")
Back to Journals » Journal of Inflammation Research » Volume 15
Inhibition of Dectin-1 Alleviates Neuroinflammatory Injury by Attenuating NLRP3 Inflammasome-Mediated Pyroptosis After Intracerebral Hemorrhage in Mice: Preliminary Study Results
Authors Ding Z , Zhong Z, Wang J, Zhang R, Shao J , Li Y, Wu G, Tu H, Yuan W, Sun H, Wang Q
Received 27 July 2022
Accepted for publication 7 October 2022
Published 19 October 2022 Volume 2022:15 Pages 5917—5933
DOI https://doi.org/10.2147/JIR.S384020
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Zhiquan Ding1 *, Zhenzhong Zhong1 *, Jun Wang1 *, Run Zhang,2 Jinlian Shao,3 Yulong Li,1 Guiwei Wu,1 Huiru Tu,1 Wen Yuan,4 Haitao Sun,1,5 Qinghua Wang1,3
1Neurosurgery Center, Department of Neurotrauma and Neurocritical Care Medicine, Guangdong Provincial Key Laboratory on Brain Function Repair and Regeneration, Zhujiang Hospital, Southern Medical University, Guangzhou, People’s Republic of China; 2Neurosurgery Center, Department of Neuro-oncological Surgery, Guangdong Provincial Key Laboratory on Brain Function Repair and Regeneration, Zhujiang Hospital, Southern Medical University, Guangzhou, People’s Republic of China; 3Department of Emergency, Zhujiang Hospital, Southern Medical University, Guangzhou, People’s Republic of China; 4Laboratory Animal Center, Zhujiang Hospital, Southern Medical University, Guangzhou, People’s Republic of China; 5Clinical Biobank Center, Microbiome Medicine Center, Department of Laboratory Medicine, Zhujiang Hospital, Southern Medical University, Guangzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Qinghua Wang; Haitao Sun, Neurosurgery Center, Department of Neurotrauma and Neurocritical Care Medicine, Guangdong Provincial Key Laboratory on Brain Function Repair and Regeneration, Zhujiang Hospital, Southern Medical University, Guangzhou, People’s Republic of China, Email [email protected]; [email protected]
Background: Neuroinflammation plays an important role following intracerebral hemorrhage (ICH). NLRP3 inflammasome-mediated pyroptosis contributes to the mechanism of neuroinflammation. It has been reported that dendritic cell-associated C-type lectin-1 (Dectin-1) activation triggers inflammation in neurological diseases. However, the role of Dectin-1 on NLRP3 inflammasome-mediated pyroptosis after ICH remains unclear. Here, we aimed to explore the effect of Dectin-1 on NLRP3 inflammasome-mediated pyroptosis and neuroinflammation after ICH.
Methods: Adult male C57BL/6 mice were used to establish the ICH model. Laminarin, an inhibitor of Dectin-1, was administered for intervention. Expression of Dectin-1 was evaluated by Western blot and immunofluorescence. Brain water content and neurobehavioral function were tested to assess brain edema and neurological performance. Western blot was conducted to evaluate the level of GSDMD-N. ELISA kits were used to measure the levels of IL-1β and IL-18. qRT–PCR and Western blot were performed to evaluate the expressions of NLRP3 inflammasome, IL-1β, and IL-18.
Results: The expression of Dectin-1 increased following ICH, and Dectin-1 was expressed on microglia. In addition, inhibition of Dectin-1 by laminarin decreased brain edema and neurological impairment after ICH. Moreover, inhibition of Dectin-1 decreased the expression of pyroptosis-related protein, GSDMD-N, and inflammatory cytokines (IL-1β and IL-18). Mechanistically, Dectin-1 blockade inhibits NLRP3 inflammasome activation, thereby alleviating neuroinflammatory injury by attenuating NLRP3 inflammasome-mediated pyroptosis both in vivo and in vitro.
Conclusion: Our study indicates that the inhibition of Dectin-1 alleviates neuroinflammation by attenuating NLRP3 inflammasome-mediated pyroptosis after ICH.
