")
Back to Journals » Journal of Inflammation Research » Volume 14
Inhibition of MALT1 Alleviates Spinal Ischemia/Reperfusion Injury-Induced Neuroinflammation by Modulating Glial Endoplasmic Reticulum Stress in Rats
Authors Zhang S, Yan Y, Wang Y, Sun Z, Han C , Qian X, Ren X, Feng Y, Cai J , Xia C
Received 17 May 2021
Accepted for publication 28 July 2021
Published 2 September 2021 Volume 2021:14 Pages 4329—4345
DOI https://doi.org/10.2147/JIR.S319023
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Professor Ning Quan
Shutian Zhang,1,2,* Yufeng Yan,3,* Yongze Wang,1,2 Zhaodong Sun,1,2 Chengzhi Han,1,2 Xinyi Qian,1,2 Xiaorong Ren,1,2 Yi Feng,4 Jian Cai,5 Chunmei Xia1
1Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Fudan University, Shanghai, 200032, People’s Republic of China; 2Department of Clinical Medicine, Shanghai Medical College, Fudan University, Shanghai, 200032, People’s Republic of China; 3Experimental Teaching Center of Basic Medicine, School of Basic Medical Sciences, Fudan University, Shanghai, 200032, People’s Republic of China; 4Department of Integrative Medicine and Neurobiology, School of Basic Medical Sciences, Fudan University, Shanghai, 200032, People’s Republic of China; 5Department of Neurology, Renji Hospital, School of Medicine, Shanghai Jiaotong University, Shanghai, 200240, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Chunmei Xia
Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Fudan University, Shanghai, 200032, People’s Republic of China
Tel +86 21 54237612-805
Email [email protected]
Jian Cai
Department of Neurology, Renji Hospital, School of Medicine, Shanghai Jiaotong University, Shanghai, 200240, People’s Republic of China
Email [email protected]
Purpose: Glial activation and the disorders of cytokine secretion induced by endoplasmic reticulum stress (ERS) are crucial pathogenic processes in establishing ischemia/reperfusion (I/R) injury of the brain and spinal cord. This present study aimed to investigate the effects of mucous-associated lymphoid tissue lymphoma translocation protein 1 (MALT1) on spinal cord ischemia/reperfusion (SCI/R) injury via regulating glial ERS.
Methods: SCI/R was induced by thoracic aorta occlusion-reperfusion in rats. The MALT1-specific inhibitor MI-2 or human recombinant MALT1 protein (hrMALT1) was administrated for three consecutive days after the surgery. Immunofluorescent staining was used to detect the localization of MALT1 and ERS profiles in activated astrocyte and microglia of spinal cord. The ultrastructure of endoplasmic reticulum (ER) was examined by transmission electron microscopy. Blood-spinal cord barrier (BSCB) disruption and noninflammatory status were assessed. The neuron loss and demyelination in the spinal cord were monitored, and the hindlimb motor function was evaluated in SCI/R rats.
Results: Intraperitoneally postoperative MI-2 treatment down-regulated phos-NF-κB (p65) and Bip (ERS marker protein) expression in the spinal cord after SCI/R in rats. Intraperitoneal injection MI-2 attenuated the swelling/dilation of ER of the glia in SCI/R rats. Furthermore, MI-2 attenuated I/R-induced Evans blue (EB) leakage and microglia M1 polarization in spinal cord, implying a role for MALT1 in the BSCB destruction and neuroinflammation after SCI/R in rats. Furthermore, intrathecal injection of hrMALT1 aggravated the fragmentation of neuron, loss of neurofibrils and demyelination caused by I/R, while 4-PBA, an ERS inhibitor, co-treatment with hrMALT1 reversed these effects in SCI/R rats. hrMALT1 administration aggravated the motor deficit index (MDI) scoring, while 4-PBA co-treatment improved SCI/R-induced motor deficits in rats.
Conclusion: Inhibition of MALT1 alleviates SCI/R injury-induced neuroinflammation by modulating glial endoplasmic reticulum stress in rats.
Keywords: MALT1, blood-spinal cord barrier, astrocytes, microglia, endoplasmic reticulum stress, spinal cord ischemia/reperfusion injury
Introduction
Aortic aneurysm/dissection undermines the circulation of segmental artery of the spinal cord, leading to spinal cord ischemia, while restoration of spinal cord blood supply after aortic surgery or endovascular therapy also aggravates spinal cord injury, which causes serious neurological complication.1 Despite improved approaches in surgical techniques, as hypothermia, intraoperative cerebrospinal fluid drainage, and a series of clinical trials with reactive oxygen scavengers/anti-inflammatory agents, there has been no fundamental improvement in the prevention and treatment of post-aortic-surgery spinal cord injury.2
Blood-spinal cord barrier (BSCB) destruction and neuroinflammation are the two key pathophysiological mechanisms of SCI/R. Neuroinflammation is a complex and well-orchestrated process by various groups of glial cells in the central nervous system and peripheral immune cells. The glial component primarily comprises astrocytes and microglia, which are the main cells that are responsible for the regulation of inflammation.3 Chronic glial activation promotes neurodegeneration via the secretion of inflammatory factors involved in neuronal loss.4 It is not yet completely clear how the regulation of inflammation is carried out and what the consequences in pathological events of spinal cord ischemia reperfusion (SCI/R).
As the main site responsible for protein synthesis and folding, the endoplasmic reticulum (ER) serves a vital role in maintaining homeostasis within the cellular microenvironment.5–7 Persistent endoplasmic reticulum stress (ERS) responses induced by the surrounding neuroglia may coordinate damaging inflammatory responses, which help fuel a neurotoxic milieu. Nevertheless, it is not well understood regarding the cell-specific mechanisms by which ERS mediates neuroinflammation. Studies have shown that ERS inhibitor sodium 4-phenylbutyrate (PBA) significantly inhibits hindlimb motor function loss caused by SCI/R and saves pro-apoptotic motor neurons.8 However, the molecular mechanism remains unknown.
Reactive oxygen species (ROS) and various pro-inflammatory factors lead to the pathological activation of astrocytes, promoting the expression of matrix decomposition signaling such as matrix metalloproteinase-9 (MMP9).9 Studies have shown that ERS induces production of MMP9,10 ROS11 and destroys the tight junction12 between endothelium, which is an important destructive mechanism of the blood–brain barrier. The blood-derived damage mediators and inflammatory cells into the spinal cord, thereafter aggravating spinal cord injury. Furthermore, microglia undergo M1 polarization, releasing a large number of pro-inflammatory factors, which aggravates disruption of BSCB and neuronal injury.5 Therefore, it is essential to regulate and maintain the proper ERS in SCI/R settings.
It was noted that mucous-associated lymphoid tissue lymphoma translocation protein 1 (MALT1) is a multi-domain cytosolic signaling molecule, which was originally identified as the target of recurrent translocations in a large fraction of MALT lymphomas. Early studies suggested that MALT1 is an essential intermediate in antigen receptor activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) in response to stimulation of a subset of G protein-coupled receptors.13–15 Recent studies have shown that changes in MALT1 activity are related to ERS induction. ERS activates the NLRP3 inflammasome via NF-κB to promote IL-1β secretion.16 So whether MALT1 regulating SCI/R via NF-κB-ERS signaling is deserved to investigate. MALT1 inhibitor MI-2 has high selectivity and non-toxic,17 which makes it an ideal drug to intervene MALT1 in vivo. Our current research hypothesized that MI-2, the MALT1 inhibitor, attenuates BSCB destruction and neuroinflammation via suppressing NF-κB-ERS signaling, thereby decreasing loss of neurons, and protecting hindlimb motor function after SCI/R.
Materials and Methods
Reagents and Antibodies
MI-2 (S7429) was purchased from Selleckchem (Houston, Texas, USA); Human recombinant MALT1 protein (hrMALT1) (ab271604) and Luxol Fast Blue (LFB) Staining Kit (ab150675) were purchased from Abcam (Cambridge, UK). Nissl Staining Solution (C0117) and Dihydroethidium (DHE) (S0063) were purchased from Beyotime (Shanghai, China). Silver Staining Kit (G1052) was purchase from Servicebio (Wuhan, China). Evans blue (DK0001) was purchase from Leagene (Beijing, China). Antibodies: p-p65 (phos-NF-κB, ab194726), Bip (ab21685), CD31 (ab64543), GFAP (ab279290), NeuN (ab104224), AQP4 (ab259318), MMP9 (ab76003), IL-1β (ab254360), ZO-1 (ab221547), Donkey Anti-Goat IgG H&L (Cy5) (ab6566), Donkey Anti-Rabbit IgG H&L (Alexa Fluor 647) (ab150075), Donkey Anti-Goat IgG H&L (Alexa Fluor 488) (ab150129), Donkey Anti-Rabbit IgG H&L (HRP) (ab6802) and Donkey Anti-Rabbit IgG H&L (Alexa Fluor 488) (ab150073) were purchased from Abcam (Cambridge, UK). MALT1 (GB11790) and Iba-1 (GB12105) were purchased from Servicebio (Wuhan, China). CD86 (NBP2-67417) was purchased from Novus (Minneapolis, USA).
Experimental Design
Figure S1 shows the outline of the experimental design. A total of 95 rats were randomly subdivided into five groups (n = 19 for each group): (1) a Sham underwent thoracotomy surgery but without transient (10 min) descending aorta occlusion, (2) an SCI/R modeling group receiving thoracotomy surgery and 10 min transient descending aorta occlusion for modeling SCI/R, (3) an SCI/R + MI-2 receiving modeling surgery following 3 days of intraperitoneal injections of MI-2 [25 mg/(kg·d)], and (4) an SCI/R + hrMALT1 receiving modeling surgery following 3 days of intrathecal injection of hrMALT1 (100 ng/day), (5) SCI/R + hrMALT1 + 4-PBA group receiving the same intrathecal co-injection of 100 ng hrMALT1 and 4-PBA (a ESR inhibitor 100 μg/days), for consecutive 3 days after modeling surgery. Equivalent volume of vehicle was delivered for the sham and I/R groups.
