")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 17
Inhibition of NOD1 Attenuates Neonatal Hypoxia-Ischemia Induced Long-Term Cognitive Impairments in Mice Through Modulation of Autophagy-Related Proteins
Authors Liu F, Shao M, Xu F, Rong F
Received 8 April 2021
Accepted for publication 12 July 2021
Published 14 August 2021 Volume 2021:17 Pages 2659—2669
DOI https://doi.org/10.2147/NDT.S314884
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Yuping Ning
Fang Liu,1 Mingyu Shao,1 Feng Xu,2 Fang Rong3
1Department of Child Health Care, Zibo Central Hospital, Zibo, 255000, Shandong, People’s Republic of China; 2Department of Pediatrics, Zibo Central Hospital, Zibo, 255000, Shandong, People’s Republic of China; 3The Community Clinic of Overseas Chinese Town, Zibo Central Hospital, North Gate of Zhongrun Overseas Chinese Town, Zibo, 255000, Shandong, People’s Republic of China
Correspondence: Fang Rong
The Community Clinic of Overseas Chinese Town, Zibo Central Hospital, North Gate of Zhongrun Overseas Chinese Town, Zhangdian District, Zibo, 255000, Shandong, People’s Republic of China
Tel +86-18678186178
Email [email protected]
Background: Autophagy is implicated in neonatal hypoxia-ischemia (HI) induced cognitive impairment. The nucleotide-oligomerizing domain-1 (NOD1), a protein involved in inflammatory responses, has been shown to activate autophagy to promote progression of other diseases. We aimed to investigate whether and how NOD1 is involved in HI-induced brain injury using an HI mouse model.
Methods: We induced HI in neonatal mice and examined levels of NOD1 and genes associated with autophagy. We then inhibited NOD1 by intracerebroventricular injection of si-NOD1 following HI induction and tested the effects on autophagy, inflammatory responses and long-term behavioral outcomes through Morris water maze and open field tests.
Results: We found that HI induction significantly elevated mRNA levels of NOD1 (3.54 folds change) and autophagy-related genes including Atg5 (3.89 folds change) and Beclin-1 (3.34 folds change). NOD1 inhibition following HI induction suppressed autophagy signaling as well as HI induced proinflammatory cytokine production. Importantly, NOD1 inhibition after HI improved long-term cognitive function, without impacting exploratory and locomotor activities.
Conclusion: We show here that NOD1 is involved in the pathogenesis of HI-induced brain injury through modulation of autophagy-related proteins and inflammatory responses. Our findings suggest that NOD1 may be a potent target for developing therapeutic strategies for treating HI-induced brain injury.
Keywords: neonatal, hypoxia-ischemia, cognitive impairment, NOD1, autophagy
Introduction
A common cause of human neonate disability and mortality, neonatal hypoxia-ischemia (HI) affects approximately 1 million infants each year, leading to 23% of infant deaths around the world.1,2 Among the patients who survive from neonatal HI, many exhibit long-term behavioral deficits, causing devastating outcomes to the individuals impacted and huge burden to families and the society.2 While the incidence of HI stays high, even in developed countries, the available protective or prevent treatments for HI-induced brain injury are scarce, with hypothermia being the only available licensed treatment currently and animal studies have revealed insufficient neuroprotection by immediate hypothermia.2,3 While the brain damage caused by HI is highly variable, three main related signaling pathways have been shown to be activated by neonatal HI including inflammatory response, oxidative stress and excitotoxicity.4–8
Inflammation is implicated in the pathogenesis of HI-induced brain damage. Following HI stimulation, multiple cytokines and chemokines including IL-6 and macrophage inflammatory protein-2 (MIP-2) are induced and secreted by a variety of cells that mediate inflammatory responses.9 Neuroinflammation has been shown to be a major factor contributing to the development of brain injury subsequent to HI and neuroprotective effects have been observed through suppression of proinflammatory mediators.10,11
Autophagy, a process that removes cellular junk that as denatured proteins and damages organelles, is closely involved in inflammation and is implicated in the pathogenesis of various inflammatory diseases.12 Multiple studies have revealed activation of autophagy upon HI stimulation, which contributes to the long-term cognitive impairment induced by HI.13,14 In a previous study using a rat model of neonatal HI, the authors found upregulation of microtubule-associated protein 1 light chain 3-II (LC3II) and suppression of P62 following HI induction and inhibition of autophagy by 3-methyladenine restored the expressions of proteins involved in autophagic process and attenuated HI induced cognitive impairment.13 Similar results were found in oxygen-glucose deprived neonatal hippocampal slice cultures including increased LC3II expression and reduced neuronal cell death was observed upon inhibition of autophagy.15
Therefore, strategies targeting autophagy and inflammation may be a potential therapy for HI induced long-term cognitive impairment. The nucleotide-oligomerizing domain-1 (NOD1) is such a candidate. NOD1 can sense bacterial products and is activated by γ-D-glutamyl-meso-diaminopimelic acid that is present in some Gram positive and many Gram negative bacteria.16 A cytosolic pattern recognition receptor, NOD1 triggers inflammation upon activation, resulting in production of inflammatory chemokines and cytokines including MIP-2 and IL-6.17 Previous studies have revealed a regulatory role of NOD1 in autophagy and activation of NOD1 plays a critical role in aggravating hepatic ischemia/reperfusion injury through activation of autophagy.18 NOD1 is expressed in the brain and is important for gut-brain axis signaling.19 In cultured rat cortical neurons, NOD1 level has been shown to be upregulated upon oxygen-glucose deprivation and reperfusion stimulation which triggered autophagy and silencing of NOD1 suppressed autophagy and increased cell viability.20 Interestingly, a connection between NOD1 and HI-induced brain injury has not been identified previously. Therefore, we explored the potential link between NOD1 and HI-induced autophagy and explored whether modulation of NOD1 expression had any impact on HI induced long-term cognitive impairment.
Materials and Methods
Inducing HI Injury in Neonatal Mice
Male C57BL6 mice at postnatal day 9 (P9) were subjected to HI injury according to a previously described procedure.21 Briefly, mice were exposed to 3.5% isoflurane anesthesia induction and maintained with 1.5% isoflurane in nitrous oxide and oxygen at a ratio of 1:1. An incision was made in the neck and the left main carotid artery was exposed and ligated permanently. After returning to the dam for 1 hour, the mice were kept in a humidified chamber with regular air for 10 minutes, exposed to 10% oxygen mixed with nitrogen for half an hour and regular air for 10 minutes. The temperature of the chamber was maintained at 36°C. Following the procedure, mice were returned to the original cage with the dam. For sham operation, mice at P9 were anesthetized and an incision was made and sutured in the neck. Sham mice were kept in a tray with regular oxygen at 36°C away from the dam for the same duration as mice undergoing HI surgery. Mice were weaned at P21 and housed together with littermates of the same gender at a maximum density of 5 mice per cage. Approximately 15% of the mice undergoing HI surgery died during the procedure and 162 mice survived the surgery were used for this study.
