")
Back to Journals » Drug Design, Development and Therapy » Volume 15
Inhibition of Lipopolysaccharide-Induced Inflammatory Bone Loss by Saikosaponin D is Associated with Regulation of the RANKL/RANK Pathway
Authors Wu X, Zhao K, Fang X, Lu F , Zhang W, Song X, Chen L , Sun J, Chen H
Received 20 August 2021
Accepted for publication 5 November 2021
Published 23 November 2021 Volume 2021:15 Pages 4741—4757
DOI https://doi.org/10.2147/DDDT.S334421
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Professor Anastasios Lymperopoulos
Xinhui Wu,1,2,* Kangxian Zhao,1,2,* Xiaoxin Fang,3,4 Feng Lu,3,4 Weikang Zhang,1,2 Xiaoting Song,1,2 Lihua Chen,5 Jiacheng Sun,1,2 Haixiao Chen1,2,4
1Wenzhou Medical University, Wenzhou, People’s Republic of China; 2Department of Orthopedics, Taizhou Hospital of Zhejiang Province Affiliated to Wenzhou Medical University, Linhai, People’s Republic of China; 3Zhejiang University School of Medicine, Hangzhou, People’s Republic of China; 4Taizhou Hospital of Zhejiang Province, Zhejiang University, Linhai, People’s Republic of China; 5Enze Medical Research Center, Taizhou Hospital of Zhejiang Province Affiliated to Wenzhou Medical University, Linhai, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Haixiao Chen
Department of Orthopedics, Taizhou Hospital of Zhejiang Province Affiliated to Wenzhou Medical University, Linhai, 317000, People’s Republic of China
Email [email protected]
Background: Osteolytic diseases such as osteoporosis are featured with accelerated osteoclast differentiation and strong bone resorption. Considering the complications and other limitations of current drug treatments, it is necessary to develop a safer and more reliable drug to deal with osteoclast-related diseases. Saikosaponin D (SSD) is the active extract of Bupleurum, which has anti-inflammation, anti-tumor and liver protection functions. However, the role of SSD in regulating the differentiation and function of osteoclasts is not clear.
Purpose: To explore whether SSD could prevent osteoclast differentiation and bone resorption induced by M-CSF and RANKL, and further evaluate the potential therapeutic properties of SSD in LPS-induced inflammatory bone loss mouse models.
Methods: BMMs were cultured in complete medium stimulated by RANKL with different concentrations of SSD. TRAP staining, bone resorption determination, qRT-PCR, immunofluorescence and Western blotting were performed. A mouse model of LPS-induced calvarial bone loss was established and treated with different doses of SSD. The excised calvaria bones were used for TRAP staining, micro-CT scan and histological analysis.
Results: SSD inhibited the formation and bone resorption of osteoclasts induced by RANKL in vitro. SSD suppressed LPS-induced inflammatory bone loss in vivo.
Conclusion: SSD inhibited osteoclastogenesis and LPS-induced osteolysis in mice both which served as a new potential agent for the treatment of osteoclast-related conditions.
Keywords: saikosaponin D, osteoclastogenesis, NF-κB, MAPKs, PI3K-AKT, therapy
Introduction
Osteoclasts, a type of giant multinucleated cells are derived from the macrophage/mononuclear lineage in the bone marrow.1 Bone homeostasis involves sustaining a balance between bone formation of osteoblasts and bone resorption of osteoclasts.2,3 Excessive osteoclast activity can lead to osteoporosis.4 Bone abnormalities caused by postmenopausal osteoporosis, rheumatoid arthritis, periodontitis and other bone diseases are closely related to the overactivity of osteoclasts.5 The differentiation and formation of osteoclast precursor cells are governed by two important factors, namely, receptor activator of nuclear factor kappa B ligand (RANKL) and macrophage colony-stimulating factor (M-CSF).6 M-CSF is involved in the survival and division of bone marrow-derived mononuclear macrophages, whereas RANKL promotes the fusion of osteoclast precursor cells and differentiation into the lineage of osteoclasts.4 During differentiation, a unique ring is formed by the actin cytoskeleton at the contact location of bones to ensure the integrity of the osteoclast structure.7 RANKL secreted by osteoblasts belongs to the tumor necrosis factor (TNF) super family. Binding of RANKL with RANK on the surface of osteoclasts triggers the release of the endothelial cytokine TNF receptor-associated factor 6 (TRAF 6) in the cytoplasm that activates various signaling pathways in the precursor cells, including the mitogen-activated protein kinase (MAPK), phosphoinositide 3-kinase/AKT (PI3K/AKT) and nuclear factor kappa B (NF-κB) pathways.8,9 These pathways increase the expression levels of osteoclast-related genes such as NFATc1, Dcstamp, c-Fos, V-ATPase d2, MMP9 and Acp 5 and initiate osteoclast differentiation and bone resorption.10,11 Therefore, suppressing osteoclast activity by inhibiting the downstream pathways activated by RANKL is a feasible approach to prevent and treat osteoclastogenesis.
LPS (Lipopolysaccharide) is a pivotal constituent lying in the outer membrane of G- bacteria.12 LPS can stimulate the secretion of various cytokines, including IL (interleukin)-1β, TNF (tumor necrosis factor)-α, and other inflammatory factors, promoting the expression of RANKL, stimulating the resorption of bone and inhibiting bone formation.13,14 Therefore, inhibiting LPS-induced inflammatory bone loss is one of the treatment methods to combat osteoclast-related diseases.
In the recent years, several studies have shown that agents extracted from plants can ameliorate bone loss by inhibiting the formation of osteoclasts.15 Saikosaponin D (SSD) is a triterpene saponin derived from the dried root of Bupleurum falcatum, a plant that has been used for the treatment of liver diseases in China for thousands of years. In addition, SSD has anti-inflammatory and anti-tumor properties and decreases depressive behaviors. SSD improves dextran sulphate sodium-induced intestinal inflammation by inhibiting the activation of NF-κB.16 A previous study showed that SSD suppressed tumor growth and promoted apoptosis of pancreatic cancer cells by targeting the MAPK kinase 4 (MKK4)–Janus kinase (JNK) signaling pathway.17 A study by Chen et al showed that SSD alleviated acute liver injury in carbon tetrachloride (CCl4)-induced hepatitis by suppressing oxidative stress and activation of the NLRP3 inflammasome.18 Su etal demonstrated that SSD effectively improved lipopolysaccharide (LPS)-induced inflammation-related depressive behaviors.19 However, the function of SSD in lytic bone disease remains unclear. This study aimed to determine whether SSD could prevent osteoclast differentiation induced by M-CSF and RANKL and evaluate the potential therapeutic properties of SSD in mouse models of LPS-induced inflammatory bone loss.
Materials and Methods
Reagents and Media
SSD with >98% purity was obtained from MedChemExpress (NJ, USA). Dimethyl sulfoxide (DMSO) (Meilunbio, Dalian, China) was used to dissolve SSD at a concentration of 1 mmol/L and was stored at −20°C. The culture medium was used for further dilutions in the in vitro experiments, and phosphate-buffered saline (PBS) was used for in vivo experiments. Alpha-modified Eagle medium (α-MEM) was provided by HyClone Laboratories (Logan, UT, USA). Foetal bovine serum (FBS) was obtained from Gibco-BRL (Gaithersburg, USA). Cell counting kit-8 (CCK-8) was obtained from Beyotime (Shanghai, China). M‐CSF (mouse recombinant) and RANKL were provided by R&D Systems (Minneapolis, USA). Primary antibodies, including β‐actin (#4970), P65 (#8242), p-P65 (#3033), P38 (#8690), p-P38 (#4511S), AKT (#4691), p-AKT(#4060), ERK(#4695), p-ERK(#4370), IκBα(#4814), p-IκBα (#2859), PI3K(#4295), p-PI3K (#17366), JNK (#9252) and p-JNK(#9255), were provided by Cell Signaling Technology (1:1000, Danvers, USA). In addition, c-Fos(ab222699) and NFATc1(ab2722) were obtained from Abcam (1:1000, Cambridge, UK), and the dilution of the primary antibodies was purchased from Beyotime Biotechnology.
Bone Marrow-Derived Macrophage Culture
All C57BL/6 male mice (8 weeks old) were obtained from the Shanghai Laboratory Animal Center (SLAC). All animal experiments were approved by the Institutional Animal Ethics Committee of Taizhou Hospital. As previously described,20 we collected primary murine bone marrow-derived macrophages (BMMs) from the bone marrow of 8-week-old male C57BL/6 mice. The BMMs were cultured in 10-cm dishes with complete medium (30ng/mL M‐CSF + α-MEM + 1% penicillin/streptomycin + 10% FBS) in an incubator (5% CO2, 37°C). The complete medium was changed every other day until the cells reached a 90% confluency.
Determination of Cell Viability
The viability of BMMs was determined using CCK-8 assays. BMMs (density, 8000 cells/well) were seeded in triplicates in 96-well plates for a day. The cells then were incubated with multiple concentrations of SSD (0, 0.5, 1, 2, 4, 8 and 16 μmol/L) for 48 and 96 h. The CCK‐8 reagent (10%) in complete medium was added in 96-well plates and BMMs were incubated with CCK‐8 reagent for 3 h at 37°C. A microplate photometer (Thermo Fisher Scientific, Waltham, USA) was used to calculate the absorbance at 450 nm.
