")
Back to Journals » OncoTargets and Therapy » Volume 11
Inhibition of EZH2 and EGFR produces a synergistic effect on cell apoptosis by increasing autophagy in gastric cancer cells
Authors Yang Y, Zhu F , Wang Q, Ding Y, Ying R , Zeng L
Received 5 September 2018
Accepted for publication 17 October 2018
Published 29 November 2018 Volume 2018:11 Pages 8455—8463
DOI https://doi.org/10.2147/OTT.S186498
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Arseniy Yuzhalin
Youping Yang,1,2 Feng Zhu,3 Qingmei Wang,3 Yan Ding,2 Rongbiao Ying,2,* Linghui Zeng3,*
1Department of Pathology, The First People’s Hospital of Wenling City, Wenling City, Zhejiang Province 317500, China; 2Department of Surgical Oncology, The Taizhou Cancer Hospital, Wenling City, Zhejiang Province 317500, China; 3Department of Pharmacology, Zhejiang University City College, Hangzhou City, Zhejiang Province 310015, China
*These authors contributed equally to this work
Background: Numerous reports have shown that a combination of two or more drugs leads to better cancer treatment. Inhibitors of zeste homology 2 and epidermal growth factor receptor have been widely used in cancer treatments. However, the mechanisms of the combined use of these two drugs remain elusive.
Methods: Sulforhodamine B assays and Alexa Fluor®-488 Annexin V/Dead Cell Apoptosis Kit were used to detect the cell proliferation and cell apoptosis in vitro, respectively. Western blotting analysis was used to detect the relative protein expression, and xenografted tumor was generated in nude mice to evaluate the effect in vivo.
Results: Treatment with either Gefitinib ranging from 0 to 12.5 µM or GSK126 ranging from 0 to 8.3 µM caused a dose-dependent decrease in the cell survival fraction, and the combination of Gefitinib at 12.5 µM and GSK126 at 8.3 µM caused further significant decrease. The combination indexes were 0.061, 0.591, 0.713, and 0.371 for MGC803, A549, PC-3, and MDB-MA-231, respectively. In MGC803 cells, the combination of GSK126 and Gefitinib synergistically induced cell apoptosis (56.2%), which was markedly higher as compared to either drug alone (7.6% and 10.6%, P<0.05). Treatment with either Gefitinib or GSK126 alone induced a significant increase in cell apoptosis in LC3-II and p-ULK, whereas the combination of the two induced a further increase. Pretreatment with an autophagy inhibitor, 3-methyladenine, prevented the apoptosis induced by the combined use of Gefitinib and GSK126. In addition, the combined use of Gefitinib and GSK126 also inhibited the activation of mammalian target of rapamycin signaling pathway. Furthermore, the combined use of GSK126 and Gefitinib synergistically inhibited xenografted tumor proliferation.
Conclusion: The combined use of GSK126 and Gefitinib exerts a synergic effect on tumor growth inhibition both in vitro and in vivo through inducing autophagy and promoting apoptosis. Therefore, GSK126 and Gefitinib in combination may be considered as a potential strategy in treating solid tumor clinically.
Keywords: EZH2, EGFR, autophagy, mTOR signaling, gastric cancer
Introduction
Cancer is the second leading cause of death globally and one of the biggest challenges facing the biomedical scientists. Despite the considerable progression made in the therapy of advanced cancers, failure to various treatments remains inevitable in most patients. Specially, gastric cancer is the fourth most common cause of cancer-related death in the world and it remains difficult to cure primarily due to the persistent presence of the advanced disease.1 New therapeutic strategies, therefore, are urgently needed to improve the treatment of gastric cancer.
Enhancer of zeste homology 2 (EZH2), a catalytic subunit of polycomb repressive complex 2, is highly expressed in numerous cancers. EZH2 silences the gene expression by methylating the histone H3 at Lys27 and regulates the expression of numerous tumor suppressor genes that are associated with cancer initiation, progression, metastasis, and prognosis.2–4 GSK126, a newly synthesized S-adenosylmethionine competitor, is a highly selective EZH2 inhibitor.5 We have previously reported that GSK126 inhibits cell migration and angiogenesis in solid tumor cell lines by downregulating the expression of vascular endothelial growth factor-A.6 Several other reports have also demonstrated the potential anticancer activity of GSK126.7–9 However, we found limited effect of GSK126 when used alone in mice xenografts, and varieties of efficacy were also reported in other studies.
Epidermal growth factor receptor (EGFR) is a member of the ErbB family of receptor tyrosine kinases and is upregulated in a variety of cancers, such as breast cancer, gastric cancer, and head and neck squamous cell carcinoma.10 EGFR tyrosine kinase inhibitors (EGFR-TKIs) have been well developed to target the mutated EGFR in different types of tumors.11,12 Gefitinib is one of the first generations of EGFR-TKIs and can effectively inhibit the EGFR pathway by blocking the intracellular tyrosine kinase domain, which is used mostly in the treatment of advanced EGFR-mutated non-small-cell lung cancer13,14 as well as gastric cancers (Phase II trial).15 Despite its effectiveness, monotherapy of Gefitinib can produce a high level of resistance and other unsatisfied therapeutic outcomes.16–18 Thus, the combination of Gefitinib with other anticancer agents is proposed to attenuate the acquisition of TKI resistance and to enhance therapeutic efficacy.19
In the present study, we investigated the interaction between the inhibition of EZH2 by GSK126 and the inhibition of EGFR by Gefitinib, and the underlying mechanisms. We show that inhibition of both EZH2 and EGFR produces a synergistic effect on reducing the survival of gastric cancer cells by enhancing autophagy.