Mice in HI and sham groups were divided into three subsets. Mice in the first subset were euthanized at 6 h, 24 h, 48 h and 72 h, respectively, for mRNA analysis of indicated genes in the hippocampus. Mice in the second subset were divided into four groups: sham, HI, HI+si-NC and HI+si-NOD1, with the mice in the latter two groups treated with siRNA (negative control or si-NOD1, respectively) by intracerebroventricular injection for three times at 1 h, 24 h and 48 h after the HI, and the hippocampus was isolated at 72 h for mRNA and protein analysis. Mice in the third subset were injected with siRNA (negative control and si-NOD1) for three times at 1 h, 24 h and 48 h after the HI or sham operation and were subjected to open field test at P55 and Morris water maze test at P60, respectively. si-NOD1 sequences were 3’-GAGCUGCACUCAGACUUCGACAUGA-5’ and 5’-UCAUGUCGAAGUCUGAGUGCAGCUC-3’.
All mouse procedures were conducted in strict accordance with the NIH guidelines for the care and use of laboratory animals (8th edition, NIH), and were approved by the ethical committee of Zibo Central Hospital.
Quantitative Real Time Polymerase Chain Reaction (qRT-PCR)
mRNA levels in mouse hippocampus were assessed by qRT-PCR with total RNA extracted from relevant tissues by the TRIzol reagent (Thermo Fisher) according to a previous procedure.13 For hippocampus extraction, we decapitated the mice after sacrificing the mice under anesthesia, opened the skull, dissected out the brain and isolated the hippocampus. Tissues from 8 mice of the same group were mixed and each experiment was repeated 3 times. cDNA generated from the total RNA through advantage Superscript II RT-PCR kit (Thermo Fisher) was used for RT-PCR. Primer sequences were obtained through the open resource: https://pga.mgh.harvard.edu/primerbank/index.html. The following primers, produced by GenScript (Nanjing, China), were used: GAPDH: 5’-AGGTCGGTGTGAACGGATTTG-3’ and 5’-TGTAGACCATGTAGTTGAGGTCA-3’; IL-6: 5’-TCCAGTTGCCTTCTTGGGACTGAT-3’ and 5’-AGCCTCCGACTTGTCAAGTGGTAT-3’; TNF-α: 5’-CATCTTCTCAAAATTCGAGTGACAA-3’ and 5’- TGGGAGTAGACAAGGTACAACCC-3’; MIP-2: 5’-CCAACCACCAGGCTACAGG-3’ and 5’-GCGTCACACTCAAGCTCTG-3’; NOD1: 5’‐GAAGGCACCCCATTGGGTT-3’and 5’‐AATCTCTGCATCTTCGGCTGA-3’; Atg5: 5’-TGTGCTTCGAGATGTGTGGTT-3’ and 5’-ACCAACGTCAAATAGCTGACTC-3’; Beclin-1(Becn1): 5’-ATGGAGGGGTCTAAGGCGTC-3’ and 5’-TGGGCTGTGGTAAGTAATGGA-3’; p62 (Sqstm1): 5’-GAGGCACCCCGAAACATGG-3’ and 5’-ACTTATAGCGAGTTCCCACCA-3’. Relative changes in gene expression were calculated using the 2−ΔΔCT method. GAPDH was employed as an internal control.
Western Blot
Western blot was carried out to determine relevant protein expression in the hippocampus of indicated mice as described previously.22 Briefly, hippocampus was isolated and homogenized in radioimmunoprecipitation assay lysis buffer. Tissues from 8 mice of the same group were mixed and each experiment was repeated 3 times. After resolving in SDS-PAGE gel, proteins were then transferred to PVDF membrane, blocked with 5% non-fat milk, incubated in primary antibodies at 4°C for overnight. Proteins were exposed with an enhanced chemiluminescence kit from Thermo Fisher Scientific after incubation in secondary antibodies. The following antibodies were used: anti-NOD1 (1:1000), anti-GAPDH (1:2000), anti-Atg5 (1:1000), anti-Beclin-1 (1:1000), anti-LC3 (1:1000), and anti-p62 (1:1000). All antibodies were purchased from Abcam. The results of Western blot were analyzed by calculating the gray-scale ratio in Image J. The ratio of the target band to GAPDH was first calculated and then normalized to the sham group.
Enzyme-Linked Immunosorbent Assay (ELISA)
IL-6, TNF-α and MIP-2 levels in the hippocampus of indicated mice were determined by ELISA using respective ELISA kits (R&D Biosystem, Minneapolis, MN, USA) according to manufacturer’s instruction.
Open Field Test
Motor function and exploratory activity of indicated mice at P55 were assessed by the open field test in a plastic box with the size of 50 cm × 50 cm × 50 cm as described previously.21 Briefly, an experimental mouse was placed into the apparatus and allowed to explore freely for 10 min. The activity of the mouse was recorded and analyzed, including duration and distance in the center, total traveling distance, total duration of locomotion, average speed of traveling, and total activity. A total of 11–13 mice in each group were tested.
Morris Water Maze Test
A subset of mice was subjected to Morris water maze test at P60 to assess cognitive function according to a previous procedure.23 Briefly, a 120 cm-diameter pool with and a height of 60 cm was filled with opaque water at 24±1°C, and a round platform with a diameter of 10 cm was placed in the center of one quadrant 1 cm from the water surface. The test included 4 days of training sessions and 1 day of probe trial session. Each training session was composed of four trials. During the trial, each mouse started exploration from four difference positions for a maximum of 60 s and was allowed 10 s to stay on the platform. The mice were guided to the platform if they were unable to locate it within 60 s. In each training session, the duration to locate the platform (escape latency) was recorded for each mouse. On the fifth day, the mouse was placed in the pool with the platform removed and was allowed to swim freely for 60 s. The activity of each mouse was recorded and analyzed including the swimming speed, time spent in the target quadrant where the platform was located and the number of times each mouse crossed the location of the platform (platform crossing). A total of 11–13 mice in each group were tested.