Keywords: intracerebral hemorrhage, Dectin-1, NLRP3 inflammasome, pyroptosis, microglia
Introduction
Intracerebral hemorrhage (ICH), a common sub-type of stroke, is a fatal disease with poor prognosis.1,2 Brain injury following ICH has two main mechanisms: One is primary injury due to the hematoma formation and enlargement, and the other is secondary injury caused by toxicity of hematoma metabolites, inflammation, oxidative stress, and neuronal damage.3,4 Increasing studies have focused on secondary injury following ICH,5 in which neuroinflammation is a significant contributor, resulting in neuronal death, brain edema, and blood-brain barrier disruption.6,7 The regulation of inflammation can alleviate brain injury and is considered a potential treatment strategy for ICH.
Inflammasome-mediated pyroptosis, a form of cell-programmed death, is believed to be an important mechanism of neuroinflammation after stroke.8–10 The NOD-like receptor, pyrin domain-containing 3 (NLRP3) inflammasome, contributes to the triggering of neuroinflammation in microglia.11,12 Activation of NLRP3 results in the cleavage of effector protein procaspase-1 into activated caspase-1,13,14 following which precursors of interleukin (IL)-1β and IL-18 (proIL-1β and proIL-18) are, respectively, cleaved into IL-1β and IL-18 by activated caspase-1.15,16 In addition, active caspase-1 promotes the formation of the pyroptosis executor N-terminal fragment of Gasdermin D (GSDMD-N), which results in the perforation of the plasma membrane and the release of inflammatory cytokines.10 Recent research on traumatic brain injury and ischemic stroke has shown that NLRP3 inflammasome blockade reduces neuroinflammation and pyroptosis.17,18 However, the underlying mechanism of inflammasome-mediated pyroptosis after ICH remains incompletely understood.
Dendritic cell-associated C-type lectin-1 (Dectin-1), a pattern recognition receptor (PRR), contributes to the triggering of inflammation in fungal infectious diseases, myocardial injury, optic nerve crush injury, ischemic stroke, and ICH.19–23 A recent study suggested that Dectin-1 contributes to regulating microglial polarization after ICH.22 However, the regulatory mechanism of Dectin-1 in neuroinflammation after ICH is still poorly understood. Interestingly, previous studies have shown that activated Dectin-1 induces the recruitment and phosphorylation of spleen tyrosine kinase (SYK), aggravating inflammation.24,25 In addition, in ischemic stroke mice, suppression of SYK alleviates neuroinflammation via NLRP3 inflammasome-mediated pyroptosis.10 Moreover, inhibition of Dectin-1 has been shown to downregulate the transcription of IL-1β and IL-18 after myocardial infarction, as well as the protein level of NLRP3.26 However, it is unclear whether inhibition of Dectin-1 alleviates neuroinflammatory injury by attenuating NLRP3 inflammasome-mediated pyroptosis following ICH.
Here, we assumed that the NLRP3 inflammasomes might be downstream of the Dectin-1 signal following ICH. We aimed to explore the roles of Dectin-1 in NLRP3 inflammasome-mediated pyroptosis and neuroinflammation after ICH.
Materials and Methods
Animals
A total of 129 adult male C57BL/6 mice, 10–12 weeks old (25–30 g), were purchased from Sja Biotechnology (Guangdong, China) and housed in the Laboratory Animal Center of Zhujiang Hospital, Southern Medical University. Those in which ICH induction failed or those that died during the study (n = 11) were excluded from the final data analysis. Animal care was approved by the Ethics Committee of Zhujiang Hospital of Southern Medical University (Approval No. LAEC-2020-146) and complied with the guidelines of the National Institute of Health. Mice were housed in a 12-h light/dark cycle room at 25°C and 55% relative humidity, and allowed free access to food and water.
Experimental Protocol
The experimental design and the number of animals used are as follows.
Experiment 1
To determine the temporal pattern of the expression of Dectin-1 after ICH, 20 mice were randomly divided into five groups: sham (n = 4), 12 h after ICH (n = 4), 1 d after ICH (n = 4), 3 d after ICH (n = 4), and 7 d after ICH (n = 4). Western blot was used to determine the expression of Dectin-1 in the perihematomal zone of each group. The spatial location of Dectin-1 was evaluated via double immunostaining labeling to co-localize Dectin-1 with ionized calcium binding adapter molecule 1 (Iba-1), glial fibrillary acidic protein (GFAP), and neuronal nuclear antigen (NeuN) at 3 d after ICH (n = 2).