There are two reasons for intraperitoneal administration MI-2 in rats: firstly, it has been well accepted that SCI/R causes the destruction of BSCB,3,5 so intraperitoneal MI-2 injection can exert its effect on spinal cord. Secondly, our preliminary experiment showed that intraperitoneal injection of MI-2 has protective roles on motor functions in SCI/R rats. The rats were euthanized using pentobarbital, and their L4–6 segments of spinal cord were obtained for BSCB disruption evaluation, histological analysis and immunofluorescent staining after 72hrs (3 days) of the surgery. In each group, 6 rats were used for hindlimb locomotor functional assessment, histopathological interpretation, and immunofluorescent staining; 5 rats were used for evaluation of Evans blue (EB) extravasation, 3 rats were used for evaluation of ER ultrastructural damage under transmission electron microscope and the other 5 rats were used for calculating water content.
Animals and Spinal Cord Ischemia Reperfusion Injury Model
Male Sprague–Dawley rats, with mass ranging from 250 to 270 g, were acquired from Animal Laboratory Center of Fudan University. Four rats per cage were accommodated in a standard 24 °C temperature-controlled, 12-hours light/dark cycle room and they were supplied with food and tap water ad libitum. Greatest possible efforts were made to minimize the number of animals used and their sufferings. This study was approved by the Animal Care Committee of the Fudan University, Shanghai, China. Animal Care and all experimental procedures were conformed to the guidelines by the Institutional Ethics Committee.
The SCI/R model was established in the rats according to the previously reported method with slightly modification.3 Specifically, the rats were anesthetized using intraperitoneal administration of 4% sodium pentobarbital (50 mg/kg). The rectal temperature was measured and maintained at 37 ± 0.5 °C with a heated operating table. Catheters were advanced into the left femoral artery and the right carotid artery in order to detect the distal and proximal arterial blood pressure (BP), respectively. We then exposed the rat’s descending aorta by performing left lateral thoracotomy. After systemic application of heparin (200 IU/kg), the descending aorta was occluded at a site just distal to the left subclavian artery via a cross-clamp. A diminution of the distal BP to less than 20 mmHg was defined as ischemia (see Figure S2). After a 10 min ischemia, the cross-clamp was removed to allow reperfusion. After surgery, the rats were placed in a 37 °C incubator for 4 h for recovery, and then they were subsequently put in separate cages with free access to food and water. Manual urinary bladder expression was performed on the rats twice daily until the rats’ autonomic function had fully recovered.
Motor Deficit Index (MDI) Scoring Neurological Behavior
Rat neurologic assessment was done before and 3 days after SCI/R according to the previously reported method.18 The motor deficit index (MDI) score (sum of scores from ambulation and placing-stepping reflex) was recorded. The utmost deficiency was demonstrated by a score of six. Rats with MDI <3 were marked as nonparaplegic while rats with MDI ≥3 were classified as paraplegic. The raters evaluated the behavior using a double-blind method.
Pathohistological Analysis
Nissl staining was performed for evaluation of neuron loss in spinal cord of SCI/R rats. The formaldehyde-fixed spinal cord specimens were embedded in paraffin followed by sectioning of tissues into 4-μm-thick transverse sections. The sections were incubated with Nissl staining solution (C0117, Beyotime) for 5 min, and then mounted with neutral balsam. The normal neurons showed a relatively large and full cell body, with spherical, prominent nuclei, while the apoptotic ones had shrunken cytoplasm or contained vacuoles. Five separated areas were randomly selected to be examined with an inverted microscope (Leica, Wetzlar, Germany) by researchers who were blinded to the experimental groups. Silver staining was performed for evaluation of neurofilament damage with the Silver Staining Kit (G1052, Servicebio), which was strictly followed the manufacturer’s instructions. Serial transverse cryosections (12 μm thickness) were stained by Luxol fast blue (LFB) (ab150675, Abcam) according to the manufacturer’s instruction. In brief, the slides were incubated in 0.1% LFB (Sigma, St. Louis, MO, USA) in acidified 95% ethanol overnight at 60 °C. The slides were then differentiated with 0.05% lithium carbonate and subsequently counterstained with cresyl violet solution. The spared myelin in white matter in LFB-stained sections was analyzed by Image pro-plus (Media Cybernetics, Silver Spring, USA).
Blood-Spinal Cord Barrier (BSCB) Disruption Evaluation
The evaluation of BSCB integrity was assessed by the degree of Evans blue (EB) extravasation after EB injection. First, we administrated EB dye (20g/L) at a dose of 10mL/kg into the rat’s tail vein and waited 60 mins for dye distribution. Then, the rats were anesthetized and transcardially perfused with 0.9% saline containing 10 IU/mL heparin, until the colorless fluid had discharged from the right atrium, representing removal of EB dye without leaking into the interstitium. The L4–6 spinal cord tissue was weighed, then immersed in methanamide for 24 h (60 °C) and then centrifuged, the fluorescent absorption of the supernatant was detected at 632 nm with a microplate reader (BioTek, Winooski, USA) and presented as EB amount per tissue weight (μg/g). Furthermore, the spinal cord tissue was subsequently fixation using 4% paraformaldehyde. The transverse sections (10 μm) were examined under a Zeiss (Carl Zeiss, Germany) fluorescent microscope. The extent of EB extravasation was evaluated by analyzing the fluorescent density of the positive labeling in the lumbar spinal cord tissue (Image Pro Plus 6.0). With reference to the method of previous reports,19 morphometric identification of IgG leakages was performed by two investigators, both were blinded to the experimental groups. The signal intensity at the core and border was used to define the severity of BSCB leakages. A series of slices were examined to identify and localize leakages of >150 kDa circulating IgG. Maximal intensity projection of the z stacks was performed per channel, and the areas of the dextran and IgG signals were obtained by confocal microscopy (x=175; y=175; z=20 µm) and analyzed by ImageJ to quantify the leakage size in mm2. The spinal cord edema was evaluated by calculating the percentage of water content via a wet-dry method with formula of (wet weight-dry weight)/wet weight × 100.
Immunofluorescent Staining and Analysis
The protocol is similar to our previous study.20 After the rat was deeply anesthetized, the left ventricle was perfused by 300mL of 0.01 M PBS (pH 7.4) and was subsequently perfused by 300mL of 4% paraformaldehyde solution in 0.1M PBS. The L4-6 spinal cord was collected and removed to post-fixation for 4 h, then they were placed in 20 and 30% sucrose at 4 °C to allow overnight dehydration, respectively. Cryostat (Microm, Germany) was used to cut and produce free-floating 30-μm coronal sections containing the spinal cord. The coronal sections of the spinal cord were subsequently washed in PBS and then permeabilized with 0.3% Triton X-100 for 30 min followed by incubation with 5% horse serum for 1 h at 37 °C to block non-specific protein. The sections were incubated with primary antibody to MALT1, p-NF-κB (p65), Bip, MMP9, ZO-1, AQP4, CD86, IL-1β, GFAP, Iba-1, NeuN, and/or CD31 for overnight at 4 °C. The Donkey Anti-Goat IgG H&L (Alexa Fluor 488), Donkey Anti-Goat IgG H&L (Cy5), Donkey Anti-Rabbit IgG H&L (Alexa Fluor 647), and Donkey Anti-Rabbit IgG H&L (Alexa Fluor 488) secondary antiserum were utilized as secondary antibody. The fluorescence signal was observed under Fluoview FV300 laser scanning confocal microscope (Zeiss, Carl Zeiss, Germany); immunoreactivity manifested specific green, red or pink fluorescence. Totally 3 slides form the L4-L6 spinal cord sections were selected by every consecutive four sections. And five separated areas of each slide were randomly selected to be examined with a confocal microscope (Leica, Wetzlar, Germany) by 2 pathologists who were blinded to the experimental groups. The immunofluorescent images were processed using ImageJ software, and noise signals were filtered in order to identify the foci of the pictures. These foci were then automatically segmented by thresholding, and the colocalization event of the segmented points from the dual channels was evaluated using the ImageJ plugin Just Another Colocalization Plugin (JACoP), which calculated Pearson coefficients, indicating the percentage of thresholded pixels in the green channel that were occupied by corresponding thresholded pixels in the red channel. To provide further information regarding statistical analysis of colocalization, ANOVA analysis was carried out using JMP 12 software.
Transmission Electron Microscopy
Samples of fresh spinal cord (1 mm × 1 mm × 1 mm) were carefully collected from the rats, then primarily fixed in 2.5% glutaraldehyde for 24 hours. Then, they were washed with Sorenson’s Phosphate Buffer (pH=7.4) and post-fixed in 1% osmium tetroxide for 1 hour. After post-fixation and dehydration, propylene oxide was used to wash the tissues, and then the tissues were embedded in epoxy resin embedding media. Following this procedure, the tissue blocks were sectioned using the ultramicrotome (LKB Nova, Sweden) to produce section around 60 nanometers in thickness. These sections were en bloc stained with uranyl acetate and lead citrate and examined using transmission electron microscope (HITACHI, Japan).
Detection of ROS in vivo
The in-situ generation of ROS in the spinal cord was detected using dihydroethidium (DHE) fluorescent probe assay. Accordingly, the rat was anesthetized, and the lumbar spinal cord was collected as above-mentioned. The spinal cord segments were sectioned to produce 40 μm thick transverse sections and then placed on a glass slide (DHE, Molecular Probes). The sections were treated with 1 mmol/L DHE in a dark, humidified chamber at 37 °C for 30 min. DHE is oxidized by superoxide to ethidium bromide, which binds to the DNA and emits red fluorescence. DHE fluorescence can thereby be detected by fluorescence microscope (Zeiss Axioscope, Hallbergmoos, Germany) and digitally captured using cooled CCD camera (Micromax Kodak 1317; Princeton Instruments, AZ).