Statistical Analysis
Data were analyzed by SPSS software. Differences among groups were determined by one-way ANOVA followed by Dunn’s test. Differences among groups over time were assessed by two-way ANOVA followed by Sidak’s test. Data were presented as mean ± SD. Statistical significance was reached with p<0.05.
Results
HI Increases NOD1 Expression and Modulates Autophagy-Related Gene Expression in the Hippocampus of Neonatal Mice
To investigate whether NOD1 was involved in the pathogenesis of HI-induced brain damage, we first determined whether HI led to a change in NOD1 expression. We examined the mRNA levels of NOD1 in the hippocampus of neonatal mice at 6 h, 24 h, 48 h and 72 h following HI induction. We found that mRNA expression was significantly increased after HI surgery compared to mice undergoing sham procedure (Figures 1A, 3.54-fold changes at 72 h following HI induction).
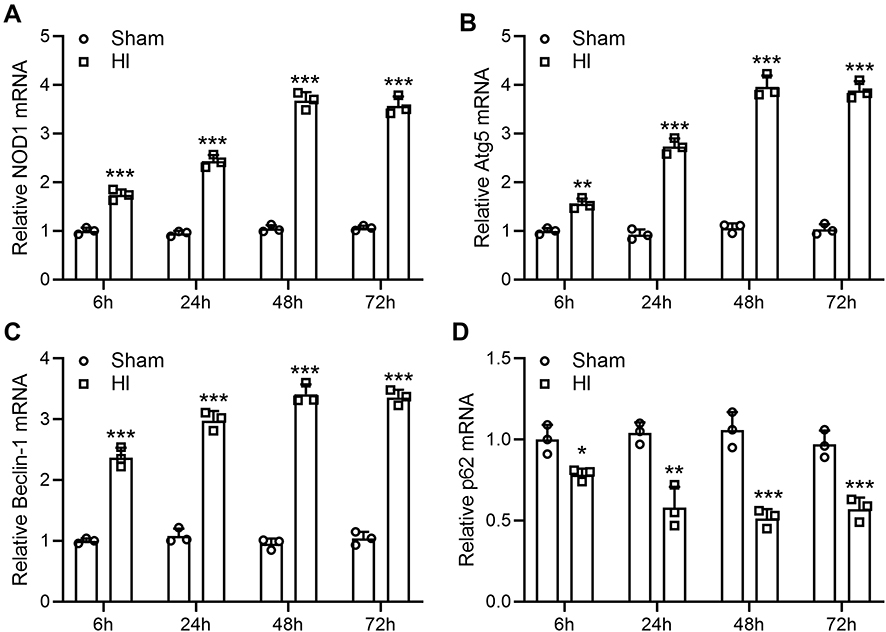 |
Figure 1 Neonatal HI increases NOD1 expression and activates autophagy in the mouse hippocampus. NOD1 (A), Atg5 (B), Beclin-1 (C) and p62 (D) mRNA levels by qRT-PCR at 6, 24, 48, and 72 h after sham surgery or hypoxia–ischemia treatment. N = 3 from 8 mice for each group at each time. *p < 0.05, **p < 0.01, ***p < 0.001 compared to sham. Two-way ANOVA followed Sidak’s multiple comparisons test. |
We then examined the expression of genes involved in autophagy. qRT-PCR showed that Atg5 (Figures 1B, 3.89-fold changes at 72 h following HI induction) and Beclin-1 (Figures 1C, 3.34-fold changes at 72 h following HI induction) mRNA levels were significantly elevated, while p62 mRNA (Figures 1D, 0.57-fold changes at 72 h following HI induction) was significantly decreased following HI induction.
Si-NOD1 Treatment in Mice Following HI Suppressed NOD1 Expression
To investigate whether inhibition of NOD1 had any impact on HIinduced brain damage, we treated mice with siRNA targeting NOD1 by intracerebroventricular injection at 1 h, 24 h and 48 h following HI surgery and examined NOD1 expression in the hippocampus of mice at 72 h after HI induction. Consistent with the above results, HI surgery significantly increased NOD1 mRNA expression. Mice treated with negative control siRNA (si-NC) showed similar mRNA level after HI in the hippocampus as HI mice without siRNA treatment (Figure 2A). Importantly, mice treated with si-NOD1 showed significantly reduced NOD1 mRNA expression following HI (0.19-fold changes to HI group), with the mRNA level similar to mice undergoing sham operation. To confirm our findings, we examined NOD1 protein expression by Western blot analysis (Figure 2B). Quantification showed that HI significantly enhanced NOD1 protein expression in the hippocampus compared to sham operation (Figures 2C, 0.36-fold changes to HI group). While si-NC showed no impact on HI-induced NOD1 expression, and si-NOD1 significantly suppressed NOD1 protein expression following HI.
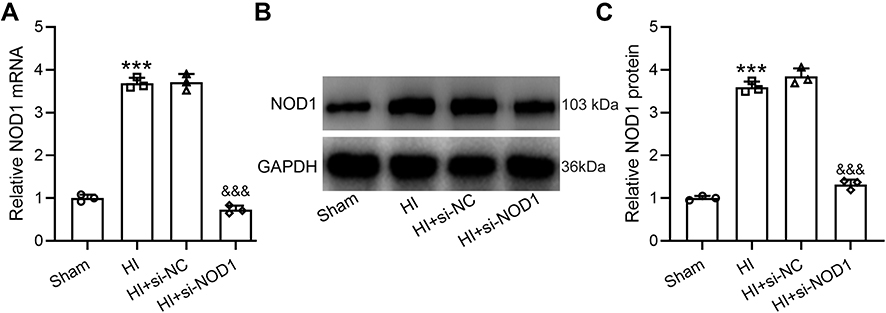 |
Figure 2 Effects of NOD1 inhibition on the expressions of hippocampal NOD1 at 72 h following HI. si-NOD1 and si-NC were administered at 1 h, 24 h and 48 h after HI through intracerebroventricular injection. NOD1 mRNA level was determined by qRT-PCR (A) and NOD1 protein level was determined by Western blot (B). Data were normalized to sham group (C). N = 3 from 8 mice for each group. ***p < 0.001 compared to sham, &&&p < 0.001 compared to HI. One-way ANOVA followed Dunn’s multiple comparisons test. |
NOD1 Inhibition Suppressed HI-Induced Autophagy in Mouse Hippocampus Following HI
We next investigated whether NOD1 inhibition had any impact on HI-induced autophagy. qRT-PCR analysis showed that si-NOD1 treatment significantly suppressed HI-induced Atg5 (Figure 3A) and Beclin-1 (Figure 3B) expression and enhanced p62 expression (Figure 3C) in the hippocampus of HI mice. We did not detect any changes in the mRNA levels of Atg5 (Figure S2A) or Beclin-1 (Figure S2B) by si-NC treatment in HI mice. We then examined proteins involved in autophagy in the hippocampus of HI mice after si-NOD1 treatment (Figure 3D). Quantification of Western blot results showed that HI surgery significantly increased protein levels of Atg5 (Figures 3E, 0.43-fold changes to HI group), Beclin-1 (Figures 3F, 0.62-fold changes to HI group) and LC3II (Figures 3G, 0.49-fold changes to HI group), which were suppressed by si-NOD1 treatment. Analysis of LC3 I and II expression showed that HI significantly increased the ratio of LC3 II/I compared to sham operation, which was suppressed by si-NOD1 treatment (Figure S3). Conversely, HI surgery significantly suppressed p62 protein expression which was restored by si-NOD1 treatment (Figures 3H, 3.95-fold changes to HI group). These results suggested that HI-induced autophagy in the hippocampus of mice undergoing HI surgery was suppressed by inhibiting NOD1.