Experiment 2
To evaluate the effect of Dectin-1 after ICH, laminarin, an inhibitor of Dectin-1, was administered through intraperitoneal injection. Neurobehavioral function and brain water content were assessed at 3 d and 7 d after ICH. To obtain the optimum dose of laminarin, 30 mice were divided into five groups: sham (n = 6), ICH + vehicle (n = 6), ICH + laminarin 50 mg/kg (n = 6), ICH + laminarin 150 mg/kg (n = 6), and ICH + laminarin 450 mg/kg (n = 6). Based on the results of the assessment of neurobehavioral function and brain water content at 3 d after ICH, 450 mg/kg of laminarin was the optimum dose. For the effect of laminarin at 7 d after ICH, 18 mice were assigned into three groups: sham (n = 6), ICH + vehicle (n = 6), and ICH + laminarin 450 mg/kg (n = 6).
Experiment 3
To determine whether Dectin-1 amplifies pyroptosis and the potential mechanism of Dectin-1 triggering pyroptosis after ICH, 45 mice were randomly divided into three groups: sham, ICH + vehicle, and ICH + laminarin. Western blot (n = 4 each group), ELISA (n = 5 each group), and PCR (n = 6 each group) were carried out at 3 d after ICH. In addition, three mice of the ICH group were used to perform double immunostaining labeling to co-localize Dectin-1 with SYK.
Experiment 4
To determine whether Dectin-1 triggers microglial pyroptosis in vitro, BV2 microglia cells were divided into three groups: control (n = 4), LPS (n = 4), and LPS + laminarin (n = 4). BV2 microglia cells were exposed to LPS for 24 h, and then Western blot was carried out.
ICH Model
The ICH model was established by intracranial injection of bacterial collagenase, as described in our previous work.27 Briefly, mice were anesthetized with tribromoethanol (1.25%, 0.02 mL/g) through intraperitoneal injection and placed in a mouse stereotaxic apparatus. A cranial drill was used to create a small burr hole before a microsyringe (Gauge, Shanghai, China) was stereotactically inserted into the right striatum (coordinates 0.2 mm anterior, 2.0 mm lateral to the bregma, and 3.5 mm below the dural surface). Next, collagenase IV (0.04 U; Sigma-Aldrich, St. Louis, MI, USA) dissolved in 0.5 μL sterile normal saline was injected at a rate of 0.25 μL/min; the needle was pulled out 5 min after injection. According to our previous work,28 the sham group was subjected to a similar procedure without collagenase infusion, but with an equal volume of normal saline.
Cell Culture
BV2 microglia cells were purchased from Procell Life Science & Technology (Wuhan, China). The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) glucose containing 100 U/mL penicillin-streptomycin and 10% fetal bovine serum, and incubated in an incubator containing 5% CO2 at 37°C. The cells were suspended on a 6-well plate at a density of approximately 5×105 cells per well. The BV2 microglia cells were randomly distributed into a control group, lipopolysaccharide (LPS) group, and LPS+ laminarin group. The cells in the LPS + laminarin group were treated with laminarin for 1 h and then exposed to LPS, while the LPS group was exposed to LPS without laminarin pretreatment. Then, the cells were cultured for 24 h under normal culture conditions before harvesting for the following experiments.
Cell Viability Assay
The cell viability was determined using the CCK-8 assay (Fdbio Science, China). Briefly, BV2 microglia cells were seeded on a 96-well plate at a density of 1×104 cells per well and treated with different concentrations of laminarin (0, 10, 100, 300, 500, and 1000 μg/mL) for 24 h. Then, 10 μL/well of CCK-8 solution was added to each well. After incubation for 2 h at 37°C, the optical density (OD) was measured using a microplate reader (SYNERGY H1 BioTek, USA) at 450 nm. Cell viability is represented by the OD value.
Drug Administration
Laminarin (L9634, Sigma-Aldrich), an inhibitor of Dectin-1, was dissolved in normal saline. In the in vivo experiments, laminarin was first administered at 1 h after surgery by intraperitoneal injection and then once a day for the next 3 days. Based on previous studies,23,29 in the experiments of neurobehavioral function tests and brain water content measurement, three doses (50 mg/kg, 150 mg/kg, and 450 mg/kg) of laminarin were evaluated to obtain the optimum dose. LPS, a prominent cell wall component of gram-negative bacteria, is a strong stimulator of microglial activation.30,31 Therefore, LPS was used to activate microglia to induce neuroinflammation in this study. Thus, in the in vitro experiments, the optimal dose of laminarin and LPS was based on previous studies.23,32 Then, microglia were treated with laminarin (300 μg/mL) for 1 h and then exposed to LPS (1 μg/mL).
