogenase in cancer. Int J Biol Sci. 2020;16(9):1495–1506. doi:10.7150/ijbs.41051
16. Yang Y, Adebali O, Wu G, et al. Cisplatin-DNA adduct repair of transcribed genes is controlled by two circadian programs in mouse tissues. Proc Natl Acad Sci U S A. 2018;115(21):E4777–E4785. doi:10.1073/pnas.1804493115
17. Yimit A, Adebali O, Sancar A, Jiang Y. Differential damage and repair of DNA-adducts induced by anti-cancer drug cisplatin across mouse organs. Nat Commun. 2019;10(1):309. doi:10.1038/s41467-019-08290-2
18. Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol. 2000;10(15):886–895. doi:10.1016/S0960-9822(00)00610-2
19. Xu W, Zhou B, Zhao X, et al. KDM5B demethylates H3K4 to recruit XRCC1 and promote chemoresistance. Int J Biol Sci. 2018;14(9):1122–1132. doi:10.7150/ijbs.25881
20. Elia MC, Bradley MO. Influence of chromatin structure on the induction of DNA double strand breaks by ionizing radiation. Cancer Res. 1992;52(6):1580–1586.
21. Costes SV, Ponomarev A, Chen JL, Nguyen D, Cucinotta FA, Barcellos-Hoff MH. Image-based modeling reveals dynamic redistribution of DNA damage into nuclear sub-domains. PLoS Comput Biol. 2007;3(8):e155. doi:10.1371/journal.pcbi.0030155
22. Xu Y, Price BD. Chromatin dynamics and the repair of DNA double strand breaks. Cell Cycle. 2011;10(2):261–267. doi:10.4161/cc.10.2.14543
23. Miller KM, Tjeertes JV, Coates J, et al. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat Struct Mol Biol. 2010;17(9):1144–1151. doi:10.1038/nsmb.1899
24. Huyen Y, Zgheib O, DiTullio RA, et al. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature. 2004;432(7015):406–411. doi:10.1038/nature03114
25. Botuyan MV, Lee J, Ward IM, et al. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127(7):1361–1373. doi:10.1016/j.cell.2006.10.043
26. Lukas J, Lukas C, Bartek J. More than just a focus: the chromatin response to DNA damage and its role in genome integrity maintenance. Nat Cell Biol. 2011;13(10):1161–1169. doi:10.1038/ncb2344
27. Mattaini KR, Sullivan MR, Vander Heiden MG. The importance of serine metabolism in cancer. J Cell Biol. 2016;214(3):249–257. doi:10.1083/jcb.201604085
28. Davis JL, Fallon HJ, Morris HP. Two enzymes of serine metabolism in rat liver and hepatomas. Cancer Res. 1970;30(12):2917–2920.
29. Chen J, Chung F, Yang G, et al. Phosphoglycerate dehydrogenase is dispensable for breast tumor maintenance and growth. Oncotarget. 2013;4(12):2502–2511. doi:10.18632/oncotarget.1540
30. Song Z, Feng C, Lu Y, Lin Y, Dong C. PHGDH is an independent prognosis marker and contributes cell proliferation, migration and invasion in human pancreatic cancer. Gene. 2018;642:43–50. doi:10.1016/j.gene.2017.11.014
31. Tabatabaie L, Klomp LW, Berger R, de Koning TJ. L-serine synthesis in the central nervous system: a review on serine deficiency disorders. Mol Genet Metab. 2010;99(3):256–262. doi:10.1016/j.ymgme.2009.10.012
32. Jing Z, Heng W, Xia L, et al. Downregulation of phosphoglycerate dehydrogenase inhibits proliferation and enhances cisplatin sensitivity in cervical adenocarcinoma cells by regulating Bcl-2 and caspase-3. Cancer Biol Ther. 2015;16(4):541–548. doi:10.1080/15384047.2015.1017690
33. Mattaini KR, Sullivan MR, Lau AN, Fiske BP, Bronson RT, Vander Heiden MG. Increased PHGDH expression promotes aberrant melanin accumulation. BMC Cancer. 2019;19(1):723. doi:10.1186/s12885-019-5933-5
34. Zhu J, Ma J, Wang X, et al. High expression of PHGDH predicts poor prognosis in non-small cell lung cancer. Transl Oncol. 2016;9(6):592–599. doi:10.1016/j.tranon.2016.08.003
35. Xian Y, Zhang S, Wang X, Qin J, Wang W, Wu H. Phosphoglycerate dehydrogenase is a novel predictor for poor prognosis in gastric cancer. Onco Targets Ther. 2016;9:5553–5560. doi:10.2147/OTT.S105787
