")
Back to Journals » Drug Design, Development and Therapy » Volume 12
Inhibition of ghrelin o-acyltransferase attenuated lipotoxicity by inducing autophagy via AMPK–mTOR pathway
Authors Zhang S , Mao Y, Fan X
Received 6 December 2017
Accepted for publication 7 March 2018
Published 18 April 2018 Volume 2018:12 Pages 873—885
DOI https://doi.org/10.2147/DDDT.S158985
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Shaoren Zhang, Yuqing Mao, Xiaoming Fan
Department of Gastroenterology and Hepatology, Jinshan Hospital of Fudan University, Shanghai, China
Background: Nonalcoholic fatty liver disease (NAFLD) has been considered the most commonly occurring chronic hepatopathy in the world. Ghrelin o-acyltransferase (GOAT) is an acylation enzyme which has an acylated position 3 serine on ghrelin. Recent investigation revealed that activated autophagy could attenuate liver steatosis. The aim of this study was to explore therapeutic roles that inhibit GOAT exerted in NAFLD, and its potential association with autophagy.
Materials and methods: Human LO2 cells were pretreated with siRNA-GOAT to induce liver steatosis using free fatty acids (FFAs). A chronic NAFLD model was established by feeding male mice C57bl/6 with high-fat diet (HFD) for 56 days with GO-CoA-Tat administrated subcutaneously. Lipid droplets were identified by Oil Red O stains. Body weight (BW) of mice was measured every week. Autophagy, tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), serum biochemical indicators (glucose [Glu], total cholesterol [TC], triglyceride [TG], aspartate aminotransferase [AST], alanine aminotransferase [ALT]) and signaling pathway proteins of phosphorylated AMPK–mTOR were measured.
Results: The TG contents of the FFA and HFD groups were decreased by the inhibition of GOAT. Among mice treated with GO-CoA-Tat and siRNA-GOAT, IL-6 and TNF-α concentrations were remarkably decreased. Indicators of liver injury such as ALT and AST were also remarkably decreased among mice treated with GO-CoA-Tat. Likewise, GO-CoA-Tat significantly reduced the BW of mice and serum TG, TC and Glu. Autophagy was induced along with reduced lipids in the cells of the FFA and HFD groups. The inhibition of GOAT upregulated autophagy via AMPK–mTOR restoration.
Conclusion: These results indicate that the inhibition of GOAT attenuates lipotoxicity by autophagy stimulation via AMPK–mTOR restoration and offers innovative evidence for using GO-CoA-Tat or siRNA-GOAT in NAFLD clinically.
Keywords: ghrelin o-acyltransferase, GOAT, high-fat diet, HFD, free fatty acid, FFA, nonalcoholic fatty liver disease, NAFLD
Introduction
Nonalcoholic fatty liver disease (NAFLD) is the most prevalent chronic liver disease worldwide and is an important risk factor for the development of liver fibrosis and cirrhosis.1 Currently, it is estimated that more than 46% of adults in the US are suffering from liver steatosis.2 Liver steatosis is closely associated with a group of metabolic disorders, including, type 2 diabetes and obesity.3 Increased lipids is not offset by increased secretion of lipoproteins or by mitochondrial oxidation.4 Human fatty liver results from reduced turnover of liver fat granules.5 In addition, currently, NAFLD cannot be cured effectively by any one therapy alone if the patients do not modify their lifestyles.6 Consequently, innovative and effective therapies need to be developed urgently.
Ghrelin o-acyltransferase (GOAT), a member of membrane-bound O-acyltransferases family, is essential for octanoylation of ghrelin, which is required for active ghrelin to bind to and activate its receptor.7 GOAT inhibitor GO-CoA-Tat, a peptide-based bisubstrate analog, can reduce weight gain and improve glucose (Glu) tolerance in wild-type mice or reduce food intake.8,9 Several other effects of GOAT have been reported, including an involvement in Glu homeostasis, bile acid reabsorption and altered responsiveness to salt and lipid taste.10–12 GOAT may be involved in insulin resistance (IR), inflammatory reaction and lipid metabolism disorders. Each of these aspects exerts an essential role in pathogenic process and mechanism of NAFLD.13,14 Consequently, further investigation of the role exerted by GOAT in NAFLD as well as the mechanism of this process, are urgently needed.
Autophagy, known as a degrading process in lysosomes, can regulate cellular lipid metabolism, ameliorate the state of IR and mediate overactive innate immunoreaction.15 Recently, a few studies revealed its roles in pathogenic process of NAFLD.15,16 The association between NAFLD and autophagy was further uncovered by progress made on autophagy. Lipid metabolism was probably stimulated by autophagy which exhibited potential in the development of innovative therapies.16 It is reported by many studies that autophagy was extremely likely to be associated with variations in pathology and physiology during NAFLD and autophagic activity was decreased by high-fat diet (HFD) or chronically accumulated lipids.15–17
However, how GOAT regulates metabolisms of lipids is unclear. Related studies on NAFLD models of rodents are rare. Whether GOAT is related to autophagic process which ameliorates hepatic damage resulting from NAFLD is still unrevealed. The current study is aimed at investigating whether the inhibition of GOAT decreased triglyceride (TG) levels and inflammatory damage to the liver during NAFLD and whether autophagy participated in this physiological activity via AMPK–mTOR pathway.
Our results showed that the inhibition of GOAT increased lipid droplet turnover of hepatocytes via pathway mediated by autophagy. It was demonstrated by a mouse NAFLD model in the current study that autophagy was related to the inhibition of lipid removal in the liver induced by GOAT, which explained that the inhibition of GOAT was capable of relieving steatosis. Furthermore, autophagy induced by the inhibition of GOAT was proved to be immediately linked to in vivo and in vitro metabolisms of lipids by utilizing pharmacological and genetic inhibitors of autophagy. In the end, it was found that AMPK–mTOR signaling pathway revealed the possible mechanism of the inhibition of GOAT promoting autophagy.
Materials and methods
Reagents
GO-CoA-Tat was provided by Phoenix Biotech (Beijing, China). Rapamycin, 3-methyladenine (3-MA), palmitic acid (PA), oleic acid (OA) and bovine serum albumin (BSA) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). The antibodies provided by Kaiji (Nanjing, China) included the following: mTOR, phosphorylated mTOR (p-mTOR), total AMPK, phosphorylated AMPK (p-AMPK) at Thr172, GOAT, Beclin-1 and LC3.
Cell culture and treatment
Human LO2 cells (obtained from Chinese Academy of Science Committee Type Culture Collection Cell Bank, Shanghai, China) were kept in Dulbecco’s Modified Eagle’s Medium (DMEM; Thermo Fisher Scientific, Waltham, MA, USA) containing 1% of penicillin–streptomycin (Thermo Fisher Scientific) and 10% of fetal bovine serum (Thermo Fisher Scientific) under the condition of 5% CO2 at 37°C. LO2 cells were exposed to 1 mM free fatty acid (FFA) mixture (oleate: palmitate, 2:1) for 24 hours to induce steatosis.
siRNA-GOAT was utilized to pretreat the cells with a final concentration of 10 nM for 1 hour, and then the cells were stimulated by FFA. The cells were grouped as follows: 1) normal control (NC) group, exclusive phosphate buffer saline (PBS) treatment as vehicle; 2) siRNA-GOAT group, treatment containing PBS dilution of siRNA-GOAT under the final concentration of 10 nm; 3) FFA group, 24 hours of treatment using 1 mM mixed FFA and 10 nm siRNA-GOAT.