Neurobehavioral Function Test
As previously described,7 the modified Garcia score, forelimb placement test, and corner turn test were conducted at 3 and 7 days after ICH to evaluate neurological performance. The modified Garcia score includes seven independent tests to assess spontaneous activity, axial sensation, tactile proprioception, symmetry of limb movement, lateral turning, forelimb walking, and climbing. Each sub-test was scored on a scale of 0 to 3, and the highest overall score was 21 (without neurobehavioral dysfunction). For the corner turn test, mice were arranged to advance into a 30° corner and were free to turn left or right to leave. The corner turn was tested 10 times, expressed as a percentage of choosing left to turn. The forelimb placement test was used to evaluate the response to vibrissae stimulation. The left forelimb was tested 10 times, expressed as a percentage of placing the left forelimb precisely on the edge of the test bench.
Brain Water Content
As previously described,33 brain water contents were measured using an electronic balance at 3 and 7 days following ICH. After the mice were euthanized, the brain tissue was immediately removed and divided into three parts (ipsilateral hemisphere, contralateral hemisphere, and cerebellum). An electronic balance was used to measure the weight of each part to obtain the wet weight before drying for 24 h at 100°C. After drying, the brain tissue was measured to obtain the dry weight. Brain water content was calculated as follows: (wet weight − dry weight)/wet weight × 100%.
Quantitative Real-Time PCR
Total RNA was isolated from the perihematomal area 3 days after ICH using an RNA Extraction Kit (AG21102, Accurate Biotechnology, Hunan, China), according to the manufacturer’s instructions. cDNA was synthesized using the Evo M-MLV RT Kit with gDNA Clean for qPCR II (AG11711, Accurate Biotechnology, Hunan, China) according to the manufacturer’s protocol. cDNA was amplified with SYBR Green (AG11701, Accurate Biotechnology, Hunan, China). Table 1 shows the primer sequences. The relative expression levels of the target genes were analyzed using the 2−ΔΔCt method, with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as the internal control. The sham group was used as the reference group.
 |
Table 1 Primers Used for qRT-PCR |
Immunofluorescence Staining
Immunostaining was performed as previously described.34 Briefly, paraffin sections (4 μm) of the brain were blocked with a solution containing 5% bovine serum albumin (BSA) and 0.3%Triton-X at room temperature for 1 h. After blocking, the sections were incubated with the corresponding primary antibody at 4°C overnight according to the experimental purpose. The following primary antibodies were used: Dectin-1 (1:100, ab140039, Abcam), Iba-1 (1:300, GTX632426, GnenTex), GFAP (1:300, G3893, Sigma), NeuN (1:100, 66836-1-Ig, Proteintech), and SYK (1:50, sc-1240, Santa Cruz Biotechnology). Sections were rewarmed at room temperature for 30 min, then washed with phosphate-buffered saline (PBS) three times, and incubated with secondary antibody at 37°C for 1 h. Then, sections were washed with PBS three times, stained with DAPI for 15 min, and then washed with PBS. Finally, the brain sections were imaged using a fluorescence microscope (Nikon, Japan). The field of interest is the striatum around the hematoma. Three images were taken of each animal at a camera setting of 1206×1208 pixels.
Enzyme-Linked Immunosorbent Assay (ELISA)
After the mice were euthanized, the brain tissue of the perihematomal area was removed and homogenized. ELISA kits (EMC001b and EMC011, Neobioscience) were used to detect the concentrations of IL-1β and IL-18, according to the manufacturer’s instructions.
Western Blot
The Western blot was conducted as previously described.7 Briefly, protein samples were obtained from the perihematomal area and BV2 cells using RIPA lysis buffer. Then, protein lysate was centrifuged for 20 min at 12,000 g at 4°C. After centrifuging, the supernatant protein solution was collected. After loading an equal amount of protein onto the gel, the electrophoresis procedure was initiated. The proteins were then transferred to PVDF membranes and blocked with a solution containing 5% bovine serum albumin (BSA) for 1 h at room temperature. After blocking, membranes were incubated with primary antibodies at 4°C overnight. The following primary antibodies were used: Dectin-1 (1:1000; ab140039, Abcam), NLRP3 (1:1000; ab263899, Abcam), ASC (1:2000; sc-33958, Santa Cruz Biotechnology), phospho-SYK (1:1000; AF3315, Affinity Biosciences), IL-18 (1:1000; 60070-1-Ig, Proteintech), GSDMD-N (1:1000; AF4012, Affinity Biosciences), caspase-1 (1:1000; 22915-1-AP, Proteintech), β-actin (1:1000; AF5003, Beyotime Biotechnology), and IL-1β (1:1000; 26048-1-AP, Proteintech). After washing three times with TBST, the membranes were incubated with secondary antibody at room temperature for 1 h and then washed a further three times. Finally, a Bio-Rad Molecular Imager was used to detect protein signals, which were quantified by Image J.