Differentiation of Osteoclasts in vitro
BMMs (8000 cells/well) were seeded in 96-well plates. The cells were incubated in complete medium supplemented with different concentrations of SSD (0, 0.5, 1 and 2 μmol/L) with or without RANKL (50 ng/mL). The culture medium was changed every other day for 7 days. Additionally, BMMs, which were incubated with RANKL and M-CSF, were treated with 2 μmol/L of SSD on days 1–3, 3–5 or 5–7. The cells stimulated by RANKL were regarded as a positive control group (control group) and untreated M-CSF-dependent cells were regarded as a negative control group (vehicle group). When multinucleated pancake‐shaped large osteoclasts were identified, the cells were fixed in 4% paraformaldehyde (PFA) for 30 min. PBS was used to wash the cells for 3 times and tartrate-resistant acid phosphatase staining (TRAP) staining was conducted as per the manufacturer’s instructions.21,22 Osteoclasts were identified as TRAP-positive cells with >3 nuclei. Pictures of the cells were taken using a microscope, and the area and number of TRAP-positive osteoclasts were measured using the ImageJ software.
Bone Resorption Determination
M-CSF-dependent BMMs (8000 cells/well) were seeded in bovine bone discs. The cells were cultured in complete medium supplemented with or without 50 ng/mL of RANKL for 5 days and were treated with or without various concentrations of SSD (0.5, 1 and 2 μmol/L) for another 3 days. Subsequently, sodium hypochlorite solution (6%) was used to remove the cells. Scanning electron microscopy (SEM) analysis was performed using the prepared bone discs. A firing error indicator (FEI) scanning electron microscope (Quanta 250; Thermo Fisher Scientific) was used to acquire SEM images. The area of bone resorption was measured using the ImageJ software.
Actin Ring Immunofluorescence
M-CSF dependent BMMs were seeded in 96-well plates at 8000 cells/well. The cells were incubated with multiple concentrations of SSD (0, 0.5, 1 and 2 μmol/L) with or without RANKL. The complete culture medium was changed every other day for 7 days. BMMs were fixed using PFA (4%) for 30 min and permeabilized using Triton X‐100 (0.1%) in PBS for 5 min. After the cells were washed thrice with PBS, F-actin staining kit (AmyJet Scientific, Wuhan, China) was used to stain the F-actin rings for 30 min at room temperature in the dark and the nucleus was stained using 4´,6-diamidino-2-phenylindole (DAPI).22 Images of the actin rings were observed using immunofluorescence microscopy (Zeiss, Japan), and the number of actin rings were counted using the ImageJ software.
Extraction of RNA and Subsequent Quantitative Reverse-Transcription Polymerase Chain Reaction Assay
M‐CSF dependent BMMs (density, 3×105 cells/well) were seeded in 6‐well plates. The cells were treated with multiple concentrations of SSD to determine the dose-dependent effect of SSD (0, 0.5, 1 and 2 μmol /L) on osteoclast formation. Additionally, M‐CSF dependent BMMs were treated with RANKL alone or RANKL plus SSD (2 μmol/L). The cells were treated for 1, 3, 5 or 7 days, respectively. TRIzol (Thermo Fisher Scientific) was used to extract total RNA from the cells according to the manufacturer’s instructions. Then the First Strand cDNA Synthesis Kit (RevertAid; Thermo Fisher Scientific) was used to synthesize cDNA. Quantitative reverse-transcription polymerase chain reaction (qRT-PCR) assay was conducted using the FastStart Universal SYBR Green Master (Roche, Indianapolis, IN, USA) and Real-Time PCR System (ABI 7500; Applied Biosystems, Foster City, CA, USA). The primer sequences of this study are shown in Table 1. β-actin was regarded as an internal control.
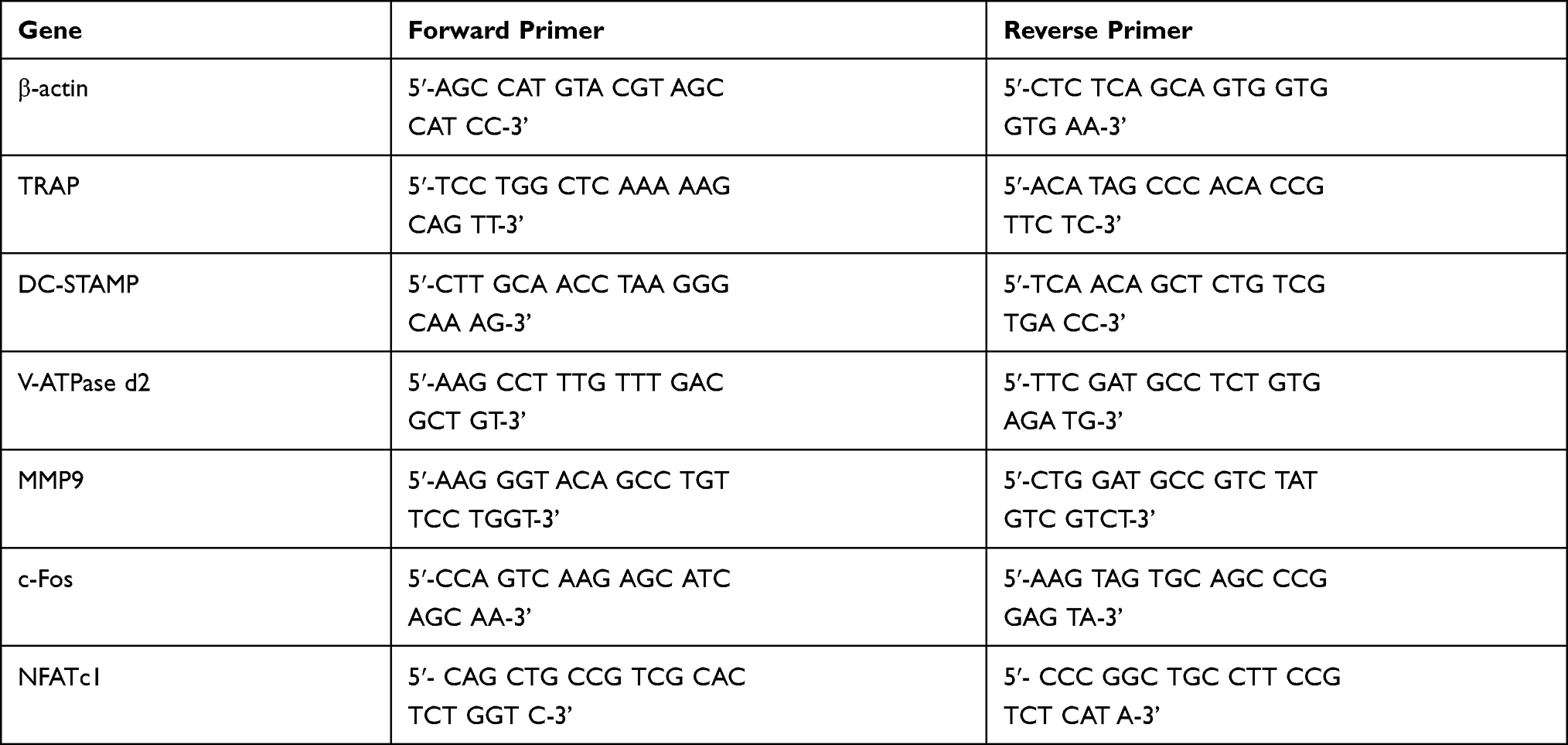 |
Table 1 Mouse Primers for qPCR |
Western Blotting Analysis
To analysis the early signaling pathways induced by RANKL, M-CSF dependent BMMs were cultured in 6‐well plates for 4 days (3 × 105 cells/well). Subsequently, the cells were incubated without or with various concentrations of SSD (0.5, 1 and 2 μmol/L) for 1 h and were stimulated by RANKL for 15 min. BMMs without any treatment were regarded as a negative control group, and BMMs treated with RANKL alone were regarded as a positive control group. For determining the late signaling pathways induced by RANKL, M-CSF-dependent BMMs treated with or without 2 μmol/L SSD were induced by RANKL for 1, 3 and 5 days, respectively. Total proteins were extracted from the cells by using radioimmunoprecipitation assay (RIPA) buffer (AMEKO, Shanghai, China) supplemented with phosphatase inhibitors and phenylmethanesulfonyl fluoride (PMSF). A refrigerated centrifuge was used to centrifuge the lysate at 12,000g for 15 min. The collected protein was separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). The separated proteins were transferred onto polyvinylidene difluoride (PVDF) membranes and were blocked using Immunol Staining Blocking Buffer (QuickBlock™; Beyotime) for 25 min under room temperature. Subsequently, the membranes were incubated with primary antibodies overnight at 4°C by rocking. The following day, the membranes were rinsed thrice with tris-buffered saline with Tween 20 (TBST) and were incubated with secondary antibodies (Beyotime) for 1 h at room temperature. Chemiluminescent detection substrates (Luminata; MilliporeSigma, MA, USA) were used to detect antibody reactivity according to the protocol. Image Quant LAS500 (GE Health Care, Chicago, IL, USA) was used to obtain images. Densitometric analyses were measured using the ImageJ software.
Nuclear Translocation of NF-κB P65
Whether SSD can influence the nuclear translocation of P65 after RANKL stimulation was determined as previously described.23 M-CSF dependent BMMs seeded in 12-well plates at a density of 4×105 cells/well were cultured in a complete medium for a day. The cells were incubated with 2μmol/L SSD for 1 h, followed by stimulation with RANKL for 30 min. The cells were fixed using PFA (4%) for 15 min, permeabilized using Triton X-100 (0.1%) in PBS and blocked with Immunol Staining Blocking Buffer (Beyotime). After washing extensively with PBS for 3 times, the cells were incubated with anti-p65 antibody at 4°C overnight. The following day, the cells were incubated with Alexa Fluor 488-labelled secondary antibody (Beyotime) at 37°C in the dark and washed with PBS. Finally, counterstaining of the nuclei was performed using DAPI for 5 min. Images of p65 nuclear translocation were observed using an immunofluorescence microscope.