Animals and treatment
Male mice C57bl/6 (8–10 weeks old, 23.0±2.0 g) were purchased from Shanghai Lingchang Biology Technology Co. Ltd. (Shanghai, China). HFD (D12492) (Shanghai QF Biosciences, Shanghai, China) was utilized with the aim of inducing steatohepatitis and obesity. The experimental animals were kept under the condition of 23°C, 50% humidity and a 12-hour cycle of light and dark. The approval of the Animal Care and Use Committee of Shanghai Fudan University was obtained for all protocols involved in animals and according to the “Guidelines for the Care and Use of Laboratory Animals” published by the National Academy Press (NIH Publication No 85-23, revised 1996).
A total of 24 mice were grouped equally at random as follows: 1) saline solution group, the animals received normal mice diet and intraperitoneal injection of saline solution every day from the third to fourth week; 2) GO-CoA-Tat group, the animals received normal mice diet throughout the 8 weeks and intraperitoneal injection of GO-CoA-Tat with the dosage of 96 μg/kg from the third to the fourth week; 3) HFD group, the animals received HFD throughout the 8 weeks; 4) HFD+GO-CoA-Tat group, the animals received HFD throughout the 8 weeks and intraperitoneal injection of GO-CoA-Tat with the dosage of 96 μg/kg from the third to fourth week. On the eighth week, all subject animals were subjected to euthanasia with blood and liver specimens collected for molecular, serum and histological analyses and tissues for TG and histological estimations and reverse transcriptase polymerase chain reaction (RT-PCR), Western blot and electron microscope analyses.
Biochemical assays
The serous levels of Glu, total cholesterol (TC), aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were determined using kits for microplate tests from Jiancheng (Nanjing, China). Enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems, Inc., Minneapolis, MN, USA) were applied for the detection of proinflammatory factors including interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) of LO2 cells. Kits for microplate tests from Jiancheng were used to determine TG levels of LO2 cells and mice livers.
Histopathological sections
In each subject animal, the intermediate part of the left lobe of the liver was collected and sliced before perfusion using 4% paraformaldehyde for 24 hours at least. Subsequently, paraffin was used to embed the fixed tissues, and Oil Red O (Sigma-Aldrich Co.) was used to stain the slices with the thickness of 5 μm to observe the intracellular lipid droplets with a light microscope.
Oil Red O stains
The levels of lipids were assessed by Oil Red O stains on cells. Initially, 4% paraformaldehyde was used to fix the cells for 20 minutes. Subsequently, Oil Red O was utilized to stain them for 15 minutes under ambient temperature, and then the cells were washed twice with PBS. Finally, an optical microscope was utilized to observe the lipid droplets inside cells.
Immunofluorescence stains
Cells were fixed with 4% paraformaldehyde for 20 minutes. About 5% of BSA was utilized to block nonspecific sites, and anti-LC3II antibody with the dilution ratio of 1:50 was applied to the cell incubation for 12 hours under the temperature of 4°C. After that, PBS was utilized to wash the cells thrice for 5 minutes, and anti-rabbit secondary antibody was used to incubate them for 30 minutes. Subsequently, nuclei of the cells were stained by 4′,6-diamidino-2-phenylindole (DAPI; Thermo Fisher Scientific).
RNA isolation and real-time quantitative reverse transcriptase polymerase chain reaction (qRT-PCR)
After isolation of total RNAs, QuantiTect SYBR Green PCR Kit (Qiagen NV, Venlo, the Netherlands) was utilized to perform qRT-PCR based on the manufacturer’s instruction. qRT-PCR was utilized to evaluate the expression profiles of LO2 cells and liver tissues. TRIzol reagent (Tiangen Biotech, Beijing, China) was applied to total RNA extraction from cells and frozen liver tissue specimens, and a 7900HT Fast Real-Time PCR system (Thermo Fisher Scientific) was used to perform qPCR using SYBR Green Quantitative RT-PCR to profile the expressions of relevant genes based on the instructions of SYBR Premix EX Taq (TaKaRa Biotechnology, Dalian, China). Sequences of primers are available on request. The primer sequences are summarized in Table 1.
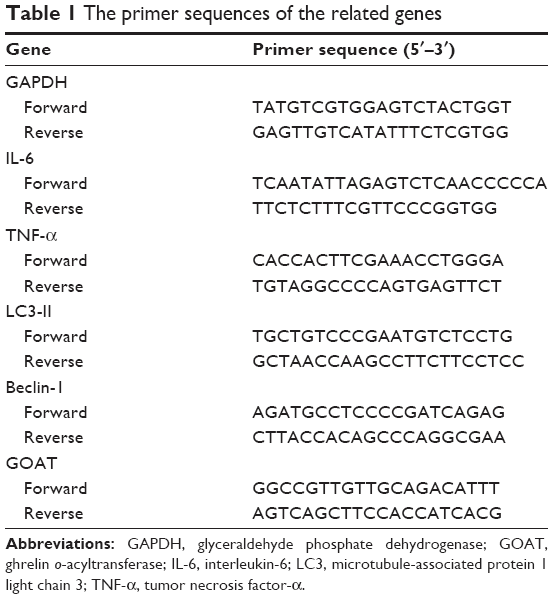 | Table 1 The primer sequences of the related genes |
siRNA transfection
Specific siRNAs against objective genes or negative control siRNAs (AllStars Negative Control siRNA; Qiagen NV) with no silencing activity to any anthropic genes were used to treat the LO2 cells. In the current study, to characterize the role of siRNA-targeting GOAT in LO2 cells, we designed three siRNA-GOATs based on the sequence deposited in Shanghai GenePharma Co., Ltd (Shanghai, China) (Table 2) and synthesized three DNA fragments encoding the siRNA. All three siRNA-GOATs were shown to cause a significant decrease in GOAT mRNA and protein expression (data shown behind), so, we used the mixture of the three siRNA-GOATs to treat LO2 cells.
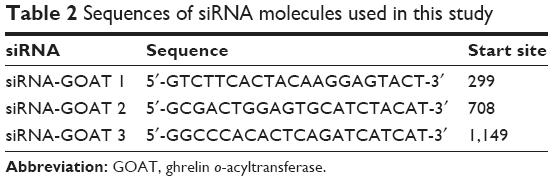 | Table 2 Sequences of siRNA molecules used in this study |
siRNAs were transfected according to the manufacturer’s protocol (Santa Cruz Biotechnology Inc., Dallas, TX, USA). The transfection medium was utilized to dilute all the siRNAs till the final concentration of 10 nM. Subsequently, 5 μL of Lipofectamine 2000 transfection reagent was added into the siRNA dilution for 20 minutes of incubation under the ambient temperature. After being washed by PBS twice, the cells were treated by siRNA mixture for 8 hours without penicillin or streptomycin. Subsequently, the treatment medium was switched into growth medium again, and the cells were cultured for 48 hours before the treatment of NaHs and fat emulsion.
Western blot
RIPA containing protease inhibitor was used to lyse the collection of LO2 cells and liver tissue specimens, and bicinchoninic acid (BCA) protocol was applied to measure protein concentrations. Initially, 8%–12% of sodium dodecyl sulfate (SDS)-polyacrylamide gel was utilized to isolate protein mixture. Subsequently, the protein spots in gel were transferred onto polyvinylidene difluoride (PVDF) membranes (ISEQ00010 0.22 mm; EMD Millipore, Billerica, MA, USA), which were successively incubated using relevant first antibody (LC3 [1:500], Beclin-1 [1:1,000], total AMPK [1:500], mTOR [1:500], p-AMPK [1:500]) and secondary antibody. In the end, the Odyssey two-color infrared laser imaging system (LI-COR Biosciences, Lincoln, NE, USA) was utilized to detect the protein levels.