Materials and methods
Chemicals
GSK126 with a purity of 99% was obtained from Shanghai Hanxiang Life Technology Ltd. (Shanghai, China). Gefitinib with a purity of 99.93% was obtained from Selleck Chemicals (Huston, TX, USA). 3-Methyladenine (3-MA) was purchased from Sigma-Aldrich (St Louis, MO, USA). GSK126 and Gefitinib were dissolved in dimethyl sulfoxide to prepare stock solutions for the in vitro studies, and dissolved in polyoxyethylenesorbitan monooleate (Tween 80) to prepare the stock solution for the in vivo studies. All other chemicals were of reagent grade.
Cell culture
Human gastric cancer cell line MGC803, human lung cancer cell line A549, human prostate cancer cell line PC-3, and human breast cancer cell line MDB-MA-231 were purchased from Shanghai Institute of Biochemistry and Cell Biology, China Academy of Sciences (Shanghai, China). MGC803 and MDB-MA-231 cells were cultured in Roswell Park Memorial Institute 1640 (RPMI 1640) medium, A549 cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM), and PC-3 cells were cultured in DMEM: F12 (1:1) medium. All media were supplemented with 10% heat-inactivated fetal calf serum (Life Technologies, Grand Island, NY, USA), 100 U/mL penicillin G, and 100 μg/mL streptomycin, and were cultured in a 37°C incubator under an atmosphere of 95% O2 and 5% CO2.
Sulforhodamine B assays (SRB)
SRB assays were performed to detect cell proliferation. Cancer cells in logarithmic growth phase were seeded into 96-well plates at 1×104 cells/well and cultured overnight before treatment with GSK126 and/or Gefitinib at the indicated concentrations. After 48 hours of exposure, the medium was removed and cells were incubated with 50 μL of cold 10% trichloroacetic acid (Sigma-Aldrich) for 30 minutes at 4°C. The cells were then washed with phosphate-buffered saline (PBS) and stained with 0.4% SRB for 30 minutes at room temperature. After rinsed with 1% acetic acid and dried, cells were incubated with 100 μL Tris-base (Biosharp, Hefei, China) at 10 mM and violently shaken for 10 minutes. Absorbance was measured at 510 nm using a Spectramax M5 spectrophotometer (Molecular Devices, San Jose, CA, USA). The combination index (CI) of GSK126 and Gefinitib was calculated and their synergy quantification was performed using the Chou-Talalay method as described previously.20 CI <1, CI =1, and CI >1 indicate synergism, additive effect, and antagonism in drug combinations, respectively.
Apoptosis assays
Cell apoptosis was determined using the Alexa Fluor®-488 Annexin V/Dead Cell Apoptosis Kit (Life Technologies). MGC803 cells were harvested and washed in cold PBS. After the cell density was determined, cells were diluted in 1× Annexin-binding buffer to 1×106 cells/mL. Each 100 μL of cell suspension was added into 5 μL of Alexa Fluor-488 Annexin V and 1 μL of 100 μg/mL working solution and incubated at room temperature for 15 minutes. Then, 400 μL of 1× annexin-binding buffer was added into the solution. Flow cytometry was performed to measure the fluorescence emission at 530 nm and >575 nm. The data were analyzed using FlowJo software (Tree Star Inc., Ashland, OR, USA).
Western blotting
MGC803 cells were treated with Gefitinib and/or GSK126 for 48 hours. Total protein was extracted using Cell Extraction Buffer (Biosource, Camarillo, CA, USA) supplemented with protease and phosphatase inhibitors. Western blotting was routinely conducted.14 In brief, protein lysates were separated by SDS-PAGE and transferred to nitrocellulose membrane, followed by incubation with the rabbit-derived primary antibodies against phosphorylated protein kinase B (Akt), phosphorylated mammalian target of rapamycin (mTOR), phosphorylated S6, phosphorylated Unc-51-like autophagy activating kinase (ULK), and microtubule-associated protein 1A/1B-light chain 3 (LC3) (1:1,000, all from Cell Signaling Technology, Danvers, MA, USA). After being stripped, the membranes were reprobed with antibodies against total Akt, mTOR, S6, ULK, and GAPDH (1:1,000, all from Cell Signaling Technology). The peroxidase-conjugated anti-rabbit secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used for the development of signals, which were visualized with ECL reagent (Pierce, Rockford, IL, USA). GAPDH was used as the internal standard of total target proteins, and phosphorylated proteins were normalized to their total proteins, respectively. Signals from triplicates (n=3) were quantified by image software ImageJ (NIH, Bethesda, MD, USA) and the intensity of the control was set to 1.
Xenografts in nude mice
Male Balb/c nude mice with a body weight of 18–20 g were purchased from Shanghai SLAC Laboratory Animal Co. (Shanghai, China) and housed at Zhejiang University City College Animal Care Facility according to the institutional guidelines for laboratory animals. All animal experiments were performed in accordance with guidelines and approved by Zhejiang University Institutional Animal Care and Use Committee. MGC803 cells at 1×106 cells were injected subcutaneously into nude mice. When tumors grew to a volume of 50–150 mm3, the mice were randomly divided into four groups. The control group of mice were intraperitoneally (i.p.) administered with vehicle containing 0.1% DMSO and 7% cremophor/ethanol (3:1) in PBS, and the other three groups of mice were administered i.p. with 200 mg/kg GSK126, 30 mg/kg Gefitinib, or their combination once every other days for 4 weeks. Body weight and tumor volume were monitored once every other day. The tumor volume was calculated by the formula V = a2× b/2 (V: xenograft volume; a: short diameter; b: long diameter). After 4-week treatment, the mice were sacrificed and the xenografts were harvested for weighing and serum samples were collected for the determination of alanine transaminase (ALT) and aspartate transaminase (AST) using commercial kits purchased from the Institute of Biological Engineering of Nanjing Jiancheng (Nanjing, China).
