")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Inhibition Of JNK Phosphorylation By Curcumin Analog C66 Protects LPS-Induced Acute Lung Injury
Authors Xiao Z, Xu F, Zhu X, Bai B, Guo L, Liang G, Shan X, Zhang Y, Zhao Y, Zhang B
Received 14 May 2019
Accepted for publication 22 October 2019
Published 10 December 2019 Volume 2019:13 Pages 4161—4171
DOI https://doi.org/10.2147/DDDT.S215712
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Qiongyu Guo
Zhongxiang Xiao,1,2 Fengli Xu,3 Xiaona Zhu,1 Bin Bai,1 Lu Guo,4 Guang Liang,1 Xiaoou Shan,3 Yali Zhang,2 Yunjie Zhao,2 Bing Zhang1,2
1Affiliated Yueqing Hospital, Wenzhou Medical University, Wenzhou, Zhejiang 325600, People’s Republic of China; 2Chemical Biology Research Center, School of Pharmaceutical Sciences, Wenzhou Medical University, Wenzhou, Zhejiang, People’s Republic of China; 3Department of Pediatrics, The Second Affiliated Hospital and Yuying Children’s Hospital of Wenzhou Medical University, Wenzhou, Zhejiang 325000, People’s Republic of China; 4Department of Pharmacy, The First People’s Hospital of Huzhou, Huzhou, Zhejiang 313000, People’s Republic of China
Correspondence: Bing Zhang
Affiliated Yueqing Hospital, Wenzhou Medical University, Wenzhou, Zhejiang 325600, People’s Republic of China
Tel +86-577-61575506
Email [email protected]
Yunjie Zhao
Chemical Biology Research Center, School of Pharmaceutical Sciences, Wenzhou Medical University, Wenzhou 325035, People’s Republic of China
Tel +86-577-86699892
Email [email protected]
Background: Acute lung injury (ALI) is characterized by high prevalence and high mortality. Thus far, no effective pharmacological treatment has been made for ALI in clinics. Inflammation is critical to the development of ALI. Curcumin analog C66, having reported as an inhibitor of c-Jun N-terminal kinase (JNK), exhibits anti-inflammatory property both in vitro and in vivo. However, whether C66 is capable of reducing lipopolysaccharide (LPS)-induced ALI through the inhibition of inflammation by targeting JNK remains unknown.
Methods: Intratracheal injection of LPS was employed to build a mouse ALI model. H&E staining, wet/dry ratio, immunofluorescence staining, inflammatory cell detection, and inflammatory gene expression were used to evaluate lung injury and lung inflammation. In vitro, LPS was used to induce the expression of inflammatory cytokines both in protein and gene levels.
Results: The results of our studies showed that the pretreatment with C66 and JNK inhibitor SP600125 was capable of attenuating the LPS-induced ALI by detecting pulmonary edema, pathological changes, total protein concentration, and inflammatory cell number in bronchoalveolar lavage fluid (BALF). Besides, C66 and SP600125 also suppressed LPS-induced inflammatory cytokine expression in BALF, serum, and lung tissue. In vitro, LPS-induced production of TNF-α and IL-6 and gene expression of TNF-α, IL-6, IL-1β, and COX-2 could be inhibited by the pretreatment with C66 and SP600125. It was found that C66 and SP600125 could inhibit LPS-induced phosphorylation of JNK both in vitro and in vivo.
Conclusion: In brief, our results suggested that C66 protects LPS-induced ALI through the inhibition of inflammation by targeting the JNK pathway. These findings further confirmed the pivotal role of JNK in ALI and implied that C66 is likely to serve as a potential therapeutic agent for ALI.
Keywords: acute lung injury, lipopolysaccharide, JNK, C66, inflammation
Corrigendum for this paper has been published.
Introduction
Acute lung injury (ALI) is one of the common clinical critical diseases. A great number of clinical studies have confirmed that ALI is one of the main causes of respiratory failure and multiple organ dysfunction, and the mortality rate reaches up to 35–40%.1 ALI is characterized by lung edema, hemorrhage, bronchiole epithelial desquamation, marked thickening of the alveolar wall, and neutrophil infiltration that leads to respiratory failure.2 The commonest cause of ALI is septicemia induced by endotoxin lipopolysaccharide (LPS), a component of Gram-negative bacteria. With a better understanding of the pathogenesis of ALI, it has been widely evidenced that the release of pro-inflammatory cytokines is critical to LPS-induced lung injury.3 Though great progress has been achieved in the understanding of the pathogenesis as well as the pathophysiology of ALI, the treatments have still been limited.
c-Jun N-terminal kinase (JNK) belongs to the family of mitogen-activated protein kinases (MAPKs) that participate in the regulation of the production of inflammatory mediators.4 JNK is phosphorylated and activated AP-1 after LPS stimulation to induce the transcription of inflammatory cytokines.5 Numerous studies have reported that JNK participated in the development of LPS-induced ALI. As Zheng et al reported, JNK inhibitor SP600125 alleviated LPS-induced ALI by inhibiting the expression of JNK.6 Many researchers have reported that LPS-induced ALI can be alleviated by a considerable amount of natural products or synthesized compounds by inhibiting MAPK pathway (e.g., JNK).7–9 Thus, JNK is an attractive therapeutic target for LPS-induced ALI.