Electron microscopy
LO2 cells were treated as described above. A transmission electron microscope (Tecnai; FEI, Portland, OR, USA) was used to examine the cell sections under the voltage of 160 kV, and an Electron Microscopy Film 4489 (Kodak, New York, NY, USA, ESTAR thick base) was used to obtain the electronic micrographs and print them on photographic paper.
Statistical analyses
The expression pattern of all data are mean ± SD. Kruskal–Wallis test was used to compare multiple groups with non-normal distribution. Mann–Whitney U-test was used to compare two groups with non-normal distribution. Student’s t-test was used to assess the differences between two groups, and one-way analysis of variance (ANOVA) and Tukey’s t-test were utilized to perform multiple comparison to normal distribution. Graphpad Prism 6.0 software (GraphPad Software, La Jolla, CA, USA) was applied to the conduction of statistical analyses. A P-value of <0.05 was considered statistically significant.
Results
siRNA-GOAT decreased GOAT mRNA and protein expression in LO2 cells
LO2 cells were subjected to the treatment of negative control siRNA or siRNAs specific for GOAT genes. RT-PCR was used to quantify the GOAT mRNA 48 hours after the cells were transfected with respective reagents. We found that siRNAs caused a significant decrease in GOAT mRNA expression (Figure 1A). It was very important whether relevant protein expressions were also significantly decreased after their genes were silenced by siRNA treatment. Therefore, we measured GOAT protein levels in LO2 cells transfected with siRNAs. The proteins isolated from cells in tests were measured using Western blot with specific GOAT antibody. We found that levels of GOAT protein were reduced remarkably among cells subjected to siRNA-GOAT treatment (Figure 1B). Consequently, it was concluded that siRNA-GOAT resulted in decreased mRNA transcription accompanied by GOAT protein.
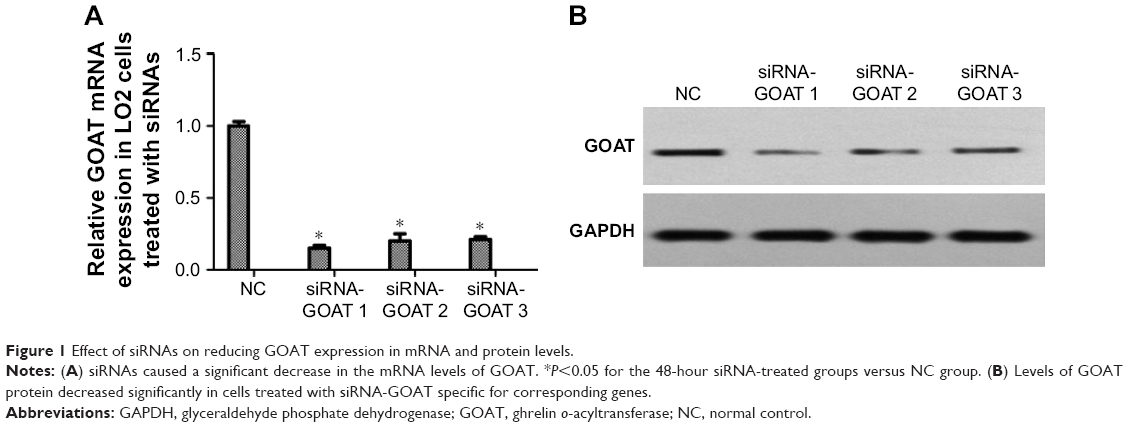 | Figure 1 Effect of siRNAs on reducing GOAT expression in mRNA and protein levels. |
Inhibition of GOAT reduced body weight (BW) of mice and maintained Glu homeostasis
C57bl/6 mice fed with HFD exhibited markedly higher BW than those fed with normal diet during the fourth to eighth week (Figure 2A). After 4 weeks of treatment with GO-CoA-Tat, BW of mice started decreasing remarkably in comparison to animals fed with HFD, and this tendency sustained throughout the rest of intervention (Figure 2A). The fasting blood Glu among animals subjected to HFD detected at week 8 was higher than that of the saline solution group. Animals subjected to GO-CoA-Tat exhibited markedly better tolerance to Glu than HFD-fed mice (Figure 2B).
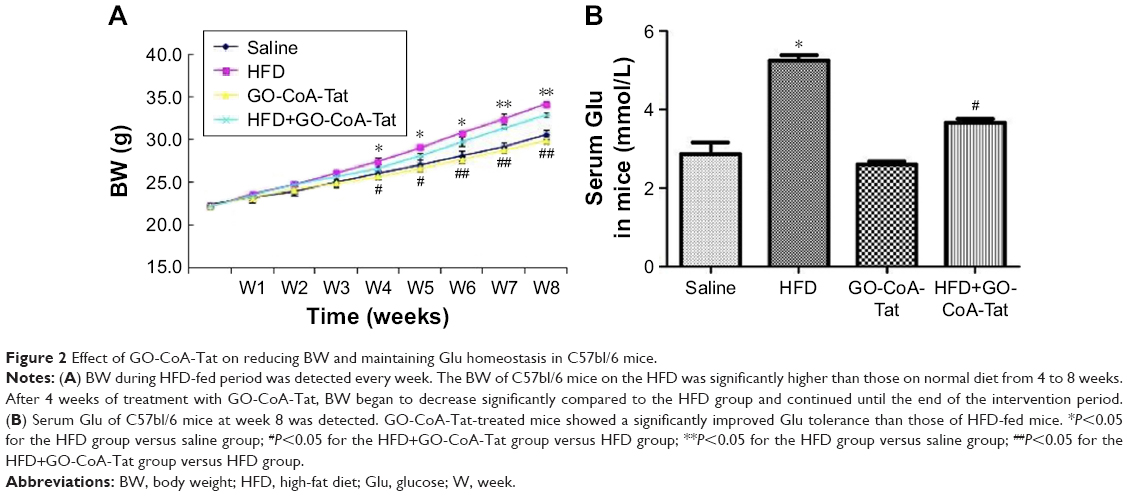 | Figure 2 Effect of GO-CoA-Tat on reducing BW and maintaining Glu homeostasis in C57bl/6 mice. |
Inhibition of GOAT resulted in reduction in accumulated lipids
It was revealed by Oil Red O stains and determined quantitatively by TG test kits as well as mice liver tissues that the accumulated lipids inside the cells were remarkably decreased by siRNA-GOAT before the stimulation with FFA (Figure 3A–C). In fact, animals fed with HFD were subjected to two dosages of GO-CoA-Tat treatment (32 μg/kg, 96 μg/kg) in pre-experiment, and the results revealed that animals subjected to higher dosage exhibited more significant effects. Consequently, the dosage of 96 μg/kg GO-CoA-Tat was utilized during the next experiment. HFD led to a higher liver TG level, higher serum TC and TG level in comparison to those of animals subjected to saline treatment, which were remarkably decreased due to GO-CoA-Tat treatment (Figure 3D).