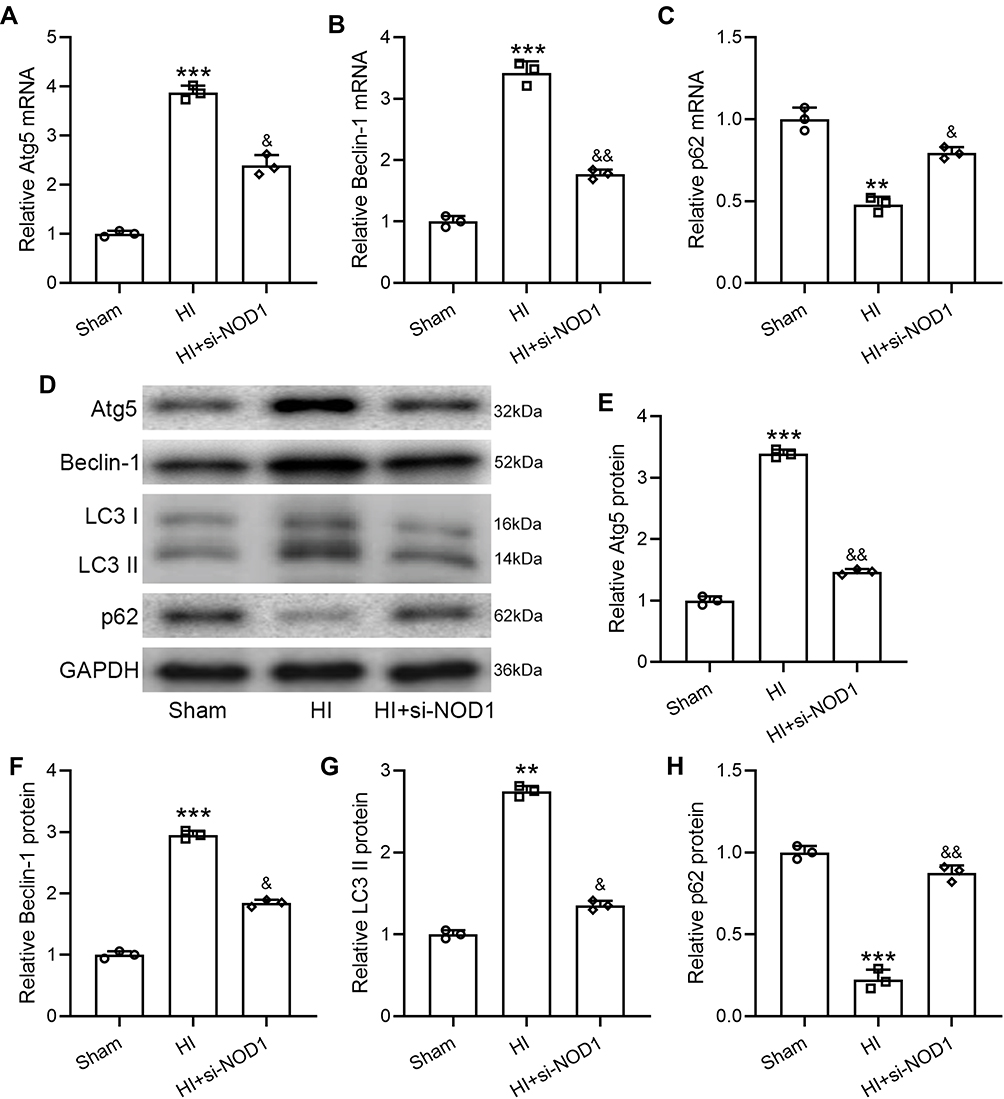 |
Figure 3 NOD1 inhibition attenuates HI induced autophagy in the hippocampus at 72 h following HI. si-NOD1 and si-NC were administered at 1 h, 24 h and 48 h after HI through intracerebroventricular injection. Atg5 (A), Beclin-1 (B) and p62 (C) mRNA levels were determined by qRT-PCR. Atg5, Beclin-1, LC3 and p62 in hippocampus (D) were measured by Western blot. Data were normalized to sham (E–H). N = 3 from 8 mice for each group. **p < 0.01, ***p < 0.001 compared to sham, &p < 0.05, &&p < 0.01 compared to HI. One-way ANOVA followed Dunn’s multiple comparisons test. |
NOD1 Inhibition Suppressed HI-Induced Inflammatory Response in Mouse Hippocampus Following HI
We then investigated the effects of NOD1 inhibition on HI-induced inflammatory response. Examination of cytokine and chemokine levels in the hippocampus of HI and sham mice by ELISA showed that HI significantly increased IL-6 (Figure 4A), TNF-α (Figure 4B) and MIP-2 (Figure 4C) levels, while mice treated with si-NOD1 showed significantly reduced IL-6, TNF-α and MIP-2 following HI compared to mice undergoing HI surgery alone (a, Sham: 34.65 ± 7.50; HI: 138.82 ± 21.22; HI+si-NOD1: 75.90 ± 23.91. b, Sham: 12.87 ± 6.65; HI: 48.13 ± 11.20; HI+si-NOD1: 26.66 ± 6.98. c. Sham: 79.54 ± 20.03; HI: 207.74 ± 51.06; HI+si-NOD1: 146.24 ± 23.97.). Additionally, we also examined the mRNA levels of cytokines and chemokines, and found consistently that HI significantly increased mRNA levels of IL-6 (Figure 4D), TNF-α (Figure 4E) and MIP-2 (Figure 4F), which were suppressed by si-NOD1 treatment. We did not detect any changes in the mRNA levels of IL-6 (Figure S2C) or TNF-α (Figure S2D) by si-NC treatment in HI mice. These results suggested that, consistent with suppressing HI-induced autophagy by NOD1 inhibition, NOD1 inhibition also suppressed HI-induced inflammatory response.