Western Blot
Total L4-L6 spinal cord tissue from each rat was homogenized in lysis buffer with 1% NP40 and 1 mmol/L PMSF. Protein samples of the same amount from each rat were extracted from RVLM homogenates to analyze protein expression by Western blot. In brief, the protein samples (20 μg each) were subjected to SDS/PAGE in 8–12% gradient gel (Invitrogen, Carlsbad, CA) and transferred to PVDF membrane. Primary antibody was incubated and followed by incubation with horseradish peroxidase-conjugated goat anti-rabbit IgG or goat anti-mouse IgG. The amount of protein was assessed by ECL detection reagents (WBKLS0050; Millipore) and the immunostaining band was visualized and quantitated by an automatic chemiluminescence image analysis system (Tanon-5200; Tanon Science & Technology, Shanghai, China). The data were normalized by developing the β-actin as loading control. The concentration of all the antibodies was 1:1000 except for β-actin (1:5000).
Statistical Analysis
SPSS 11.0 was applied to analyze the data. Data were presented as mean ± standard error of the mean (SEM). Treatment group means were assessed by variance analysis followed by Tukey’s multiple comparison post hoc test (if variance was equal) or Dunnett T3 (if variance was not equal) using GraphPad Prism Software. P-values <0.05 were considered significant for all tests.
Results
Spinal NF-κB Was Activated and MALT1 Was Upregulated in the Glia of Spinal Cord After SCI/R in Rats, Which Could Be Reversed by MI-2, a MALT1 Inhibitor
Figure 1A and B showed at days 3 after SCI/R, MALT1 in the SCI/R group increased compared to the sham group (P < 0.05). Western blot analysis showed the increase in MALT1 protein expression in the spinal cord L4-6 of SCI/R rats compared with that of the sham group, while intraperitoneal administration of MI-2, the MALT1 inhibitor for three consecutive days significantly inhibited the MALT1 increase (Figure S3A and B, P < 0.05). Compared to the sham group, the protein expression of phosphorylated NF-κB [phos-NF-κB (p65)] was increased in spinal cord tissues measured by immunofluorescent staining (Figure 1C and D, P < 0.05). Furthermore, the immunofluorescent staining showed that the increased MALT1 and phos-NF-κB (p65) mainly expressed in astrocytes (Figure 2A-C, P < 0.05) and microglia (Figure 2D-F, P < 0.05) instead of neurons (Figure 2G-I) and endothelial cells (Figure 2J-L). Intraperitoneal administration of MI-2, the MALT1 inhibitor for three consecutive days significantly inhibited the activation of NF-κB induced by I/R (Figure 1, P < 0.05). These results suggest that I/R induces MALT1-dependent activation of NF-κB in the spinal cord.
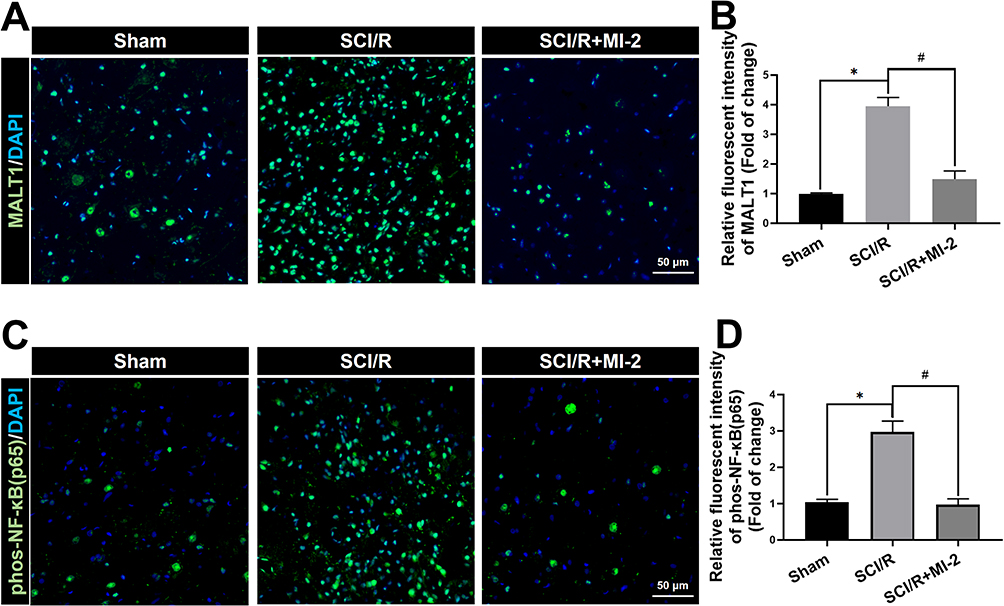 |
Figure 1 Effects of MI-2 on the spinal MALT1 expression and phos-NF-κB (p65) activation in SCI/R rats. (A) Immunofluorescent staining of MALT1 in the anterior horn of rat’s spinal cord. Scale bar = 50μm. (B) Immunofluorescent density analysis to quantify the expression of MALT1. (C) Immunofluorescent staining of [phos-NF-κB (p65)]-positivities. Scale bar = 50μm. (D) Immunofluorescent density analysis to quantify the expression of phos-NF-κB (p65). Data are presented as the mean ± SEM. n = 6 per group. *P < 0.05 vs sham group, #P < 0.05 vs SCI/R group. |
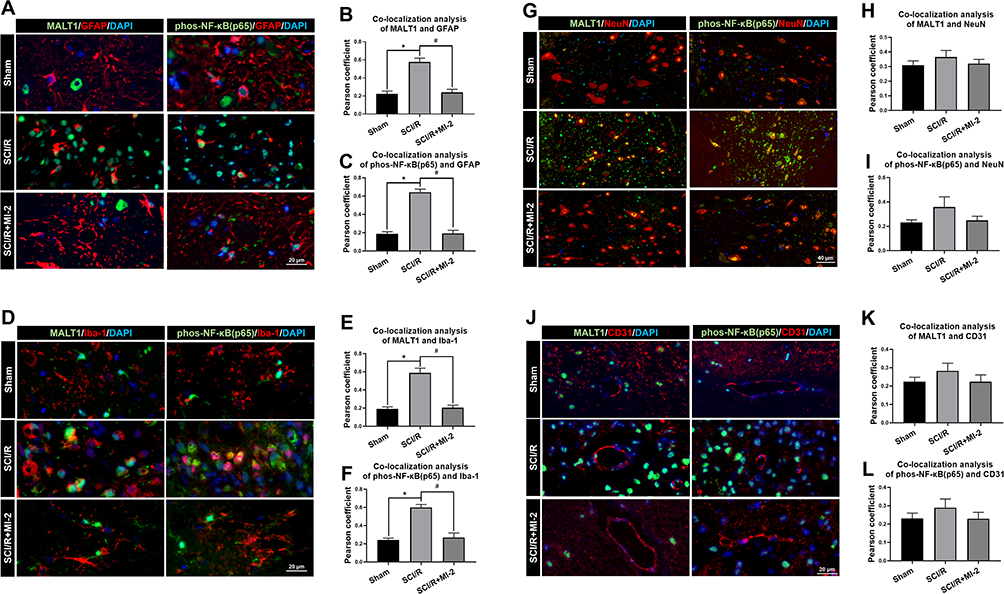 |
Figure 2 Localization of MALT1 and phos-NF-κB (p65) in different type of cells in the spinal cord of rats. (A) Double immunofluorescent staining showed colocalization of MALT1 or phos-NF-κB (p65) expression in astrocytes (GFAP positive cells) in the anterior horn of spinal cord. Scar bar = 20μm. (B, C) The quantified colocalization of MALT1 or phos-NF-κB (p65) with GFAP were assessed using the Pearson coefficient. (D) Double immunofluorescent staining to check colocalization of MALT1 or phos-NF-κB (p65) expression in microglia (Iba-1 positive cells) in the anterior horn of spinal cord. Scar bar = 20μm. (E, F) The quantified colocalization of MALT1 or phos-NF-κB (p65) with Iba-1 were assessed using the Pearson coefficient. (G) Double immunofluorescent staining showed MALT1 or phos-NF-κB (p65) expression in neurons (NeuN positive cells) in the anterior horn of spinal cord. Scar bar = 40μm. (H, I) The quantified colocalization of MALT1 or phos-NF-κB (p65) with NeuN were assessed using the Pearson coefficient. (J) Double immunofluorescent staining to colocalize MALT1 or phos-NF-κB (p65) with endothelial cells (CD31 positive cells) in the anterior horn of spinal cord. Scar bar = 20μm. (K, L) The quantified colocalization of MALT1 or phos-NF-κB (p65) with CD31 were assessed using the Pearson coefficient. Data are presented as the mean ± SEM. n = 6 per group. *P < 0.05 vs sham group, #P < 0.05 vs SCI/R group. |
MALT1 Activated ERS in the Glia of Spinal Cord After SCI/R in Rats
Numerous physiological and pathological factors can cause ER environment disturbance, ERS is a condition which is accelerated by aggregation of unfolded/misfolded proteins. We found that there was a significant increase in spinal Bip (the representative protein of ERS) expression at 3 days after SCI/R (Figure 3, P < 0.05), which corroborated with previous reports. The double immunofluorescent staining and co-localization analysis showed that increased Bip mainly present in astrocytes (Figure 4A and B, P < 0.05) and microglia (Figure 4C and D, P < 0.05) instead of neurons (Figure 4E and F) and/or endothelial cells (Figure 4G and H). The MALT1 inhibitor, MI-2 downregulated the ERS in the spinal cord of SCI/R rats (Figure 3, P < 0.05). Three consecutive days of intrathecal injection of human recombinant MALT1 protein (hrMALT1) further aggravated I/R-induced ERS (Figure 3, P < 0.05). Western blot verified the increased Bip expression in L4-6 of SCI/R rats compared with that of the sham group (Figure S4 and B, P < 0.05). Furthermore, as presented in Figure S4A and B, MI-2 treatment resulted in a decreased Bip protein expression, while intrathecal injection of hrMALT1 resulted in higher level of Bip expression, which indicated that MALT1 activation aggravates ERS (P < 0.05). The ultrastructure of the ER in the spinal glial cells was further investigated using transmission electron microscopy. It was found that ischemia/reperfusion induced the swelling of the ER and the dilatation of the lumen, which could be reversed by MI-2. While to the contrary, the hrMALT1 further aggravated the ultra-pathological injury of the ER induced by I/R (Figure 5). These results suggested that MALT1 activated ERS in the SI/R rats.