C66, (2E,6E)-2,6-bis(2-(trifluoromethyl)benzylidene)cyclohexanone, is a synthetic analog of natural active curcumin (Figure 1A). It was reported in one of our previous studies that C66 could inhibit the LPS-induced production of inflammatory cytokines (TNF-α and IL-6) in J774A.1 macrophages.10 The other two studies reported that C66 reduced the high glucose-induced inflammatory response in renal and myocardial cells and mitigated the diabetic kidney and heart injury by targeting JNK.11,12 In the present study, C66 and the specific JNK inhibitor SP600125 (Figure 1B) was adopted as a positive control to investigate their effects on LPS-induced ALI by targeting JNK and to further confirm the significant role of JNK in the regulation of ALI.
 |
Figure 1 Chemical structure of C66 (A) and JNK inhibitor SP600125 (B). |
Materials And Methods
Reagents And Antibodies
LPS and JNK inhibitor SP600125 were purchased from Sigma-Aldrich (St. Louis, MO, USA). Compound C66, provided by our own lab with HPLC purity >98.36%, was identified using ESI-MS as described in our previous publication.10 We dissolved C66 and SP600125 in dimethyl sulfoxide (DMSO) for cell experiments and in carboxymethylcellulose sodium (0.5%) for animal experiments, respectively. ELISA kits of TNF-α and IL-6 were provided by eBioscience, Inc. (San Diego, CA, USA). Anti-CD68 and Anti-p-JNK antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Cell Culture
Before being sacrificed for mouse peritoneal macrophages (MPMs) harvest, C57BL/6 mice were stimulated by intraperitoneal injection of 6% starch broth (0.3 g beef extract, 1 g tryptone, 0.5 g NaCl and 6 g starch dissolved in 100 mL H2O, 2.5 mL/mouse) for 3 days. Subsequently, MPMs were centrifuged and suspended in RPMI-1640 medium (Gibco, Eggenstein, Germany), and then supplemented with 10% FBS and 1% penicillin/streptomycin. The cells were incubated overnight at 37°C in a 5% CO2-humidified air.
Animals And ALI Model
Male C57BL/6 mice weighing 18–22 g were purchased from the Animal Center of Wenzhou Medical University (Wenzhou, China). All animals were housed at a constant ambient temperature in a 12:12-hr light-dark cycle and had free access to standard rodent feed and tap water. All animal care and experimental procedures were approved by the Wenzhou Medical University Animal Policy and Welfare Committee (Approval Document No. wydw2016-0124) and all animals received humane care according to the National Institutes of Health (USA) guidelines.
Forty-two C57BL/6 mice were randomly split into seven groups (n = 6 in each group): three sham groups: only treated with vehicle group (CON group), treated with C66 group (C66 10 group) and treated with SP600125 group (SP 10 group); and four LPS-treated groups: treated with vehicle plus LPS group (LPS group), treated with different doses of C66 plus LPS groups (C66 5+LPS group and C66 10+LPS group), and treated with SP600125 plus LPS group (SP 10+LPS group). Before being challenged by intratracheal instillation of LPS (5 mg/kg, 0.9% saline) or equal volume 0.9% saline, all animals were orally administrated vehicle, C66, or SP600125 for 7 consecutive days. The animals were killed with an overdose of chloral hydrate at 6 hrs after instillation LPS or 0.9% saline. Subsequently, the serum, lung tissues and bronchoalveolar lavage fluid (BALF) were taken out.
BALF Analysis
BALF was conducted thrice using a tracheal cannula with 0.5 mL 0.9% saline solution. After being centrifuged at 1000 rpm for 5 mins, the supernatant of BALF was immediately collected to ascertain the total protein concentration and cytokine levels of TNF-α and IL-6. The sediment was applied for cell counting. The protein concentration of BALF was measured using the Bradford protein assay kit (Bio-Rad Laboratories, Hercules, CA, USA). The total number of cells in BALF was counted using a standard hemocytometer, and neutrophil cells were confirmed by counting 200 cells on a smear prepared under Wright–Giemsa staining (Nanjing Institute of Biological Engineering, Nanjing, China).
Lung Wet/Dry Ratio
The middle lobe of the right lung was collected, and the wet weight was recorded. Subsequently, the lung was then heated in a thermostatic oven at 65°C for 72 hrs and then weighed to determine the baseline lung dry mass levels.
Determination Of TNF-α And IL-6
To ascertain cytokine levels in the cell medium, BALF, mice serum, and lung tissues, cytokine-specific ELISA kits (Bioscience, San Diego, CA, USA) were employed following the manufacturer's instructions. The total amounts of TNF-α and IL-6 in the cell medium were normalized to the total protein amount from the viable cell pellets.
Real-Time Quantitative PCR (qPCR)
MPMs were treated with 5 μM and 10 μM C66 and 10 μM SP600125 for 30 mins and followed by 0.5 μg/mL LPS for 6 hrs. Total RNAs were isolated from cells or lung tissues (10–20 mg) using the TRIzol method (Invitrogen, Carlsbad, CA, USA). Then, they underwent reverse transcription using a two-step M-MLV kit (Invitrogen). The Eppendorf Mastercycler ep realplex detection system (Eppendorf, Hamburg, Germany) was applied to conduct real-time qPCR analysis. The primers of genes (e.g., TNF-ɑ, IL-6, IL-1β, COX-2, CD80, CD64, CD86, and β-actin) were synthesized from Invitrogen (Shanghai, China). The amount of each gene was determined and normalized to the amount of β-actin. The primer sequences used are listed as follows: mouse TNF-α sense, 5ʹ-TGATCCGCGACGTGGAA-3ʹ, mouse TNF-α antisense, 5ʹ-ACCGCCTGGAGTTCTGGAA-3ʹ; mouse IL-6 sense,5ʹ-GAGGATACCACTCCCAACAGACC-3ʹ, mouse IL-6 antisense, 5ʹ-AAGTGCATCATCGTTGTTCATACA-3ʹ; mouse IL-1β sense, 5ʹ-ACTCCTTAGTCCTCGGCCA-3ʹ, mouse IL-1β antisense, 5ʹ-CCATCAG-AGGCAAGGAGGAA-3ʹ; mouse COX-2 sense, 5ʹ-TGGTGCCTGGTCTGATGATG-3ʹ, mouse COX-2 antisense, 5ʹ-GTGGTAACCGCTCAGGTGTTG-3ʹ; mouse CD80 sense, 5ʹ-ACCCCCAACATAACTGAGTCT-3ʹ; mouse CD80 antisense, 5ʹ-TTCCAACCAAGAGAAGCGAGG-3ʹ; mouse CD64 sense, 5ʹ-AGGTTCCTCAATGCCAAGTGA-3ʹ; mouse CD64 antisense, 5ʹ-GCGACCTCCGAATCTGAAGA-3ʹ; mouse CD86 sense, 5ʹ-TGTTTCCGTGGAGACGCAAG-3ʹ; mouse CD86 antisense, 5ʹ-TTGAGCCTTTGTAAATGGGCA-3ʹ; mouse β-actin sense, 5ʹ-CCGTGAAA-AGATGACCCAGA-3ʹ, mouse β-actin antisense, 5ʹ-TACGACCAGAGGCATACAG-3ʹ.
