")
Back to Journals » Journal of Inflammation Research » Volume 14
Inhibition of Long Non-Coding RNA KCNQ1OT1 Attenuates Neuroinflammation and Neuronal Apoptosis Through Regulating NLRP3 Expression via Sponging miR-30e-3p
Authors Song A, Yang Y, He H, Sun J, Chang Q, Xue Q
Received 9 November 2020
Accepted for publication 14 April 2021
Published 5 May 2021 Volume 2021:14 Pages 1731—1742
DOI https://doi.org/10.2147/JIR.S291274
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Aixia Song,1 Yuying Yang,2 Hongmei He,1 Jian Sun,1 Qing Chang,1 Qian Xue1
1Department of Neurology, The First Affiliated Hospital of Hebei North University, Zhangjiakou, Hebei, 075000, People’s Republic of China; 2Stroke Office, The First Affiliated Hospital of Hebei North University, Zhangjiakou, Hebei, 075000, People’s Republic of China
Correspondence: Qian Xue
Department of Neurology, The First Affiliated Hospital of Hebei North University, No. 12 Changqing Road, Qiaoxi District, Zhangjiakou, Hebei, 075000, People’s Republic of China
Email [email protected]
Background: Neuroinflammation and neuronal apoptosis are considered as the critical factors in the pathogenesis of multiple neurological diseases. Recent studies have shown that long non-coding RNA (lncRNA) plays a crucial part in neuroinflammation and neuronal apoptosis.
Methods: The expression levels of lncRNA KCNQ1OT1, miR-30e-3p and NLRP3 in lipopolysaccharide (LPS)-induced HMC3 cells were analyzed using RT-qPCR. MTT assay, LDH release assay and ELISA were used to assess the effect of KCNQ1OT1 and miR-30e-3p on neuroinflammation and neuronal apoptosis. The targeted regulatory relationships among KCNQ1OT1, miR-30e-3p and NLRP3 were evaluated by bioinformatics analysis, dual-luciferase reporter gene assay, RT-qPCR and Western blot.
Results: In LPS-induced HMC3 cells, the expression levels of KCNQ1OT1 and NLRP3 were increased, while the expression level of miR-30e-3p was reduced. Knockdown of KCNQ1OT1 alleviated LPS-induced apoptosis and neuroinflammation of HMC3 cells, accompanied by increased cell viability, low LDH release and reduced cell apoptosis rate, and reduced levels of TNF-α, IL-1β and IL-6. Overexpression of miR-30e-3p had a similar effect. Additionally, KCNQ1OT1 could bind with miR-30e-3p and repress its expression in HMC3 cells, and KCNQ1OT1 overexpression counteracted miR-30e-3p’s inhibitory effect on LPS-induced neuronal damage and inflammatory response in HMC3 cells. Furthermore, KCNQ1OT1 could positively regulate the expression of NLRP3 via repressing miR-30e-3p.
Conclusion: Inhibition of KCNQ1OT1 could reduce neuroinflammation and neuronal apoptosis induced by LPS in HMC3 cells by regulating miR-30e-3p/NLRP3 pathway, suggesting that KCNQ1OT1 and miR-30e-3p could serve as promising therapeutic targets for treating neurological diseases.
Keywords: NLRP3, KCNQ1OT1, miR-30e-3p, neuroinflammation
Introduction
Multiple neurological diseases such as Parkinson’s disease (PD), Alzheimer’s disease (AD) and stroke are associated with neuroinflammation and neuronal apoptosis.1–5 Microglia, as an important part of the brain's immune system, exerts a vital effect on maintaining the homeostasis of the brain's microenvironment.6 Taking the pathogenesis of PD as an example, damaged or dead dopaminergic (DA) neurons induce microglial activation, and activated microglia produces excessive inflammatory cytokines, aggravating the degeneration of DA neurons.7,8 Previous studies have shown that inflammatory cytokines, such as tumor necrosis factor α (TNF-α), interleukin-1β (IL-1β) and interferon-γ (IFN-γ) in the serum of PD patients, are at high levels.9,10 Nod-like receptor protein 3 (NLRP3) is a central component of the inflammasome and is highly expressed in microglia, which is crucial in regulating neuroinflammation.11 Inflammasome containing NLRP3 is activated in various neurological diseases, such as AD and amyotrophic lateral sclerosis.12 Controlling microglial activation can probably contribute to the survival of neurons.13 Therefore, NLRP3 is a potential target for treating these neurological diseases.14 Nevertheless, the regulatory mechanism of NLRP3 in microglia is obscure.
Long non-coding RNAs (lncRNAs), which cannot be translated into proteins, are a class of transcripts with a length of more than 200 nucleotides.15 LncRNAs participate in regulating a lot of biological processes. Increasing studies show that lncRNAs have a close relation with the pathogenesis of neurological diseases.16–18 For example, in mice with PD and MPP+ induced SH-SY5Y cells, lncRNA-UCA1 accelerates the pathogenesis by up-regulating SNCA expression.17 In recent years, lncRNA potassium voltage-gated channel subfamily q member 1 overlapping transcript 1 (KCNQ1OT1) has been linked to multiple human diseases, including cancers and myocardial injury.19,20 Interestingly, it is reported that KCNQ1OT1 promotes oxygen-glucose-deprivation/reoxygenation-induced neuron injury.21,22 However, the role of KCNQ1OT1 in neuroinflammation and neuronal apoptosis and its molecular mechanism have not been fully clarified.
MicroRNA (miRNA) can degrade mRNA or inhibit translation by binding to 3ʹUTR of mRNA.23 Increasing evidence shows that miRNA is of substantial significance in the pathogenesis of neurological diseases.24–27 For example, in PD, miR-7 mediates inflammation by regulating α-Syn and NLRP3 inflammasome in DA neurons.25,26 Previous studies have shown that miR-30e-3p exerts tumor-suppressive functions.28 MiR-30e-3p also participates in myocardial injury induced by coronary microembolization by regulating autophagy.29 Importantly, miR-30e-3p can suppress neuroinflammation in PD model by repressing NLRP3.30
In this study, we found that KCNQ1OT1 was up-regulated and miR-30e-3p was down-regulated in lipopolysaccharide (LPS)-induced HMC3 cells. Gain-of-function and loss-of-function assays confirmed that KCNQ1OT1 could aggravate neuroinflammation and neuronal injury via regulating miR-30e-3p/NLRP3. The present study suggested that KCNQ1OT1 was a promising therapy target for neurological diseases.
Materials and Methods
Cell Culture
HMC3 microglia were obtained from the Cell Bank of Chinese Academy of Medical Science (Shanghai, China). HMC3 cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) (Gibco, Carlsbad, CA, USA) containing 10% fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA, USA), cultured at 37°C, in 5% CO2 in a humidified incubator. HMC3 cells were treated with different concentrations of LPS (0, 0.1, 0.2, 0.5 and 1 μg/mL) for 24 h, or treated with 1 μg/mL LPS for different times (0, 1, 6, 12 and 24 h). In subsequent experiments, HMC3 cells were treated with 1 μg/mL LPS for 24 h.