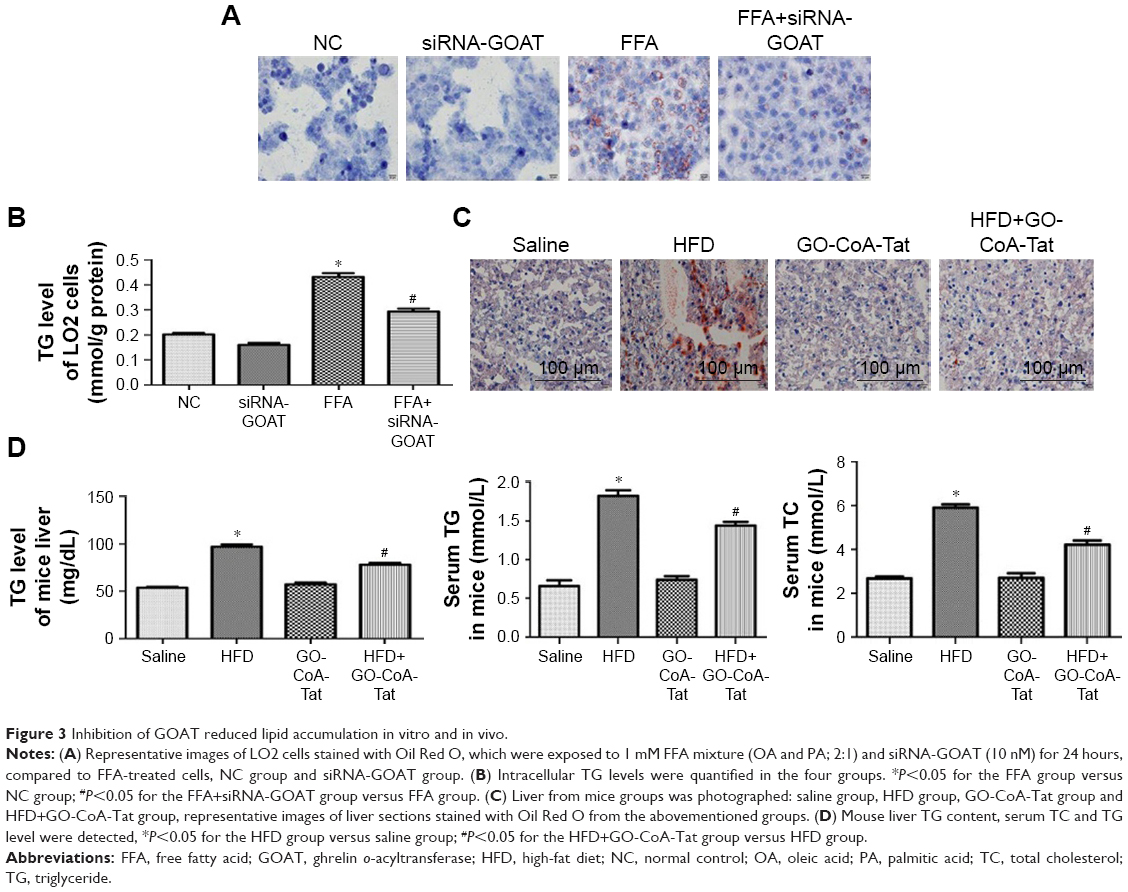 | Figure 3 Inhibition of GOAT reduced lipid accumulation in vitro and in vivo. |
Inhibition of GOAT relieved the livers inflammatory reaction resulting from the toxicity of lipids
Results of RT-PCR and ELISA showed that, after FFA treatment, proinflammatory cytokine transcriptions and expressions (IL-6 and TNF-α) were remarkably elevated. The quantities of protein and mRNA were both decreased after siRNA-GOAT treatment (Figure 4A and B). Among animals in the HFD group, the increased serous ALT and AST levels indicated that hepatic damages were remarkably decreased by the treatment of GO-CoA-Tat (Figure 4C). It was revealed that mRNA quantities of IL-6 and TNF-α were remarkably elevated among animals fed with HFD and decreased among those subjected to HFD and GO-CoA-Tat (Figure 4D).
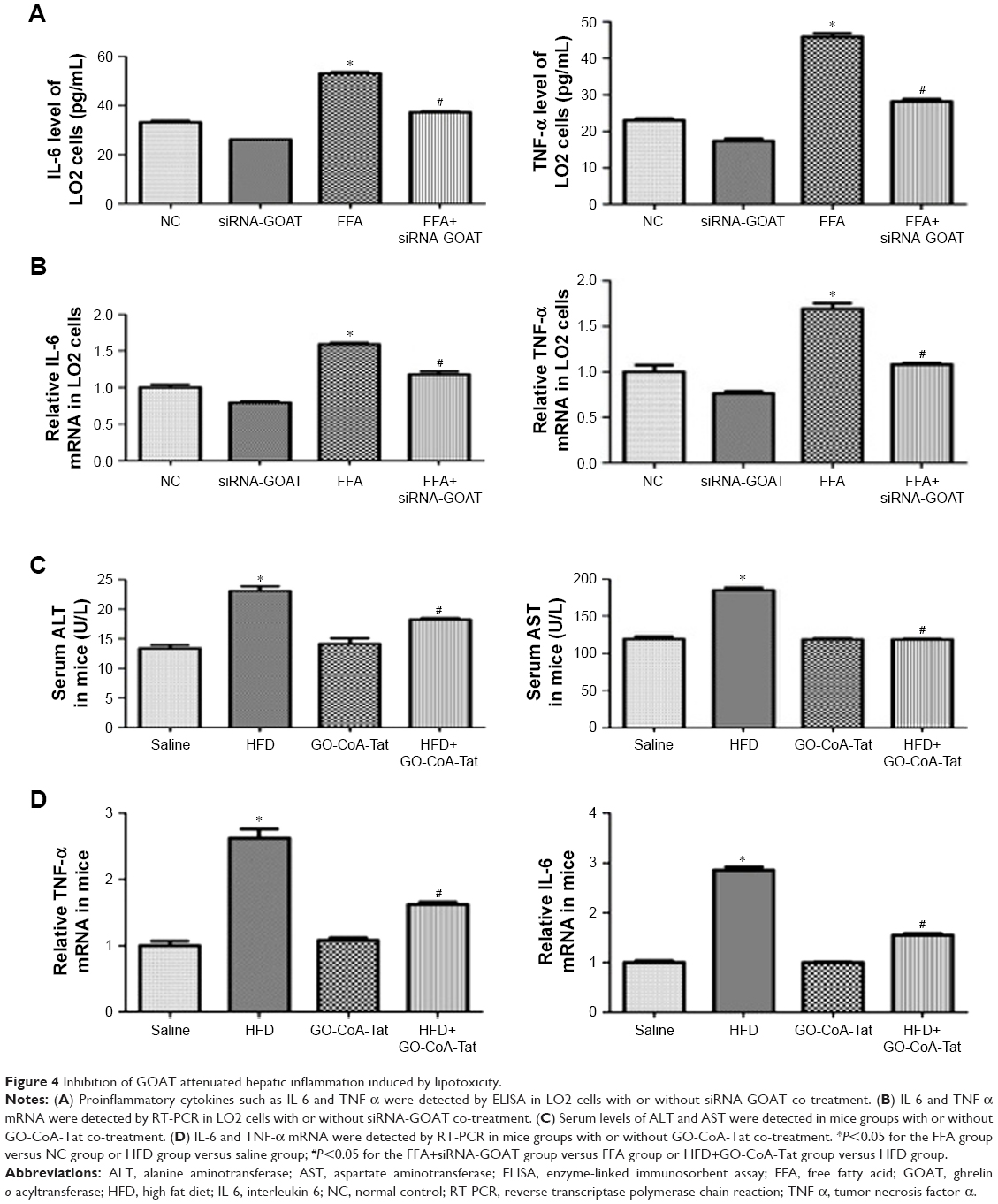 | Figure 4 Inhibition of GOAT attenuated hepatic inflammation induced by lipotoxicity. |
Clearance of lipids resulting from inhibited GOAT was accompanied by enhanced autophagic activity
The association between autophagic activity and clearance of lipids was examined in four ways: electron microscopy, immunofluorescence, RT-PCR and Western blot. Elevated mRNA transcription level of Beclin-1 and LC3-II existed in animals treated by FFA or HFD, which were subsequently induced by siRNA-GOAT in vitro or GO-CoA-Tat in vivo (Figure 5A and B). The protein levels of Beclin-1 and LC3-II were similar to the results of their mRNA transcription level (Figure 5C and D). Immunofluorescence of LC3-II revealed elevated autophagic flux among the FFA group, and siRNA-GOAT in vitro treatment further activated it (Figure 5E). It was revealed by electron microscopy that cells subjected to siRNA-GOAT co-treatment in vitro exhibited increasing autophagosome quantity (Figure 5F).