Statistical analysis
Numerical data were expressed as mean ± SD, and statistical analyses were performed by one-way ANOVA and SNK-Q tests (SPSS version 16.0; SPSS Inc., Chicago, IL, USA). P<0.05 was considered statistically significant. Experiments were repeated at least three times and the representative results were shown.
Results
Combined use of GSK126 and Gefitinib synergistically inhibited the survival of cancer cells
The cell survival fraction of four cancer cell lines – human gastric cancer cell line MGC803, human lung cancer cell line A549, human prostate cancer cell line PC-3, and human breast cancer cell line MDB-231 – were determined by SRB assays after 48 hours of treatments with Gefitinib and GSK126 alone or in combination. In the MGC803 cells, either Gefitinib ranging from 0 to 12.5 μM or GSK126 ranging from 0 to 8.3 μM strongly inhibited cell survival in a dose-dependent manner, while their combination caused further inhibition in the survival of cells, with a CI of 0.061 (Figure 1A). Moreover, in the A549 and PC-3 cells, treatment with either Gefitinib or GSK126 caused a minor decrease in the cell survival, whereas the combination of Gefitinib at 12.5 μM and GSK126 at 8.3 μM caused further decrease in cell survival, with CIs of 0.591 and 0.713, respectively (Figure 1B and C). In the MDB-MA-231 cells, Gefitinib or GSK126 alone exhibited a modest inhibition in the survival of cells, and their combination caused a further significant inhibition in the survival of cells, with a CI of 0.371 (Figure 1D). Thus, these results suggest that the combination of GSK126 and Gefitinib exerts a synergistic effect against the survival of cancer cells, especially in MGC803 and MDB-MA-231 cells.
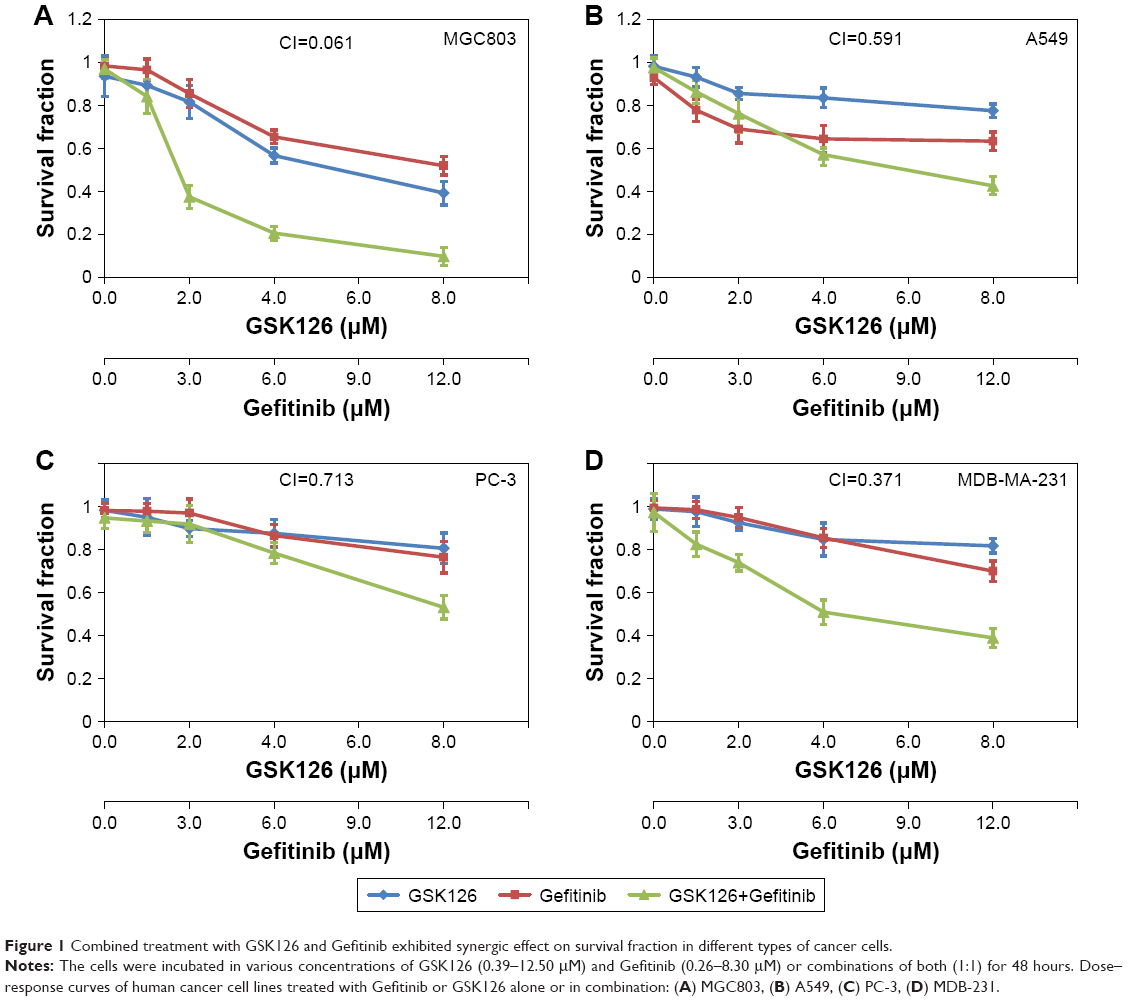 | Figure 1 Combined treatment with GSK126 and Gefitinib exhibited synergic effect on survival fraction in different types of cancer cells. |
Combination of GSK126 and Gefitinib synergistically induces cell apoptosis
To determine whether the combination of GSK126 and Gefitinib decreases the survival of cancer cells due primarily to the increase of cell apoptosis, we treated MGC803 cells with GSK126 at 12.5 μM, with Gefinitib at 8.3 μM, and with their combination for 48 hours and examined the cell apoptosis by flow cytometry. The apoptotic rates were 7.6%, 10.6%, and 10.8% in cells treated with vehicle, GSK126, and Gefinitib, respectively, whereas the apoptotic rate was 56.2% in the cells treated with the combination of GSK126 and Gefitinib (Figure 2A and B). Thus, the combination of GSK126 and Gefitinib synergistically induces the cell apoptosis in MGC803 cells.