A Mouse Model of LPS-Induced Bone Loss
To detect the role of SSD in osteolytic diseases, a mouse model of LPS-induced calvarial bone loss was conducted as previously described.16,24,25 The study protocol was approved by the Institutional Animal Ethics Committee of Taizhou Hospital (ethic code: tzy-2021048). We performed the animal experiments in accordance with National Institutes of Health Guide for Care and Use of Laboratory Animals (NIH Pub No 85–23, USA). We randomly assigned 24 8-week-old healthy C57/BL6 male mice to four groups (n = 6): sham (treated with PBS), LPS (treated with 5 mg/kg of LPS), low‐dose SSD (treated with 5 mg/kg of LPS and 2 mg/kg of SSD) and high‐dose SSD (treated with 5 mg/kg of LPS and 4 mg/kg of SSD). Anesthesia was induced with an intraperitoneal injection of pentobarbital (40 mg/kg); subsequently, LPS dissolved in PBS was subcutaneously injected into the middle cranial suture of mice in all groups, except the sham group. During a 21‐day period, LPS was subcutaneously injected every other day for 7 days. PBS and SSD were injected intraperitoneally a day before the injection of LPS and then injected every other day until the end of the experiment.
TRAP Staining of the Calvaria
The mice were sacrificed at the end of the experiment. We excised the calvarial bones and removed other tissues. Subsequently, we randomly selected 3 calvarial bones from each group, placed them in a 6-well plate and immersed them in the TRAP staining solution at room temperature for a day. We took pictures of these bones using a camera and compared the results of the staining around the middle suture of the calvaria as previously described.20 Finally, all bones were fixed with PFA (4%) for further micro-computed tomography (µCT) scanning and histological analysis.
μCT Scan
High-resolution μCT (μCT-100; SCANCO Medical AG, Switzerland) was used to examine the fixed bones. The scanning protocol was set as follows: isometric resolution at 20 µm, 200 μA and 80 kV. After three-dimensional (3D) images were reconstructed, a nummular region of interest (ROI; 3×3 × 1 mm) around the middle suture of the calvaria was selected to be used in further analysis.26 A software (SCANCO Medical AG) was used to analyze the CT images and measure the number of pores, area of pores in the ROI and bone volume to tissue volume (BV/TV, %).
Histomorphometric and Histological Analysis
After fixing, the calvarial bones were decalcified using 10% ethylenediaminetetraacetic acid (EDTA) for 3 weeks and then paraffin embedding was performed. As previously described,21,25,26 TRAP and H&E staining were performed for morphometric examination. A high-quality microscope (Olympus, Japan) was used to obtain the photographs of the sections. The BV/TV (%) and the number of TRAP-positive cells (number of osteoclasts on the bone surface) were measured using The Image ProPlus 6.0 software (Media Cybernetics, Rockville, MD, USA).
Statistical Analysis
Data were expressed as mean ± standard deviation (SD). All in vitro experiments were performed independently at least 3 times. There were 6 randomly assigned male mice in each group. Statistical analysis and calculation were conducted by Student’s t test or Kruskal Wallis test using the IBM SPSS Statistics for Windows, version 20.0 (IBM Corp, Armonk, NY, USA). The differences between groups were compared using one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test. P values <0.05 were considered statistically significant.
Results
SSD Attenuated RANKL-Induced Osteoclast Formation
The structure of SSD is shown in Figure 1A. The potential cytotoxicity of SSD to BMMs was detected using the CCK8 cell viability assay. BMMs were incubated with various concentrations of SSD (0, 0.5, 1, 2, 4, 8 and 16 μmol/L) for 48 and 96 h. The viability of BMMs remained normal when the concentration of SSD was below 2 μmol/L at both time points (Figure 1B). Subsequently, BMMs were cultured in osteoclast complete medium and treated with RANKL and 0, 0.5, 1 or 2 μmol/L SSD for 7 days to determine the effect of SSD on the formation of osteoclasts. On day 7, mature osteoclasts were identified by observing the morphology (large multinucleated pancake shape) and increased TRAP activity. The number and area of TRAP-positive osteoclasts markedly decreased after treatment with various concentrations of SSD (Figure 1C and D). In addition, we detected the stage at which SSD exerted its inhibitory effect. RANKL was used to continuously stimulate M-CSF dependent BMMs followed by treatment with 2 μmol/L of SSD from day 1 to 3 (early phase), day 3 to 5 (mid phase) or day 5 to 7 (late phase). SSD exerted significant inhibitory effects on osteoclast formation in the late stage (Figure 1E and F). Therefore, our results revealed that SSD strongly inhibited the differentiation and formation of osteoclasts.
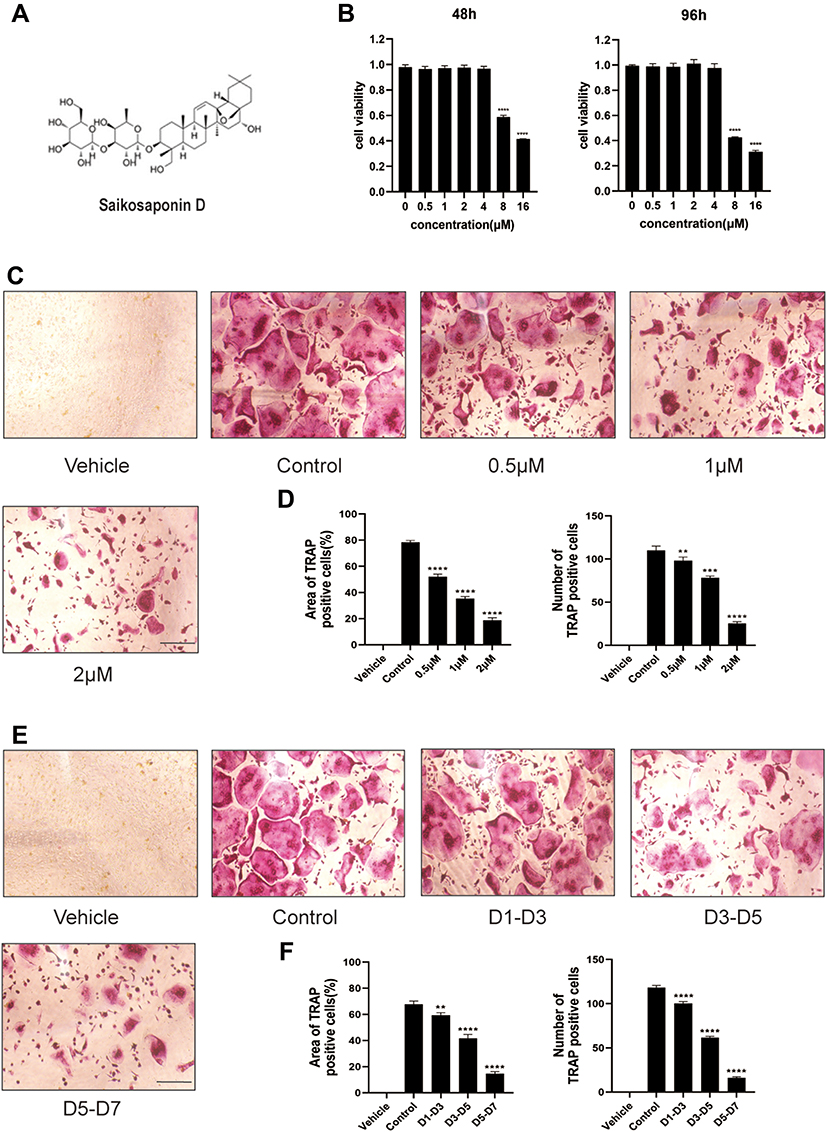 |
Figure 1 Saikosaponin D attenuated the formation of osteoclasts induced by RANKL. (A) Chemical structure of SSD. (B) Cell viability of BMMs (bone marrow-derived macrophages) was evaluated by CCK-8 at 48, 96h (OD 450nm). (C) The role of SSD in osteoclast formation in a dose-dependent manner. BMMs stimulated by RANKL (50ng/mL) were treated with or without various concentrations of SSD for 7 days. After fixing, the cells subjected to TRAP staining. (D) The number and area of TRAP-positive multinucleated cells (≥3 nuclei). (E) The effect of SSD on osteoclast formation in a time-dependent manner. BMMs which were stimulated by 50ng/mL RANKL and treated with 2μM SSD from day 1 to day 3, from day 3 to day 5, or from day 5 to day 7 were fixed with 4% PFA and subjected to TRAP staining. (F) The number and area of TRAP-positive multinucleated cells (≥3 nuclei). Original magnification, ×100. Scale bar, 200 μm. Mean ± SD is used to describe the data. Above experiments were conducted independently at least 3 times. ****P <0.0001, ***P<0.001, **P <0.01. |
SSD Attenuated F-Actin Formation and Bone Resorption
Our results demonstrated that SSD significantly inhibited the formation of multinucleated large osteoclasts (Figure 1C–F). Therefore, we investigated its role in bone resorption and actin ring formation. We examined the effects of SSD on the formation of F-actin rings. The actin rings were visible after BMMs stimulated by RANKL alone, and their size and number were analyzed. However, the formation of actin rings markedly decreased in a dose-dependent manner after treatment with different concentrations of SSD (Figure 2A and B). In addition, the area of bone resorption was remarkably reduced after treatment with different concentrations of SSD. There were few changes detected on the bovine bone slices when BMMs were treated with SSD at a concentration of 2 μmol/L (Figure 2C and D). Therefore, our results suggested that SSD suppressed the formation of F-actin rings and the function of bone resorption.