Lung Histopathology
We fixed a portion of lung tissues in 4% paraformaldehyde solution and embedded this portion in paraffin. To estimate the degree of lung injury under a light microscopy (200× amplification; Nikon, Tokyo, Japan), the paraffin sections (5 μm) were stained with H&E (Beyotime Biotech, Nantong, China). Lung injury was graded from 0 (normal) to 4 (severe) in four types, i.e., interstitial inflammation, neutrophil infiltration, congestion, and edema. By adding the individual scores for each type, we calculated the lung injury score. Grading was conducted by blinded pathologists. Lung injury score for each animal was calculated as the mean of 10 lung sections. Paraffin sections were obtained from each mouse at 6 hrs after undergoing intratracheal instillation.
Immunohistochemistry
Tissue sections (5 μm thickness) were prepared, deparaffinized in xylene, and then hydrated with an ethanol gradient. A pressure cooker was used to achieve heat-induced antigen retrieval (10 mM sodium citrate buffer, pH 6.5). After being treated with 30% of hydrogen peroxide, all sections were blocked in 5% BSA and then incubated with primary anti-CD68 or anti-p-JNK antibody (1:200) at 4°C overnight. Next, the slides were incubated with HRP-labeled (for CD68) or fluorophore-conjugated (for p-JNK and JNK) secondary antibody (1:200) for 10 mins. Tissues were counterstained with hematoxylin or DAPI. We captured the images under a light microscope (CD68 staining; 200× amplification; Nikon, Tokyo, Japan) and confocal microscopy (p-JNK and JNK staining; 200× amplification; Leica-TCS SP8, Leica, Germany).
Statistical Analysis
Data are expressed as the mean ± standard error of the mean (SEM). Student’s t-test was employed to find the differences between sets of data. Statistics were performed using GraphPad Pro (GraphPad, San Diego, CA, USA). P values below 0.05 (P < 0.05) are considered to be statistical significant.
Results
C66 And SP600125 Protected Mice From LPS-Induced Lung Injury
Pulmonary edema, as an index of lung injury, was determined in accordance with the lung wet/dry ratio. Data from Figure 2A suggest that lung wet/dry ratio increased significantly after LPS injection, and pretreatment with C66 or SP600125 almost reversed the LPS effects. However, mice that received only C66 or SP600125 did not show obvious variations in lung wet/dry ratio in comparison with the CON group. LPS instillation also leads to an obvious increase in the total protein concentration in BALF. Besides, the administration of C66 and SP600125 evidently eliminated the increase in protein concentration induced by LPS (Figure 2B). To determine the histological variations, lung tissues underwent H&E staining. Results from Figure 2C suggest that normal pulmonary histology was observed in CON, C66 10, and SP 10 groups, suggesting that C66 and SP600125 showed no obvious tissue toxicity. Tissues from the LPS group showed infiltration of inflammatory cells in lung interstitium and alveolar spaces as well as alveolar wall thickening and congestion, while C66 or SP600125 pretreatment significantly ameliorated these pathological variations attributed to LPS (Figure 2C). The lung injury score, assessed by a blinded pathologist, further shows the protective effects of C66 and SP600125 on LPS-induced lung injury (Figure 2D). These results imply that C66 and SP600125 can protect mice against LPS-induced lung injury.
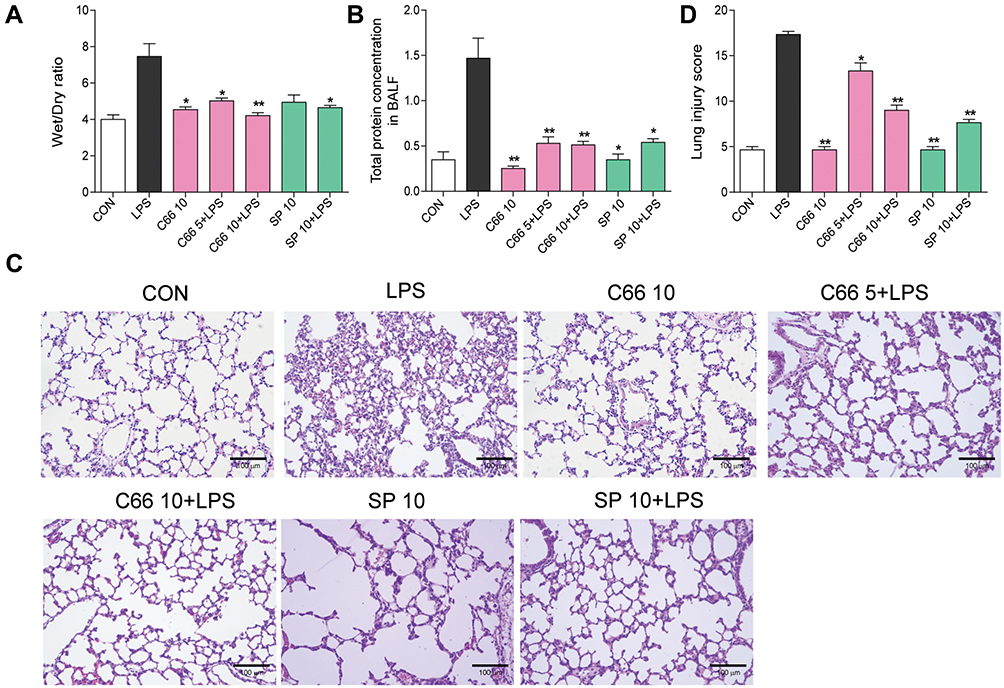 |
Figure 2 C66 and SP600125 protect mice from LPS-induced lung injury. Before LPS administration, mice underwent gavage once a day for 7 consecutive days of C66 (5 and 10 mg/kg) and SP600125 (10 mg/kg). After 6 hrs of LPS challenge, we euthanized mice and then took out the lung tissue and BALF. (A) Lung wet/dry ratio. (B) Total protein concentration in BALF. (C) Histopathological variations in lung tissues determined by H&E staining. (D) Lung injury score was ascertained. Columns represent the mean ± SEM of four to six separate animals. *P < 0.05, **P < 0.01, vs LPS group. |
C66 And SP600125 Attenuated LPS-Induced Inflammatory Cells Infiltration
LPS exposure increased the influx of total cells (Figure 3A) and neutrophils (Figure 3B) into BALF, whereas C66 or SP600125 administration inhibited the LPS-mediated inflammatory cell infiltration in BALF. Using immunohistochemical staining, we also detected the CD68, a marker of macrophages, -positive cells. CD68-positive macrophages significantly increased in lung interstitial areas of ALI mice, whereas such an increase was markedly mitigated by the treatment with either C66 or SP600125, as shown in Figure 3C. Lung tissues from CON, C66 10, and SP 10 groups did not show any CD68-positive cells. Meanwhile, C66 and SP600125 inhibited LPS-induced several inflammatory cell markers – CD80 (Figure 3D), CD64 (Figure 3E), and CD86 (Figure 3F) – and gene expression in lung tissues. These results imply that C66 and SP600125 are capable of attenuating LPS-induced inflammatory cell infiltration.