Cell Transfection
KCNQ1OT1 overexpression plasmid (pc-KCNQ1OT1), KCNQ1OT1 siRNA (si-KCNQ1OT1), NLRP3 overexpression plasmid (pc-NLRP3), miR-30e-3p mimics, and corresponding negative controls (pc-NC, si-NC, and NC mimics) were all obtained from GenePharma (Shanghai, China). According to the manufacturer’s instructions, they were transfected into HMC3 cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). The total RNA of the cell was extracted at 48 h after the transfection to measure the transfection efficiency, by real-time quantitative polymerase chain reaction (RT-qPCR).
RT-qPCR
HMC3 cells were collected and the total RNA of the cells was extracted using TRIzol reagent (Invitrogen, Shanghai, China) according to the manufacturer’s instructions. RNA was reverse transcribed into cDNA using PrimeScriptTM RT Reagent Kit (Takara, Shanghai, China). SYBR®Premix-Ex-Taq (Takara, Shanghai, China) and ABI7300 systems were employed to conduct RT-qPCR. GAPDH was the internal reference for the expression levels of lncRNA KCNQ1OT1, NLRP3, TNF-α, IL-1β and IL-6. U6 was the internal reference for miR-30e-3p. Relative quantification was conducted by using the 2(-ΔΔCt) method. The primer sequences are shown in Table 1.
 |
Table 1 The Primer Sequences for qRT-PCR |
Enzyme-Linked Immunosorbent Assay (ELISA)
The contents of TNF-α, IL-1β and IL-6 in HMC3 cell culture supernatant were determined by the corresponding ELISA kits (Abcam, Shanghai, China) according to the manufacturer’s instructions.
Cell Viability Assay
MTT assay was employed to assess the viability of HMC3 cells. Briefly, after the cells were inoculated into the 96-well plate (2 × 103 cell/well) and cultured for 12 h, the cells were incubated with 20 μL of MTT solution (Beyotime, Shanghai, China) for 8 h. Finally, 150 μL of DMSO (Beyotime, Shanghai, China) was added to dissolve the crystals and terminate the reaction. Finally, the absorbance of the cells in each well at 450 nm wavelength was measured by a microplate reader (Molecular Devices, CA, USA).
Lactate Dehydrogenase (LDH) Assay
Cell membrane integrity of HMC3 cells was evaluated by measuring LDH in the supernatants of cells. LDH Cytotoxicity Test Kit (Jiancheng Bioengineering Institute, Nanjing, China) was employed to evaluate the level of LDH released by HMC3 cells. In short, the cell culture supernatant was collected, and absorbance was measured at 440 nm using a microplate reader according to the manufacturer’s instructions.
Measurement of Nitric Oxide (NO)
Levels of nitric oxide (NO) production were determined indirectly by measuring the stable NO catabolite, nitrite, in the medium using the Griess method. Briefy, 50 μL of culture supernatant of HMC3 cells was collected. Then, 100 μL of Griess reagent (Sigma-Aldrich, Deisenhofen, Germany) was added. After 20 min of incubation, the absorbance was measured at 540 nm. Nitrite concentrations were calculated according to the standard curve.
Measurement of Reactive Oxygen Species (ROS)
ROS levels in HMC3 cells were determined using 2,7-dichlorodihydrofluorescein (DCFH-DA, Sigma-Aldrich, Deisenhofen, Germany). HMC3 cells were seeded in 6-well plates and treated with LPS. 24 h later, the cells were harvested and then re-suspended in 10 μM of DCFH-DA solution, and incubated at 37°C for 30 min. Intracellular ROS were then quantified according to the manufacturer’s instruction. Fluorescence absorbance was detected on a FL800-BioTek spectrofluorometer (Bio-Tek Instruments INC, Germany) with excitation/emission wavelengths of 480 nm/525 nm.
Dual-Luciferase Reporter Assay
The targeting relationship between miR-30e-3p and KCNQ1OT1 or NLRP3 3ʹUTR was examined by luciferase reporter gene assay. The wild type (WT) reporters (KCNQ1OT1-WT or NLRP3-WT) and mutant type (MUT) reporters (KCNQ1OT1-MUT or NLRP3-MUT) containing the predicted miR-30e-3p binding sites were subcloned to pmirGLO dual-luciferase miRNA target expression vector (Promega Corp., Madison, WI, USA). Subsequently, they were co-transfected into HMC3 cells with miR-30e-3p mimics or NC mimics, respectively. After 48 h of transfection, the luciferase activity was assessed by the dual-luciferase reporter gene analysis system (Promega, Madison, WI, USA).
Western Blot
HMC3 cells were lysed in RIPA lysis buffer (Beyotime Biotechnology, Shanghai, China) to extract the total protein. The protein samples were mixed with loading buffer and then denatured in the boiling water for 10 min. Next, the total protein was separated by SDS-PAGE and transferred to PVDF membrane (Millipore, Bedford, MA, USA). After that, the membranes were blocked in 5% defatted milk, and then incubated with the primary antibodies: anti-NLRP3 (1:500, ab214185, Abcam, Shanghai, China) and anti-β-actin (1:1000, ab8226, Abcam, Shanghai, China), respectively, overnight at 4°C. After the membranes were rinsed by TBST, the membrane was incubated with horseradish peroxidase coupled secondary antibody (Beyotime, Shanghai, China) at room temperature for 1 h. Finally, the protein bands were developed with ECL kit (Beyotime, Shanghai, China).
Statistical Analysis
All experiments were performed in triplicate, and each experiment was repeated three times. All the experimental data were expressed as “mean ± standard deviation”. SPSS 19.0 software (SPSS, Inc., Chicago, IL, USA) was employed for statistical analysis. One-way ANOVA and Student’s t-test were used to make the comparison. P < 0.05 was considered to show a statistically significant difference.
Results
KCNQ1OT1 Was Up-Regulated and miR-30e-3p Was Down-Regulated in LPS-Induced HMC3 Cells
Firstly, LPS was added (concentrations of 0, 0.1, 0.2, 0.5 or 1 μg/mL) to stimulate HMC3 cells, respectively, and incubated for 24 h; meanwhile, 1 μg/mL LPS was used to treat HMC3 cells for different times (0 h, 2 h, 8 h, 12 h and 24 h). Then, RT-qPCR was employed to analyze the relative expression levels of KCNQ1OT1 and miR-30e-3p in HMC3 cells, the results of which showed that the expression of KCNQ1OT1 was increased with the elevation of LPS concentration (Figure 1A), and its expression was gradually increased with the increase in treatment time (Figure 1B). In addition, the relative expression of miR-30e-3p showed opposite trend compared with KCNQ1OT1 in a concentration- and time-dependent manner (Figure 1C and D). These results suggested that KCNQ1OT1 and miR-30e-3p might be involved in LPS-induced neuroinflammation and neuronal apoptosis.