36. Locasale JW, Grassian AR, Melman T, et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet. 2011;43(9):869–874. doi:10.1038/ng.890
37. Zaal EA, Wu W, Jansen G, Zweegman S, Cloos J, Berkers CR. Bortezomib resistance in multiple myeloma is associated with increased serine synthesis. Cancer Metab. 2017;5:7. doi:10.1186/s40170-017-0169-9
38. Ross KC, Andrews AJ, Marion CD, Yen TJ, Bhattacharjee V. Identification of the serine biosynthesis pathway as a critical component of BRAF inhibitor resistance of melanoma, pancreatic, and non-small cell lung cancer cells. Mol Cancer Ther. 2017;16(8):1596–1609. doi:10.1158/1535-7163.MCT-16-0798
39. Dong JK, Lei HM, Liang Q, et al. Overcoming erlotinib resistance in EGFR mutation-positive lung adenocarcinomas through repression of phosphoglycerate dehydrogenase. Theranostics. 2018;8(7):1808–1823. doi:10.7150/thno.23177
40. Yoshino H, Nohata N, Miyamoto K, et al. PHGDH as a key enzyme for serine biosynthesis in HIF2α-targeting therapy for renal cell carcinoma. Cancer Res. 2017;77(22):6321–6329. doi:10.1158/0008-5472.CAN-17-1589
41. Wei L, Lee D, Law C-T, et al. Genome-wide CRISPR/Cas9 library screening identified PHGDH as a critical driver for Sorafenib resistance in HCC. Nat Commun. 2019;10(1). doi:10.1038/s41467-019-12606-7.
42. Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13(10):714–726. doi:10.1038/nrc3599
43. Khanna A. DNA damage in cancer therapeutics: a boon or a curse? Cancer Res. 2015;75(11):2133–2138. doi:10.1158/0008-5472.CAN-14-3247
44. Turgeon MO, Perry NJS, Poulogiannis G. DNA damage, repair, and cancer metabolism. Front Oncol. 2018;8:15. doi:10.3389/fonc.2018.00015
45. Li X, Egervari G, Wang Y, Berger SL, Lu Z. Regulation of chromatin and gene expression by metabolic enzymes and metabolites. Nat Rev Mol Cell Biol. 2018;19(9):563–578. doi:10.1038/s41580-018-0029-7
46. Sulli G, Di Micco R, d’Adda Di Fagagna F. Crosstalk between chromatin state and DNA damage response in cellular senescence and cancer. Nat Rev Cancer. 2012;12(10):709–720. doi:10.1038/nrc3344
47. Kalhan SC, Hanson RW. Resurgence of serine: an often neglected but indispensable amino acid. J Biol Chem. 2012;287(24):19786–19791. doi:10.1074/jbc.R112.357194
48. Clarke TL, Tang R, Chakraborty D, et al. Histone lysine methylation dynamics control EGFR DNA copy number amplification. Cancer Discov. 2019;10:306–325.
49. Xiang Y, Yan K, Zheng Q, et al. Histone demethylase KDM4B promotes DNA damage by activating long interspersed nuclear element-1. Cancer Res. 2019;79(1):86–98. doi:10.1158/0008-5472.CAN-18-1310
50. Young LC, McDonald DW, Hendzel MJ. Kdm4b histone demethylase is a DNA damage response protein and confers a survival advantage following γ-irradiation. J Biol Chem. 2013;288(29):21376–21388. doi:10.1074/jbc.M113.491514
51. Pappa S, Padilla N, Iacobucci S, et al. PHF2 histone demethylase prevents DNA damage and genome instability by controlling cell cycle progression of neural progenitors. Proc Natl Acad Sci U S A. 2019;116(39):19464–19473. doi:10.1073/pnas.1903188116
52. Pippa S, Mannironi C, Licursi V, et al. Small molecule inhibitors of KDM5 histone demethylases increase the radiosensitivity of breast cancer cells overexpressing JARID1B. Molecules. 2019;24(9):1739. doi:10.3390/molecules24091739
53. Chatterjee S, Senapati P, Kundu TK. Post-translational modifications of lysine in DNA-damage repair. Essays Biochem. 2012;52:93–111. doi:10.1042/bse0520093
54. Jha PK, Khan MI, Mishra A, Das P, Sinha KK. HAT2 mediates histone H4K4 acetylation and affects micrococcal nuclease sensitivity of chromatin in Leishmania donovani. PLoS One. 2017;12(5):e0177372. doi:10.1371/journal.pone.0177372
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.