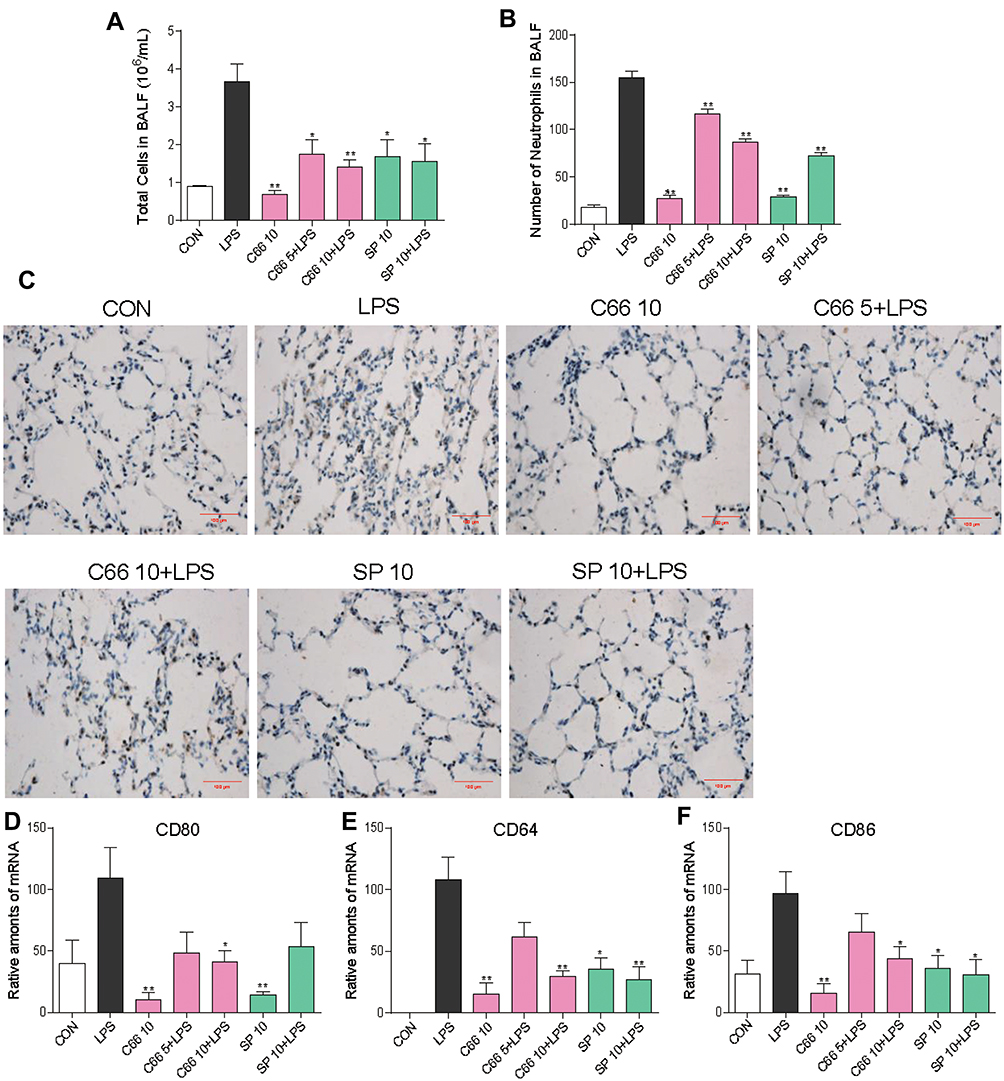 |
Figure 3 C66 and SP600125 attenuate LPS-induced inflammatory cell infiltration. Before LPS administration, mice were given by gavage once a day for 7 consecutive days of C66 (5 and 10 mg/kg) and SP600125 (10 mg/kg). After 6 hrs of LPS challenge, we euthanized mice and then took out lung tissue and BALF. (A) Total number of cells in BALF was ascertained using a hemocytometer. (B) Number of neutrophils was measured by Wright-Giemsa staining. (C) Macrophages infiltration in lung tissue was detected by performing CD68 immunohistochemical assay. The gene expression of inflammatory cell markers, CD80 (D), CD64 (D), CD86 (D), was detected by real-time quantitative PCR assay. Data are mean ± SEM of four to six separate animals. *P < 0.05, **P < 0.01 vs LPS group. |
C66 And SP600125 Inhibited The Inflammatory Cytokine Expression In Vivo
Pro-inflammatory cytokines, including TNF-α, IL-6, IL-1β, etc., serve as main mediators involved in the LPS-induced pulmonary injury. The TNF-α protein levels in BALF and serum were first determined by ELISA. LPS treatment led to an increase in the concentration of TNF-α in both BALF and serum, while pretreatment with C66 and SP600125 significantly reduced the LPS-induced TNF-α levels, as shown in Figure 4A and B. By performing real-time qPCR assay, we also tested the mRNA levels of inflammatory cytokines in mouse lung tissues. C66 and SP600125 decreased the overexpression of TNF-α, IL-6, IL-1β, and COX-2 mRNA in LPS-challenged mouse lung tissues, as shown in Figure 4C–F. Finally, we detected the protein level of TNF-α and IL-6 in lung tissue using ELISA. As shown in Figure 4G and H, LPS significantly induced TNF-α and IL-6 protein level in lung tissues, and C66 and SP600125 reduced the protein level of TNF-α and IL-6 in lung tissues. These results suggest that C66 and SP600125 reduced the expression of proinflammatory cytokines, which in turn attenuates the lung damage attributed to LPS-induced ALI.