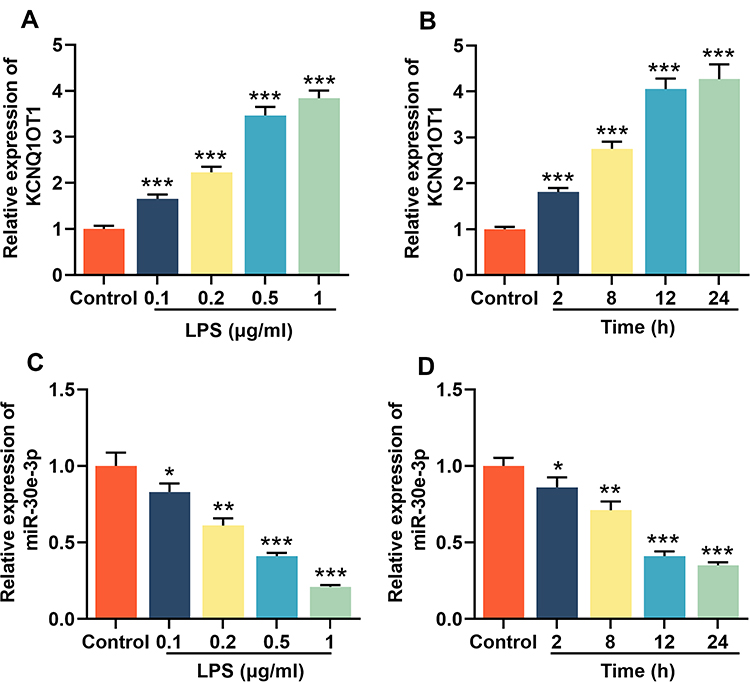 |
Figure 1 KCNQ1OT1 was up-regulated and miR-30e-3p was down-regulated in LPS-induced HMC3 cells. HMC3 cells were treated with different concentrations of LPS (0, 0.1, 0.2, 0.5 and 1 μg/mL) for 24 h, or treated with 1 μg/mL of LPS for different times (0, 1, 6, 12 and 24 h). RT-qPCR was used to detect the expression levels of KCNQ1OT1 (A and B) and miR-30e-3p (C and D) in HMC3 cells. All experiments were performed in triplicate. *P<0.05, **P<0.01, and ***P<0.001. Abbreviation: LPS, lipopolysaccharide. |
Knocking Down KCNQ1OT1 Suppressed LPS-Induced Neuroinflammation and Neuronal Apoptosis in HMC3 Cells
For further exploring the function of KCNQ1OT1, we transfected HMC3 cells with pc-KCNQ1OT1 or si-KCNQ1OT1. The expression of KCNQ1OT1 was evaluated by RT-qPCR. Compared with control cells (cells transfected with pc-NC or si-NC), the expression of KCNQ1OT1 in HMC3 cells transfected with pc-KCNQ1OT1 was significantly increased, while the expression of KCNQ1OT1 in cells transfected with si-KCNQ1OT1 was significantly decreased (Figure 2A). Subsequently, we used RT-qPCR and ELISA kits to examine the expressions of TNF-α, IL-1β and IL-6 to evaluate the inflammatory response. The results indicated that LPS treatment markedly promoted the expression levels of inflammatory cytokines TNF-α, IL-1β and IL-6 in HMC3 cells; overexpression of KCNQ1OT1 promoted the expressions of inflammatory cytokines, while KCNQ1OT1 knockdown inhibited their expression (Figure 2B and C). The expression of M1 polarization marker CD86 was significantly increased with LPS treatment, and M2 polarization marker CD206 decreased notably; the overexpression of KCNQ1OT1 further promoted the expression of CD86 and inhibited CD206 expression, while KCNQ1OT1 knockdown had the opposite effects (Figure 2D and E). In addition, MTT and LDH assays were used to detect the cell survival and apoptosis. The results showed that after LPS treatment, the cell viability was markedly reduced and the LDH release was remarkably elevated; overexpression of KCNQ1OT1 promoted these effects, while knocking down KCNQ1OT1 reversed these LPS-induced effects (Figure 2F and G). In addition, KCNQ1OT1 knockout significantly reduced the generation of NO and ROS of HMC3 cells (Figure 2H and I). These results indicated that the inhibition of KCNQ1OT1 could inhibit LPS-induced activation of microglia, neuroinflammation and neuronal apoptosis in HMC3 cells.
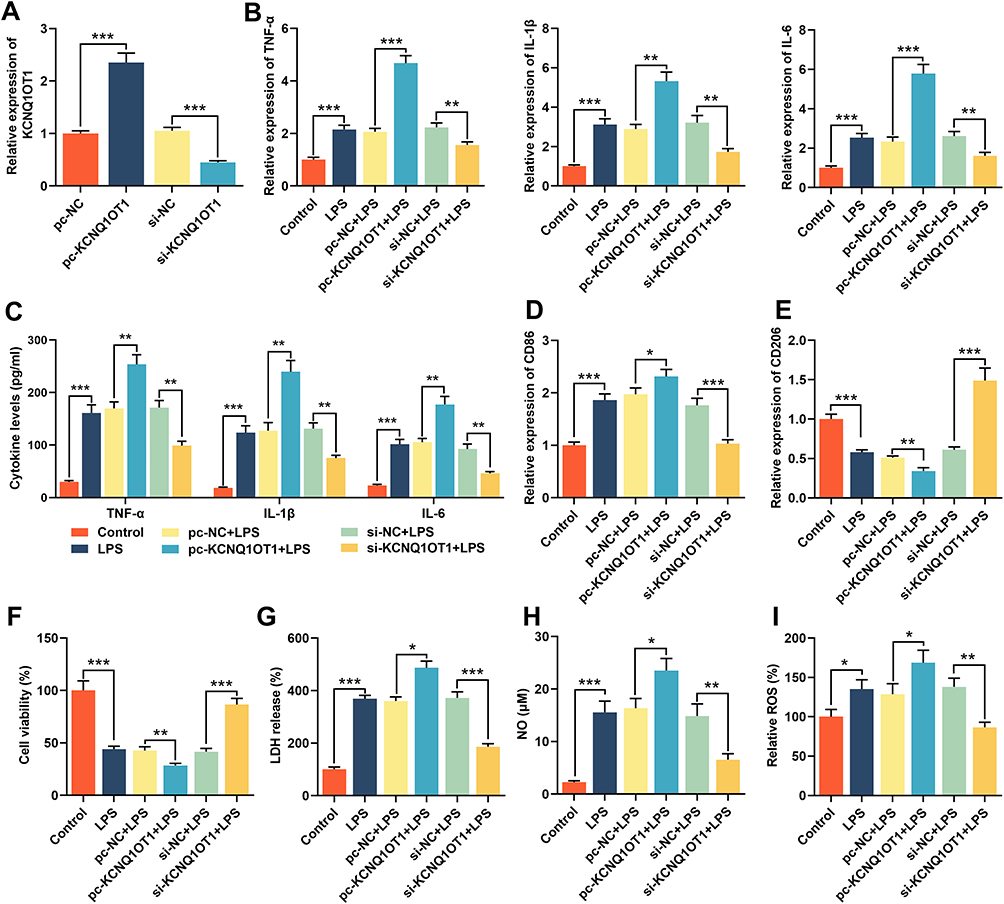 |
Figure 2 Knocking down KCNQ1OT1 inhibited LPS-induced neuroinflammation and neuronal apoptosis in HMC3 cells. HMC3 cells were transfected with pc-NC, pc-KCNQ1OT1, si-NC or si-KCNQ1OT1, respectively, and HMC3 cells were treated with 1 μg/mL LPS for 24 h after transfection. (A) RT-qPCR was used to detect KCNQ1OT1 expression. (B) RT-qPCR was used to detect the expression level of TNF-α, IL-1β and IL-6 mRNA. (C) ELISA was used to detect TNF-α, IL-1β and IL-6 in the cell culture supernatant of HMC3 cells. (D and E) RT-qPCR was used to detect the expression level of CD86 and CD206. (F) MTT assay was used to detect the viability of HMC3 cells. (G) The neuronal injury was detected using LDH cytotoxicity detection kit. (H and I) The levels of NO and ROS were detected. All experiments were performed in triplicate. *P<0.05, **P<0.01, and ***P<0.001. Abbreviations: pc-NC, pc-DNA negative controls; pc-KCNQ1OT1, KCNQ1OT1 overexpression plasmid; si-NC, negative controls siRNA; si-KCNQ1OT1, KCNQ1OT1 siRNA; LPS, lipopolysaccharide; LDH, lactate dehydrogenase; ELISA, enzyme-linked immunosorbent assay; NO, nitric oxide; ROS, reactive oxygen species. |