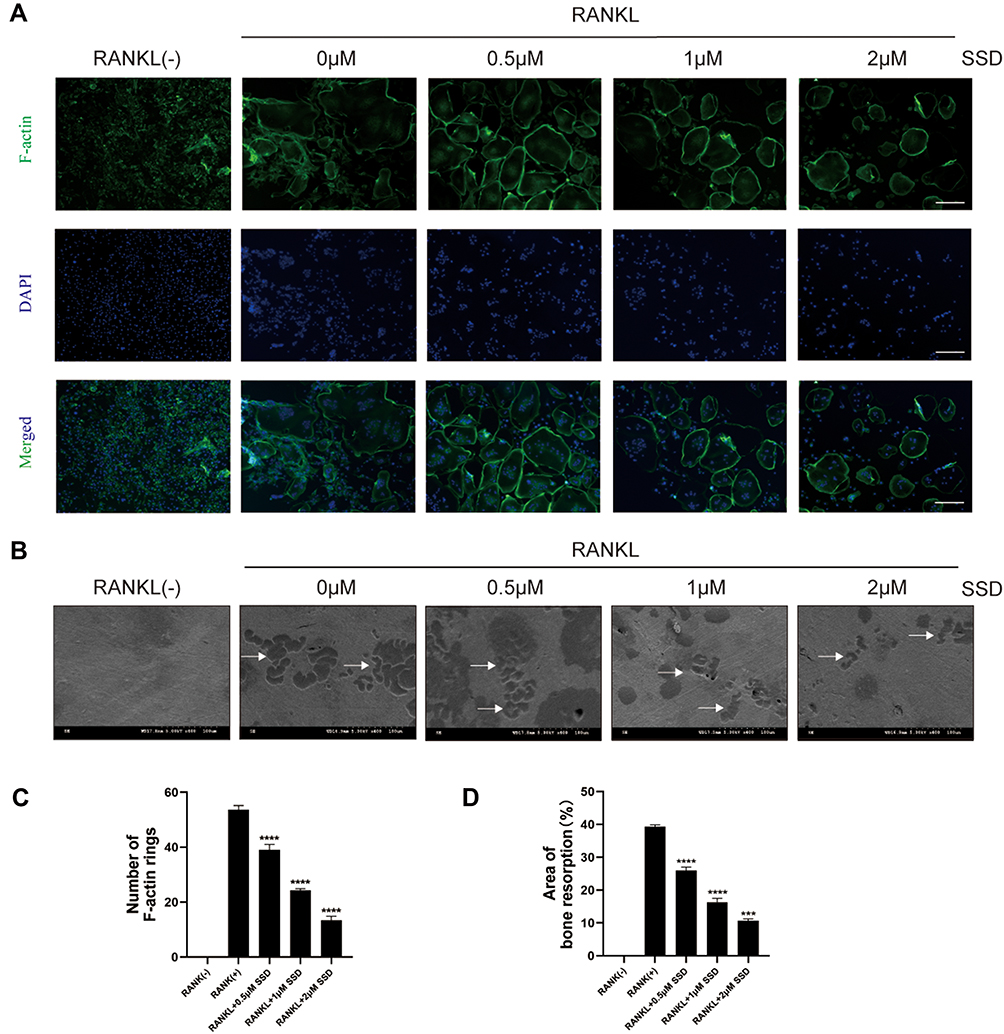 |
Figure 2 Saikosaponin D attenuated the formation of F-actin and bone resorption. (A) M-CSF-dependent BMMs stimulated by RANKL were treated with SSD of different concentrations. Mature osteoclasts were stained with F-actin Staining Kit and DAPI respectively. (B) The number of osteoclasts with F-actin rings. (C) An equal number of M-CSF-dependent BMMs stimulated by RANKL were seeded on bone slices in the presence of different concentrations of SSD. After 7 days, bone resorption lacunae were observed by SEM. Bone resorption pits were highlighted with white arrows. (D) The proportion of resorption pit areas. Original magnification, × 100. Scale bar, 200 μm. Mean ± SD is used to describe the data. Above experiments were conducted independently at least 3 times. ****P <0.0001, ***P<0.001. |
SSD Reduced the RANKL-Induced Expression Level of Osteoclast-Related Genes in vitro
To investigate the molecular mechanism underlying the inhibition of osteoclastogenesis by SSD, we determined the mRNA expression level of genes related to osteoclast formation (Dcstamp, NFATc1 and c-Fos), bone resorption (Acp 5 and V-ATPase d2) and inflammatory cytokines (MMP9). We found an obvious and time-dependent increase in the expression levels of these genes after treatment with M-CSF and RANKL. However, this increase was suppressed by treatment with SSD (Figure 3A). In addition, treatment with SSD decreased the expression levels of these genes in a dose-dependent manner (Figure 3B). Therefore, our results revealed that SSD effectively suppressed the expression level of osteoclast-related genes.
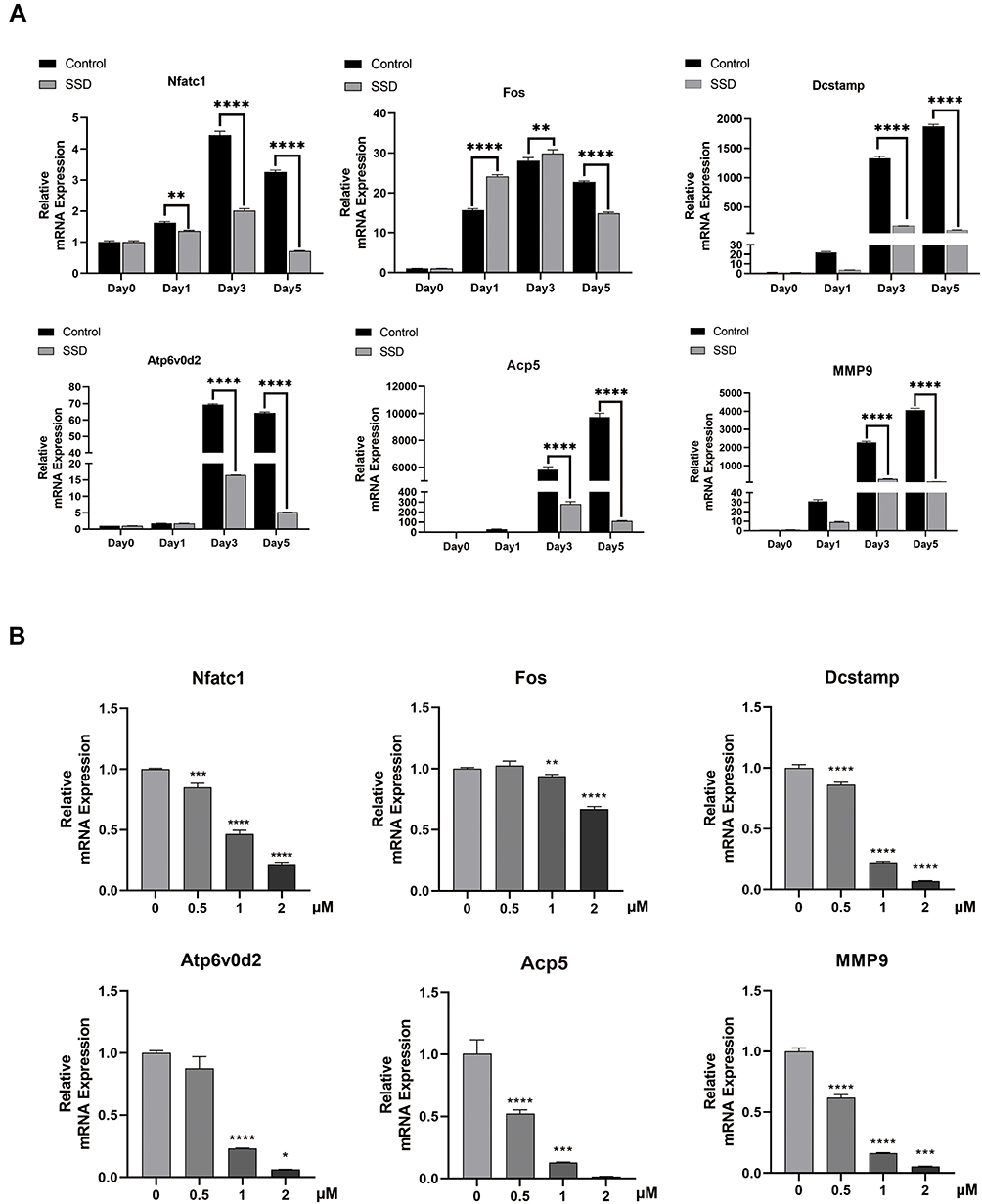 |
Figure 3 Saikosaponin D reduced the expression level of osteoclast-related genes induced by RANKL in vitro. (A) mRNA expression profile of OMG (osteoclast marker genes) (time-dependent). RT-qPCR (real-time quantitative PCR) was performed on total RNA extracted from cells stimulated by RANKL (50ng/mL) and treated with 2μM SSD for the indicated time points during the 5 day osteoclast culture. With β-actin as the reference, RANKL-induced related mRNA expressions of Dcstamp, MMP9, NFATc1, c-FOS, Atp6V0d2, and Acp5 were normalized. (B) mRNA expression profile of OMG (dose-dependent). RT-qPCR was performed on total RNA extracted from cells stimulated by RANKL (50ng/mL) and treated with various concentrations of SSD for 5 days. With β-actin as the reference, RANKL-induced related mRNA expressions of Dcstamp, MMP9, NFATc1, c-FOS, V-ATPase-d2, and Acp 5 were normalized. Mean ± SD is used to describe the data. Above experiments were conducted independently at least 3 times. ****P <0.0001, ***P<0.001, **P <0.01, *P <0.05. |
SSD Block Multiple Downstream Pathways Regulated by RANK/RANKL Pathway
Signaling pathways activated by RANKL in the early stages play a crucial role in osteoclast formation. After treating M-CSF dependent BMMs with RANKL, the most pivotal and the earliest activated signaling cascade was the NF-κB signaling pathway. Our results revealed that IκBα was degraded during RANKL-induced osteoclastogenesis, and the degradation of IκBα was inhibited after treatment with SSD. P65 is mainly located in the cytoplasm of BMMs, and IκBα degradation may contribute to phosphorylation and nuclear translocation of P65.27 We observed that P65 phosphorylation decreased in a dose-dependent manner after SSD treatment (Figure 4A and B). In addition, the results of immunofluorescence staining revealed that P65 was translocated to the nucleus after phosphorylation induced by RANKL in the positive control group. BMMs treated with SSD significantly suppressed nuclear translocation of P65 (Figure 4C and D). Therefore, SSD significantly attenuated RANKL-induced activation of NF-κB pathway.