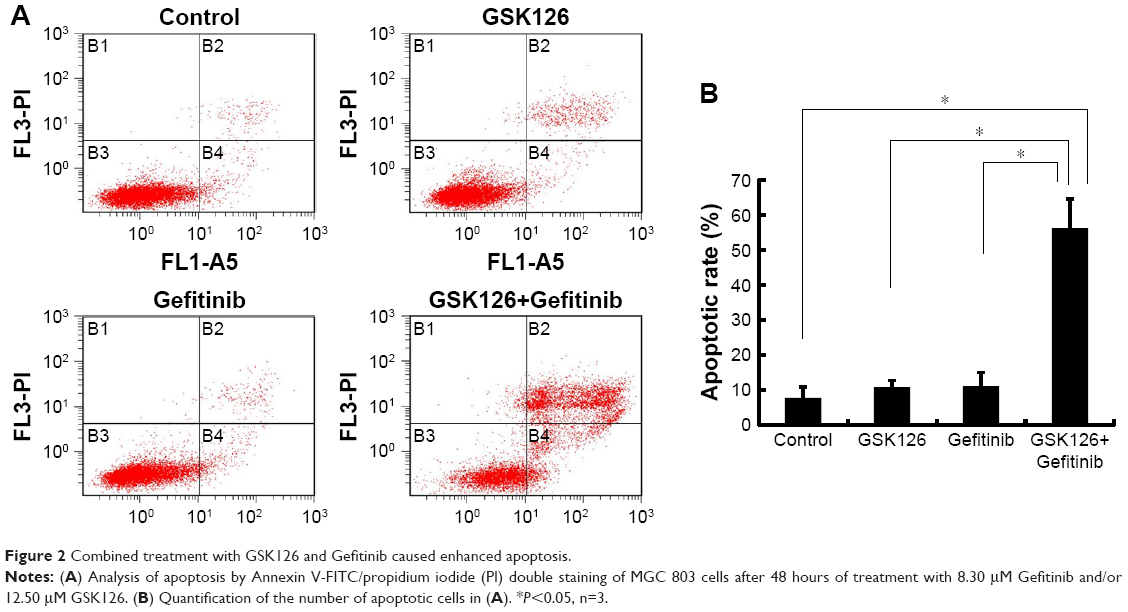 | Figure 2 Combined treatment with GSK126 and Gefitinib caused enhanced apoptosis. |
Combination of GSK126 and Gefitinib increased autophagy
Autophagy, a caspase-independent intracellular degradation system, is closely involved in cell apoptosis and cell death. We next treated the MGC803 cells with GSK126 at 12.5 μM, with Gefitinib at 8.3 μM, and with their combination for 48 hours and examined cell autophagy by detecting the expression of LC3 protein. As reflected by the ratio of LC3-II to LC3-I, treatment with either Gefitinib or GSK126 alone induced evident conversion of cytoplasmic LC3-I into membrane-bound LC3-II, while treatment with the combination of Gefitinib and GSK126 induced more significant conversion of LC3-I into LC3-II (Figure 3A and B). ULK, another marker related to autophagy, was also detected. Similarly, treatment with either Gefitinib or GSK126 alone induced a notable decrease in the p-ULK, whereas treatment with the combination of Gefitinib and GSK126 induced a further decrease in p-ULK in MGC803 cells (Figure 3A and B).