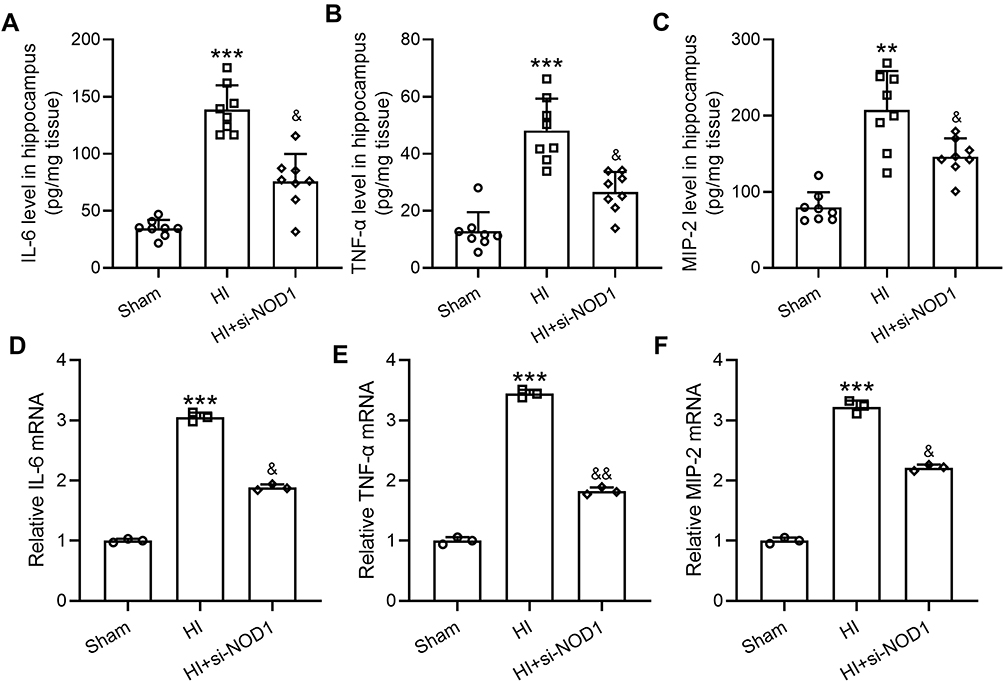 |
Figure 4 NOD1 inhibition attenuates HI induced inflammatory response in the hippocampus at 72 h following HI. si-NOD1 and si-NC were administered at 1 h, 24 h and 48 h after HI through intracerebroventricular injection. Hippocampal IL-6 (A), TNF-α (B) and MIP-2 (C) levels were determined by ELISA. N = 8 (8 mice) for each group. IL-6 (D), TNF-α (E) and MIP-2 (F) mRNA levels were determined by qRT-PCR. N = 3 from 8 mice for each group. **p < 0.01, ***p < 0.001 compared to sham, &p < 0.05, &&p < 0.01 compared to HI. One-way ANOVA followed Dunn’s multiple comparisons test. |
NOD1 Inhibition or HI Procedure Showed No Impact on the Locomotor and Exploratory Activity of Adult Mice
We then explored the long-term impact of NOD1 inhibition and HI surgery on the behavioral outcome of adult mice. Analysis of the open field test results showed that neither HI procedure nor si-NOD1 treatment had any significant long-term impact on the locomotor and exploratory activity of adult mice. Mice undergoing HI surgery or HI surgery and si-NOD1 administration had similar average speed (Figure 5A, Sham: 4.87 ± 1.42; HI: 4.62 ± 1.25; HI+si-NOD1: 4.68 ± 0.9691), total activity (Figure 5B, Sham: 53.55 ± 10.49; HI: 48.89 ± 8.85; HI+si-NOD1: 51.06 ± 12.76), total locomotion time (Figure 5C, Sham: 278.56 ± 68.54; HI: 255.71 ± 59.48; HI+si-NOD1: 273.83 ± 63.72), total distance travelled (Figure 5D), distance in center (Figure 5E) and time in center (Figure 5F), compared to mice undergoing sham operation alone.
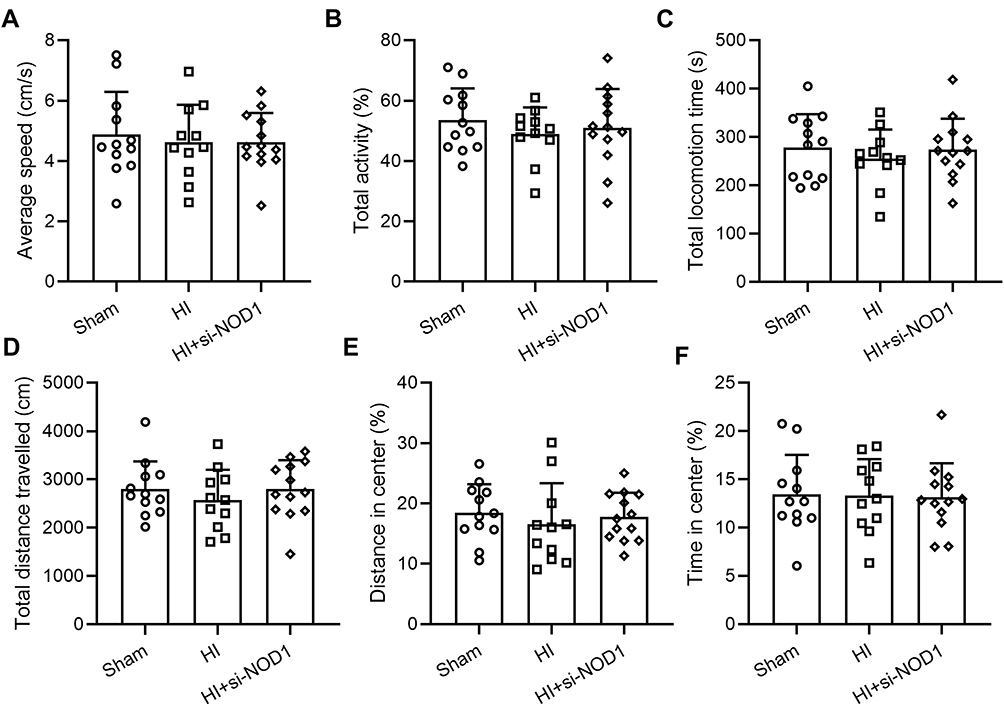 |
Figure 5 Locomotion and activity are not impacted by HI insult or NOD1 inhibition. si-NOD1 and si-NC were administered at 1 h, 24 h and 48 h after HI through intracerebroventricular injection. (A) Average speed, (B) total activity, (C) total locomotion time, (D) total distance travelled, (E) distance travelled in the center and (F) time spent in the center in open field test at P55. N = 10–14 per group. One-way ANOVA followed Dunn’s multiple comparisons test. |
NOD1 Inhibition Alleviated HI-Induced Cognitive Impairment in Mice
Finally, since HI has been shown to induce long-term cognitive impairment, we examined the cognitive function of mice at P60 by Morris water maze test. Consistent with previous studies,24,25 we found that HI surgery resulted in long-term cognitive impairment in mice. Compared with sham operation, HI procedure led to reduced spatial learning as indicated by significantly increased escape latency during the training session of the test (Figure 6A), while si-NOD1 treatment following HI significantly reduced escape latency compared to mice undergoing HI surgery alone. We found that these differences were not caused by differences in swimming speed since mice undergoing HI surgery or HI surgery and si-NOD1 treatment exhibited similar