Statistical Analysis
The SPSS 22.0 software was used for statistical analysis. Differences among multiple groups were compared using one-way analysis of variance (ANOVA); this was followed by the least significant difference (LSD) test for data of equal variances and Tamhane’s T2 test for data of unequal variances. Data are described as the mean ± SEM. P-values < 0.05 were taken to indicate statistical significance.
Results
Temporal Patterns and Spatial Expressions of Dectin-1 Receptor Following ICH
To determine the potential role of Dectin-1 after ICH, Western blot and double immunostaining were conducted to evaluate the temporal and spatial expressions of Dectin-1. We found that the protein level of Dectin-1 increased significantly at 12 h after surgery, peaked at 3 days, and then began to decrease compared with the sham group (F = 10.530; 12 h after ICH, p = 0.019; 1 d after ICH, p = 0.004; 3 d after ICH, p < 0.001; 7 d after ICH, p < 0.001) (Figure 1A and Figure 1B). Based on the protein expression results, double immunofluorescence staining was performed 3 days after ICH. We observed that the Dectin-1 receptor co-localized with microglia (Iba-1), but was not expressed in astrocytes (GFAP) or neurons (NeuN) (Figure 1C).
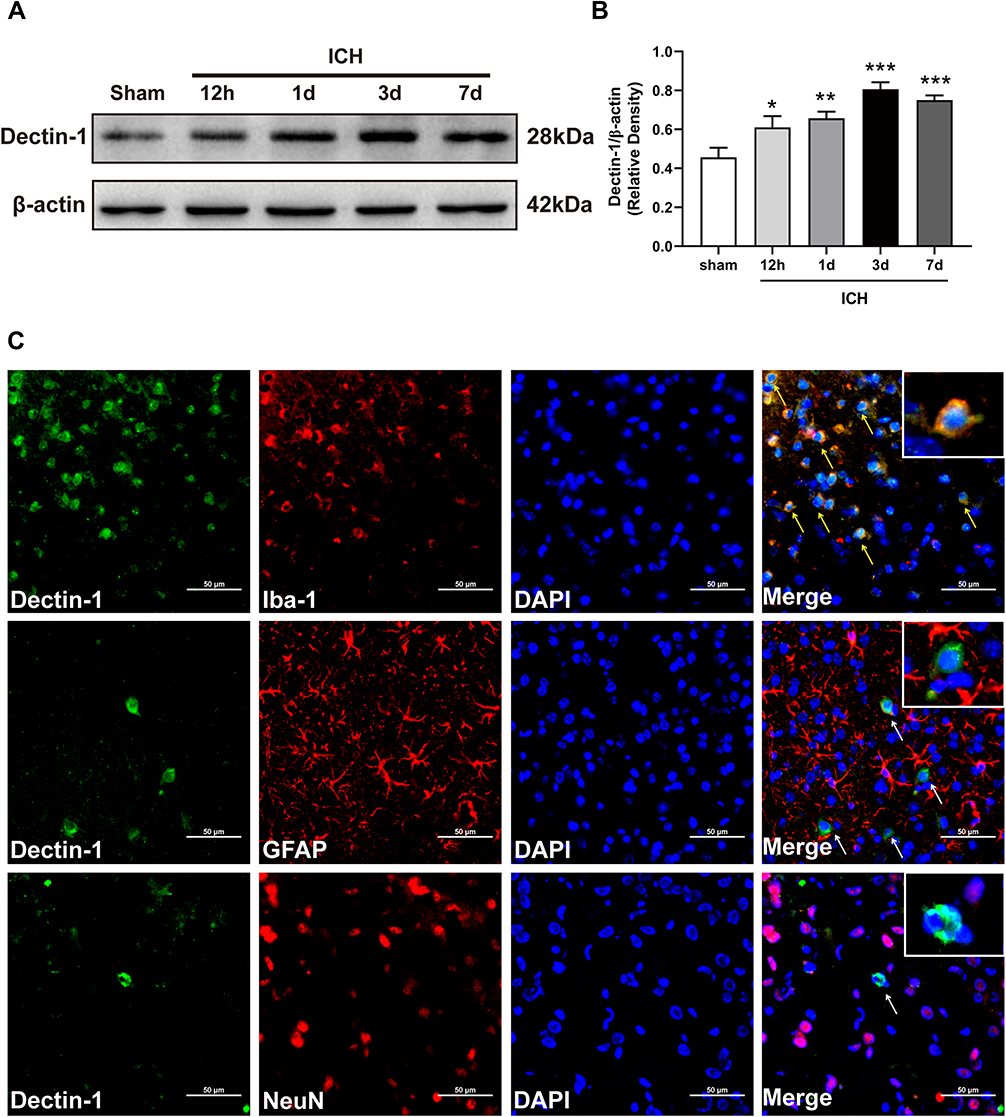 |
Figure 1 Temporal pattern of the expression and location of Dectin-1 following ICH. (A) Protein bands of temporal expression of Dectin-1 following ICH. (B) Protein quantitative analyses of Dectin-1; n = 4. Data are expressed as the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 vs sham. (C) Double immunofluorescence staining images of Dectin-1 co-located with microglia (Iba-1), astrocytes (GFAP), and neurons (NeuN) at 3 days following ICH. Nuclei: DAPI; n = 2. Scale bar = 50 μm. |
Inhibition of Dectin-1 Reduced Neurological Impairment and Brain Edema Following ICH
To confirm the effect of Dectin-1 by ICH, laminarin, an inhibitor of Dectin-1, was administered by intraperitoneal injection. Three doses (50 mg/kg, 150 mg/kg, and 450 mg/kg) of laminarin were used to determine the optimum dose for alleviating ICH-induced brain damage. The results of the modified Garcia score, forelimb placement test, and corner turn test indicated that ICH induced significant neurological impairment, compared with the sham group at 3 days. Low dose (50 mg/kg) laminarin administration