MiR-30e-3p Was the Target of KCNQ1OT1
Additionally, expression level of miR-30e-3p in HMC3 cells transfected with pc-KCNQ1OT1 or si-KCNQ1OT1 was detected by RT-qPCR. In comparison with the control group, the expression of miR-30e-3p in cells transfected pc-KCNQ1OT1 was inhibited, while the expression of miR-30e-3p in the cells transfected with si-KCNQ1OT1 was significantly up-regulated (Figure 3A). Bioinformatics analysis showed that KCNQ1OT1 3ʹUTR contained a potential binding site for miR-30e-3p (Figure 3B). Dual-luciferase report assay showed that miR-30e-3p mimics was capable of markedly suppressing the luciferase activity of KCNQ1OT1-MUT reporter, but showed no remarkable effect on the luciferase activity of KCNQ1OT1-WT reporter (Figure 3C). These data suggested that miR-30e-3p was a target gene of KCNQ1OT1, which was capable of negatively regulating the expression of miR-30e-3p in microglia.
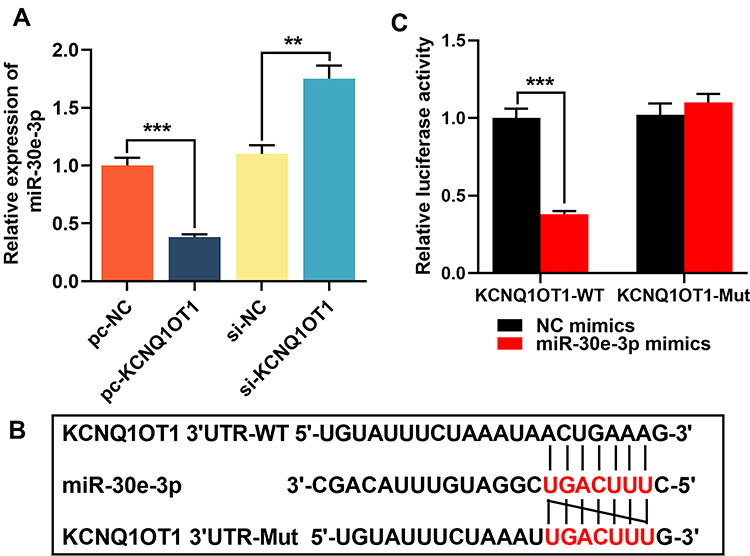 |
Figure 3 miR-30e-3p was the target of KCNQ1OT1. (A) Lncbase database was used to predict the binding site for miR-30e-3p in KCNQ1OT1 sequence. (B) HMC3 cells were co-transfected with miR-30e-3p mimics or control mimics and luciferase reporter vector KCNQ1OT1-WT or KCNQ1OT1-MUT, and luciferase activity of the cells in different group was detected after 48 h. (C) RT-qPCR was used to detect the expression of miR-30e-3p in HMC3 cells transfected with pc-KCNQ1OT1 or si-KCNQ1OT1. All experiments were performed in triplicate. **P<0.01 and *** P <0.001. Abbreviations: pc-NC, pc-DNA negative controls; pc-KCNQ1OT1, KCNQ1OT1 overexpression plasmid; si-NC, negative controls siRNA; si-KCNQ1OT1, KCNQ1OT1 siRNA; KCNQ1OT1-WT, KCNQ1OT1 wild-type reporter vector; KCNQ1OT1-MUT mutant-type reporter vector. |
KCNQ1OT1 Counteracted the Inhibitory Effects of miR-30e-3p on LPS-Induced Neuroinflammation and Neuronal Apoptosis in HMC3 Cells
To validate whether KCNQ1OT1 exhibited an effect in neuroinflammation and neuronal apoptosis through regulating miR-30e-3p, miR-30e-3p mimics was transfected into HMC3 cells with KCNQ1OT1 overexpression. RT-qPCR showed that the transfection of miR-30e-3p mimics remarkably increased the expression of miR-30e-3p, and had no effect on the expression of miR-30e-5p (Figure 4A; Supplementary Figure 1). In addition, the overexpression of KCNQ1OT1 remarkably suppressed the up-regulation of miR-30e-3p (Figure 4A). After LPS treatment, RT-qPCR and ELISA indicated that overexpression of miR-30e-3p markedly suppressed the expression of TNF-α, IL-1β, IL-6 and CD86 but increased CD206 expression; but the inhibitory effects of miR-30e-3p on LPS-induced neuroinflammation of HMC3 cells was remarkably reversed by overexpression of KCNQ1OT1 (Figure 4B-E). Furthermore, MTT and LDH assays displayed that overexpression of miR-30e-3p remarkably facilitated cell viability, decreased LDH release, reduced the generation of NO and ROS; and these effects were reversed by overexpression of KCNQ1OT1 (Figure 4F-I). These results indicated that KCNQ1OT1 promoted the activation of microglia, neuroinflammation and neuronal apoptosis in HMC3 cells by negatively regulating the expression of miR-30e-3p.