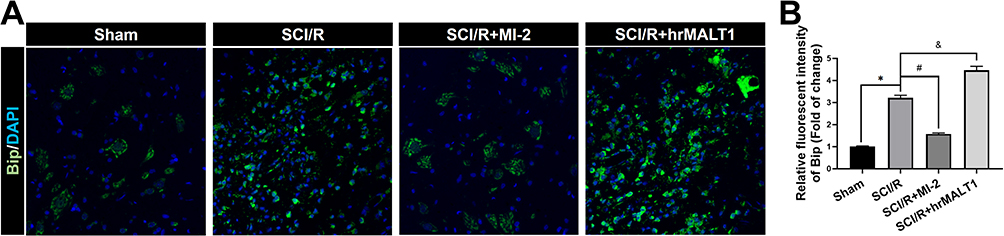 |
Figure 3 The expression and localization of ER stress marker protein Bip in spinal cord of rats. (A) Immunofluorescent staining of Bip in the anterior horn of rat’s spinal cord. Scale bar = 50μm. (B) Immunofluorescent density analysis quantifies the expression of Bip. Data are presented as the mean ± SEM. n = 6 per group. *P < 0.05 vs sham group, #P < 0.05 vs SCI/R group, &P < 0.05 vs SCI/R group. |
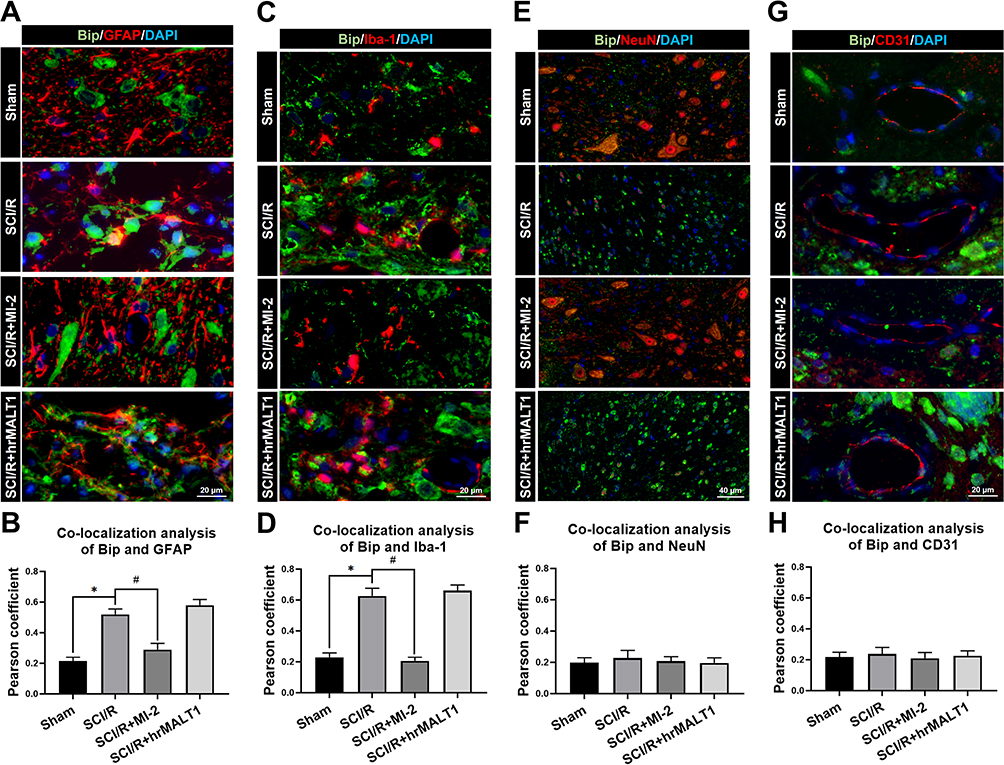 |
Figure 4 The colocalization of Bip with different cells in spinal cord. (A) Double immunofluorescent staining to check the colocalization of Bip with astrocytes (GFAP positive cells) in the anterior horn of spinal cord. Scar bar = 20μm. (B) Quantified colocalization of Bip with GFAP were assessed using the Pearson coefficient. (C) Double immunofluorescent staining showed colocalization of Bip with microglia (Iba-1 positive cells) in the anterior horn of spinal cord. Scar bar = 20μm. (D) Quantified colocalization of Bip with Iba-1 were assessed using the Pearson coefficient. (E) Double immunofluorescent staining showed colocalization of Bip with neuron (NeuN positive cells) in the spinal cord. Scar bar = 40μm. (F) Quantified colocalization of Bip with NeuN were assessed by using the Pearson coefficient. (G) Double immunofluorescent staining showed colocalization of Bip with endothelial cells (CD31 positive cells) in the spinal cord. Scar bar = 20μm. (H) Quantified colocalization of Bip with CD31 were assessed using the Pearson coefficient. Data are presented as the mean ± SEM. n = 6 per group. *P < 0.05 vs sham group, #P < 0.05 vs SCI/R group. |
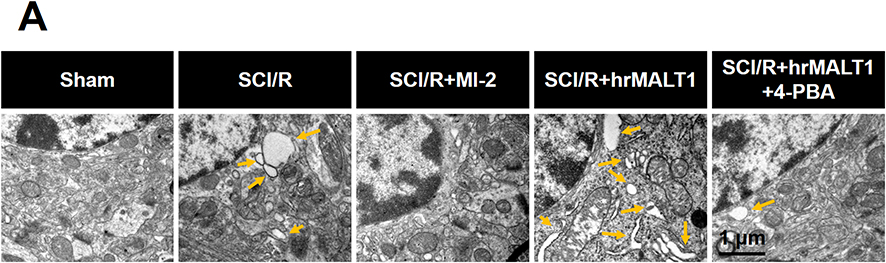 |
Figure 5 Transmission electron microscopy showed the morphology of endoplasmic reticulum of glial cells in the spinal cord. Yellow arrow indicates swallowed and dilated endoplasmic reticulum. n = 3. Scale bar = 1 μm. |
ERS Exacerbated Disruption of Blood-Spinal Cord Barrier and Loss of Neurovascular Units in the Spinal Cord After SCI/R in Rats
It has been noted that normal astrocytes are essential to contribute the blood–brain barrier integrity, extracellular homeostasis, neurotransmitter release and so on. We found that intrathecal injection of hrMALT1 upregulated the expression of MMP9 in astrocytes (Figure 6A and B, P < 0.05), and it also decreased the expression of endothelial tight junction protein ZO-1 (Figure 6C and D, P < 0.05) in SCI/R rats, which could be abolished by co-treatment 4-phenylbutyric acid (4-PBA), the ERS inhibitor in SCI/R rats. Furthermore, intrathecal injection of hrMALT1 aggravated increased EB leakage (Figure 6E and F, P < 0.05) and extravascular IgG leakage (Figure 6G and H, P < 0.05) in the anterior horn of spinal cord compared to that of SCI/R group, while 4-PBA co-treatment abolished its effects. Also, quantitative analysis of EB content in spinal cord tissue was performed. The results further confirmed that MI-2 inhibited SCI/R induced EB leakage in the spinal cord (Figure S5, P < 0.05). As presented in Figure S5, intrathecal injection of hrMALT1 increased EB levels in the spinal cord, while 4-PBA blocked the effect of hrMALT1 (P < 0.05). Furthermore, hrMALT1 injection significantly downregulated the expression of AQP4 in astrocytes (Figure 7A and B, P < 0.05) and decreased water content (Figure 7C, P < 0.05) in the spinal cord induced by I/R, while 4-PBA co-treatment abolished these effects. Antibody of NeuN (Neuron), GFAP (astrocyte) and CD31 (endothelial marker) was used to perform triple immunofluorescent staining, which evaluated their colocalization and reflected the profile of vascular-nerve unit. I/R not only led to the loss of spinal neurons (Figure 8A and B, P < 0.05) but also resulted in the reduction of perivascular astrocytes (Figure 8A and C, P < 0.05). MI-2 significantly inhibited the loss of neurons and perivascular astrocytes. Furthermore, 4-PBA can alleviate their loss when it was co-administrated with hrMALT1to SCI/R rats (Figure 8, P < 0.05). These results suggest that ERS might result in the destruction of the blood-spinal barrier and loss of neurovascular units in the spinal cord after SCI/R in rats.