did not attenuate the neurological deficits, while the administration of laminarin (150 mg/kg and 450 mg/kg) significantly improved neurological outcomes. However, the high dose of laminarin (450 mg/kg) more effectively improved neurological outcomes than the medium dose of laminarin (150 mg/kg) (modified Garcia score: F = 43.523; ICH + laminarin 50 mg/kg vs ICH + vehicle, p = 0.185; ICH + laminarin 150 mg/kg vs ICH + vehicle, p = 0.018; ICH + laminarin 450 mg/kg vs ICH + vehicle, p < 0.001; ICH + laminarin 450 mg/kg vs ICH + laminarin 150 mg/kg, p = 0.003) (corner turn test: F = 26.086; ICH + laminarin 50 mg/kg vs ICH + vehicle, p = 0.088; ICH + laminarin 150 mg/kg vs ICH + vehicle, p = 0.020; ICH + laminarin 450 mg/kg vs ICH + vehicle, p < 0.001; ICH + laminarin 450 mg/kg vs ICH + laminarin 150 mg/kg, p = 0.009) (forelimb placement test: F = 61.450; ICH + laminarin 50 mg/kg vs ICH + vehicle, p = 0.395; ICH + laminarin 150 mg/kg vs ICH + vehicle, p = 0.004; ICH + laminarin 450 mg/kg vs ICH + vehicle, p < 0.001; ICH + laminarin 450 mg/kg vs ICH + laminarin 150 mg/kg, p = 0.395) (Figure 2A–C). Meanwhile, the results of brain tissue water content suggested that the water content of the ipsilateral hemisphere significantly increased after 3 days in the ICH + vehicle groups compared to the sham group, suggesting that ICH generated severe edema. Treatment with low and medium doses of laminarin failed to reduce brain edema, while a high dose of laminarin (450 mg/kg) significantly reduced brain edema (F = 15.962; ICH + laminarin 50 mg/kg vs ICH + vehicle, p = 0.988; ICH + laminarin 150 mg/kg vs ICH + vehicle, p = 0.650; ICH + laminarin 450 mg/kg vs ICH + vehicle, p = 0.023) (Figure 2D). To further determine the treatment efficacy of laminarin (450 mg/kg), neurological function and brain water content assessment were also conducted at 7 days following ICH. Consistently, ICH induction caused significant neurological damage and edema of the ipsilateral hemisphere, both of which were reduced by laminarin treatment (450 mg/kg) (ICH + laminarin 450 mg/kg vs ICH + vehicle—modified Garcia score: F = 52.711, p < 0.001; corner turn test: F = 12.318, p = 0.020; forelimb placement test: F = 45.032, p = 0.012; brain water content: F = 39.477, p < 0.001) (Figure 3A–D). Therefore, a dose of 450 mg/kg was selected for the following studies.