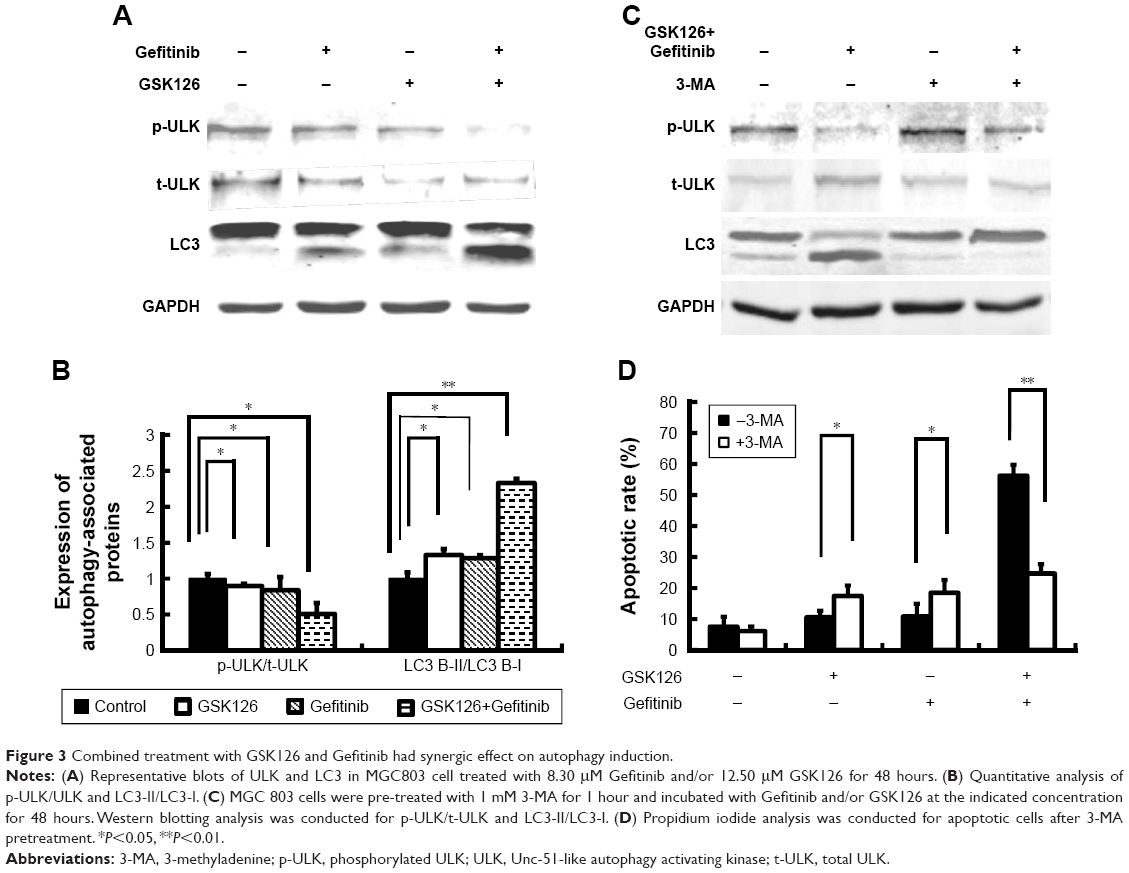 | Figure 3 Combined treatment with GSK126 and Gefitinib had synergic effect on autophagy induction. |
We next determined whether inhibition of autophagy could attenuate the apoptosis in MGC803 cells after the combined use of GSK126 and Gefitinib. Autophagy inhibitor 3-MA at 1 mM was administered 1 hour before the cells were exposed to GSK126 and Gefitinib. Administration of 3-MA had limited effect on the conversion of LC3-I to LC3-II and p-ULK expression in cells that were not treated with GSK126 or Gefinitib, but conversion of LC3-I to LC3-II was almost stopped and p-ULK decreased in cells that were treated with the combination of both drugs (Figure 3C). Notably, 3-MA increased the GSK126- and Gefitinib-induced cell apoptosis by 7% and 9%, respectively; in contrast, it decreased the apoptosis by 26% in the cells treated with the two drugs (Figure 3D). These data indicate that synergic effect of the combination of GSK126 and Gefitinib on apoptosis was due to the increase of autophagy.
Combination of GSK126 and Gefitinib strongly inhibited the activity of mTOR signaling pathway
Despite the critical role in protein synthesis and cell proliferation, the mammalian target of rapamycin complex 1 (mTORC1) has emerged as a key regulator of autophagy. To determine the potential upstream signaling governing the activation of ULK by the combination of GSK126 and Gefitinib, we next examined the activity of mTOR signaling pathway reflected by protein phosphorylation. Gefitinib at 8.3 μM or GSK126 at 12.5 μM slightly downregulated the expression of p-S6, p-mTOR, and p-Akt, whereas their combination significantly attenuated the expression of these phosphorylated proteins (Figure 4A–D). Thus, these data indicate that the downregulation of mTOR signaling and subsequent activation of ULK is possibly involved in cell apoptosis when GSK126 and Gefitinib are used in combination.