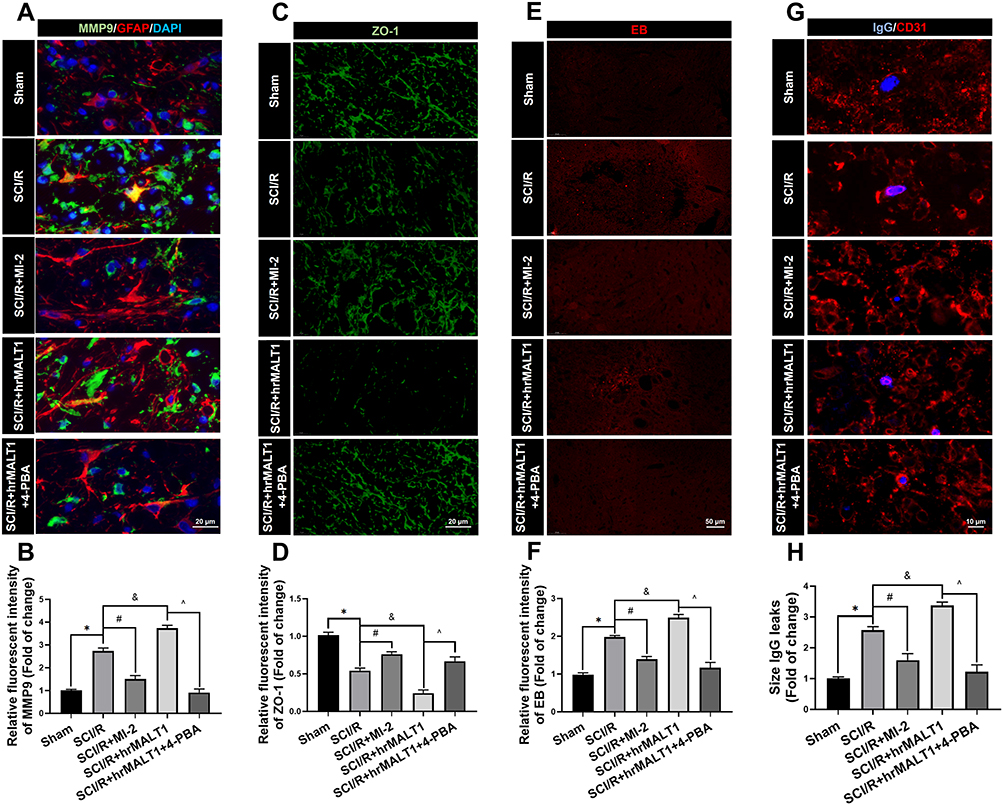 |
Figure 6 Evaluation of the blood-spinal cord barrier permeability. (A) Double immunofluorescent staining showed the MMP9 expression in astroglia of spinal cord. Scale bar = 20 μm. (B) Immunofluorescent density analysis quantifies the expression of MMP9. (C) Immunofluorescent staining of ZO-1 in the anterior horn of spinal cord in rats. Scale bar = 20μm. (D) Immunofluorescent density analysis quantifies the expression of ZO-1. (E) Evans Blue (EB) dye extravasation (red fluorescence) from the blood vessel in the spinal cord. (F) Immunofluorescent density analysis quantifies the extravasation of EB. n = 5 per group. (G) Representative graphs of blood-spinal cord barrier permeability identified by IgG-positive leakages (blue) outside of the endothelium. Scale bar = 10 µm. (H) Average size calculation based on IgG+ leakages and fold change in different groups. Data are presented as the mean ± SEM. n = 6 per group. *P < 0.05 vs sham group, #P < 0.05 vs SCI/R group, &P < 0.05 vs SCI/R group, ^P < 0.05 vs SCI/R + hrMALT1 group. |
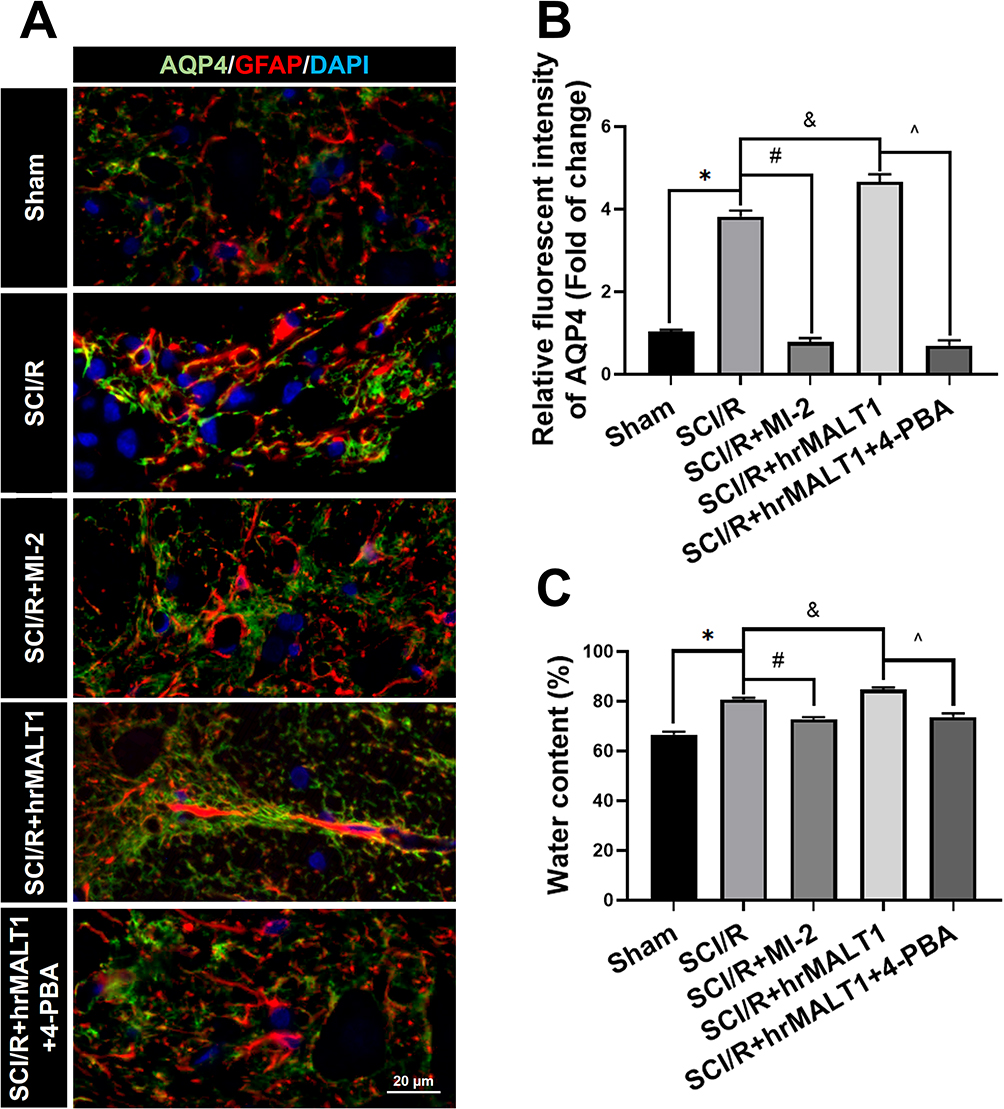 |
Figure 7 Spinal cord edema and water content evaluation. (A) Double immunofluorescent staining showed astroglial AQP4, a water channel protein, expression in spinal cord. Scale bar = 20 μm. (B) Immunofluorescent density analysis quantifies the expression of AQP4 in spinal cord. Data are presented as the mean ± SEM. n = 6 per group. (C) Water content of the spinal cord tissue was evaluated by calculating the percentage of water content via a wet-dry method with formula of (wet weight-dry weight)/wet weight × 100. n = 5 per group. Data are presented as the mean ± SEM. *P < 0.05 vs sham group, #P < 0.05 vs SCI/R group, &P < 0.05 vs SCI/R group, ^P < 0.05 vs SCI/R + hrMALT1 group. |
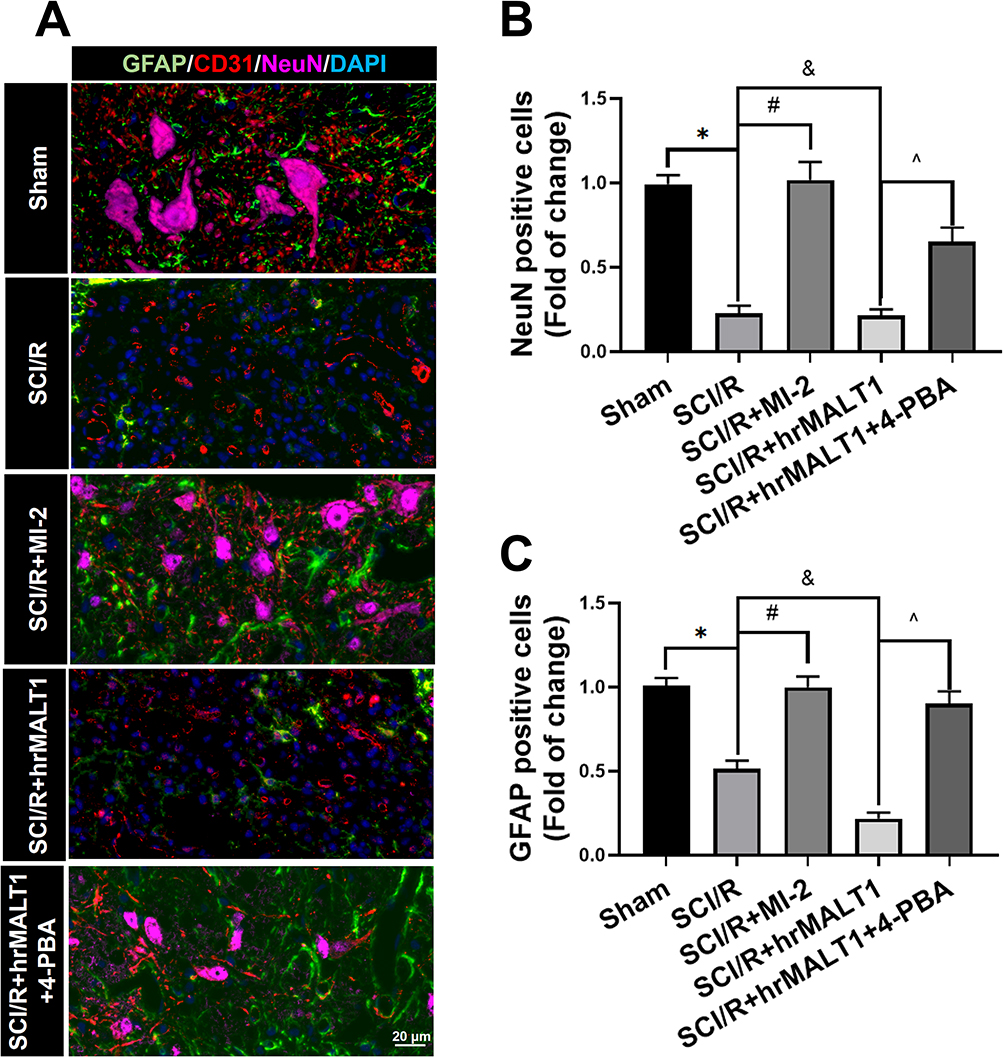 |
Figure 8 Evaluation of the neurovascular units in spinal cord. (A) The neurons, astrocytes and endothelial cells co-localization in spinal cord anterior horn were detected by triple immunofluorescent staining. Scale bar = 20 μm. (B, C) The number of NeuN and/or GFAP positivities was quantitatively analyzed. Data are presented as the mean ± SEM. n = 6 per group. *P < 0.05 vs sham group, #P < 0.05 vs SCI/R group, &P < 0.05 vs SCI/R group, ^P < 0.05 vs SCI/R + hrMALT1 group. |
ERS Triggered the Inflammatory Response and Oxidative Stress in the Spinal Cord After SCI/R in Rats
It was reported that mild ERS diminished LPS-induced neuroinflammation and shifted the microglia polarization from M1/2b to M2a subtype in the hippocampus. However, very little is known about the involvement of ER hormesis in SCI/R-induced neuroinflammation until now. We found that hrMALT1 administration aggravated the I/R-induced M1 polarization, while 4-PBA with hrMALT1 co-treatment alleviated M1 polarization (Figure 9A and B, P < 0.05) and the pro-inflammatory (IL-1β) releasing (Figure 9C and D, P < 0.05) in the spinal cord after SCI/R in rats. Furthermore, DHE superoxide anion probe detection revealed that hrMALT1 administration aggravated the I/R-induced oxidative stress indicated by ROS production, while 4-PBA with hrMALT1 co-treatment attenuated the ROS production in the spinal cord after SCI/R in rats (Figure 9E and F, P < 0.05).