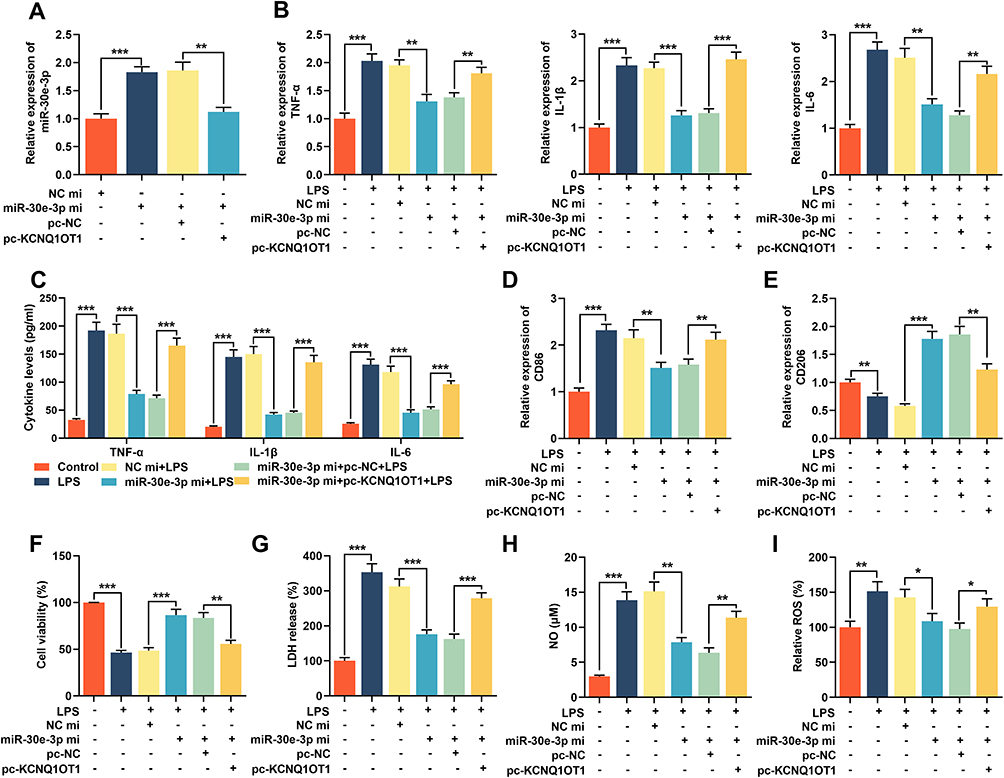 |
Figure 4 KCNQ1OT1 reversed the inhibitory effect of miR-30e-3p on LPS-induced neuroinflammation and neuronal apoptosis in HMC3 cells. MiR-NC, miR-30e-3p mimics, miR-30e-3p mimics + pc-NC or miR-30e-3p mimics + pc-KCNQ1OT1 were transfected into HMC3 cells, respectively. (A) The expression level of miR-30e-3p was detected by RT-qPCR. (B) RT-qPCR was used to detect the expression level of TNF-α, IL-1β and IL-6 mRNA. (C) ELISA was used to detect TNF-α, IL-1β and IL-6 in the cell culture supernatant of HMC3 cells. (D and E) RT-qPCR was used to detect the expression level of CD86 and CD206. (F) MTT assay was used to detect the viability of HMC3 cells. (G) The neuronal injury was detected using LDH cytotoxicity detection kit. (H and I) The levels of NO and ROS were detected. All experiments were performed in triplicate. *P<0.05, **P<0.01 and ***P<0.001. Abbreviations: miR-NC, miRNA negative controls; miR-30e-3p mi, miR-30e-3p mimics; pc-NC, pc-DNA negative controls; pc-KCNQ1OT1, KCNQ1OT1 overexpression plasmid; LPS, lipopolysaccharide; LDH, lactate dehydrogenase; ELISA, enzyme-linked immunosorbent assay; NO, nitric oxide; ROS, reactive oxygen species. |
KCNQ1OT1 Indirectly Promoted NLRP3 Expression by Inhibiting miR-30e-3p Expression
Bioinformatics analysis indicated that the 3ʹUTR of NLRP3 had a binding site for miR-30e-3p (Figure 5A). Dual-luciferase reporter gene experiment verified that miR-30e-3p could repress the luciferase activity of NLRP3-WT reporter, but showed no impact on the luciferase activity of NLRP3-MUT reporter (Figure 5B). In addition, RT-qPCR and Western blot indicated that LPS stimulation promoted the expression levels of NLRP3 mRNA and protein. Overexpression of miR-30e-3p markedly repressed the expression levels of NLRP3 mRNA and protein, while overexpression of KCNQ1OT1 significantly reduced the inhibitory effect of overexpression of miR-30e-3p on NLRP3 expression (Figure 5C and D), indicating that KCNQ1OT1 indirectly promoted NLRP3 expression by inhibiting miR-30e-3p expression.
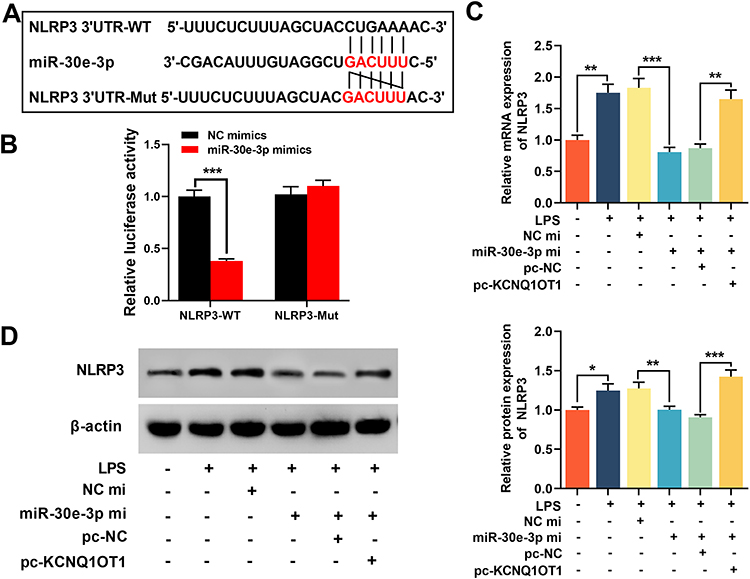 |
Figure 5 KCNQ1OT1 indirectly promoted NLRP3 expression by inhibiting miR-30e-3p expression. (A) miRanda database was used to predict the binding site between miR-30e-3p and NLRP3 3ʹUTR sequence. (B) HMC3 cells were co-transfected with miR-30e-3p mimics or control mimics and luciferase reporter NLRP3-WT or NLRP3-MUT, and the luciferase activity of the cells in different groups was detected after 48 h. (C and D) RT-qPCR and Western blot were employed to detect the expression level of NLRP3 mRNA and protein in HMC3 cells after KCNQ1OT1 and miR-30e-3p were selectively regulated. All experiments were performed in triplicate. *P<0.05, **P<0.01 and ***P<0.001. Abbreviations: pc-NC, pc-DNA negative controls; pc-KCNQ1OT1, KCNQ1OT1 overexpression plasmid; miR-NC, miRNA negative controls; miR-30e-3p mi, miR-30e-3p mimics; NLRP3-WT, NLRP3 3ʹUTR wild-type reporter vector; NLRP3-MUT, NLRP3 3ʹUTR mutant-type reporter vector; LPS, lipopolysaccharide. |
Overexpression of NLRP3 Reversed the Inhibitory Effect of Knocking Down KCNQ1OT1 on LPS-Induced Neuroinflammation and Neuronal Apoptosis in HMC3 Cells
To further elucidate the role of NLRP3 in neuroinflammation and neuronal apoptosis mediated by KCNQ1OT1, we transfected pc-NLRP3 into HMC3 cells transfected with si-KCNQ1OT1. RT-qPCR and Western blot manifested that the expression of NLRP3 was inhibited in cells transfected with si-KCNQ1OT1, and pc-NLRP3 restored the expression of NLRP3 mRNA and protein (Figure 6A and B). The expression levels of TNF-α, IL-1β, IL-6, CD86 and CD206 were analysed by RT-qPCR and/or ELISA, the results of which showed that NLRP3 overexpression significantly promoted the expressions of inflammatory cytokines TNF-α, IL-1β, IL-6 and CD86, inhibited CD206 expression compared with cells transfected with si-KCNQ1OT1 (Figure 6C-F). In addition, the overexpression of NLRP3 could markedly inhibit cell viability, increase the release of LDH and generation of NO and ROS (Figure 6E and F). These findings implied that overexpression of NLRP3 remarkably reduced the inhibitory effect of knocking down KCNQ1OT1 on LPS-induced activation of microglia, neuroinflammation and neuronal apoptosis.