swimming speed compared to mice undergoing sham surgery (Figure 6B). Additionally, we found that HI surgery significantly reduced the time of mice spent in target quadrant (Figure 6C, Sham: 34.73 ± 5.94; HI: 19.93 ± 6.27; HI+si-NOD1: 28.18 ± 5.57) and platform crossings (Figure 6D, Sham: 5.58 ± 0.98; HI: 2.46 ± 1.04; HI+si-NOD1: 4.31 ± 0.85) during the probe trial, which were both restored by si-NOD1 treatment. These results suggested that inhibiting NOD1 suppressed HI-induced long-term cognitive impairment in mice.
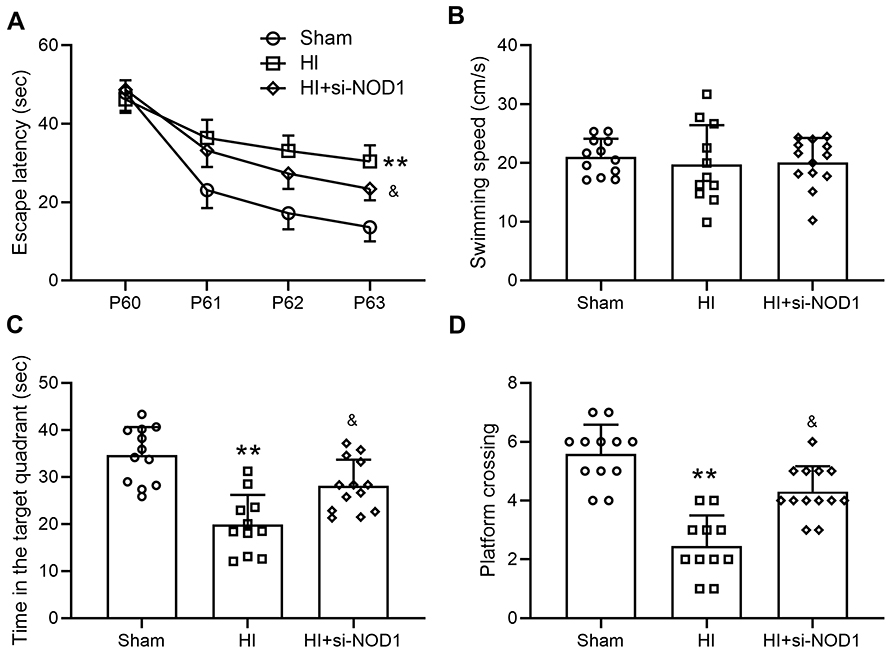 |
Figure 6 NOD1 inhibition ameliorates HI induced cognitive impairment. si-NOD1 and si-NC were administered at 1 h, 24 h and 48 h after HI through intracerebroventricular injection. Morris water maze test was started at P60. Escape latencies (A) and the average swim speed (B) were analyzed in the training sessions (4 days). Time in target quadrant (C) platform site crossings (D) were analyzed in probe trial. N = 10–14 for each group. **p < 0.01 compared to sham, &p < 0.05 compared to HI. Two-way ANOVA followed Tukey’s multiple comparisons test for a. One-way ANOVA followed Dunn’s multiple comparisons test for (B–D). |
Si-NOD1 Treatment Led to Long-Term Suppression of HI-Induced Autophagy and Inflammatory Response
To determine the long-term effects of si-NOD1 treatment on HI-induced autophagy and inflammatory response, we examined the levels of autophagy and inflammation markers in the hippocampus of mice following behavioral assays. Consistent with the results found in neonatal mice, we showed that HI led to significant upregulation of NOD1 mRNA (Figure S1A) and protein (Figures S1B and S1C) expression, which were suppressed by si-NOD1 treatment. Similarly, HI-induced long-term upregulation of Atg5 (Figure S1D), Beclin-1 (Figure S1E) and IL-6 (Figures S1F and S1G) were also suppressed by si-NOD1 treatment.
Discussion
This study investigated the potential connection between NOD1 and HI-induced brain injury and how modulating NOD1 activity might alleviate HI-induced long-term cognitive impairment. We showed that NOD1 expression in the hippocampus was significantly elevated upon HI stimulation in neonatal mice, which was accompanied with autophagy. NOD1 inhibition by si-NOD1 in the brain after HI induction suppressed NOD1 expression, which also inhibited HI-induced autophagy, both short and long term. Additionally, NOD1 inhibition significantly suppressed HI-induced inflammatory response. Importantly, while neither HI procedure nor si-NOD1 treatment in neonate mice resulted in significant changes in the locomotion and exploratory activities in adulthood, inhibiting si-NOD1 significantly alleviated HI-induced cognitive impairment in the Morris water maze test. To our knowledge, this is the first study showing a link between NOD1 and neonatal HI-induced brain injury.