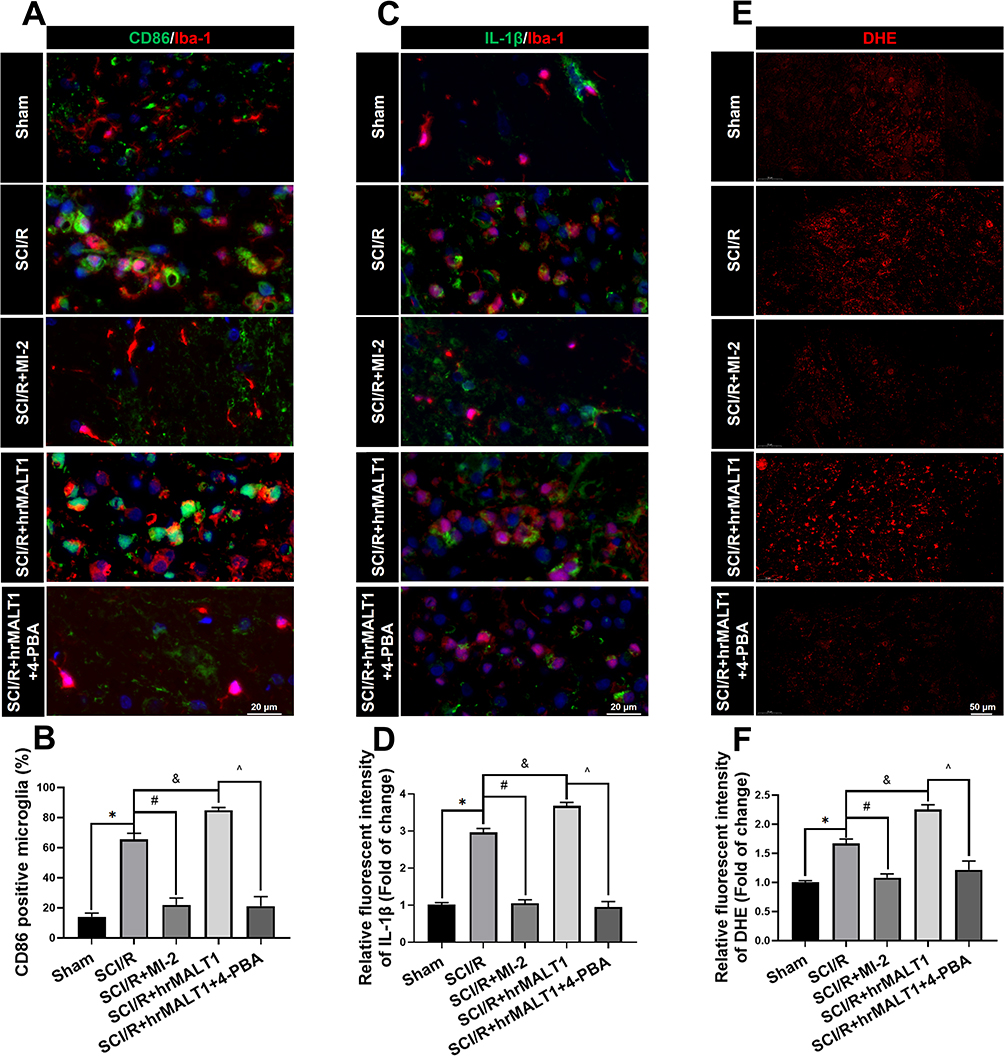 |
Figure 9 Neuroinflammation and oxidative stress profiles in the spinal cord of rats. (A) Double immunofluorescent staining detecting microglial M1 polarization (CD86 as M1 marker) in spinal cord. Scale bar = 20 μm. (B) Quantified percentage of CD86 positivities (M1 polarization). (C, D) Double immunofluorescent staining of inflammatory cytokines IL-1β in microglia and quantitative analysis of IL-1β fluorescent intensity to evaluate neuroinflammation. Scale bar=20 μm. (E) representative figures of DHE probe detecting superoxide anion in spinal cord and quantified immunofluorescent intensity values of DHE staining. (F). Scale bar = 50 μm. Data are presented as the mean ± SEM. n = 6 per group. *P < 0.05 vs sham group, #P < 0.05 vs SCI/R group, &P < 0.05 vs SCI/R group, ^P < 0.05 vs SCI/R + hrMALT1 group. |
MI-2 Treatment Improved SCI/R-Induced Motor Deficits by Attenuating the Neural Loss and Demyelination via Suppressing ERS
It was noted that blood spinal cord barrier destruction and neuroinflammation aggravate neuronal injury and axonal demyelination, which resulted in SCI/R-induced hindlimb motor function loss. We used Nissl staining, silver staining and LFB staining, respectively, to evaluate the structural damage to motor neurons and axonal ulceration in the anterior horn of the spinal cord after SCI/R in rats. We found that in the I/R group, the motor neurons in the anterior horn of the spinal cord were fragmented and lost their integrity. MI-2 treatment significantly inhibited the motor neuron fragmentation caused by I/R surgery (Figure 10A). We further evaluated the destruction of neurofibrils by silver staining. We found that MI-2 significantly inhibited the loss of neurofibrils and the axonal varicosities caused by I/R, while 4-PBA alleviated the deterioration of neurofibrils and axon breakage aggravated by hrMALT1 co-administration in the spinal cord after SCI/R in rats (Figure 10B). Luxol fast blue (LFB) staining was used to detect the demyelination of I/R spinal cord. We found that MI-2 attenuated I/R-induced demyelination in the SCI/R rats (Figure 10C and D, P < 0.05). Finally, the motor deficit index (MDI) score was used to quantify the hindlimb motor function of each group on the 3rd day after SCI/R surgery. A 10-minute aortic occlusion and 3-day reperfusion resulted in severe hind-extremity neurologic deficits in rats. By contrast, MI-2 treatment remarkably enhanced the motor function of hind limbs after spinal cord ischemia, as indicated by the significantly lower MDI scores. It was found that hrMALT1 administration aggravated the MDI score, while 4-PBA with hrMALT1 co-treatment improved SCI/R-induced motor deficits after SCI/R in rats (Figure 10E, P < 0.05).
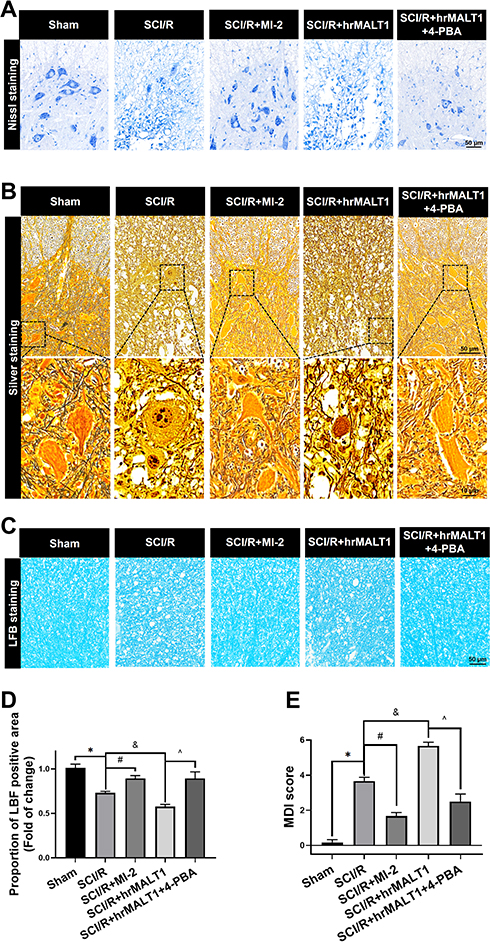 |
Figure 10 Neuronal loss and hindlimb locomotor function evaluation in rats. (A) Nissl staining and (B) silver staining were used to detect neuronal damage in the anterior horn of spinal cord. (C, D) LFB staining was used to detect demyelination of the spinal cord. (E) Neurological scores accessed by motor deficit index (MDI) scale. Data are presented as the mean ± SEM. n = 6 per group. *P < 0.05 vs sham group, #P < 0.05 vs SCI/R group, &P < 0.05 vs SCI/R group, ^P < 0.05 vs SCI/R + hrMALT1 group. |
Discussion
SCI/R is the fundamental cause of hindlimb motor function injury after aortic surgery. Previous studies have confirmed that the destruction of the BSCB and neuroinflammation are important mechanisms of motor neuron injury,21 but their initial molecular triggers are not fully understood. Our present study reveals that MALT1 activates ERS in glia and affects the function of astrocytes thereby destroys the blood spinal cord barrier; furthermore, it also promotes the polarization of microglia M1 and aggravates neuroinflammation. Spinal cord and traumatic brain injury share similar pathohistological features characterized by neuronal loss, axonal destruction, and demyelination during the secondary inflammatory-induced injury cascade.22 We found that MI-2, the MALT1-specific inhibitor, significantly reduced the BSCB leakage and neuroinflammation, thereafter inhibited the neuron loss and demyelination in the spinal cord, thus improving the hindlimb motor function of SCI/R rats. Our study suggests that MALT1 inhibitors have a potential protective effect on SCI/R.