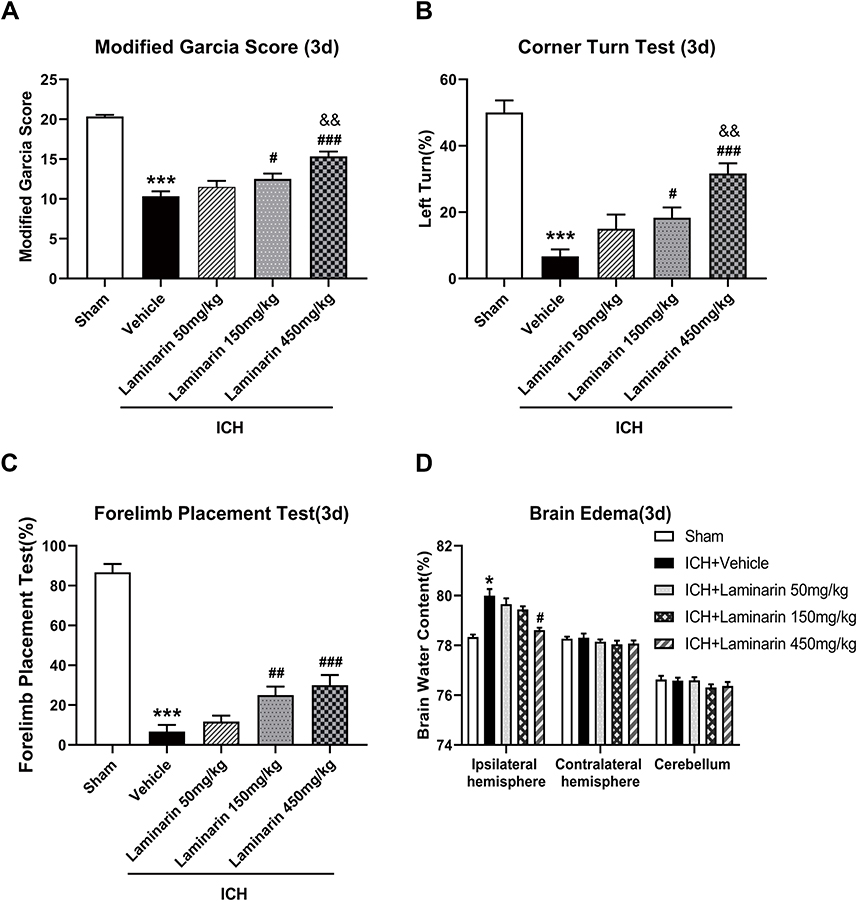 |
Figure 2 Effects of different doses of laminarin on neurological function and brain edema. (A) Modified Garcia score. (B) Corner turn test. (C) Forelimb placement test. (D) Brain water content at 3 days following ICH; n = 6. Data are expressed as the mean ± SEM. *p < 0.05, ***p < 0.001 vs sham; #p < 0.05, ##p < 0.01, ###p < 0.001 vs ICH + vehicle; &&p < 0.01 vs ICH + laminarin 150 mg/kg. |
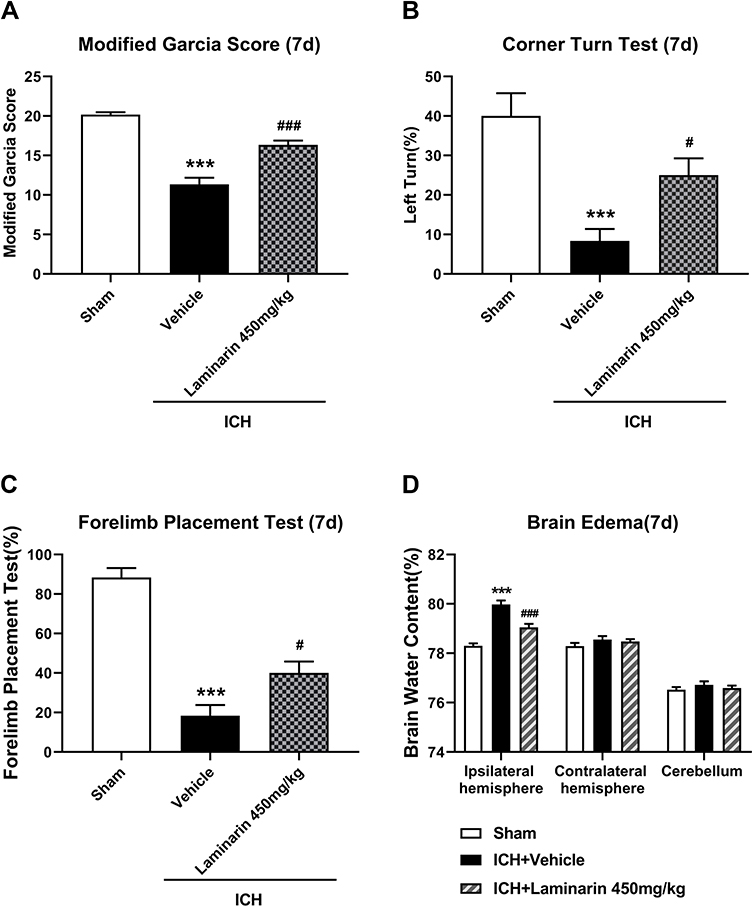 |
Figure 3 Inhibition of Dectin-1 reduced neurological impairment and brain edema at 7 days following ICH. (A) Modified Garcia score. (B) Corner turn test. (C) Forelimb placement test. (D) Brain water content at 7 days following ICH; n = 6. Data are expressed as the mean ± SEM. ***p < 0.001 vs sham. #p < 0.05, ###p < 0.001 vs ICH + vehicle. |
Inhibition of Dectin-1 Alleviated Pyroptosis and Neuroinflammation Following ICH