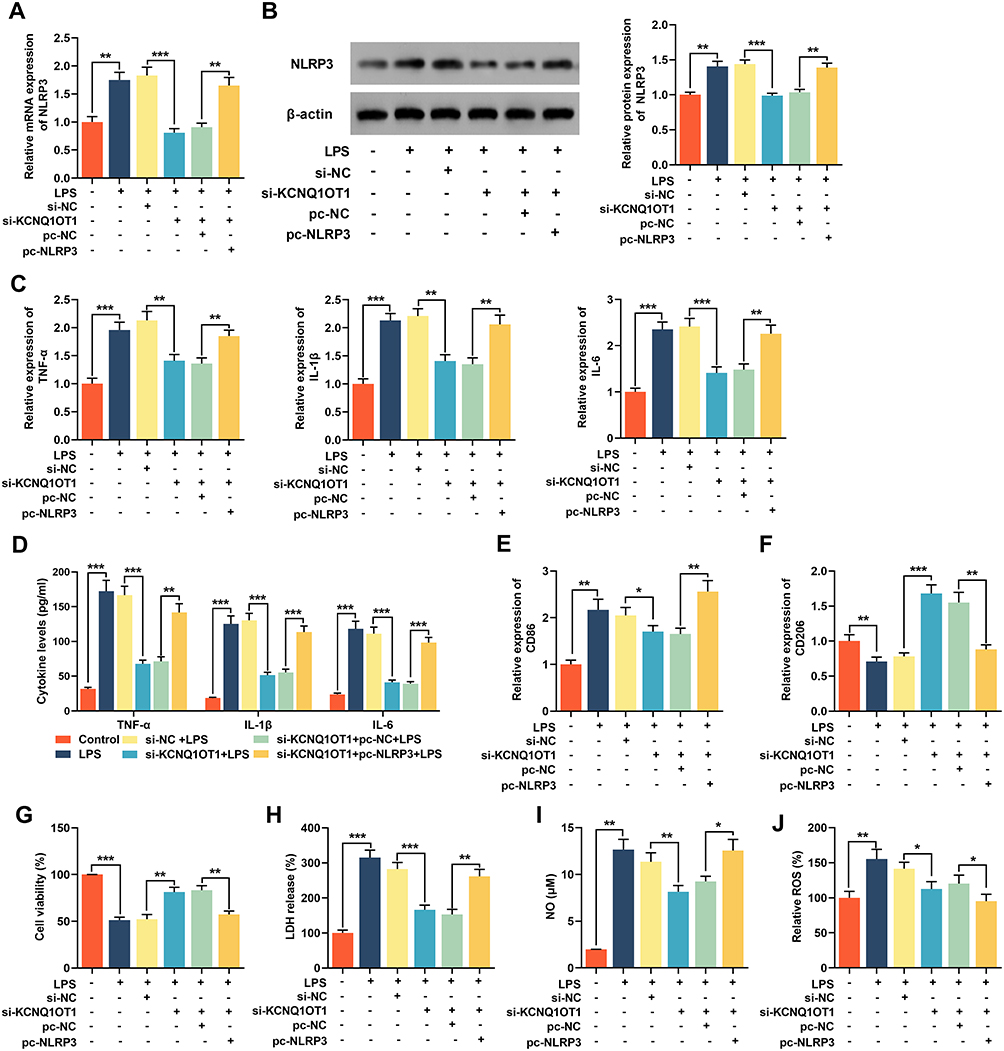 |
Figure 6 Overexpression of NLRP3 reversed the inhibitory effect of knocking down KCNQ1OT1 on LPS-induced neuroinflammation and neuronal apoptosis in HMC3 cells. The HMC3 cells were transfected with si-NC, si-KCNQ1OT1, si-KCNQ1OT1+pc-NC or si-KCNQ1OT1+pc-NLRP3, respectively. After 48 h of transfection, HMC3 cells were treated with 1 μg/mL LPS for 24 h. (A and B) The expression level of NLRP 3 was detected using RT-qPCR and Western blot. (C) RT-qPCR was used to detect the expression level of TNF-α, IL-1β and IL-6 mRNA. (D) ELISA was used to detect TNF-α, IL-1β and IL-6 in cell culture supernatant of HMC3 cells. (E and F) RT-qPCR was used to detect the expression level of CD86 and CD206. (G) MTT assay was used to detect the viability of HMC3 cells. (H) The neuronal injury was detected using LDH cytotoxicity detection kit. (I and J) The levels of NO and ROS were detected. All experiments were performed in triplicate. *P<0.05, **P<0.01 and ***P<0.001. Abbreviations: si-NC, negative controls siRNA; si-KCNQ1OT1, KCNQ1OT1 siRNA; pc-NC, pc-DNA negative controls; pc-NLRP3, NLRP3 overexpression plasmid; LPS, lipopolysaccharide; LDH, lactate dehydrogenase; ELISA, enzyme-linked immunosorbent assay; NO, nitric oxide; ROS, reactive oxygen species. |
Discussion
Increasing evidence indicates that the dysregulation of lncRNAs is associated with the pathogenesis of neurological diseases, including AD, PD, traumatic brain injury, stroke and so on.31–38 For example, lncRNA MALAT1 regulates neuronal apoptosis in ischemic stroke by sponging miR-205-3p and up-regulating PTEN expression.33 In addition, up-regulation of SNHG1 significantly increases LPS-induced activation of BV2 microglia.37 Lnc-p21 regulates MPP+ induced neuronal damage by regulating miR-625/TRPM2 axis in SH-SY5Y cells.38 In this study, it was demonstrated that KCNQ1OT1 was significantly up-regulated in LPS-induced HMC3 cells; inhibition of KCNQ1OT1 expression could significantly repress the expression of inflammatory cytokines, improve cell viability, reduce LDH release and the generation of NO and ROS, while overexpression of KCNQ1OT1 had opposite effects. Considering that up-regulation of pro-inflammatory cytokines is a marker of microglia activation,39 our results indicate that KCNQ1OT1 facilitates the activation of microglia, contributing to inflammatory response, and further resulting in the reduction of neurons’ viability. Our data suggest that KCNQ1OT1 is an injurious factor in the pathogenesis of neurological diseases, which is consistent with the previous reports.21,22
In this work, we also observed that the expression of miR-30e-3p was down-regulated in LPS-induced HMC3 cells. Furthermore, bioinformatics analysis, luciferase reporter gene assay and RT-qPCR confirmed that KCNQ1OT1 targeted miR-30e-3p and inhibited miR-30e-3p expression. Previously, accumulating studies have reported that the alterations of miRNAs are related to the pathogenesis of neurological diseases.40–43 Taking PD as an example, it is reported that miR-124 inhibits neuroinflammation in PD development by regulating MEK3/NF-κ B signaling pathway.41 In addition, miR-124 can also protect DA neurons by regulating AMPK/mTOR pathway.42 Besides, miR-155 has been proved to be closely related to α-syn-induced neuroinflammation and neurodegeneration.43 In the present work, the role of miR-30e-3p in neuroinflammation and neuronal viability was investigated. It was observed that overexpression of miR-30e-3p remarkably reduced the expressions of LPS-induced inflammatory cytokines TNF-α, IL-1β and IL-6 in HMC3 cells, promoting cell viability and reducing LDH release; further, the up-regulation of KCNQ1OT1 significantly reversed the inhibitory effect of miR-30e-3p on inflammation and neuronal apoptosis. These demonstrations imply that miR-30e-3p may be a protective factor in neurological diseases, and KCNQ1OT1 aggravates neuroinflammation and neuronal apoptosis via repressing miR-30e-3p.