MALT1 is the key platform linking pro-inflammatory G protein coupled receptor (GPCR) activation. It has been reported that AT1 receptor (AT1R) and Toll-like receptors23 can both induce the activation of MALT1 in blood vessels24 and kidneys,25 thereby triggering inflammatory injury. The activation of AT1R and TLR4 in the neurovascular units leads to increased permeability of the BSCB and the occurrence of neuroinflammation.26–28 Neuroinflammation can be seen as a counter-response against the presence of dangerous factors during SCI/R; however, it results in detrimental effect that is associated with pathomechanism of brain and spinal cord injury.29 While NF-κB signaling activation is a strong mediator of vascular injury and neuroinflammation.30,31 MALT1 is an adaptor protein that plays crucial roles in regulating the NF-κB pathway, specifically MALT1 functions as a scaffold protein and protease that initiate downstream signals. So, blocking MALT1 might be a feasible approach to inhibit inflammatory injury caused by pathogenic GPCR activation and also might be beneficial in the settings of SCI/R.
Growing evidences have highlighted that the pathophysiology of SCI/R injury is linked to cerebrovascular dysfunction.32–34 During SCI/R injury, disruption of the integrity of BSCB triggers infiltration of inflammatory cell at the injured area. Various pro-inflammatory cytokines are also produced by the neurovascular unit (for example: neurons, microglia and perivascular astrocyte) which further exacerbates neuroinflammation and BSCB damage.35,36 In our present study, we found that MALT1 was expressed in neurons, astrocytes, microglia and endothelial cells. I/R induced significant activation of MALT1 in astrocytes and microglia in the anterior horn of the spinal cord. Inhibition of MALT1 blocked the destruction of the BSCB and the occurrence of neuroinflammation caused by I/R. Our findings suggest that glial MALT1 is one of the important inducers of BSCB barrier destruction and neuroinflammation. The MALT1 inhibitor MI-2 used in this study has been proved to be non-toxic in animals by previous studies,17 so it may serve as basic research evidence for the potential protective effect of MI-2 on spinal cord in clinical practice, but the underlying mechanism needs to be further investigated.
It was noted that ERS is an important cellular and molecular mechanism for I/R injury, and inhibition of ERS is a common intervention strategy for I/R injury in the heart, kidney, brain and other important organs, but its upstream target remains unknown.37–39 ERS induces unfolded protein response to correct endoplasmic reticulum protein folding errors and activate autophagy to maintain cell homeostasis. Recent studies illustrate that the change of MALT1 activity is associated with ERS, and NF-κB signal is also one of the triggers of ERS.15 It was known that MALT1 plays a pivotal role in activating immune response by regulating and transducing NF-κB signaling.13 We found that SCI/R injury activated NF-κB signaling and ERS, which suggested that NF-κB signaling might mediate the ERS. Persistent ERS is considered to trigger the pathogenic cascades of many chronic disorders due to its ability to provoke aberrant inflammatory activity and facilitate cell death. ERS results in inflammation and cell death via overloading intracellular calcium, aggravating mitochondrial metabolic disorders, triggering further ROS damage and ATP depletion.40–42 More importantly, later cascades may lead to a series of catastrophic reactions that ultimately aggravate inflammation and death pathway activation.43
Astrocytes are necessary to maintain BBB integrity in the central nervous system.44,45 We observed that MI-2 downregulated the expression of I/R-induced MMP9 in astrocytes, and it also upregulated the expression of endothelial tight junction protein – zona occludens protein 1 (ZO-1).46 The MMP9 activation undermines BSCB integrity via degradation of ZO-1, which stimulates disruptions and opening of tight junctions, thereby modulate cascade inflammatory responses following tissue leakage via recruiting inflammatory cell infiltration.47 Our results are consistent with previous study that the upregulation of matrix metalloproteinases disrupts neurovascular permeability, which inhibits functional and structural recovery of spinal cord injury.48 Therefore, our findings provide additional insight that MALT1 inhibitor treatment may contribute to the preservation of BSCB integrity via modulation of inflammatory responses following SCI/R.
It is noted that the reactive astrocytes around neurovasculature activate endothelial MMP9 or secrete proinflammatory cytokines such as IL-1β, IL-6, and TNFα that contributes to blood–brain barrier permeability,49,50 eventually leading to blood–brain barrier disruption during inflammation. Consistent with the rest of other studies, inflammation is a subsequent event of spinal cord ischemia and a plausible pathway in SCI/R injury. Our study suggested that astrocyte and microglia both contributed the neuroinflammation and neuron loss. Yamanaka et al reported that reactive astrocytes were clearly visible at 7 days after reperfusion, and the number of activated microglia peaked at 12 and 48 h after reperfusion.51 Other researches indicate that the proliferation and activation of microglia contributes to excitotoxicity, which is an important mechanism of SCI/R injury.52 In a rabbit model of SCI/R, astrocytes were activated early (2 hours) after reperfusion in the gray matter of the lumbar spinal cord, but confined to the area where neurons started to show degeneration,53 which suggested that astrocytes might be important in the mechanism of delayed onset motor dysfunction in SCI/R. The temporal profile of the reaction of microglia, astrocytes, and macrophages in the delayed onset paraplegia after transient spinal cord ischemia in rabbits.
Zhu et al reported that the delayed spinal cord injury involves both ischemic cellular death and reperfusion injury. Inflammatory cytokines (eg, TNF- α and IL-1) may play an important role in SC/IR.5 Interestingly, our current study not only found that MALT1-mediated ERS was involved in regulation of astroglial MMP9, we also observed that MI-2 inhibited I/R-induced perivascular astrocyte loss at the anterior horn of the spinal cord. We firstly report that SCI/R is associated with astrocyte loss, suggesting that increased permeability of the blood-spinal barrier is not only associated with perivascular astrocyte activation, inflammatory cytokines, and MMP9 release but may also be associated with astrocytes loss and demyelination, while inhibition of MALT1 blocked the loss of perivascular astrocytes. Our results suggest that MALT1-specific inhibitor significantly improved vascular neural unit integrity, thus improving the lower limb motor function of SCI/R rats. However, the development of inflammatory mediators is incompletely understood, and treatments available for inflammation in SCI/R are insufficient. Improved understanding of SCI/R and the development of inflammatory cells and cytokines in these settings will provide novel therapeutic strategies for SCI/R.
There were several technical limitations in this study. First, the cellular localization of MALT1 might be microglia, astrocyte, and neurons in the spinal cord, therefore, further experiments are needed to specifically interfere MALT1 expression in different cell types to define the specific effects of MALT1 on SCI/R. Second, the interactions among cell types noted above in the regulation of SCI/R need to be clarified.
Conclusion
Collectively, inhibition of MALT1 alleviates spinal ischemia/reperfusion injury-induced neuroinflammation by modulating glial ERS in rats. MI-2 might be a new therapeutic strategy for interfering spinal cord ischemia/reperfusion injury.
Abbreviations
PBA, 4-phenylbutyrate; AQP4, Aquaporin type 4; Bcl10, B-cell lymphoma/leukemia 10; Bip, endoplasmic reticulum lumenal Ca(2+)-binding protein; BP, Blood pressure; BSCB, Blood-spinal cord barrier; CARMA, Caspase recruitment domain containing protein 11; CD31, endothelial cell adhesion molecule 1; DHE, Dihydroethidium; EB, Evans blue; ER, Endoplasmic reticulum; ERS, endoplasmic reticulum stress; GFAP, Glial fibrillary acidic protein; hrMALT1, human recombinant MALT1 protein; IL-1β, Interleukin 1 beta; I/R, ischemia/reperfusion; LFB, Luxol Fast Blue; MALT1, Mucosa-associated lymphoid tissue lymphoma translocation protein 1; MDI, Motor deficit index; MMP9, Matrix metalloproteinase-9; NeuN, neuronal nuclei; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; phos-NF-κB, phosphorylated NF-κB; ROS, Reactive oxygen species; SCI/R, spinal cord ischemia/reperfusion; ZO-1, Zona occludens protein 1.
Acknowledgments
This work was funded by grants from the Chinese National Natural Science Foundation of No.81770423 to CMX, No.81973945 to YF, and 81673766 to YF. We thank Kokwin Ooi for his contribution in improving and polishing the article.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Awad H, Ramadan ME, El Sayed HF, Tolpin DA, Tili E, Collard CD. Spinal cord injury after thoracic endovascular aortic aneurysm repair. Can J Anaesth. 2017;64(12):1218–1235.
2. Aucoin VJ, Eagleton MJ, Farber MA, et al. Spinal cord protection practices used during endovascular repair of complex aortic aneurysms by the U.S. Aortic Research Consortium. J Vasc Surg. 2021;73(1):323–330.
3. Karve IP, Taylor JM, Crack PJ. The contribution of astrocytes and microglia to traumatic brain injury. Br J Pharmacol. 2016;173(4):692–702.
4. Zhu P, Li JX, Fujino M, Zhuang J, Li XK. Development and treatments of inflammatory cells and cytokines in spinal cord ischemia-reperfusion injury. Mediators Inflamm. 2013;2013:701970.
5. Roussel BD, Kruppa AJ, Miranda E, Crowther DC, Lomas DA, Marciniak SJ. Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurol. 2013;12(1):105–118.