Autophagy and inflammation are critically implicated in the pathogenesis of HI-induced brain injury and long-term cognitive impairment. HI results in altered expression of genes involved in autophagy including increased Atg5 and Beclin-1 and decreased p62, as well as formation of autophagosomes responsible for HI-induced neuronal death. Inhibiting autophagy by 3-methyladenine or by deletion of genes essential for autophagy such as Atg7 exhibits neuroprotective effects.15,26 Consistently, our study showed that HI surgery in neonatal mice resulted in significant alterations in gene expression corresponding to an induction of autophagy. Autophagy is also critically involved in the inflammation responses of various disease conditions, and plays important roles in the survival and homeostasis of inflammatory cells.12 Along with the activation of autophagy, we also detected significant elevation of pro-inflammatory cytokines and chemokines including IL-6, TNF-α and MIP-2 in the hippocampus of HI mice. Previous studies have revealed a neuroprotective effect of inhibiting HI-induced inflammation. For example, inhibiting inflammatory response by erythropoietin in neonatal HI rats attenuated brain injury.27 We therefore searched for potential molecular targets associated with autophagy and inflammation.
NOD1 has long been recognized as an important regulator of immune response.16,28,29 Activating NOD1 induces the MAPK and NF-κB signaling pathways, leading to expression of pro-inflammatory proteins and enhanced cytokine production30 which are associated with the pathogenesis and progression of various diseases, including ocular and cardiovascular inflammations.31 Although previous studies have not identified a link between NOD1 and inflammation in the scenario of HI-induced brain injury, we show here that both NOD1 and proinflammatory cytokines and chemokines are elevated upon HI stimulation. Importantly, inhibiting NOD1 suppressed HI-induced inflammatory response. The involvement of NOD1 in the inflammatory response of HI-induced brain injury suggests that NOD1 activation may be involved in HI pathogenesis.
In addition to regulating inflammatory response, NOD1 also plays a significant role in autophagy. Activation of NOD1 has been shown to trigger autophagy in the macrophages and epithelial cells leading to the clearance of intracellular pathogens.17 In a mouse model of hepatic ischemia-reperfusion injury, NOD1 was found to be upregulated and aggravate the disease condition through activating autophagy, and inhibiting NOD1 by siRNA suppressed Atg5 and LC3II expression, consistent with a reduction in autophagy.18 Consistently, we also observed significantly upregulated NOD1 expression following HI which was accompanied by upregulated autophagy markers. Importantly, we showed here that si-NOD1 not only suppression NOD1 expression but suppressed autophagy as manifested by reduced Atg5, Beclin-1 and LC3II and increased p62 levels. Considering the important function of autophagy in mediating HI pathogenesis, our findings suggest that NOD1 is a potent target for suppressing HI-induced autophagy to inhibit disease progression.
HI is frequently associated with long-term behavioral deficits including motor dysfunction and cognitive impairment.2,32 In our HI mouse model, we did not detect any changes in the locomotion and exploratory activities of mice undergoing HI surgery in the open field test. Inhibiting NOD1 in sham-operated mice and HI-stimulated mice did not negatively impact these behavioral aspects, either. Similar findings were reported using neonatal HI rat and mouse models,21,33 with the overall motor function not impacted by HI. It is possible that only severe HI may cause long-term locomotor abnormality in the open field test as suggested by a previous study.34 The other possibility is that, the test might not be sensitive enough to detect HI induced subtle locomotor deficits. In the future, we will perform more sensitive assays to test this possibility. We showed here that neonatal HI procedure resulted in long-term cognitive impairment in adult mice. Importantly, the deficit, as indicated by the compromised spatial learning and memory ability, was significantly attenuated by NOD1 inhibition. Interestingly, a previous study also showed that NOD1 and NOD2 double knock out mice were cognitively impaired,19 suggesting that careful modulation of NOD1 activity may be required for optimal behavioral outcomes.
One limitation of this study is that we only focused on male mice. In the future studies, we will include both male and female mice for a more detailed comparison. Another limitation is that, in this study we focused on the hippocampus for biochemical analysis and did not analyze other brain regions such as the cerebral cortex or perform histological analysis. Future studies will include more detailed analysis of other relevant brain regions and histopathological manifestations.
In addition to autophagy, Beclin 1 is also able to interact with Bcl-2 to regulate apoptosis, which may play a role in HI-mediated brain injury and inhibiting NOD1 may modulate this pathway to exert its neuroprotective effects.8 We will explore this possibility in future studies.
Conclusion
In summary, our study first showed that NOD1 was significantly elevated in response to HI stimulation in neonatal mice, while inhibiting NOD1 modulated HI-induced changes of autophagy-related proteins, suppressed inflammatory responses and alleviated long-term cognitive impairment. Our findings suggest that NOD1 is a potential target for the development of therapeutic strategies to prevent or treat neonatal HI-induced brain injury.
Funding
There isno funding to report.
Disclosure
The authors declare that they have no competing interests.
References
1. Lawn JE, Cousens S, Zupan J, et al. 4 million neonatal deaths: when? Where? Why? Lancet. 2005;365(9462):891–900. doi:10.1016/S0140-6736(05)71048-5
2. Millar LJ, Shi L, Hoerder-Suabedissen A, et al. Neonatal hypoxia ischaemia: mechanisms, models, and therapeutic challenges. Front Cell Neurosci. 2017;11:78.