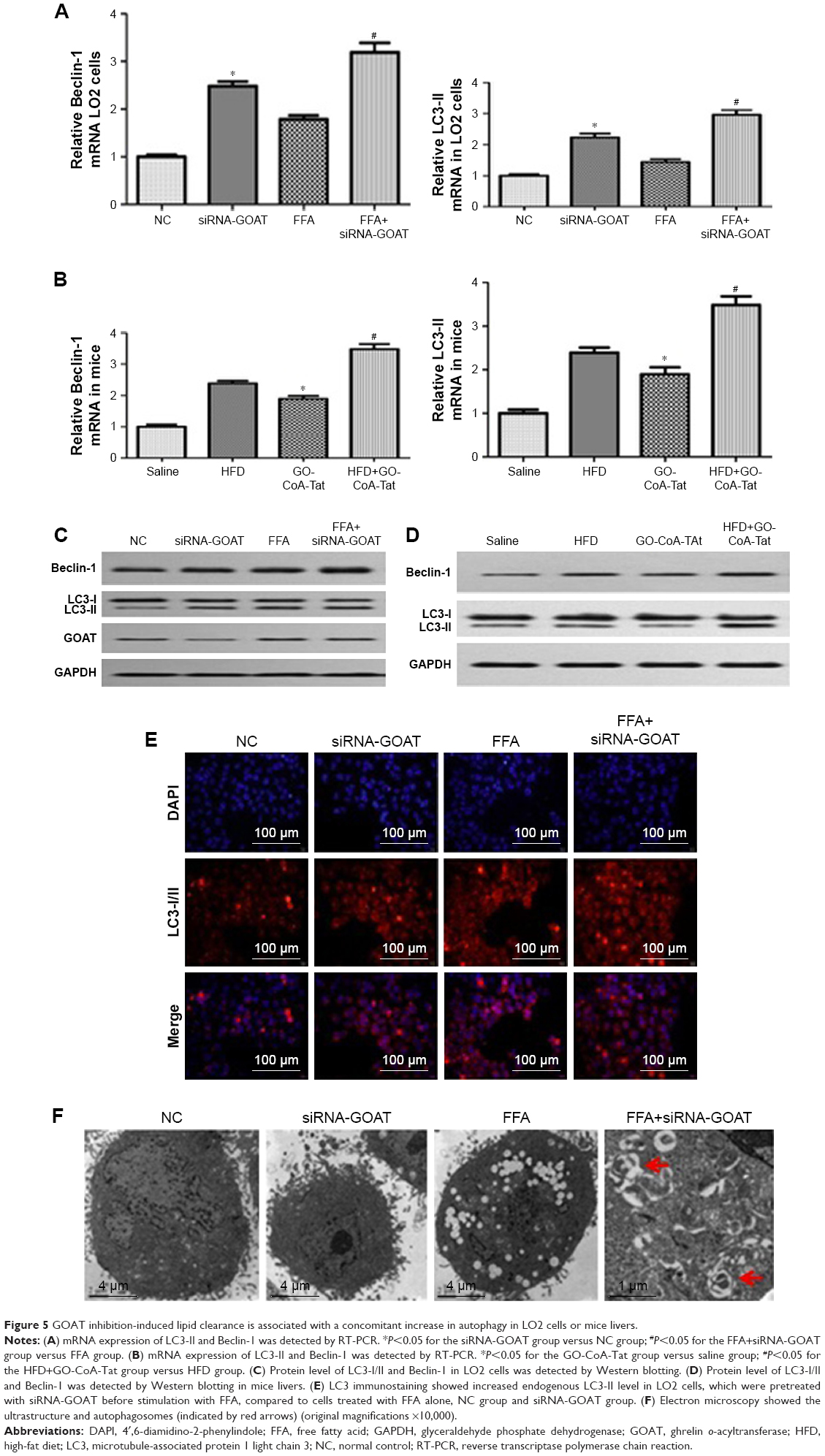 | Figure 5 GOAT inhibition-induced lipid clearance is associated with a concomitant increase in autophagy in LO2 cells or mice livers. |
The decrease in lipid droplet quantity was reversed by inhibited autophagy
With the aim of investigating whether TG reduction in cells due to siRNA-GOAT was reversed by autophagy inhibition, LO2 cells were subjected to treatment of 1 μM rapamycin (autophagic enhancer), 10 mM 3-MA (autophagic inhibitor) and 10 nM siRNA-ATG5. The intracellular lipid levels in the FFA+siRNA-ATG5 and FFA+3-MA groups were superior to those in the FFA+siRNA-GOAT group in spite of the significant reduction in accumulated lipids in LO2 cells due to rapamycin (Figure 6A). In comparison to the FFA+siRNA-GOAT group, the FFA+siRNA-ATG5 and FFA+3-MA groups exhibited decreased LC3-II levels. Conversely, the FFA+rapamycin group exhibited an increase in LC3-II level, which indicated that the enhanced autophagic activity resulting from siRNA-GOAT was restrained by siRNA-ATG5 or 3-MA and augmented by rapamycin stimulation (Figure 6B).