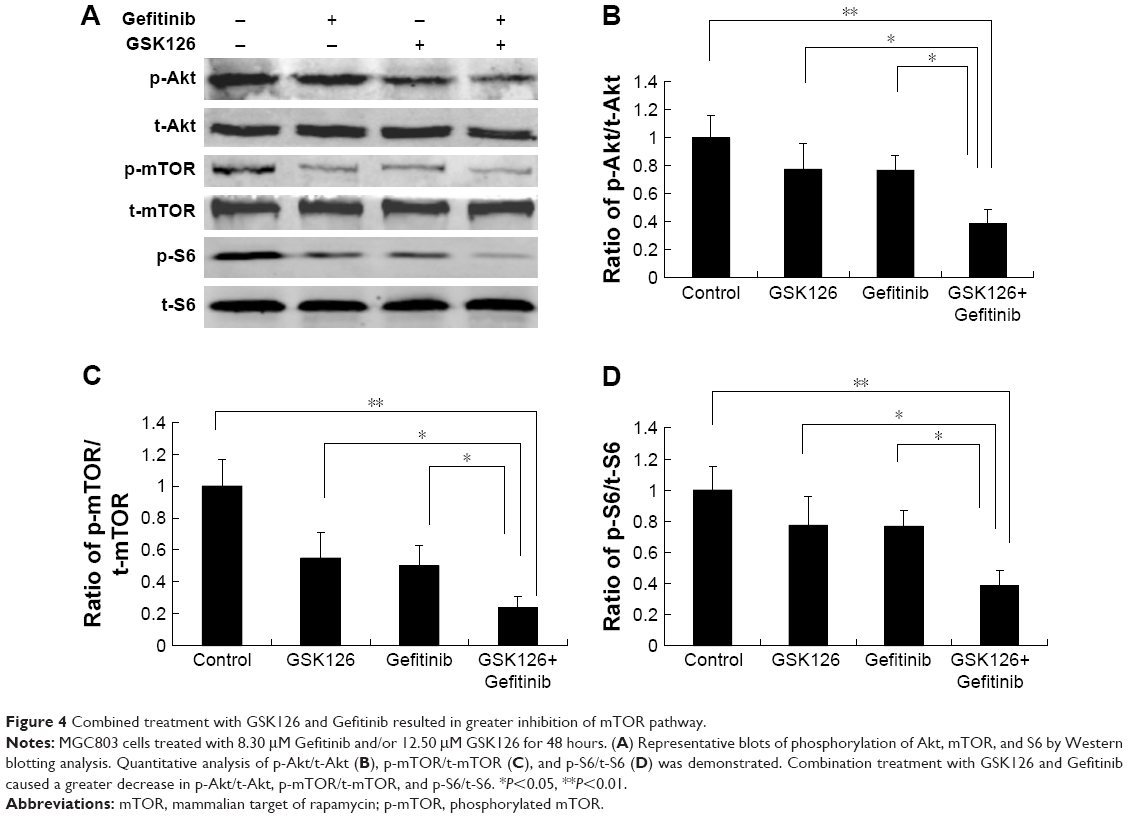 | Figure 4 Combined treatment with GSK126 and Gefitinib resulted in greater inhibition of mTOR pathway. |
Combination of GSK126 and Gefitinib synergistically suppressed the growth of MGC803 cell xenografts
To determine whether the combination of GSK126 and Gefitinib could produce a synergistic effect on the tumor growth in vivo, we generated MGC803 cell xenografts in Balb/c nude mice. The volumes of MGC803 cell xenografts treated with GSK126, Gefitinib, or their combination time dependently increased within 28 days after transplanting (Figure 5A). The volumes of either GSK126- or Gefitinib-treated xenografts did not significantly differ from those of vehicle-treated xenografts within the 28-day treatments; however, the tumor volume was significantly small in mice treated with both GSK126 and Gefitinib from day 16 to 28 (Figure 5A). On day 28, the tumor volumes in the mice treated with the combination drugs were only approximately 30% of those in either GSK126- or Gefitinib-treated xenografts (Figure 5B). In addition, neither the body weights (Figure 5C) within 28 days nor the serum levels of AST and ALT on day 28 were significantly different in the four groups (Figure 5D). Thus, these data suggest that non-cytotoxic dose of GSK126 and Gefitinib produces a synergistic effect against the growth of MGC803 cell xenografts.
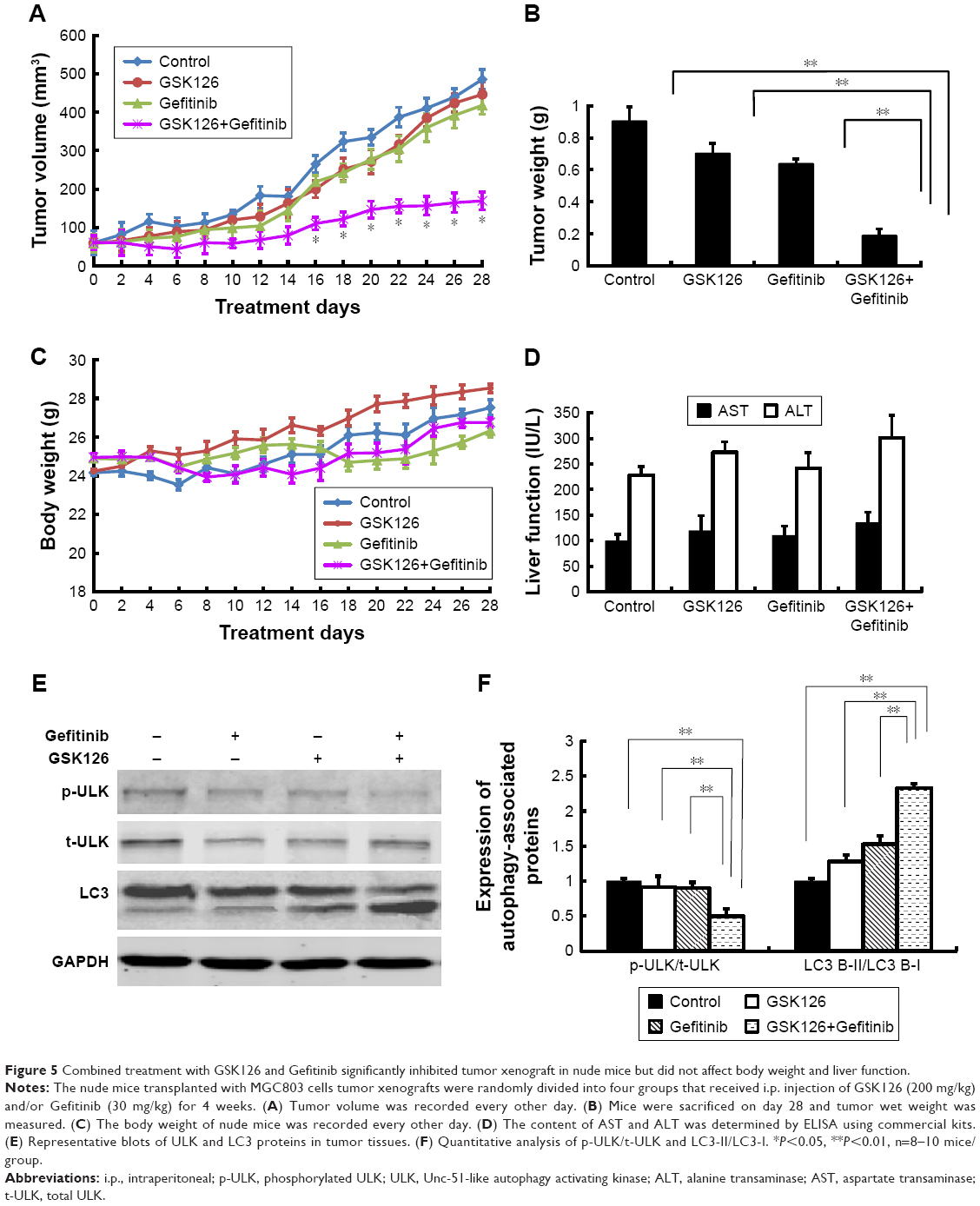 | Figure 5 Combined treatment with GSK126 and Gefitinib significantly inhibited tumor xenograft in nude mice but did not affect body weight and liver function. |
We also detected the expression of autophagy-related proteins in xenografted tumor tissues. Similar to the results obtained in cell line, treatment with either Gefitinib or GSK126 alone induced evident conversion of cytoplasmic LC3-I into membrane-bound LC3-II and a notable decrease in the p-ULK, whereas treatment with the combination of Gefitinib and GSK126 induced a further conversion or decrease in LC3-II or p-ULK, respectively (Figure 5E and F).