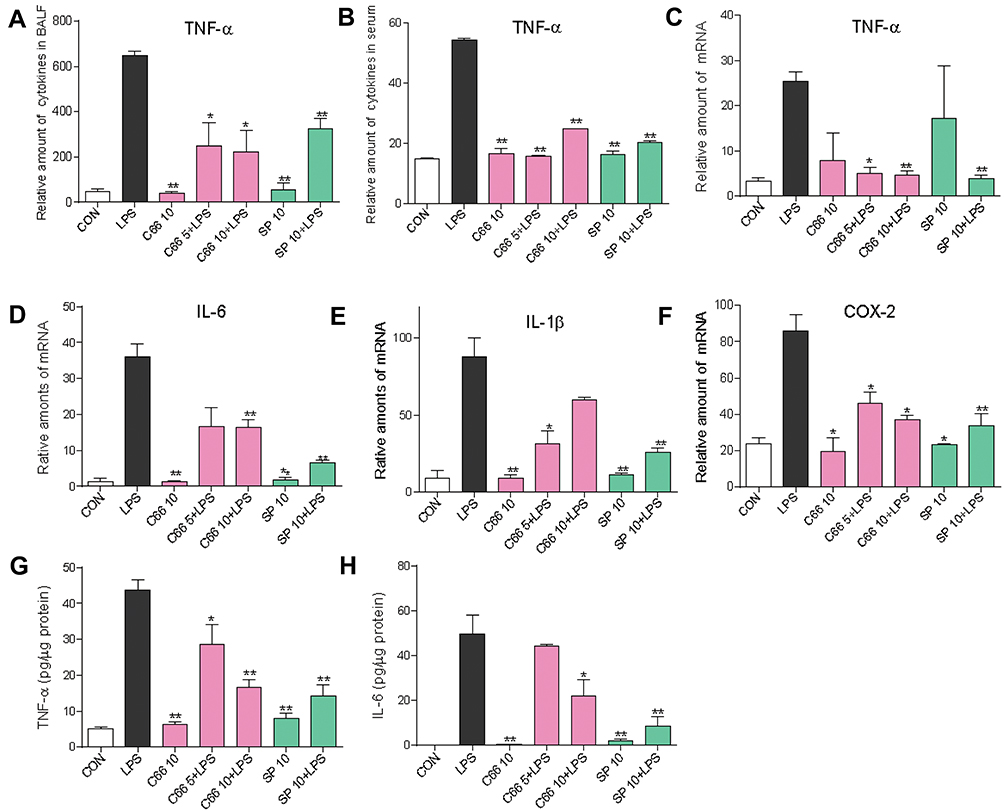 |
Figure 4 C66 and SP600125 could inhibit the expression of the inflammatory cytokine in vivo. Before LPS administration, mice were given by gavage once a day for 7 consecutive days of C66 (5 and 10 mg/kg) and SP600125 (10 mg/kg). After 6 hrs of LPS challenge, we euthanized mice and then collected lung tissue, BALF, and serum. Expression of inflammatory cytokine TNF-α in BALF (A) and serum (B) was determined using ELISA. Total mRNA was extracted from the lung tissue using TRIzol agent, and the mRNA levels of TNF-α (C), IL-6 (D), IL-1β (E), and COX-2 (F) were detected by conducting real-time quantitative PCR analysis. The protein level of TNF-α (G) and IL-6 (H) in lung tissues was detected by ELISA. Data are mean ± SEM of four to six separate animals. *P < 0.05, **P < 0.01 vs LPS group. |
C66 And SP600125 Inhibited JNK Phosphorylation In Lung Tissues
It was previously reported that C66 is an inhibitor of JNK, and the level of JNK phosphorylation in mice lung tissues was examined using immunofluorescence staining. Results from Figure 5 show that LPS injection can induce JNK phosphorylation in lung tissues, yet pretreatment with C66 and SP600125 can obviously inhibit the JNK phosphorylation. It is noteworthy that JNK phosphorylation was also observed in the lung tissue from mice only treated with 10 mg/kg C66. Meanwhile, JNK immunofluorescence staining showed no obvious changes in all groups. The results suggest that C66 and SP600125 are capable of protecting mice from LPS-induced lung injury by targeting JNK.