6. Zhu H, Zhou H. Novel insight into the role of endoplasmic reticulum stress in the pathogenesis of myocardial ischemia-reperfusion injury. Oxid Med Cell Longev. 2021;2021:5529810.
7. Park SJ, Li C, Chen YM. Endoplasmic reticulum calcium homeostasis in kidney disease: pathogenesis and therapeutic targets. Am J Pathol. 2021;191(2):256–265.
8. Mizukami T, Orihashi K, Herlambang B, et al. Sodium 4-phenylbutyrate protects against spinal cord ischemia by inhibition of endoplasmic reticulum stress. J Vasc Surg. 2010;52(6):1580–1586.
9. Amani H, Shahbazi MA, D’Amico C, Fontana F, Abbaszadeh S, Santos HA. Microneedles for painless transdermal immunotherapeutic applications. J Control Release. 2021;330:185–217.
10. Nan D, Jin H, Deng J, et al. Cilostazol ameliorates ischemia/reperfusion-induced tight junction disruption in brain endothelial cells by inhibiting endoplasmic reticulum stress. FASEB J. 2019;33(9):10152–10164.
11. Wang HF, Wang ZQ, Ding Y, et al. Endoplasmic reticulum stress regulates oxygen-glucose deprivation-induced parthanatos in human SH-SY5Y cells via improvement of intracellular ROS. CNS Neurosci Ther. 2018;24(1):29–38.
12. Lin Y, Zhang C, Xiang P, Shen J, Sun W, Yu H. Exosomes derived from HeLa cells break down vascular integrity by triggering endoplasmic reticulum stress in endothelial cells. J Extracell Vesicles. 2020;9(1):1722385.
13. Ruland J, Hartjes L. CARD-BCL-10-MALT1 signalling in protective and pathological immunity. Nat Rev Immunol. 2019;19(2):118–134.
14. Engelmann C, Haenold R. Transcriptional control of synaptic plasticity by transcription factor NF-κB. Neural Plast. 2016;2016:7027949.
15. Izumi K, Nishikori M, Yuan H, Otsuka Y, Nakao K, Takaori-Kondo A. Establishment and characterization of a MALT lymphoma cell line carrying an API2-MALT1 translocation. Genes Chromosomes Cancer. 2020;59(9):517–524.
16. Frakes AE, Dillin A. The UPR(ER): sensor and coordinator of organismal homeostasis. Mol Cell. 2017;66(6):761–771.
17. Fontan L, Yang C, Kabaleeswaran V, et al. MALT1 small molecule inhibitors specifically suppress ABC-DLBCL in vitro and in vivo. Cancer Cell. 2012;22(6):812–824.
18. Taira Y, Marsala M. Effect of proximal arterial perfusion pressure on function, spinal cord blood flow, and histopathologic changes after increasing intervals of aortic occlusion in the rat. Stroke. 1996;27(10):1850–1858.
19. Kerkhofs D, van Hagen BT, Milanova IV, et al. Pharmacological depletion of microglia and perivascular macrophages prevents Vascular Cognitive Impairment in Ang II-induced hypertension. Theranostics. 2020;10(21):9512–9527.
20. Zhang S, Hu L, Jiang J, et al. HMGB1/RAGE axis mediates stress-induced RVLM neuroinflammation in mice via impairing mitophagy flux in microglia. J Neuroinflammation. 2020;17(1):15.
21. Khachatryan Z, Haunschild J, von Aspern K, Borger MA, Etz CD. Ischemic spinal cord injury - experimental evidence and evolution of protective measures. Ann Thorac Surg. 2021;S0003–4975:21.
22. Bramlett HM, Dietrich WD. Long-term consequences of traumatic brain injury: current status of potential mechanisms of injury and neurological outcomes. J Neurotrauma. 2015;32(23):1834–1848.
23. Dong W, Liu Y, Peng J, et al. The IRAK-1-BCL10-MALT1-TRAF6-TAK1 cascade mediates signaling to NF-kappaB from Toll-like receptor 4. J Biol Chem. 2006;281(36):26029–26040.
24. McAllister-Lucas LM, Ruland J, Siu K, et al. CARMA3/Bcl10/MALT1-dependent NF-kappaB activation mediates angiotensin II-responsive inflammatory signaling in nonimmune cells. Proc Natl Acad Sci U S A. 2007;104(1):139–144.
25. Markó L, Park JK, Henke N, et al. B-cell lymphoma/leukaemia 10 and angiotensin II-induced kidney injury. Cardiovasc Res. 2020;116(5):1059–1070.
26. Güler A, Sahin MA, Ucak A, et al. Protective effects of angiotensin II type-1 receptor blockade with olmesartan on spinal cord ischemia-reperfusion injury: an experimental study on rats. Ann Vasc Surg. 2010;24(6):801–808.
27. Li XQ, Lv HW, Wang ZL, Tan WF, Fang B, Ma H. MiR-27a ameliorates inflammatory damage to the blood-spinal cord barrier after spinal cord ischemia: reperfusion injury in rats by downregulating TICAM-2 of the TLR4 signaling pathway. J Neuroinflammation. 2015;12:25.
28. Li XQ, Lv HW, Tan WF, Fang B, Wang H, Ma H. Role of the TLR4 pathway in blood-spinal cord barrier dysfunction during the bimodal stage after ischemia/reperfusion injury in rats. J Neuroinflammation. 2014;11:62.
29. Wang L, Yao Y, He R, et al. Methane ameliorates spinal cord ischemia-reperfusion injury in rats: antioxidant, anti-inflammatory and anti-apoptotic activity mediated by Nrf2 activation. Free Radic Biol Med. 2017;103:69–86.
30. Mussbacher M, Salzmann M, Brostjan C, et al. Cell type-specific roles of NF-κB linking inflammation and thrombosis. Front Immunol. 2019;10:85.
31. Shih RH, Wang CY, Yang CM. NF-kappaB signaling pathways in neurological inflammation: a mini review. Front Mol Neurosci. 2015;8:77.
32. Wang D, Chen F, Fang B, et al. MiR-128-3p alleviates spinal cord ischemia/reperfusion injury associated neuroinflammation and cellular apoptosis via SP1 suppression in rat. Front Neurosci. 2020;14:609613.
33. Bhattacharya A, Kaushik DK, Lozinski BM, Yong VW. Beyond barrier functions: roles of pericytes in homeostasis and regulation of neuroinflammation. J Neurosci Res. 2020;98(12):2390–2405.
34. Mészáros Á, Molnár K, Nógrádi B, et al. Neurovascular inflammaging in health and disease. Cells. 2020;9(7):1614.
35. Ye LX, An NC, Huang P, et al. Exogenous platelet-derived growth factor improves neurovascular unit recovery after spinal cord injury. Neural Regen Res. 2021;16(4):765–771.
36. Yuan X, Wu Q, Wang P, et al. Exosomes derived from pericytes improve microcirculation and protect blood-spinal cord barrier after spinal cord injury in mice. Front Neurosci. 2019;13:319.
37. Rashid HO, Yadav RK, Kim HR, Chae HJ. ER stress: autophagy induction, inhibition and selection. Autophagy. 2015;11(11):1956–1977.
38. Senft D, Ronai ZA. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem Sci. 2015;40(3):141–148.
39. Folch-Puy E, Panisello A, Oliva J, et al. Relevance of endoplasmic reticulum stress cell signaling in liver cold ischemia reperfusion injury. Int J Mol Sci. 2016;17(6):807.
40. Lin Y, Jiang M, Chen W, Zhao T, Wei Y. Cancer and ER stress: mutual crosstalk between autophagy, oxidative stress and inflammatory response. Biomed Pharmacother. 2019;118:109249.
41. Namgaladze D, Khodzhaeva V, Brüne B. ER-mitochondria communication in cells of the innate immune system. Cells. 2019;8(9):1088.
42. Sprenkle NT, Sims SG, Sánchez CL, Meares GP. Endoplasmic reticulum stress and inflammation in the central nervous system. Mol Neurodegener. 2017;12(1):42.
43. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13(2):89–102.
44. Liu LR, Liu JC, Bao JS, Bai QQ, Wang GQ. Interaction of microglia and astrocytes in the neurovascular unit. Front Immunol. 2020;11:1024.
45. Neuroimmunological I-KH. Implications of AQP4 in astrocytes. Int J Mol Sci. 2016;17(8):1306.
46. Sun XG, Duan H, Jing G, Wang G, Hou Y, Zhang M. Inhibition of TREM-1 attenuates early brain injury after subarachnoid hemorrhage via downregulation of p38MAPK/MMP-9 and preservation of ZO-1. Neuroscience. 2019;406:369–375.
47. Li XF, Zhang XJ, Zhang C, et al. Ulinastatin protects brain against cerebral ischemia/reperfusion injury through inhibiting MMP-9 and alleviating loss of ZO-1 and occludin proteins in mice. Exp Neurol. 2018;302:68–74.
48. Noble LJ, Donovan F, Igarashi T, Goussev S, Werb Z. Matrix metalloproteinases limit functional recovery after spinal cord injury by modulation of early vascular events. J Neurosci. 2002;22(17):7526–7535.
49. Abbott NJ. Inflammatory mediators and modulation of blood-brain barrier permeability. Cell Mol Neurobiol. 2000;20(2):131–147.
50. Cohen-Salmon M, Slaoui L, Mazaré N, et al. Astrocytes in the regulation of cerebrovascular functions. Glia. 2021;69(4):817–841.
51. Yamanaka K, Sasaki N, Fujita Y, Kawamoto A, Hirata K, Okita Y. Impact of acquired and innate immunity on spinal cord ischemia and reperfusion injury. Gen Thorac Cardiovasc Surg. 2016;64(5):251–259.
52. Tikka T, Fiebich BL, Goldsteins G, Keinanen R, Koistinaho J. Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J Neurosci. 2001;21(8):2580–2588.
53. Matsumoto S, Matsumoto M, Yamashita A, et al. The temporal profile of the reaction of microglia, astrocytes, and macrophages in the delayed onset paraplegia after transient spinal cord ischemia in rabbits. Anesth Analg. 2003;96:6.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.