NLRP3 exerts a crucial role in human immunity and pathogenesis of various inflammatory diseases.44 Recent studies have reported that NLRP3 inflammasome participates in neuroinflammation and neuronal apoptosis.14,45,46 Reportedly, activation of NLRP3 inflammasome can regulate astrocyte-mediated neuroinflammation.46 Previous data indicate that miR-7 targets NLRP3 to regulate neuroinflammation in PD.26,37 In MPTP-induced PD mouse model, miR-190 attenuates neuronal injury and inhibits inflammation by negatively regulating the expression and activation of NLRP3.47 It was reported that miR-223 targets NLRP3 to relieve inflammation and alleviate spinal cord injury.48 MiR-30a regulates retinal microglial activation and inflammation by targeting NLRP3.49 In addition, miR-30e-3p can ameliorate neuroinflammation in PD model by reducing the activity of NLRP3 inflammasome.30 In the present work, we further verified that NLRP3 was the target gene of miR-30e-3p through bioinformatics and luciferase reporter gene assay, and overexpression of miR-30e-3p remarkably suppressed the expression of NLRP3. Additionally, we demonstrated that overexpression of NLRP3 significantly promoted the expression of inflammatory cytokines TNF-α, IL-1β and IL-6, reduced cell viability, increased LDH release, and reversed the effects of KCNQ1OT1 knockdown on LPS-induced HMC3 cells. These results indicated that KCNQ1OT1 promoted LPS-induced the activation of microglia, neuroinflammation and neuronal apoptosis in HMC3 cells by promoting the expression of NLRP3 by sponging miR-30e-3p.
Collectively, we report that the expression of KCNQ1OT1 is up-regulated in LPS-treated HMC3 cells, and inhibition of KCNQ1OT1 expression reduces LPS-induced neuroinflammation and neuronal damage by regulating miR-30e-3p/NLRP3 axis in HMC3 cells. These findings help clarify the mechanism of microglia activation, neuroinflammation and neuronal injury in neurological diseases, providing clues for the treatment of these diseases. Nonetheless, our conclusions require further validation with in vivo models.
Data Sharing Statement
The data used to support the findings of this study are available from the corresponding author upon request.
Ethics Statement
Our study was approved by the Ethics Review Board of The First Affiliated Hospital of Hebei North University.
Acknowledgment
We thank Hubei Yican Health Industry Co., Ltd for its linguistic assistance during the preparation of this manuscript.
Disclosure
The authors declare that they have no competing interests.
References
1. Vezzani B, Carinci M, Patergnani S, et al. The dichotomous role of inflammation in the CNS: a mitochondrial point of view. Biomolecules. 2020;10:1437.
2. The L. Parkinson’s disease: a complex disease revisited. Lancet (London, England). 2017;390(10093):430.
3. Kalia LV, Lang AE. Parkinson’s disease. Lancet (London, England). 2015;386(9996):896–912.
4. Appel SH. Inflammation in Parkinson’s disease: cause or consequence? Movement Disorders. 2012;27(9):1075–1077.
5. Kaur K, Gill JS, Bansal PK, Deshmukh R. Neuroinflammation - A major cause for striatal dopaminergic degeneration in Parkinson’s disease. J Neurol Sci. 2017;381:308–314.
6. Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol. 2017;35:441–468.
7. Tang Y, Le, Differential W. Roles of M1 and M2 microglia in neurodegenerative diseases. Mol Neurobiol. 2016;53(2):1181–1194.
8. Koprich JB, Reske-Nielsen C, Mithal P, Isacson O. Neuroinflammation mediated by IL-1beta increases susceptibility of dopamine neurons to degeneration in an animal model of Parkinson’s disease. J Neuroinflammation. 2008;5:8.
9. Mogi M, Harada M, Kondo T, et al. Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci Lett. 1994;180(2):147–150.
10. Stojkovska I, Wagner BM, Morrison BE. Parkinson’s disease and enhanced inflammatory response. Exp Biol Med. 2015;240(11):1387–1395.
11. Heneka MT, Kummer MP, Stutz A, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493(7434):674–678.
12. Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nat Rev Immunol. 2014;14(7):463–477.
13. Lee HJ, Bae EJ, Lee SJ. Extracellular alpha–synuclein-a novel and crucial factor in Lewy body diseases. Nat Rev Neurol. 2014;10(2):92–98.
14. Wen L, Zhang QS, Heng Y, et al. NLRP3 inflammasome activation in the thymus of MPTP-induced Parkinsonian mouse model. Toxicol Lett. 2018;288:1–8.
15. Quan Z, Zheng D, Qing H. Regulatory roles of long non-coding RNAs in the central nervous system and associated neurodegenerative diseases. Front Cell Neurosci. 2017;11:175.
16. Mortezaei Z, Lanjanian H, Masoudi-Nejad A. Candidate novel long noncoding RNAs, MicroRNAs and putative drugs for Parkinson’s disease using a robust and efficient genome-wide association study. Genomics. 2017;109(3–4):158–164.
17. Lu M, Sun WL, Shen J, et al. LncRNA-UCA1 promotes PD development by upregulating SNCA. Eur Rev Med Pharmacol Sci. 2018;22(22):7908–7915.