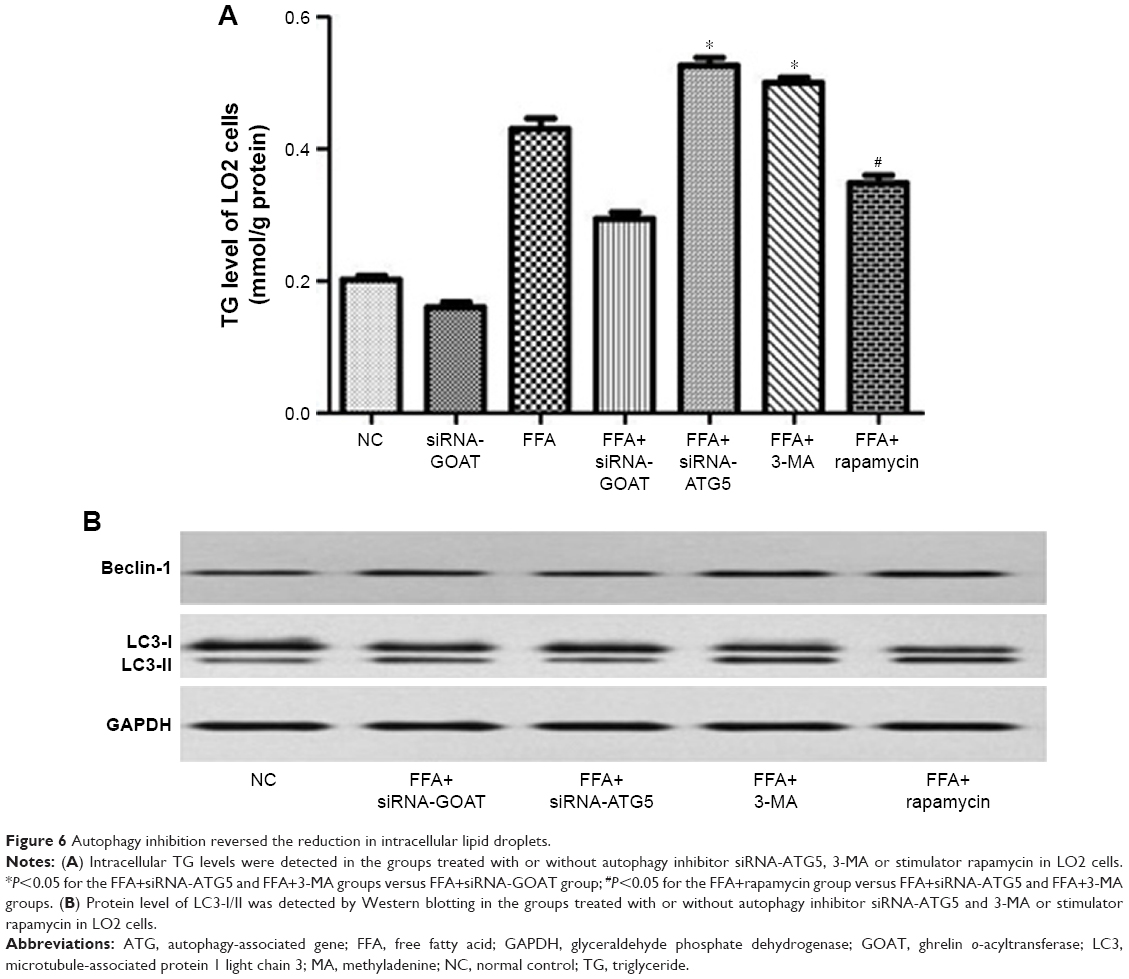 | Figure 6 Autophagy inhibition reversed the reduction in intracellular lipid droplets. |
GO-CoA-Tat or siRNA-GOAT enhanced autophagic activity partially through restoring AMPK–mTOR signaling pathway
It was revealed previously that autophagy was impaired by activated mTOR and induced by pharmacological mTOR inhibitors in most models. In the current study, p-AMPK was significantly inhibited by FFA or HFD and reconstructed by GO-CoA-Tat or siRNA-GOAT with unaffected entire AMPK level. According to Figure 7, mTOR that acted as one of the AMPK downstream molecules, accompanied by its phosphorylated form (p-mTOR), was found in LO2 cells or subject mice. The phosphorylation in subject animals and cells was restrained by GO-CoA-Tat or siRNA-GOAT. Consequently, it was verified that the toxicity of lipids was alleviated by GO-CoA-Tat or siRNA-GOAT which enhanced autophagic activity through reconstructing AMPK–mTOR signaling pathway.
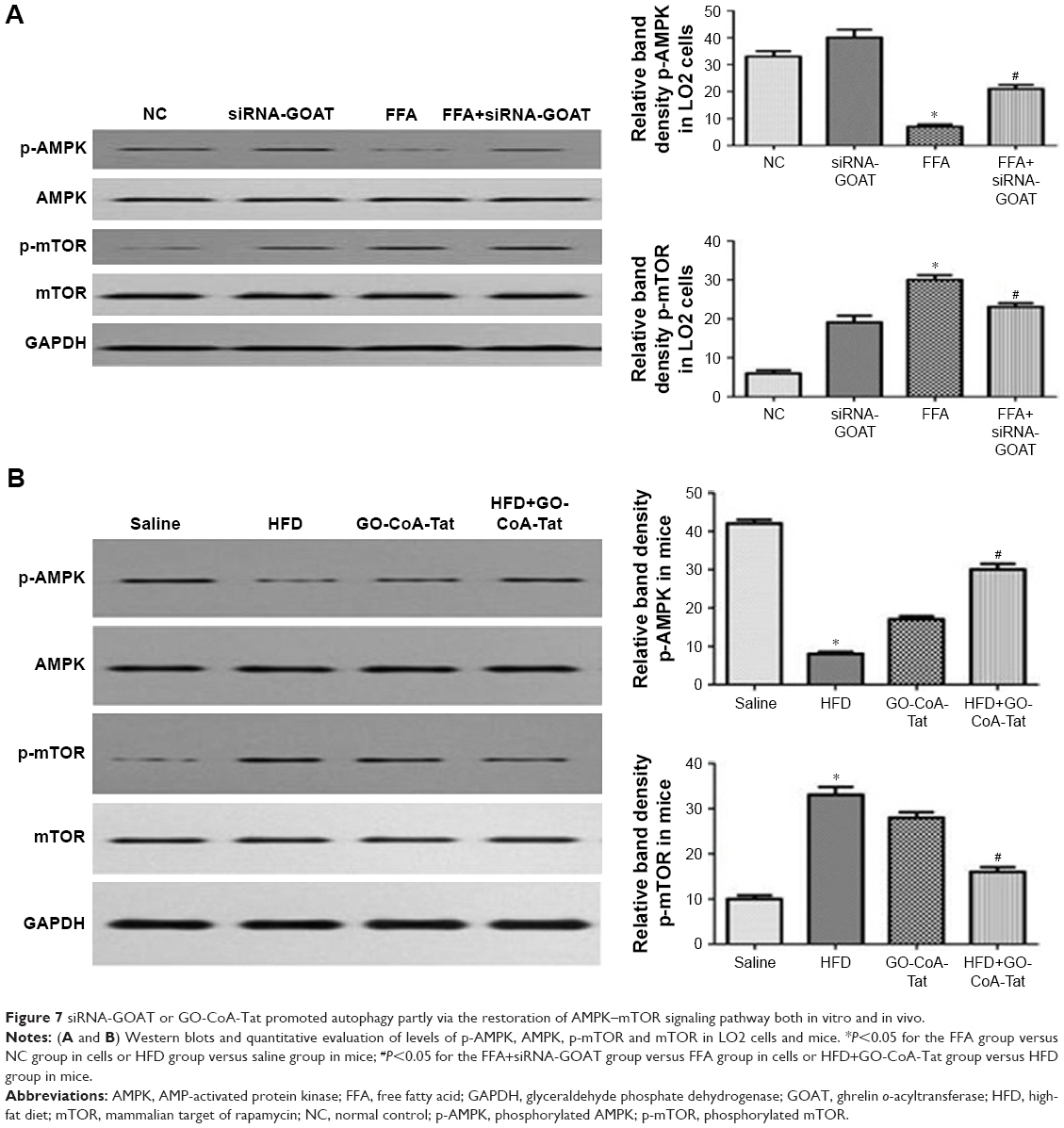 | Figure 7 siRNA-GOAT or GO-CoA-Tat promoted autophagy partly via the restoration of AMPK–mTOR signaling pathway both in vitro and in vivo. |
Discussion
Since obesity is becoming increasingly common, NAFLD has drawn much attention worldwide, and the risk of NAFLD increases with older age.18 NAFLD is initially quiet hepatopathy, which gradually turns into inflammatory reaction, fibrosis and liver cancer in the end. In addition, NAFLD exhibits close association with risk factors of coronary artery disease (CAD), for example, metabolic syndrome, diabetes and dyslipidemia, which are regarded as the major causes of death.19 Nevertheless, efficient approaches for preventing and attenuating NAFLD are extremely rare.