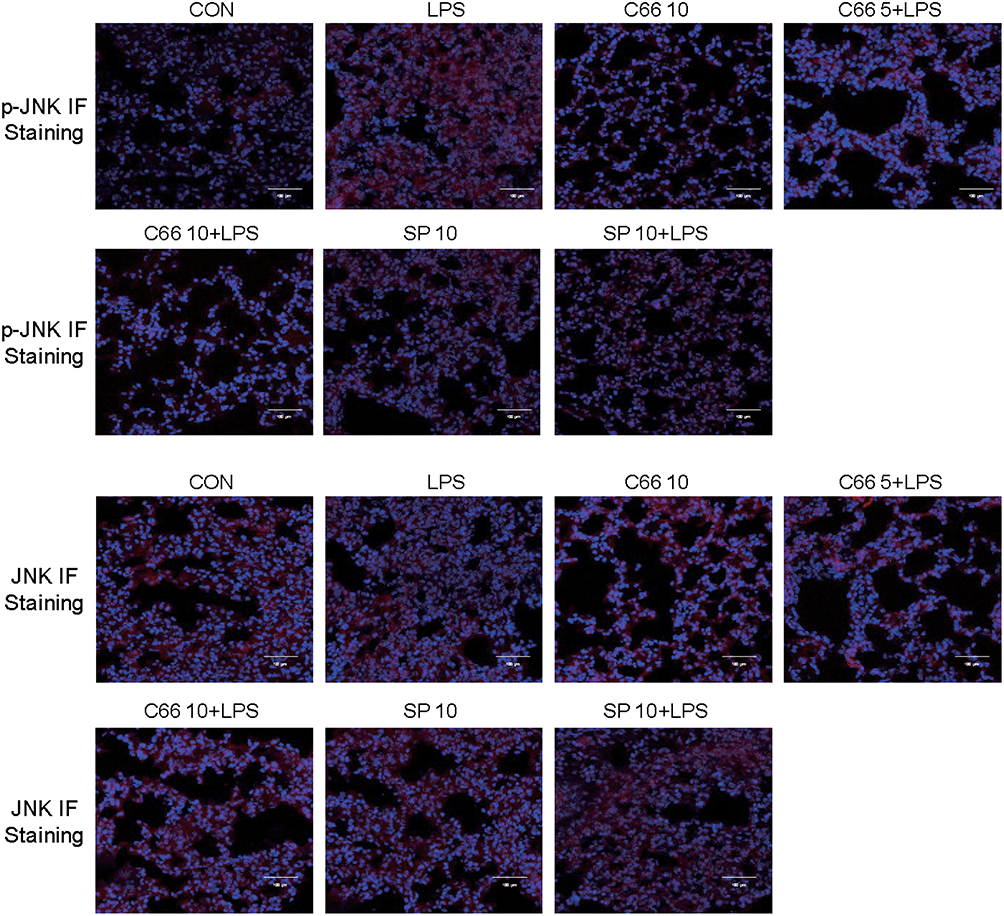 |
Figure 5 C66 and SP600125 could inhibit LPS-induced JNK phosphorylation in lung tissues. Before LPS administration, mice were given by gavage once a day for 7 consecutive days of C66 (5 and 10 mg/kg) and SP600125 (10 mg/kg). After 6 hrs of LPS challenge, we euthanized mice and then collected lung tissue. JNK phosphorylation in lung tissue was detected by performing immunofluorescence assay. |
C66 And SP600125 Inhibited LPS-Induced Inflammatory Cytokines Expression Through Inhibiting Phosphorylation Of JNK In MPMs
The protective effects of C66 and SP600125 in LPS-induced ALI through its anti-inflammatory activity in mice were detailed in the present study. Then, the anti-inflammatory effects of C66 and SP600125 in vitro are to be detected. MPMs were pretreated with DMSO, 5 or 10 μM C66, or 10 μM SP600125 for 30 mins, followed by the incubation with or without 0.5 μg/mL LPS for 24 hrs. The secretion of TNF-α (Figure 6A) and IL-6 (Figure 6B) induced by LPS was significantly inhibited by C66 and SP600125, and the inhibitory rate of C66 was dose-dependent. To detect the expression of inflammatory genes, MPMs were pretreated with DMSO, 5 or 10 μM C66, or 10 μM SP600125 for 30 mins, followed by the incubation with or without 0.5 μg/mL LPS for 6 hrs. LPS caused a significant increase in the mRNA expression of pro-inflammatory cytokines, including TNF-α (Figure 6C), IL-6 (Figure 6D), IL-1β (Figure 6E), and COX-2 (Figure 6F) as shown in Figure 6C–F. In contrast, SP600125 at 10 μM and C66 dose-dependently decreased the expression of these transcripts. Further, we detected the effects of C66 on JNK phosphorylation in MPMs. Pretreatment with C66 can inhibit LPS-induced phosphorylation of JNK at both concentrations as shown in Figure 6G. Also, the inhibitory activity of C66 at 10 μM was equivalent to SP600125 at 10 μM. These results suggest that C66 and SP600125 show their anti-inflammatory activity via the JNK pathway in vitro.