18. Wang S, Zhang X, Guo Y, Rong H, Liu T. The long noncoding RNA HOTAIR promotes Parkinson’s disease by upregulating LRRK2 expression. Oncotarget. 2017;8(15):24449–24456.
19. Wang J, Zhang H, Situ J, Li M, Sun H. KCNQ1OT1 aggravates cell proliferation and migration in bladder cancer through modulating miR-145-5p/PCBP2 axis. Cancer Cell Int. 2019;19:325.
20. Rong J, Pan H, He J, et al. Long non-coding RNA KCNQ1OT1/microRNA-204-5p/LGALS3 axis regulates myocardial ischemia/reperfusion injury in mice. Cell Signal. 2019;66:109441.
21. Wang HJ, Tang XL, Huang G, et al. Long non-coding KCNQ1OT1 promotes oxygen-glucose-deprivation/reoxygenation-induced neurons injury through regulating MIR-153-3p/FOXO3 axis. J Stroke Cerebrovasc Dis. 2020;29:105126.
22. Ren Y, Gao XP, Liang H, Zhang H, Hu CY. LncRNA KCNQ1OT1 contributes to oxygen-glucose-deprivation/reoxygenation-induced injury via sponging miR-9 in cultured neurons to regulate MMP8. Exp Mol Pathol. 2020;112:104356.
23. Liu HT, Gao P. The roles of microRNAs related with progression and metastasis in human cancers. Tumour Biol. 2016.
24. Basak I, Patil KS, Alves G, Larsen JP, Moller SG. microRNAs as neuroregulators, biomarkers and therapeutic agents in neurodegenerative diseases. Cell Mol Life Sci. 2016;73(4):811–827.
25. Junn E, Lee KW, Jeong BS, Chan TW, Im JY, Mouradian MM. Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc Natl Acad Sci U S A. 2009;106(31):13052–13057.
26. Zhou Y, Lu M, Du RH, et al. MicroRNA-7 targets Nod-like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson’s disease. Mol Neurodegener. 2016;11:28.
27. Geng L, Liu W, Chen Y. miR-124-3p attenuates MPP(+)-induced neuronal injury by targeting STAT3 in SH-SY5Y cells. Exp Biol Med. 2017;242(18):1757–1764.
28. Wang D, Zhu C, Zhang Y, et al. MicroRNA-30e-3p inhibits cell invasion and migration in clear cell renal cell carcinoma by targeting Snail1. Oncol Lett. 2017;13(4):2053–2058.
29. Wang XT, Wu XD, Lu YX, et al. Potential involvement of MiR-30e-3p in myocardial injury induced by coronary microembolization via autophagy activation. Cell Physiol Biochem. 2017;44(5):1995–2004.
30. Li D, Yang H, Ma J, Luo S, Chen S, Gu Q. MicroRNA-30e regulates neuroinflammation in MPTP model of Parkinson’s disease by targeting Nlrp3. Hum Cell. 2018;31(2):106–115.
31. Chanda K, Mukhopadhyay D. LncRNA Xist, X-chromosome Instability and Alzheimer’s Disease. Curr Alzheimer Res. 2020;17:499–507.
32. Yang L, Yang L, Zhu J, Chen J, Wang Y, Xiong K. Expression signatures of long non-coding RNA and mRNA in human traumatic brain injury. Neural Regen Res. 2019;14:632–641.
33. Gao Q, Wang Y. Long noncoding RNA MALAT1 regulates apoptosis in ischemic stroke by sponging miR-205-3p and modulating PTEN expression. Am J Transl Res. 2020;12:2738–2748.
34. Carrieri C, Forrest AR, Santoro C, et al. Expression analysis of the long non-coding RNA antisense to Uchl1 (AS Uchl1) during dopaminergic cells’ differentiation in vitro and in neurochemical models of Parkinson’s disease. Front Cell Neurosci. 2015;9:114.
35. Liu Y, Lu Z. Long non-coding RNA NEAT1 mediates the toxic of Parkinson’s disease induced by MPTP/MPP+ via regulation of gene expression. Clin Exp Pharmacol Physiol. 2018;45(8):841–848.
36. Ni Y, Huang H, Chen Y, Cao M, Zhou H, Zhang Y. Investigation of long non-coding RNA expression profiles in the substantia nigra of Parkinson’s disease. Cell Mol Neurobiol. 2017;37(2):329–338.
37. Cao B, Wang T, Qu Q, Kang T, Yang Q. Long noncoding RNA SNHG1 promotes neuroinflammation in parkinson’s disease via regulating miR-7/NLRP3 pathway. Neuroscience. 2018;388:118–127.
38. Ding XM, Zhao LJ, Qiao HY, Wu SL, Wang XH. Long non-coding RNA-p21 regulates MPP(+)-induced neuronal injury by targeting miR-625 and derepressing TRPM2 in SH-SY5Y cells. Chem Biol Interact. 2019;307:73–81.
39. Wang YL, Han QQ, Gong WQ, et al. Microglial activation mediates chronic mild stress-induced depressive- and anxiety-like behavior in adult rats. J Neuroinflammation. 2018;15(1):21.
40. Singh A, Sen D. MicroRNAs in Parkinson’s disease. Exp Brain Res. 2017;235(8):2359–2374.
41. Yao L, Ye Y, Mao H, et al. MicroRNA-124 regulates the expression of MEKK3 in the inflammatory pathogenesis of Parkinson’s disease. J Neuroinflammation. 2018;15(1):13.
42. Gong X, Wang H, Ye Y, et al. miR-124 regulates cell apoptosis and autophagy in dopaminergic neurons and protects them by regulating AMPK/mTOR pathway in Parkinson’s disease. Am J Transl Res. 2016;8(5):2127–2137.
43. Thome AD, Harms AS, Volpicelli-Daley LA, Standaert DG. microRNA-155 regulates alpha-synuclein-induced inflammatory responses in models of Parkinson's disease. J Neurosci. 2016;36(8):2383–2390.
44. He Y, Hara H, Nunez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. 2016;41(12):1012–1021.
45. Mao Z, Liu C, Ji S, et al. The NLRP3 Inflammasome is involved in the pathogenesis of Parkinson’s Disease in rats. Neurochem Res. 2017;42(4):1104–1115.
46. Qiao C, Yin N, Gu HY, et al. Atp13a2 deficiency aggravates astrocyte-mediated neuroinflammation via NLRP3 inflammasome activation. CNS Neurosci Ther. 2016;22(6):451–460.
47. Sun Q, Wang S, Chen J, et al. MicroRNA-190 alleviates neuronal damage and inhibits neuroinflammation via Nlrp3 in MPTP-induced Parkinson’s disease mouse model. J Cell Physiol. 2019;234(12):23379–23387.
48. Zhang M, Wang L, Huang S, He X. MicroRNA-223 targets NLRP3 to relieve inflammation and alleviate spinal cord injury. Life Sci. 2020;254:117796.
49. Dong N, Wang Y. MiR-30a regulates S100A12-induced retinal microglial activation and inflammation by targeting NLRP3. Curr Eye Res. 2019;44(11):1236–1243.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.