Our study found that intraperitoneal administration of GO-CoA-Tat could significantly reduce weight gain in HFD mice, and maintain Glu homeostasis (Figure 2). GOAT inhibitor GO-CoA-Tat, which was introduced by Barnett et al,8 showed GOAT was inhibited in cell lines stably expressing GOAT and preproghrelin as well as in vivo in mice. GO-CoA-Tat resulted in reduction of weight gain and improvement in Glu tolerance among wild-type mice, and it could reduce food intake.9 GOAT seems to be involved in NAFLD progression and fatty degeneration. The GOAT-ghrelin system was probably related to inflammatory reaction, dysfunction of lipid metabolism and IR, and all of which entirely exert essential roles in NAFLD pathogenic property.13,14 However, the principle of GO-CoA-Tat valued in NAFLD treatment was mostly unrevealed, particularly the effect of reducing accumulated lipids. In the current study the inhibition of GOAT was initially demonstrated to reduce accumulated lipids by promoting autophagic activity, partially through restoring the AMPK–mTOR signaling pathway.
This study verified that GO-CoA-Tat or siRNA-GOAT effectively decreased intracellular lipids (Figure 3) and increased autophagic flux (Figure 5) in LO2 cells and in mice livers. Elevated serum ALT and AST levels among HFD animals were effectively reduced after the administration of GO-CoA-Tat, indicating that the GO-CoA-Tat alleviated the chronic hepatic injury. Taking the improved analyses of histology into consideration (Figure 4C), obviously, the hepatic injury resulting from NAFLD was alleviated by GO-CoA-Tat at least in regard to histology and biochemistry. The miRNA and protein quantities of IL-6 and TNF-α induced by GO-CoA-Tat were both significantly decreased while NAFLD was developing (Figure 4A–D). The “obvious” property of GO-CoA-Tat inhibiting hepatic inflammation partially resulted from the fact that GO-CoA-Tat not only directly inhibited liver inflammatory reactions but also induced more variations in metabolism under HFD.
However, whether the inhibition of GOAT was related to autophagy when hepatic injuries induced by NAFLD were amelior not revealed. siRNA-GOAT reduced accumulated lipids in cells, while autophagy was increased accordingly both in vivo and in vitro (Figure 5). LO2 cells were subjected to pretreatment using siRNA-GOAT with gradient concentrations ranged from 5 to 20 nM, and then stimulated by mixed FFA during pre-experiments. The levels of LC3-II protein and TG inside the cells were determined although the results revealed that cells subjected to the treatment of 10 nM siRNA-GOAT exhibited lowest lipid and highest LC3-II protein levels (data not shown). Consequently, it was speculated that the induction of autophagy and reduced lipids were partially associated with siRNA-GOAT concentrations.
The mildly increased LC3-II expression among animals fed with HFD and cells treated by FFA was remarkably promoted by co-treatment of GO-CoA-Tat or siRNA-GOAT. Autophagy was promoted among cells treated by FFA and animals fed with HFD for a short period, by which the lipids were digested partially through mTOR complex. Conversely, autophagy was inhibited among cells treated by FFA and animals fed with HFD for a long period, and the accumulation of lipids was accelerated meanwhile.20 The lipid accumulation due to alcoholic fatty liver and NAFLD was reduced by autophagy.16,21,22 Autophagic vacuoles took up cholesterol and TG and transferred it to lysosomes which subsequently degraded these molecules into FFA in the lipophagic process. The redundant lipids were accumulated due to impairment of lipophagic activity.23 In the current study, it was revealed that autophagy was induced when the accumulated lipids in cells were reduced. Autophagy inhibitor and stimulator (3-MA and rapamycin, respectively) were used to demonstrate that autophagy is essential for reduction of intracellular fat accumulation (Figure 6). In accordance with expectations, TG level was elevated and autophagy was restrained by 3-MA and siRNA-ATG5. Conversely, autophagy was induced and TG quantity was obviously reduced due to rapamycin. The newly observed phenomenon that autophagy regulated the storage of lipids in cells was named as macrolipophagy which existed exclusively in the liver. The storage of lipids in cells was increased by 3-MA which inhibited autophagy pharmacologically and knocking down of ATG genetically.21 Lipids were accumulated excessively, and sequentially NAFLD developed due to ablative macrolipophagy.24 To sum up, it was demonstrated by the abovementioned literature that autophagy exerted an essential role in maintaining intra-hepatic lipid homeostasis.
AMPK pathway was subjected to the inhibition of fat accumulation and oxidative stress resulting from NAFLD,25,26 which was shown to play important roles in the initiation and progression of NAFLD. Furthermore, AMPK acts in the regulation of autophagy, which is activated by the elevation of the AMP/ATP ratio,27 according to the upregulation of autophagy under conditions of starvation, which activates autophagy. Since GOAT is an energy regulator which stimulates appetite, we hypothesized that AMPK may be the connection mechanism between GO-CoA-Tat and autophagy. In the current study, p-AMPK was significantly decreased due to NAFLD, while GO-CoA-Tat or siRNA-GOAT reestablished the quantity of phosphorylated kinases without disturbing its total form, which was in accordance with previous studies.25,28 HFD or FFA dramatically reduced p-AMPK, while GO-CoA-Tat or siRNA-GOAT recovered it without any effect on the quantity of total AMPK. As shown in Figure 7, mTOR that acted as one of the AMPK downstream molecules, accompanied by its phosphorylated form (p-mTOR), was found in LO2 cells or subject mice livers. The phosphorylation of mTOR was inhibited in vitro as well as in vivo by GO-CoA-Tat or siRNA-GOAT. It was verified that the toxicity of lipids was alleviated by GO-CoA-Tat or siRNA-GOAT which enhanced autophagic activity through reconstructing AMPK–mTOR signaling pathway.
Li et al29 have shown that ghrelin increases hepatosteatosis by increasing lipogenesis. As mentioned earlier, the inhibition of GOAT would decrease ghrelin. Our study showed that the inhibition of GOAT decreased hepatic lipotoxicity. However, other studies demonstrated that ghrelin increased lipophagy and reduced hepatic lipogenesis.25,30 We have to take into account the differences among these reports. Therefore, more studies are needed for further investigating the effect of ghrelin GOAT system on NAFLD. There are limitations in the current study. The products of oxidative reactions in mouse liver specimens and LO2 cells were not detected. Lipid dyes commonly applied to immunofluorescence experiments were probably more appropriate for manifesting the association between lipid droplets and autophagy. Other ways of inhibiting autophagy, for example, knocking out relevant genes, were probably more convincing. It is particularly worth noting that NAFLD34–37 was possibly treated by stimulating autophagy pharmacologically31,32 and hormonally.33
Conclusion
We have demonstrated that GO-CoA-Tat or siRNA-GOAT alleviated hepatic steatosis and inflammation induced by NAFLD through restoring AMPK–mTOR signaling pathway. Due to the fact that no drugs have been proved as effective in NAFLD (eg, chemicals that regulate energy metabolisms), the inhibition of GOAT, exhibit a very promising prospect in preventing and treating NAFLD.