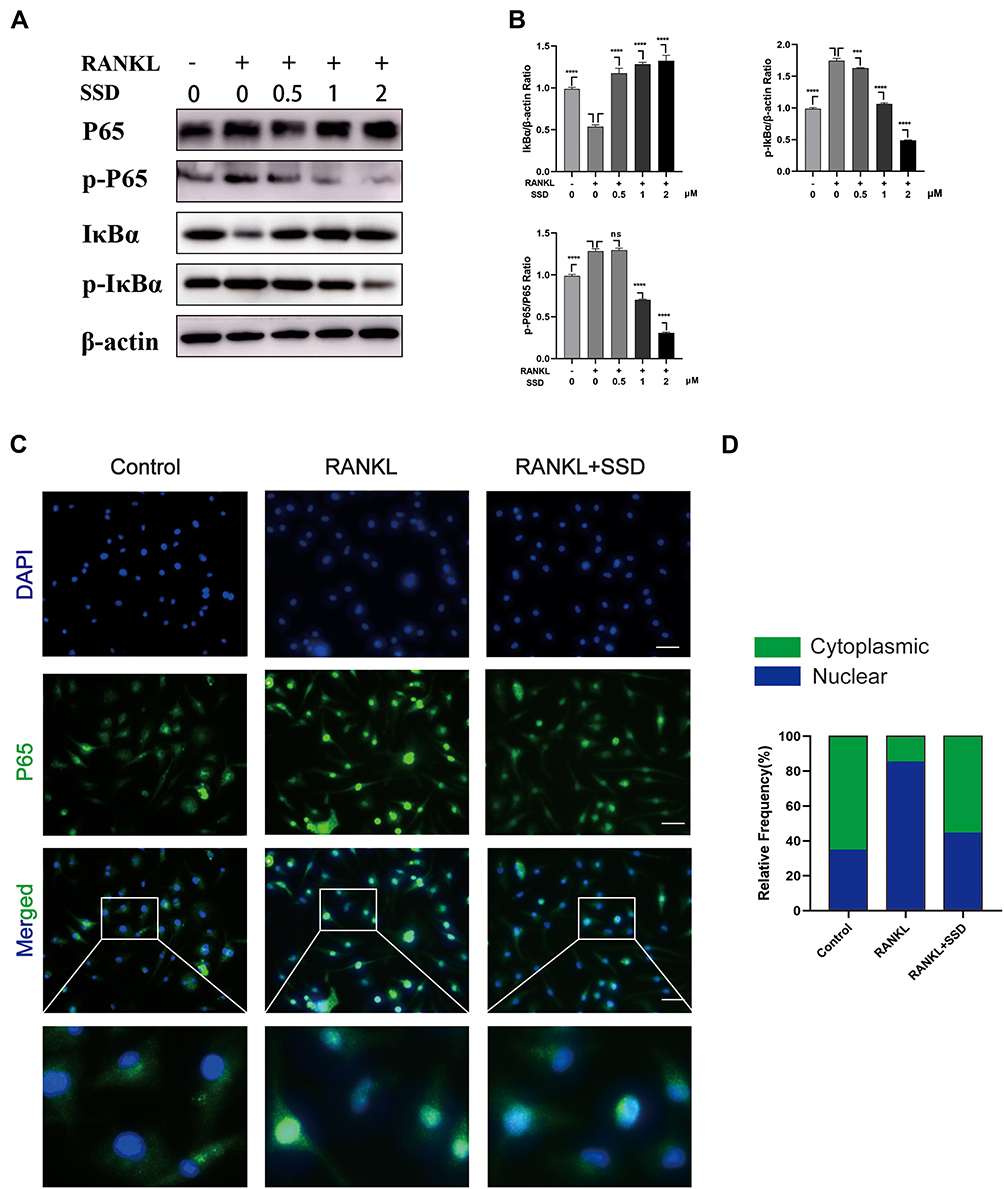 |
Figure 4 Saikosaponin D attenuated RANKL-induced activation of NF-κB pathway. (A) SSD suppressed RANKL-induced relative phosphorylation of P65 and IκBα and the degradation of IκBα. BMMs were treated without or with various concentrations of SSD (0.5, 1 and 2 μmol/L) for 1 h and then were treated with RANKL (50 ng/mL) for 15 min. Total proteins extracted from BMMs were probed for protein levels using immunoblot analyses. (B) Expressions of IκBα and p‐IκBα relative to β‐actin and p‐NFκB P65 relative to total NF-κB P65. (C) Nuclear translocation of P65 indicated by immunofluorescence. (D) The relative frequency of cytoplasmic and nuclear P65 in each experimental group was quantified using Image J. Original magnification, ×200. Scale bar, 200 μm. Mean ± SD is used to describe the data. Above experiments were conducted independently at least 3 times. ****P <0.0001, ***P<0.001. Abbreviation: ns, not significant. |
Furthermore, the MAPK and PI3K–AKT pathways play a crucial role in the formation of osteoclasts. Stimulation by RANKL induced phosphorylation of PI3K, AKT and MAPK members, including ERK, JNK and P38, which increased the expression of the downstream NFATc1.4 The results revealed that SSD significantly inhibited the activation of PI3K–AKT and MAPK signaling pathways (Figure 5A–D). We speculate that SSD targets a common protein, which connects these pathways as an adaptor, thereby resulting in their inhibition.
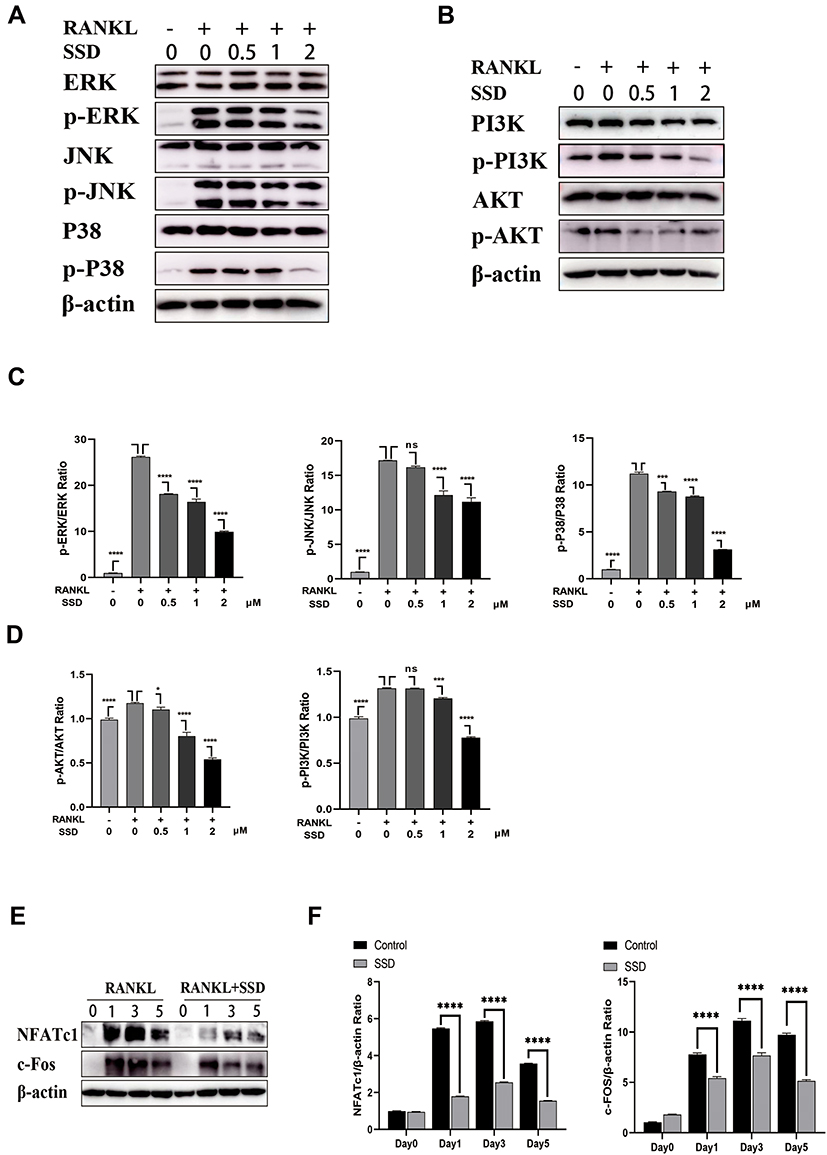 |
Figure 5 Saikosaponin D attenuated RANKL-induced activation of MAPKs and PI3K-AKT pathways. (A and B) SSD suppressed the phosphorylation and activation of JNK, ERK, P38, AKT and PI3K induced by RANKL. (C and D) The expressions of p‐ERK, p-JNK, p-P38, p-AKT and p-PI3K relative to total cellular proteins. (E) SSD suppressed RANKL-induced activation of NFATc1 and c-FOS. Immunoblot analyses were performed on total proteins extracted from BMMs treated with SSD and RANKL for 0, 1, 3, or 5 days. (F) The expressions of NFATc1 and c-FOS relative to β-actin. Mean ± SD is used to describe the data. Above experiments were conducted independently at least 3 times. ****P <0.0001, ***P<0.001, *P <0.05. Abbreviation: ns, not significant. |
Activation of the NF-κB, MAPKs and PI3K–AKT pathways after RANKL stimulation resulted in the induction of c-Fos and NFATc1, which were regarded as transcriptional regulators for osteoclastogenesis.6 The expression levels of NFATc1 and c-Fos were the highest 3 days after M-CSF and RANKL processing. However, the expression levels markedly reduced after treatment with SSD (Figure 5E and F). Therefore, these results revealed that SSD reduced the expression of NFATc1 and c-Fos by inhibiting NF-κB, MAPKs and PI3K–AKT pathways in the early stage.