Discussion
To the best of our knowledge, this is the first study demonstrating that inhibition of both EZH2 and EGFR produces a synergistic effect on the cancer cell apoptosis. In this molecular event, inhibition of both EZH2 and EGFR inactivates the Akt and mTORC1 to activate the ULK, thereby causing an increase in the autophagy that possibly contributes to the induction of cell apoptosis.
Combination chemotherapy is widely exploited for enhancing cancer treatment in clinic. However, the treatment efficacy has not always been promising. In our observations, the combination of GSK126 and Gefitinib has showed synergistic anti-cancer effect in multiple tumor cell lines (MGC803, A549, PC-3, and MDB231). The strong synergistic effect was also validated in a gastric cancer MGC803 xenograft model. Consistent with our findings, Katona et al have reported that combined use of EZH2 and EGFR inhibitor results in a significant decrease in cell number, and increased apoptosis and autophagy in colon cancer cells.21 Another report has also shown synergic effects of EZH2 inhibitor combined with histone deacetylase (HDAC) inhibitor in glioblastoma, which augments apoptosis and increases DNA damage.22 However, combination therapy may cause toxicity. Although we have not explored the detailed toxicity of the combined use of GSK126 and Gefitinib in the present study, we observed normal body weight gain and liver function in these animals, suggesting that the toxicity of the combination use is likely minimal.
Drug-induced decrease of survival rate of cancer cells may be due to the inhibition of proliferation or the induction of apoptosis. In the present study, we found a modest decrease in cell survival fraction when Gefitinib or GSK126 is administered alone; however, we observed no significant effect on apoptosis. These results indicate that effect of monotherapy on cell survival fraction likely resulted from its effect on inhibition of proliferation. Although the effect of combined use of drugs on cell proliferation has not been observed in this study. We have found the decrease in survival rate and the increase in apoptosis is approximately equal. Thus, the synergistic effect of combination use on survival inhibition is more likely resulted from its induction of apoptosis, which is identical to other reports.23
The relationship between the processes of autophagy and apoptosis is quite complicated, and sometimes contradictory, but is critical to the overall fate of the cell.24 In essence, autophagy-dependent mechanisms that contribute to the maintenance of cellular homeostasis could indirectly postpone the onset of apoptosis. Inhibiting autophagy leads to increased apoptosis and decreased clonogenic activity in cancer cells that survived.25,26 On the contrary, excessive autophagy may predispose the cells to apoptosis in order to eliminate cells.27 Many autophagic proteins tend to corroborate in induction of apoptosis in a stressful condition via upregulation of proapoptotic proteins.28–30 In this study, we have demonstrated that the combination of GSK126 and Gefitinib induced notable increase in both autophagy and apoptosis, and autophagy inhibitor pretreatment reversed the apoptosis induced by combined use of drugs. These data indicate that synergic effect of the combined use of drugs on cancer cell survival may result from the inhibition of Akt/mTORC1, which subsequently induces the activation of ULK and autophagy, and eventually increases the event of apoptosis.
Conclusion
The present study provides evidence in vitro and in vivo that combination of GSK126 and Gefitinib exerts synergic therapeutic activity, which may be considered as a potential therapeutic strategy to overcome the low efficacy of current single drug strategies.
Acknowledgment
Research reported in this publication was supported by the Foundation of Zhejiang Health Bureau (2015 KYA148, 2015RCB004) and Zhejiang Provincial Foundation of National Science (LY12H16005).
Disclosure
The authors report no conflicts of interest in this work.