3. Sabir H, Scull-Brown E, Liu X, et al. Immediate hypothermia is not neuroprotective after severe hypoxia-ischemia and is deleterious when delayed by 12 hours in neonatal rats. Stroke. 2012;43(12):3364–3370. doi:10.1161/STROKEAHA.112.674481
4. Torres-Cuevas I, Corral-Debrinski M, Gressens P. Brain oxidative damage in murine models of neonatal hypoxia/ischemia and reoxygenation. Free Radic Biol Med. 2019;142:3–15. doi:10.1016/j.freeradbiomed.2019.06.011
5. Zhang X, Peng K, Zhang X. The function of the NMDA receptor in hypoxic-ischemic encephalopathy. Front Neurosci. 2020;14:567665. doi:10.3389/fnins.2020.567665
6. Failor S, Nguyen V, Darcy DP, et al. Neonatal cerebral hypoxia-ischemia impairs plasticity in rat visual cortex. J Neurosci. 2010;30(1):81–92. doi:10.1523/JNEUROSCI.5656-08.2010
7. McQuillen PS, Ferriero DM. Selective vulnerability in the developing central nervous system. Pediatr Neurol. 2004;30(4):227–235. doi:10.1016/j.pediatrneurol.2003.10.001
8. Towfighi J, Mauger D, Vannucci RC, et al. Influence of age on the cerebral lesions in an immature rat model of cerebral hypoxia-ischemia: a light microscopic study. Brain Res Dev Brain Res. 1997;100(2):149–160. doi:10.1016/S0165-3806(97)00036-9
9. Liu F, McCullough LD. Inflammatory responses in hypoxic ischemic encephalopathy. Acta Pharmacol Sin. 2013;34(9):1121–1130. doi:10.1038/aps.2013.89
10. Borjini N, Sivilia S, Giuliani A, et al. Potential biomarkers for neuroinflammation and neurodegeneration at short and long term after neonatal hypoxic-ischemic insult in rat. J Neuroinflammation. 2019;16(1):194. doi:10.1186/s12974-019-1595-0
11. Schuffels S, Nakada S, Wu Y, et al. Effects of inter-alpha inhibitor proteins on brain injury after exposure of neonatal rats to severe hypoxia-ischemia. Exp Neurol. 2020;334:113442. doi:10.1016/j.expneurol.2020.113442
12. Qian M, Fang X, Wang X. Autophagy and inflammation. Clin Transl Med. 2017;6(1):24. doi:10.1186/s40169-017-0154-5
13. Xu Y, Tian Y, Tian Y, et al. Autophagy activation involved in hypoxic-ischemic brain injury induces cognitive and memory impairment in neonatal rats. J Neurochem. 2016;139(5):795–805. doi:10.1111/jnc.13851
14. Tao J, Shen C, Sun Y, et al. Neuroprotective effects of pinocembrin on ischemia/reperfusion-induced brain injury by inhibiting autophagy. Biomed Pharmacother. 2018;106:1003–1010. doi:10.1016/j.biopha.2018.07.026
15. Lu Q, Harris VA, Kumar S, et al. Autophagy in neonatal hypoxia ischemic brain is associated with oxidative stress. Redox Biol. 2015;6:516–523. doi:10.1016/j.redox.2015.06.016
16. Caruso R, Warner N, Inohara N, et al. NOD1 and NOD2: signaling, host defense, and inflammatory disease. Immunity. 2014;41(6):898–908. doi:10.1016/j.immuni.2014.12.010
17. Juarez E, Carranza C, Hernandez-Sanchez F, et al. Nucleotide-oligomerizing domain-1 (NOD1) receptor activation induces pro-inflammatory responses and autophagy in human alveolar macrophages. BMC Pulm Med. 2014;14(1):152. doi:10.1186/1471-2466-14-152
18. Xi J, Yan M, Li S, et al. NOD1 activates autophagy to aggravate hepatic ischemia-reperfusion injury in mice. J Cell Biochem. 2019;120(6):10605–10612. doi:10.1002/jcb.28349
19. Pusceddu MM, Barboza M, Keogh CE, et al. Nod-like receptors are critical for gut-brain axis signalling in mice. J Physiol. 2019;597(24):5777–5797. doi:10.1113/JP278640
20. Ma X, Zhang W, Xu C, et al. Nucleotide-binding oligomerization domain protein 1 enhances oxygen-glucose deprivation and reperfusion injury in cortical neurons via activation of endoplasmic reticulum stress-mediated autophagy. Exp Mol Pathol. 2020;117:104525. doi:10.1016/j.yexmp.2020.104525
21. Moran J, Stokowska A, Walker FR, et al. Intranasal C3a treatment ameliorates cognitive impairment in a mouse model of neonatal hypoxic-ischemic brain injury. Exp Neurol. 2017;290:74–84. doi:10.1016/j.expneurol.2017.01.001
22. Fu CH, Lai FF, Chen S, et al. Silencing of long non-coding RNA CRNDE promotes autophagy and alleviates neonatal hypoxic-ischemic brain damage in rats. Mol Cell Biochem. 2020;472(1–2):1–8. doi:10.1007/s11010-020-03754-2
23. He XF, Liu DX, Zhang Q, et al. Voluntary exercise promotes glymphatic clearance of amyloid beta and reduces the activation of astrocytes and microglia in aged mice. Front Mol Neurosci. 2017;10:144. doi:10.3389/fnmol.2017.00144
24. Schreglmann M, Ground A, Vollmer B, et al. Systematic review: long-term cognitive and behavioural outcomes of neonatal hypoxic-ischaemic encephalopathy in children without cerebral palsy. Acta Paediatr. 2020;109(1):20–30. doi:10.1111/apa.14821
25. Greggio S, de Paula S, de Oliveira IM, et al. NAP prevents acute cerebral oxidative stress and protects against long-term brain injury and cognitive impairment in a model of neonatal hypoxia-ischemia. Neurobiol Dis. 2011;44:152–159.
26. Koike M, Shibata M, Tadakoshi M, et al. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol. 2008;172(2):454–469. doi:10.2353/ajpath.2008.070876
27. Sun Y, Calvert JW, Zhang JH. Neonatal hypoxia/ischemia is associated with decreased inflammatory mediators after erythropoietin administration. Stroke. 2005;36(8):1672–1678. doi:10.1161/01.STR.0000173406.04891.8c
28. Trindade BC, Chen GY. NOD1 and NOD2 in inflammatory and infectious diseases. Immunol Rev. 2020;297(1):139–161. doi:10.1111/imr.12902
29. Philpott DJ, Sorbara MT, Robertson SJ, et al. NOD proteins: regulators of inflammation in health and disease. Nat Rev Immunol. 2014;14(1):9–23. doi:10.1038/nri3565
30. Mukherjee T, Hovingh ES, Foerster EG, et al. NOD1 and NOD2 in inflammation, immunity and disease. Arch Biochem Biophys. 2019;670:69–81. doi:10.1016/j.abb.2018.12.022
31. Correa RG, Milutinovic S, Reed JC. Roles of NOD1 (NLRC1) and NOD2 (NLRC2) in innate immunity and inflammatory diseases. Biosci Rep. 2012;32(6):597–608. doi:10.1042/BSR20120055
32. Lee AC, Kozuki N, Blencowe H, et al. Intrapartum-related neonatal encephalopathy incidence and impairment at regional and global levels for 2010 with trends from 1990. Pediatr Res. 2013;74(Suppl S1):50–72. doi:10.1038/pr.2013.206
33. Arteaga O, Revuelta M, Uriguen L, et al. Docosahexaenoic acid reduces cerebral damage and ameliorates long-term cognitive impairments caused by neonatal hypoxia-ischemia in rats. Mol Neurobiol. 2017;54(9):7137–7155. doi:10.1007/s12035-016-0221-8
34. Ten VS, Wu EX, Tang H, et al. Late measures of brain injury after neonatal hypoxia-ischemia in mice. Stroke. 2004;35(9):2183–2188. doi:10.1161/01.STR.0000137768.25203.df
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.