SSD Inhibited LPS-Induced Inflammatory Bone Loss in vivo
We investigated whether SSD could exert a protective role in an in vivo model of LPS-induced osteolysis. TRAP staining of the calvaria demonstrated that SSD significantly inhibited osteoclastogenesis (Figure 6A). In addition, 3D image reconstruction and µCT scanning revealed a significant decrease in BV/TV and an increased porosity percentage and pore number in the LPS treatment group. SSD (high and low dose) treatment ameliorated LPS-induced inflammatory osteolysis in a dose‐dependent manner (Figure 6B), which was consistent with the in vitro results. The quantification of the bone parameters revealed that SSD remarkably decreased BV/TV and porosity percentage (Figure 6C). Furthermore, results of the histological examination revealed that SSD exerted a protective effect against LPS-induced bone loss. Histological assessment showed that SSD decreased the number of TRAP‐positive cells and increased BV/TV in a dose‐dependent manner (Figure 7A and B).
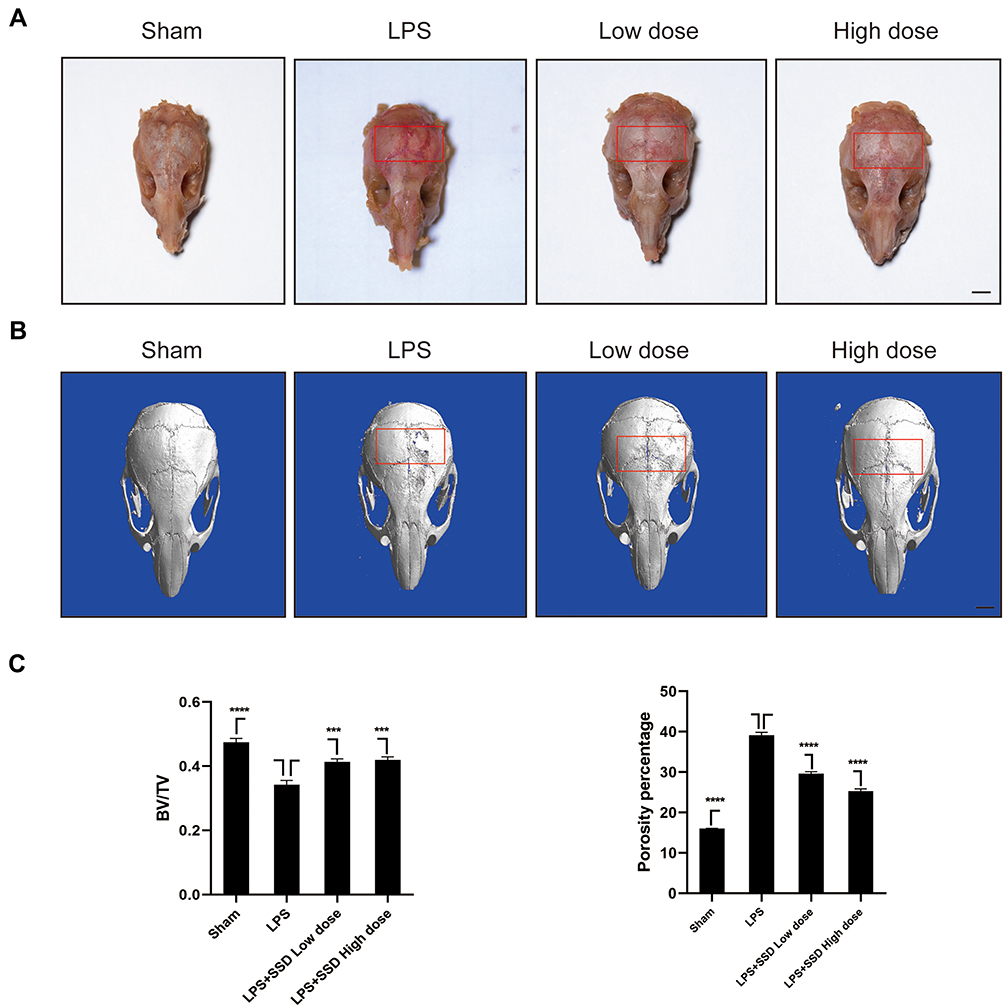 |
Figure 6 Saikosaponin D inhibited inflammatory bone loss induced by LPS. (A) The calvarial bones were subjected to TRAP staining. The region of interest was highlighted with a red box. (B) Micro‐CT scanning and subsequent 3D reconstruction of the calvarial bones from the LPS in high‐dose SSD group, LPS with low‐dose SSD group, LPS group, and Sham group (scale bar = 1mm). The region of interest was highlighted with a red box. (C) Determination of BV/TV (bone volume/total volume). Percentage porosity and the number of pores. There were 6 randomly assigned mice in each group. Mean ± SD is used to describe the data. ****P <0.0001, ***P<0.001. |
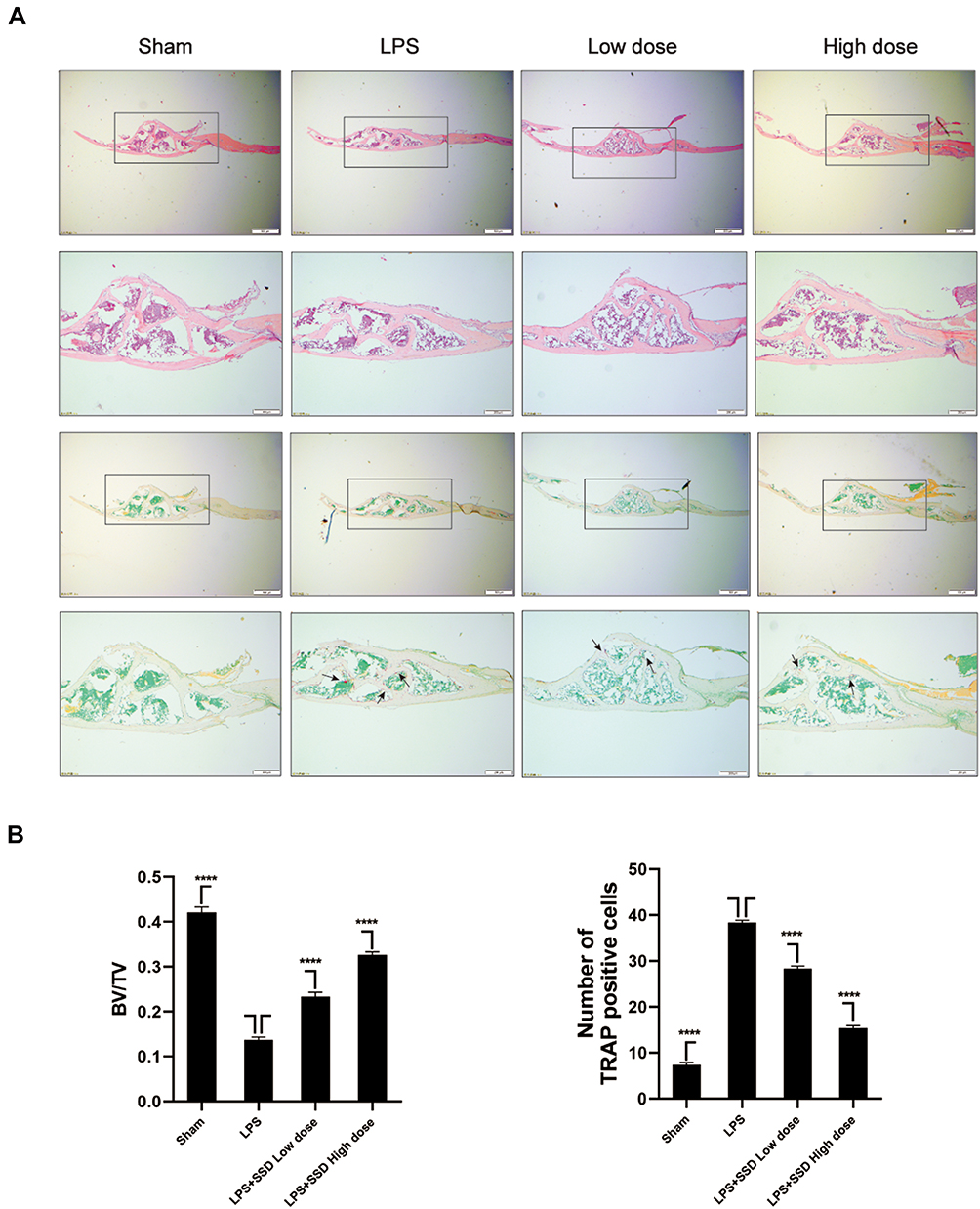 |
Figure 7 Saikosaponin D inhibited inflammatory bone loss induced by LPS. (A) H&E staining and TRAP staining of calvaria from the high-dose SSD group, low-dose SSD group, LPS group, and sham group (scale bar = 200μm). (B) Determination of TRAP (+) cells and BV/TV. There were 6 randomly assigned mice in each group. The area of TRAP (+) cells were highlighted with black arrows. Mean ± SD is used to describe the data. ****P <0.0001. |
Discussion
Excessive osteoclast activity impairs bone homeostasis, leading to osteolytic diseases such as osteoporosis.2,3,28 Currently, many agents, including bisphosphonates, targeting osteoclast formation and bone resorption are available, which provide therapeutic strategies for these diseases. However, these drugs have limitations and side effects such as development of atypical subtrochanteric fractures and osteonecrosis of the jaw.29,30 SSD is a type of triterpene saponin extracted from Bupleurum. In this study, SSD exerted an inhibitory effect on osteoclast formation and bone resorption induced by M-CSF and RANKL in vitro. The results of qRT-PCR and Western blotting revealed that SSD inhibited multiple signaling pathways, including the NF-κB, MAPKs and PI3K-AKT pathways. Furthermore, SSD decreased the expression levels of specific genes related to osteoclast differentiation and function, including V-ATPase d2, Dcstamp, Acp 5 and c-Fos, and the related inflammatory factors such as MMP9. Moreover, the expression level of downstream NFATc1 and c-FOS were decreased, which are the major transcription factors modulating the differentiation, fusion and bone resorption of osteoclasts (Figure 8). Genetic evidence indicates that the NF-κB pathway plays a crucial role in osteoclast formation.31,32 RANKL induces rapid phosphorylation of IκBα, and the degraded IκBα releases the RelA (p65)/p50 heterodimer, which causes the nuclear translocation of P65 to activate the related target genes.27,33 Our results revealed that treatment with SSD inhibited the degradation of IκBα and nuclear translocation of P65 induced by RANKL.