Acknowledgment
The study was supported by research grants from the following: Outstanding Young Talent Training Program of Jinshan District Health and Family Planning Commission, Shanghai, China (no JSYQ201606); Youth Research Project of Shanghai Municipality Health and Family Planning Commission, China (no 20154Y0096); Surface Research Project of Jinshan District Health and Family Planning Commission, Shanghai, China (no JSKJ-KTMS-2015-02); and Key Clinical Discipline Construction of Jinshan District, Shanghai, China (no JSZK2015A06).
Disclosure
The authors report no conflicts of interest in this work.
References
Adams LA, Lymp JF, St Sauver J, et al. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology. 2005;129(1):113–121. | ||
Williams CD, Stengel J, Asike MI, et al. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: a prospective study. Gastroenterology. 2011;140(1):124–131. | ||
Berlanga A, Guiu-Jurado E, Porras JA, Auguet T. Molecular pathways in non-alcoholic fatty liver disease. Clin Exp Gastroenterol. 2014;7:221–239. | ||
Kawano Y, Cohen DE. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J Gastroenterol. 2013;48(4):434–441. | ||
Greenberg AS, Coleman RA, Kraemer FB, et al. The role of lipid droplets in metabolic disease in rodents and humans. J Clin Invest. 2011;121(6):2102–2110. | ||
Ferre P, Foufelle F. Hepatic steatosis: a role for de novo lipogenesis and the transcription factor SREBP-1c. Diabetes Obes Metab. 2010;12(suppl 2):83–92. | ||
Inui A, Asakawa A, Bowers CY, et al. Ghrelin, appetite, and gastric motility: the emerging role of the stomach as an endocrine organ. FASEB J. 2004;18(3):439–456. | ||
Barnett BP, Hwang Y, Taylor MS, et al. Glucose and weight control in mice with a designed ghrelin O-acyltransferase inhibitor. Science. 2010;330(6011):1689–1692. | ||
Teuffel P, Wang L, Prinz P, et al. Treatment with the ghrelin-O-acyltransferase (GOAT) inhibitor GO-CoA-Tat reduces food intake by reducing meal frequency in rats. J Physiol Pharmacol. 2015;66(4):493–503. | ||
Zhao TJ, Liang G, Li RL, et al. Ghrelin O-acyltransferase (GOAT) is essential for growth hormone-mediated survival of calorie-restricted mice. Proc Natl Acad Sci U S A. 2010;107(16):7467–7472. | ||
Kang K, Schmahl J, Lee JM, et al. Mouse ghrelin-O acyltransferase (GOAT) plays a critical role in bile acid reabsorption. FASEB J. 2012;26(1):259–271. | ||
Cai H, Cong WN, Daimon CM, et al. Altered lipid and salt taste responsivity in ghrelin and GOAT null mice. PLoS One. 2013;8(10):e76553. | ||
Yang J, Brown MS, Liang G, Grishin NV, Goldstein JL. Identification of the acyltransferase that octanoylates ghrelin, an appetite-stimulating peptide hormone. Cell. 2008;132(3):387–396. | ||
Gualillo O, Lago F, Dieguez C. Introducing GOAT: a target for obesity and anti-diabetic drugs? Trends Pharmacol Sci. 2008;29(8):398–401. | ||
Amir M, Czaja MJ. Autophagy in nonalcoholic steatohepatitis. Expert Rev Gastroenterol Hepatol. 2011;5(2):159–166. | ||
Sinha RA, Farah BL, Singh BK, et al. Caffeine stimulates hepatic lipid metabolism by the autophagy-lysosomal pathway in mice. Hepatology. 2014;59(4):1366–1380. | ||
Park HW, Lee JH. Calcium channel blockers as potential therapeutics for obesity-associated autophagy defects and fatty liver pathologies. Autophagy. 2014;10(12):2385–2386. | ||
Noureddin M, Yates KP, Vaughn IA, et al. Clinical and histological determinants of nonalcoholic steatohepatitis and advanced fibrosis in elderly patients. Hepatology. 2013;58(5):1644–1654. | ||
Kim D, Choi SY, Park EH, et al. Nonalcoholic fatty liver disease is associated with coronary artery calcification. Hepatology. 2012;56(2):605–613. | ||
Liu HY, Han J, Cao SY, et al. Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: inhibition of FoxO1-dependent expression of key autophagy genes by insulin. J Biol Chem. 2009;284(45):31484–31492. | ||
Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature. 2009;458(7242):1131–1135. | ||
Ding WX, Li M, Chen X, et al. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology. 2010;139(5):1740–1752. | ||
Liu K, Czaja MJ. Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ. 2013;20(1):3–11. | ||
Singh R. Autophagy and regulation of lipid metabolism. Results Probl Cell Differ. 2010;52:35–46. | ||
Li Y, Hai J, Li L, et al. Administration of ghrelin improves inflammation, oxidative stress, and apoptosis during and after non-alcoholic fatty liver disease development. Endocrine. 2013;43(2):376–386. | ||
Winder WW, Hardie DG. AMP-activated protein kinase, a metabolic master switch: possible roles in type 2 diabetes. Am J Physiol. 1999;277(1 pt 1):E1–E10. | ||
Yin XM, Ding WX, Gao W. Autophagy in the liver. Hepatology. 2008;47(5):1773–1785. | ||
Barazzoni R, Semolic A, Cattin MR, Zanetti M, Guarnieri G. Acylated ghrelin limits fat accumulation and improves redox state and inflammation markers in the liver of high-fat-fed rats. Obesity (Silver Spring). 2014;22(1):170–177. | ||
Li ZR, Xu GY, Qin Y, et al. Ghrelin promotes hepatic lipogenesis by activation of mTOR-PPARγ signaling pathway. Proc Natl Acad Sci U S A. 2014;111(36):13163–13168. | ||
Mao YQ, Cheng J, Yu FJ, Li HQ, Guo CY, Fan XM. Ghrelin attenuated lipotoxicity via autophagy induction and nuclear factor-κB inhibition. Cell Physiol Biochem. 2015;37(2):563–576. | ||
Shoji-Kawata S, Sumpter R, Leveno M, et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature. 2013;494(7436):201–206. | ||
Lin CW, Zhang H, Li M, et al. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J Hepatol. 2013;58(5):993–999. | ||
Sinha RA, You SH, Zhou J, et al. Thyroid hormone stimulates hepatic lipid catabolism via activation of autophagy. J Clin Invest. 2012;122(7):2428–2438. | ||
Ni HM, Williams JA, Yang H, Shi YH, Fan J, Ding WX. Targeting autophagy for the treatment of liver diseases. Pharmacol Res. 2012;66(6):463–474. | ||
Codogno P, Meijer AJ. Autophagy in the liver. J Hepatol. 2013;59(2):389–391. | ||
Ding WX. Induction of autophagy, a promising approach for treating liver injury. Hepatology. 2014;59(1):340–343. | ||
Lomonaco R, Sunny NE, Bril F, Cusi K. Nonalcoholic fatty liver disease. current issues and novel treatment approaches. Drugs. 2013;73(1):1–14. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.