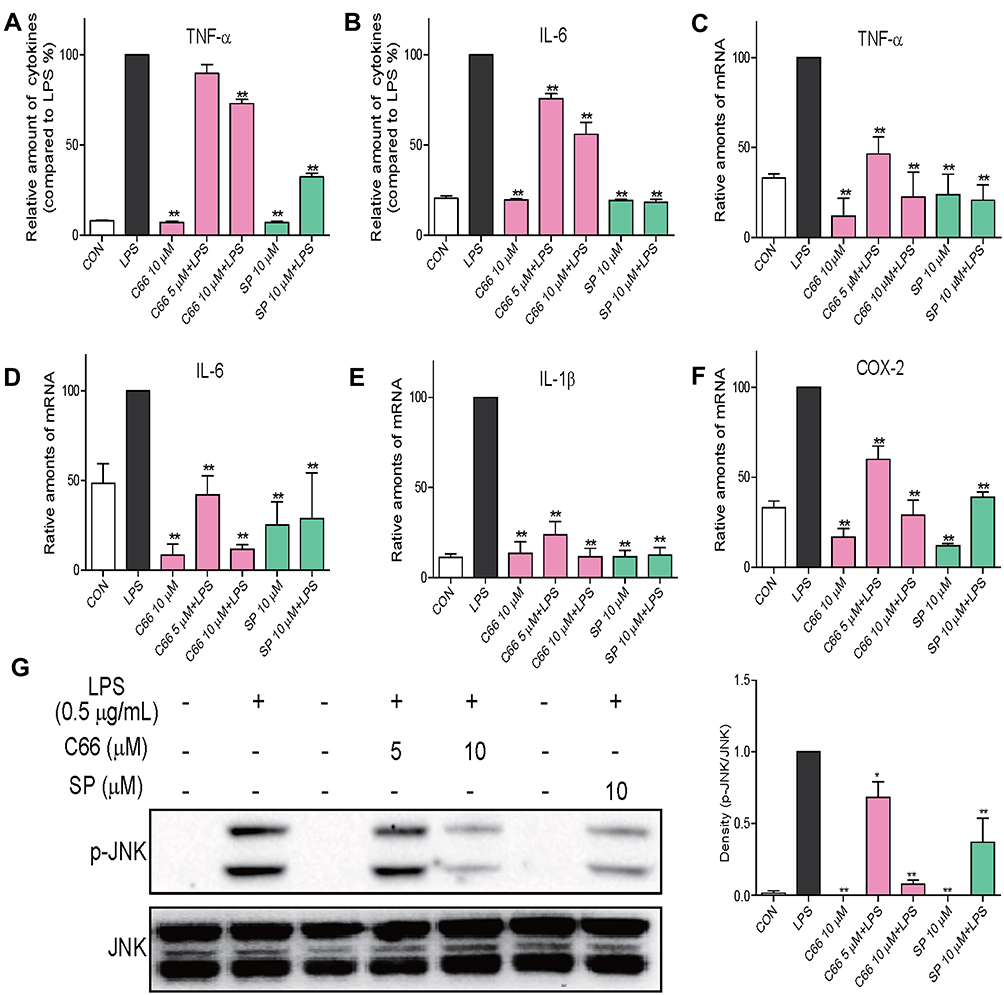 |
Figure 6 C66 and SP600125 inhibited LPS-induced inflammation via JNK pathway in macrophages. MPMs were pretreated with C66 at various doses (5, 10 μM) or SP600125 at 10 μM or vehicle (DMSO) for 30 mins before 0.5 μg/mL LPS treatment. (A, B) After 24 hrs of treatment of LPS, medium was collected. ELISA assay was performed to detect the production of inflammatory cytokines TNF-α (A) and IL-6 (B) in medium. (C–F) After 6 hrs of treatment of LPS, total mRNA was extracted using the TRIzol agent. By performing real-time quantitative PCR analysis, the mRNA levels of TNF-α (C), IL-6 (D), IL-1β (E), and COX-2 (F) were detected. (G) After 30 mins-treatment of LPS, total protein was collected. The protein levels of p-JNK were detected by Western blot assay, and JNK served as the loading control. Data are mean ± SEM of more than three independent experiments performed in duplicate. *P < 0.05, **P < 0.01 vs LPS group. |
Discussion
ALI is considered as a major cause of death in the intensive care unit. Nemours medications have been applied to treat ALI. Yet no treatment has been approved to be effective. The present study found that curcumin analog C66 could reduce LPS-induced edema, pathological variations, protein exudation, inflammatory cell infiltration, and inflammatory cytokine gene expression in mouse lung tissues. Furthermore, C66 inhibits LPS-induced inflammatory cytokines expression in MPMs. The pulmonary protective effects of C66 were found to be mediated by inhibiting the JNK pathway. These findings suggest that C66 may have clinical utility for the treatment of ALI.
The physiological hallmark of ALI is the disruption of the alveolar–capillary membrane barrier (i.e., pulmonary vascular leak).13,14 As a result, noncardiogenic pulmonary edema is developed, in which a proteinaceous exudate floods the alveolar spaces, impairs gas exchange, and precipitates respiratory failure.15 One of our previous studies showed that curcumin could inhibit LPS-induced increase of protein concentration and neutrophils in BALF.16 In a rat model of ALI, SP600125 was found being able to inhibit LPS-induced total protein and neutrophil influx into the lungs.17 In line with these studies, we show that LPS injection can increase wet/dry ratio, protein concentration, and inflammatory cells in BALF. These observations significantly suggest that curcumin analog C66 is capable of attenuating LPS-induced ALI.
JNK, a vital member of MAPK family, is involved in a larger number of physiological and pathological processes, including inflammation, apoptosis, cancer, diabetes, and metabolism.18,19 Many extracellular stimuli or chemical stimuli can activate the JNK pathway, which include LPS, cytokines, growth factors, pH, oxidation–reduction, and stress.20 When LPS stimulates the activation of TLR4 signaling, downstream JNK signaling pathway is activated, which upregulates various inflammatory cytokines and adhesion molecule expression, resulting in acute lung tissue injury.8,21 Many natural products and synthesized compounds, including berberine, resveratrol, curcumin and anisalcohol, were reported able to reduce inflammation by inhibiting activation of TLR4 and JNK pathway.22-25 C66, synthesized by our group, was reported able to attenuate diabetic cardiomyopathy and nephropathy by targeting JNK.11,12 The present study shows that C66 and JNK inhibitor, SP600125, are capable of inhibiting LPS-induced JNK phosphorylation both in vitro and in vivo. These results further confirm that JNK is critical for LPS-induced ALI.
Conclusion
Our current research shows that pretreatment with C66 and SP600125 could inhibit LPS-induced lung injury and inflammation by targeting the JNK pathway. These results imply that C66 can not only mitigate the development of diabetic complications but also inhibit LPS-induced ALI. Though there is a long way ahead to make C66 a drug, our study verifies the pivotal role of JNK in LPS-induced ALI and made C66 as one important candidate in the continuing drug development and research.
Abbreviations
ALI, acute lung injury; BALF, bronchoalveolar lavage fluid; LPS, lipopolysaccharide; JNK, c-Jun N-terminal Kinase; MAPKs, mitogen activated protein kinases; MPMs, mouse peritoneal macrophages; SEM, mean ± standard error of the mean.
Acknowledgment
The language of this manuscript has been revised by a native speaker from Zibo Yimore Translation Co. Ltd.
Author Contributions
All authors contributed to data analysis, drafting, or revising the article; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare there are no competing interests.