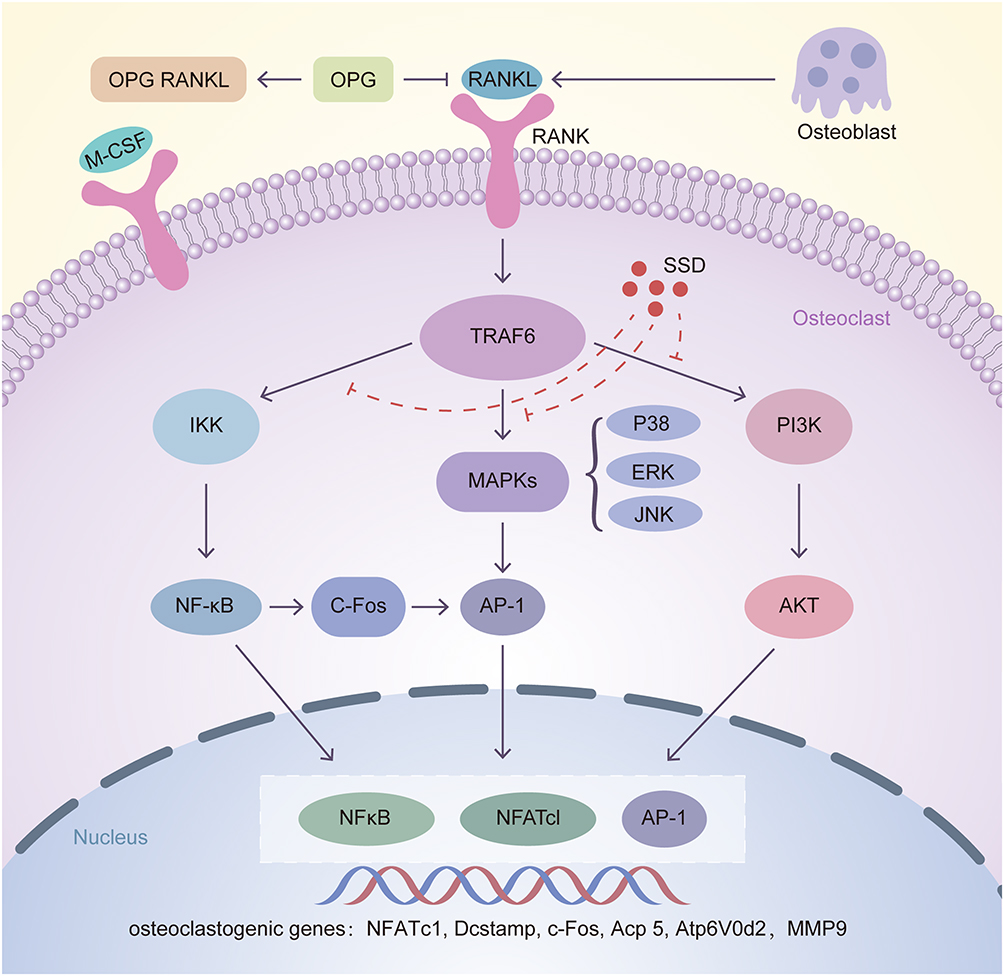 |
Figure 8 Inhibition of osteoclastogenesis by SSD is associated with regulation of RANKL/RANK pathway. After RANKL secreted by osteoblasts combined with RANKL, TRAF6 is activated in the cytoplasm of osteoclasts. Then the downstream pathways including NF-κB, MAPKs and PI3K-AKT are activated, leading to the automatic expansion of NFATc1, c-FOS and AP-1. Several osteoclast related genes such as NFATc1, Dcstamp, c-Fos, Acp 5, V-ATPase d2 and MMP9 are upregulated, which are essential for osteoclasts differentiation and formation. Our results for the first time demonstrated that SSD suppressed osteoclastogenesis via inhibiting RANKL-induced multiple downstream pathways. |
RANKL treatment activated the three MAPK signaling pathways, namely, ERK, JNK and P38.4 The P38 pathway is mainly involved in osteoclast formation, which was proved by P38 inhibitors.34 Precursor cells lacking JNK1 but not JNK2 can attenuate osteoclast differentiation. In addition, JNK plays a pivotal role in regulating the survival of osteoclasts.35 ERK is necessary for the formation of osteoclasts and the maintenance of bone resorption.8,36 In addition, RANKL activates the PI3K–AKT pathway via TRAF6 to regulate cell proliferation and differentiation. Specific PI3K inhibitors, glucocorticoids, ameliorate osteoclast formation.37,38
The activated PI3K–AKT pathway can inactivate GSK3β and NFATc1 via calcineurin. Overexpressed AKT signals enhance the expression of NFATc1 and the localization to nucleus, which results from an increased phosphorylation of GSK3β.39 NFATc1 is the main transcriptional regulator of osteoclast formation, and NFATc1-deficient BMMs cannot differentiate into osteoclasts. Furthermore, when active NFATc1 is overexpressed in osteoclast precursors, the differentiation of osteoclasts could be completed even without RANKL.40 In addition, NFATc1 can coordinate with other transcription factors in the nucleus to increase the expression levels of several osteoclast related genes.41,42 In our study, according to the responses after RANKL stimulation to the activated NF-κB, MAPKs and PI3K-AKT, SSD remarkably attenuated the expression level of NFATc1 and c-Fos and suppressed the expression of osteoclast-related mRNA in a dose- and time-dependent manner. Overall, our results reveal the anti-osteoclastogenic and anti-resorptive effects of SSD in vitro via inhibition of a variety of signaling pathways induced by RANKL.
LPS, located on the outer membrane of gram-negative bacteria, is an amphiphilic molecule. LPS activates the NF-κB pathway and produces a series of inflammatory cytokines such as TNF-α and interleukin 1 (IL-1),13,43 which play an important role in osteoclast differentiation and formation. Results of the in vivo experiments revealed that SSD inhibited LPS-induced inflammatory bone loss in a dose‐dependent manner. Our experiment model is limited to the local bone destruction of the calvaria induced by LPS. Several pro-inflammatory factors play an important role in the occurrence and development of osteolytic diseases. However, we only selected LPS as a pro-inflammatory factor in our study. In addition, we used a short-term experimental model in this study. Osteolytic diseases such as osteoporosis are often long-term systemic diseases. Therefore, additional in vivo studies such as a study in a mouse model of ovariectomy-induced osteoporosis should be performed to further establish the therapeutic efficacy of SSD in treating osteoclast-related diseases in the future.
In summary, this study for the first time proved that SSD suppressed osteoclastogenesis via inhibiting RANKL-induced multiple pathways in vitro. In addition, SSD could inhibit LPS-induced inflammatory bone destruction in vivo. Thus, SSD might be a potential drug in treating osteolytic diseases such as periprosthetic and inflammatory osteolysis.
Statement
No obvious side effects on mice are available when using Saikosaponin D according to the previous research.16,44 In our experiment, the status of the mice, such as eating and drinking, is normal after administrated. Therefore, side effects are absent for SSD.
Acknowledgments
The study was sponsored by Natural Science Foundation of Zhejiang Province (LY20H060006) and Experimental Animal Science Project of Zhejiang Province (LGD19H310001).
Disclosure
The authors declare no conflicts of interest.