References
Charalampakis N, Economopoulou P, Kotsantis I, et al. Medical management of gastric cancer: a 2017 update. Cancer Med. 2018;7(1):123–133. | ||
Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med. 2016;22(2):128–134. | ||
Rogawski DS, Grembecka J, Cierpicki T. H3K36 methyltransferases as cancer drug targets: rationale and perspectives for inhibitor development. Future Med Chem. 2016;8(13):1589–1607. | ||
Jiang T, Wang Y, Zhou F, Gao G, Ren S, Zhou C. Prognostic value of high EZH2 expression in patients with different types of cancer: a systematic review with meta-analysis. Oncotarget. 2016;7(4):4584–4597. | ||
Mccabe MT, Ott HM, Ganji G, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492(7427):108–112. | ||
Chen YT, Zhu F, Lin WR, Ying RB, Yang YP, Zeng LH. The novel EZH2 inhibitor, GSK126, suppresses cell migration and angiogenesis via down-regulating VEGF-A. Cancer Chemother Pharmacol. 2016;77(4):757–765. | ||
Koppens MA, Bounova G, Cornelissen-Steijger P, et al. Large variety in a panel of human colon cancer organoids in response to EZH2 inhibition. Oncotarget. 2016;7(43):69816–69828. | ||
Khan M, Walters LL, Li Q, et al. Characterization and pharmacologic targeting of EZH2, a fetal retinal protein and epigenetic regulator, in human retinoblastoma. Lab Invest. 2015;95(11):1278–1290. | ||
Zeng D, Liu M, Pan J. Blocking EZH2 methylation transferase activity by GSK126 decreases stem cell-like myeloma cells. Oncotarget. 2017;8(2):3396. | ||
Wieduwilt MJ, Moasser MM. The epidermal growth factor receptor family: biology driving targeted therapeutics. Cell Mol Life Sci. 2008;65(10):1566–1584. | ||
Valentini AM, Pirrelli M, Caruso ML. EGFR-targeted therapy in colorectal cancer: does immunohistochemistry deserve a role in predicting the response to cetuximab? Curr Opin Mol Ther. 2008;10(2):124. | ||
Rusnak DW, Alligood KJ, Mullin RJ, et al. Assessment of epidermal growth factor receptor (EGFR, ErbB1) and HER2 (ErbB2) protein expression levels and response to lapatinib (Tykerb, GW572016) in an expanded panel of human normal and tumour cell lines. Cell Prolif. 2007;40(4):580–594. | ||
Riely GJ. The use of first-generation tyrosine kinase inhibitors in patients with NSCLC and somatic EGFR mutations. Lung Cancer. 2008;60 Suppl 2:S19–S22. | ||
Dhillon S. Gefitinib: a review of its use in adults with advanced non-small cell lung cancer. Target Oncol. 2015;10(1):153–170. | ||
Morishita A, Gong J, Masaki T. Targeting receptor tyrosine kinases in gastric cancer. World J Gastroenterol. 2014;20(16):4536–4545. | ||
Prahallad A, Sun C, Huang S, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483(7387):100–103. | ||
Girgert R, Emons G, Gründker C. 17β-estradiol-induced growth of triple-negative breast cancer cells is prevented by the reduction of GPER expression after treatment with gefitinib. Oncol Rep. 2017;37(2):1212–1218. | ||
Song Z, Huang S, Yu H, et al. Synthesis and biological evaluation of morpholine-substituted diphenylpyrimidine derivatives (Mor-DPPYs) as potent EGFR T790M inhibitors with improved activity toward the gefitinib-resistant non-small cell lung cancers (NSCLC). Eur J Med Chem. 2017;133:329–339. | ||
Azad N, Zahnow CA, Rudin CM, Baylin SB. The future of epigenetic therapy in solid tumours – lessons from the past. Nat Rev Clin Oncol. 2013;10(5):256–266. | ||
Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70(2):440–446. | ||
Katona BW, Liu Y, Ma A, Jin J, Hua X. EZH2 inhibition enhances the efficacy of an EGFR inhibitor in suppressing colon cancer cells. Cancer Biol Ther. 2014;15(12):1677–1687. | ||
Grinshtein N, Rioseco CC, Marcellus R, et al. Small molecule epigenetic screen identifies novel EZH2 and HDAC inhibitors that target glioblastoma brain tumor-initiating cells. Oncotarget. 2016;7(37):59360–59376. | ||
Petraglia F, Singh AA, Carafa V, et al. Combined HAT/EZH2 modulation leads to cancer-selective cell death. Oncotarget. 2018;9(39):25630–25646. | ||
Oral O, Akkoc Y, Bayraktar O, Gozuacik D. Physiological and pathological significance of the molecular cross-talk between autophagy and apoptosis. Histol Histopathol. 2016;31(5):479. | ||
Rubin BP, Debnath J. Therapeutic implications of autophagy-mediated cell survival in gastrointestinal stromal tumor after treatment with imatinib mesylate. Autophagy. 2010;6(8):1190–1191. | ||
Ravegnini G, Sammarini G, Nannini M, et al. Gastrointestinal stromal tumors (GIST): Facing cell death between autophagy and apoptosis. Autophagy. 2017;13(3):452–463. | ||
Rubinstein AD, Eisenstein M, Ber Y, Bialik S, Kimchi A. The autophagy protein Atg12 associates with antiapoptotic Bcl-2 family members to promote mitochondrial apoptosis. Mol Cell. 2011;44(5):698–709. | ||
Pei J, Deng J, Ye Z, et al. Absence of autophagy promotes apoptosis by modulating the ROS-dependent RLR signaling pathway in classical swine fever virus-infected cells. Autophagy. 2016;12(10):1738–1758. | ||
Zhu J, Yu W, Liu B, et al. Escin induces caspase-dependent apoptosis and autophagy through the ROS/p38 MAPK signalling pathway in human osteosarcoma cells in vitro and in vivo. Cell Death Dis. 2017;8(10):e3113. | ||
Booth LA, Tavallai S, Hamed HA, Cruickshanks N, Dent P. The role of cell signalling in the crosstalk between autophagy and apoptosis. Cell Signal. 2014;26(3):549–555. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.