References
1. Wheeler AP, Bernard GR. Acute lung injury and the acute respiratory distress syndrome: a clinical review. Lancet. 2007;369(9572):1553–1564. doi:10.1016/S0140-6736(07)60604-7
2. Rubenfeld GD, Caldwell E, Peabody E, et al. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353(16):1685–1693. doi:10.1056/NEJMoa050333
3. Goodman RB, Pugin J, Lee JS, Matthay MA. Cytokine-mediated inflammation in acute lung injury. Cytokine Growth Factor Rev. 2003;14(6):523–535. doi:10.1016/S1359-6101(03)00059-5
4. Kaminska B. MAPK signalling pathways as molecular targets for anti-inflammatory therapy—from molecular mechanisms to therapeutic benefits. Biochim Biophys Acta Proteins Proteom. 2005;1754(1–2):253–262. doi:10.1016/j.bbapap.2005.08.017
5. Chen X, Zong C, Gao Y, et al. Curcumol exhibits anti-inflammatory properties by interfering with the JNK-mediated AP-1 pathway in lipopolysaccharide-activated RAW264. 7 cells. Eur J Pharmacol. 2014;723:339–345. doi:10.1016/j.ejphar.2013.11.007
6. Zheng Y, Zhang M, Zhao Y, Chen J, Li B, Cai W. JNK inhibitor SP600125 protects against lipopolysaccharide‑induced acute lung injury via upregulation of claudin‑4. Exp Ther Med. 2014;8(1):153–158. doi:10.3892/etm.2014.1684
7. Zhang Y, Wu J, Ying S, et al. Discovery of new MD2 inhibitor from chalcone derivatives with anti-inflammatory effects in LPS-induced acute lung injury. Sci Rep. 2016;6:25130. doi:10.1038/srep25130
8. Zhang Y, Xu T, Pan Z, et al. Shikonin inhibits myeloid differentiation protein 2 to prevent LPS‐induced acute lung injury. Br J Pharmacol. 2018;175(5):840–854. doi:10.1111/bph.v175.5
9. Li C, Yang D, Cao X, et al. LFG-500, a newly synthesized flavonoid, attenuates lipopolysaccharide-induced acute lung injury and inflammation in mice. Biochem Pharmacol. 2016;113:57–69. doi:10.1016/j.bcp.2016.05.007
10. Liang G, Zhou H, Wang Y, et al. Inhibition of LPS‐induced production of inflammatory factors in the macrophages by mono‐carbonyl analogues of curcumin. J Cell Mol Med. 2009;13(9b):3370–3379. doi:10.1111/j.1582-4934.2009.00711.x
11. Pan Y, Zhang X, Wang Y, et al. Targeting JNK by a new curcumin analog to inhibit NF-kB-mediated expression of cell adhesion molecules attenuates renal macrophage infiltration and injury in diabetic mice. PLoS One. 2013;8(11):e79084. doi:10.1371/journal.pone.0079084
12. Pan Y, Wang Y, Zhao Y, et al. Inhibition of JNK phosphorylation by a novel curcumin analog prevents high glucose-induced inflammation and apoptosis in cardiomyocytes and the development of diabetic cardiomyopathy. Diabetes. 2014;63(10):3497–3511.
13. Bhattacharya J, Matthay MA. Regulation and repair of the alveolar-capillary barrier in acute lung injury. Annu Rev Physiol. 2013;75:593–615. doi:10.1146/annurev-physiol-030212-183756
14. Ware, Lorraine. (2006). Pathophysiology of Acute Lung Injury and the Acute Respiratory Distress Syndrome. Seminars in respiratory and critical care medicine. 27. 337–49. doi:10.1055/s-2006-948288.
15. Johnson ER, Matthay MA. Acute lung injury: epidemiology, pathogenesis, and treatment. J Aerosol Med Pulm Drug Deliv. 2010;23(4):243–252. doi:10.1089/jamp.2009.0775
16. Zhang Y, Liang D, Dong L, et al. Anti-inflammatory effects of novel curcumin analogs in experimental acute lung injury. Respir Res. 2015;16(1):43. doi:10.1186/s12931-015-0199-1
17. Lee HS, Kim HJ, Moon CS, Chong YH, Kang JL. Inhibition of c-Jun NH 2-terminal kinase or extracellular signal-regulated kinase improves lung injury. Respir Res. 2004;5(1):23. doi:10.1186/1465-9921-5-23
18. Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Cell Biol. 2007;19(2):142–149. doi:10.1016/j.ceb.2007.02.001
19. Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Genet Dev. 2002;12(1):14–21. doi:10.1016/S0959-437X(01)00258-1
20. Zeke A, Misheva M, Reményi A, Bogoyevitch MA. JNK signaling: regulation and functions based on complex protein-protein partnerships. Microbiol Mol Biol Rev. 2016;80(3):793–835.
21. Kesharwani SS, Ahmad R, Bakkari MA, et al. Site-directed non-covalent polymer-drug complexes for inflammatory bowel disease (IBD): formulation development, characterization and pharmacological evaluation. J Control Release. 2018;290:165–179. doi:10.1016/j.jconrel.2018.08.004
22. Wang Y. Attenuation of berberine on lipopolysaccharide-induced inflammatory and apoptosis responses in β-cells via TLR4-independent JNK/NF-κB pathway. Pharm Biol. 2014;52(4):532–538. doi:10.3109/13880209.2013.840851
23. Zhong L-M, Zong Y, Sun L, et al. Resveratrol inhibits inflammatory responses via the mammalian target of rapamycin signaling pathway in cultured LPS-stimulated microglial cells. PLoS One. 2012;7(2):e32195. doi:10.1371/journal.pone.0032195
24. Xiang B, Xiao C, Shen T, Li X. Anti-inflammatory effects of anisalcohol on lipopolysaccharide-stimulated BV2 microglia via selective modulation of microglia polarization and down-regulation of NF-κB p65 and JNK activation. Mol Immunol. 2018;95:39–46. doi:10.1016/j.molimm.2018.01.011
25. Meng Z, Yan C, Deng Q, Gao D-F, Niu X-L. Curcumin inhibits LPS-induced inflammation in rat vascular smooth muscle cells in vitro via ROS-relative TLR4-MAPK/NF-κB pathways. Acta Pharmacol Sin. 2013;34(7):901. doi:10.1038/aps.2013.24
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.