References
1. Asagiri M, Takayanagi H. The molecular understanding of osteoclast differentiation. Bone. 2007;40(2):251–264. doi:10.1016/j.bone.2006.09.023
2. Kim JM, Lin C, Stavre Z, Greenblatt MB, Shim JH. Osteoblast-osteoclast communication and bone homeostasis. Cells. 2020;9(9):2073. doi:10.3390/cells9092073
3. Ono T, Nakashima T. Recent advances in osteoclast biology. Histochem Cell Biol. 2018;149(4):325–341. doi:10.1007/s00418-018-1636-2
4. Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423(6937):337–342. doi:10.1038/nature01658
5. Wang X, Yamauchi K, Mitsunaga T. A review on osteoclast diseases and osteoclastogenesis inhibitors recently developed from natural resources. Fitoterapia. 2020;142:104482. doi:10.1016/j.fitote.2020.104482
6. Park JH, Lee NK, Lee SY. Current Understanding of RANK Signaling in Osteoclast Differentiation and Maturation. Mol Cells. 2017;40(10):706–713. doi:10.14348/molcells.2017.0225
7. Kodama J, Kaito T. Osteoclast Multinucleation: review of Current Literature. Int J Mol Sci. 2020;21(16):5685. doi:10.3390/ijms21165685
8. Kim B, Lee KY, Park B. Icariin abrogates osteoclast formation through the regulation of the RANKL-mediated TRAF6/NF-kappaB/ERK signaling pathway in Raw264.7 cells. Phytomedicine. 2018;51:181–190. doi:10.1016/j.phymed.2018.06.020
9. Walsh MC, Lee J, Choi Y. Tumor necrosis factor receptor- associated factor 6 (TRAF6) regulation of development, function, and homeostasis of the immune system. Immunol Rev. 2015;266(1):72–92. doi:10.1111/imr.12302
10. Yagi M, Miyamoto T, Sawatani Y, et al. DC-STAMP is essential for cell-cell fusion in osteoclasts and foreign body giant cells. J Exp Med. 2005;202(3):345–351. doi:10.1084/jem.20050645
11. Kim K, Lee SH, Ha kim J, Choi Y, Kim N. NFATc1 induces osteoclast fusion via up-regulation of Atp6v0d2 and the dendritic cell-specific transmembrane protein (DC-STAMP). Mol Endocrinol. 2008;22(1):176–185. doi:10.1210/me.2007-0237
12. Bandow K, Maeda A, Kakimoto K, et al. Molecular mechanisms of the inhibitory effect of lipopolysaccharide (LPS) on osteoblast differentiation. Biochem Biophys Res Commun. 2010;402(4):755–761. doi:10.1016/j.bbrc.2010.10.103
13. Islam S, Hassan F, Tumurkhuu G, et al. Bacterial lipopolysaccharide induces osteoclast formation in RAW 264.7 macrophage cells. Biochem Biophys Res Commun. 2007;360(2):346–351. doi:10.1016/j.bbrc.2007.06.023
14. Guo C, Yuan L, Wang JG, et al. Lipopolysaccharide (LPS) induces the apoptosis and inhibits osteoblast differentiation through JNK pathway in MC3T3-E1 cells. Inflammation. 2014;37(2):621–631. doi:10.1007/s10753-013-9778-9
15. An J, Hao D, Zhang Q, et al. Natural products for treatment of bone erosive diseases: the effects and mechanisms on inhibiting osteoclastogenesis and bone resorption. Int Immunopharmacol. 2016;36:118–131. doi:10.1016/j.intimp.2016.04.024
16. Li P, Wu M, Xiong W, et al. Saikosaponin-d ameliorates dextran sulfate sodium-induced colitis by suppressing NF-kappaB activation and modulating the gut microbiota in mice. Int Immunopharmacol. 2020;81:106288. doi:10.1016/j.intimp.2020.106288
17. Lai M, Ge Y, Chen M, Sun S, Chen J, Cheng R. Saikosaponin D Inhibits Proliferation and Promotes Apoptosis Through Activation of MKK4-JNK Signaling Pathway in Pancreatic Cancer Cells. Onco Targets Ther. 2020;13:9465–9479. doi:10.2147/OTT.S263322
18. Chen Y, Que R, Lin L, Shen Y, Liu J, Li Y. Inhibition of oxidative stress and NLRP3 inflammasome by Saikosaponin-d alleviates acute liver injury in carbon tetrachloride-induced hepatitis in mice. Int J Immunopathol Pharmacol. 2020;34:2058738420950593. doi:10.1177/2058738420950593
19. Su J, Pan YW, Wang SQ, Li XZ, Huang F, Ma SP. Saikosaponin-d attenuated lipopolysaccharide-induced depressive-like behaviors via inhibiting microglia activation and neuroinflammation. Int Immunopharmacol. 2020;80:106181. doi:10.1016/j.intimp.2019.106181
20. Kim HS, Nam ST, Mun SH, et al. DJ-1 controls bone homeostasis through the regulation of osteoclast differentiation. Nat Commun. 2017;8(1):1519. doi:10.1038/s41467-017-01527-y
21. Zhai Z, Qu X, Li H, et al. The effect of metallic magnesium degradation products on osteoclast-induced osteolysis and attenuation of NF-kappaB and NFATc1 signaling. Biomaterials. 2014;35(24):6299–6310. doi:10.1016/j.biomaterials.2014.04.044
22. Chen K, Qiu P, Yuan Y, et al. Pseurotin A Inhibits Osteoclastogenesis and Prevents Ovariectomized-Induced Bone Loss by Suppressing Reactive Oxygen Species. Theranostics. 2019;9(6):1634–1650. doi:10.7150/thno.30206
23. Peng J, Zhao K, Zhu J, et al. Sarsasapogenin Suppresses RANKL-Induced Osteoclastogenesis in vitro and Prevents Lipopolysaccharide-Induced Bone Loss in vivo. Drug Des Devel Ther. 2020;14:3435–3447. doi:10.2147/DDDT.S256867
24. Zhang T, Zhao K, Han W, et al. Deguelin inhibits RANKL-induced osteoclastogenesis in vitro and prevents inflammation-mediated bone loss in vivo. J Cell Physiol. 2019;234(3):2719–2729. doi:10.1002/jcp.27087
25. Shao S, Fu F, Wang Z, et al. Diosmetin inhibits osteoclast formation and differentiation and prevents LPS-induced osteolysis in mice. J Cell Physiol. 2019;234(8):12701–12713. doi:10.1002/jcp.27887
26. Yang H, Xu Y, Zhu M, et al. Inhibition of titanium-particle-induced inflammatory osteolysis after local administration of dopamine and suppression of osteoclastogenesis via D2-like receptor signaling pathway. Biomaterials. 2016;80:1–10. doi:10.1016/j.biomaterials.2015.11.046
27. Jung Y, Kim H, Min SH, Rhee SG, Jeong W. Dynein light chain LC8 negatively regulates NF-kappaB through the redox-dependent interaction with IkappaBalpha. J Biol Chem. 2008;283(35):23863–23871. doi:10.1074/jbc.M803072200
28. Reginster JY, Burlet N. Osteoporosis: a still increasing prevalence. Bone. 2006;38(2 Suppl 1):S4–9. doi:10.1016/j.bone.2005.11.024
29. Black DM, Bauer DC, Schwartz AV, Cummings SR, Rosen CJ. Continuing bisphosphonate treatment for osteoporosis–for whom and for how long? N Engl J Med. 2012;366(22):2051–2053. doi:10.1056/NEJMp1202623
30. Harsløf T, Langdahl BL. New horizons in osteoporosis therapies. Curr Opin Pharmacol. 2016;28:38–42. doi:10.1016/j.coph.2016.02.012
31. Yamashita T, Yao Z, Li F, et al. NF-kappaB p50 and p52 regulate receptor activator of NF-kappaB ligand (RANKL) and tumor necrosis factor-induced osteoclast precursor differentiation by activating c-Fos and NFATc1. J Biol Chem. 2007;282(25):18245–18253. doi:10.1074/jbc.M610701200
32. Iotsova V, Caamaño J, Loy J, Yang Y, Lewin A, Bravo R. Osteopetrosis in mice lacking NF-kappaB1 and NF-kappaB2. Nat Med. 1997;3(11):1285–1289. doi:10.1038/nm1197-1285
33. Novack DV. Role of NF-kappaB in the skeleton. Cell Res. 2011;21(1):169–182. doi:10.1038/cr.2010.159
34. Kim K, Kim JH, Kim I, Seong S, Kim N. Rev-erbalpha Negatively Regulates Osteoclast and Osteoblast Differentiation through p38 MAPK Signaling Pathway. Mol Cells. 2020;43(1):34–47. doi:10.14348/molcells.2019.0232
35. Ma Z, Yu R, Zhao J, et al. Constant hypoxia inhibits osteoclast differentiation and bone resorption by regulating phosphorylation of JNK and IkappaBalpha. Inflamm Res. 2019;68(2):157–166. doi:10.1007/s00011-018-1209-9
36. Koga Y, Tsurumaki H, Aoki-Saito H, et al. Roles of Cyclic AMP Response Element Binding Activation in the ERK1/2 and p38 MAPK Signalling Pathway in Central Nervous System, Cardiovascular System, Osteoclast Differentiation and Mucin and Cytokine Production. Int J Mol Sci. 2019;20(6):1346. doi:10.3390/ijms20061346
37. Adapala NS, Barbe MF, Langdon WY, Tsygankov AY, Sanjay A. Cbl-phosphatidylinositol 3 kinase interaction differentially regulates macrophage colony-stimulating factor-mediated osteoclast survival and cytoskeletal reorganization. Ann N Y Acad Sci. 2010;1192:376–384. doi:10.1111/j.1749-6632.2009.05346.x
38. Fu L, Wu W, Sun X, Zhang P. Glucocorticoids Enhanced Osteoclast Autophagy Through the PI3K/Akt/mTOR Signaling Pathway. Calcif Tissue Int. 2020;107(1):60–71. doi:10.1007/s00223-020-00687-2
39. Wang D, Lu X, Wang E, Shi L, Ma C, Tan X. Salvianolic acid B attenuates oxidative stress-induced injuries in enterocytes by activating Akt/GSK3β signaling and preserving mitochondrial function. Eur J Pharmacol. 2021;909:174408. doi:10.1016/j.ejphar.2021.174408
40. Takayanagi H, Kim S, Koga T, et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3(6):889–901. doi:10.1016/S1534-5807(02)00369-6
41. Feng H, Cheng T, Steer JH, et al. Myocyte enhancer factor 2 and microphthalmia-associated transcription factor cooperate with NFATc1 to transactivate the V-ATPase d2 promoter during RANKL-induced osteoclastogenesis. J Biol Chem. 2009;284(21):14667–14676. doi:10.1074/jbc.M901670200
42. Song I, Kim JH, Kim K, Jin HM, Youn BU, Kim N. Regulatory mechanism of NFATc1 in RANKL-induced osteoclast activation. FEBS Lett. 2009;583(14):2435–2440. doi:10.1016/j.febslet.2009.06.047
43. Lv L, Lv L, Zhang Y, Kong Q. Luteolin prevents LPS-induced TNF-alpha expression in cardiac myocytes through inhibiting NF-kappaB signaling pathway. Inflammation. 2011;34(6):620–629. doi:10.1007/s10753-010-9271-7
44. Qin T, Yuan Z, Yu J, et al. Saikosaponin-d impedes hippocampal neurogenesis and causes cognitive deficits by inhibiting the survival of neural stem/progenitor cells via neurotrophin receptor signaling in mice. Clin Transl Med. 2020;10(8):e243. doi:10.1002/ctm2.243
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.