To determine whether Dectin-1 amplifies pyroptosis after ICH, we next studied the expression of pyroptosis-related molecules in the perihematomal zone at 3 days following ICH. The results demonstrated that GSDMD-N showed a higher expression following ICH, whereas inhibition of Dectin-1 significantly reduced the ICH-induced elevation of GSDMD-N (ICH + laminarin vs ICH + vehicle: F = 4.609, p = 0.042) (Figure 4A). Notably, the results of ELISA indicated that the expression of IL-1β and IL-18 significantly increased following ICH, both of which were alleviated by inhibition of Dectin-1 (ICH + laminarin vs ICH + vehicle—IL-1β: F = 106.320, p = 0.002; IL-18: F = 53.796, p = 0.002) (Figure 4B). The above results confirmed that the inhibition of Dectin-1 alleviated pyroptosis and neuroinflammation after ICH.
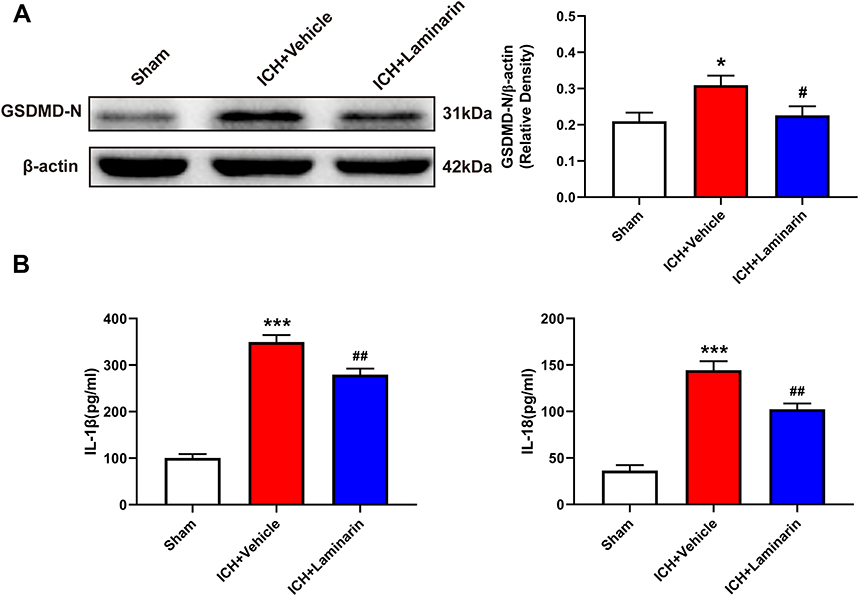 |
Figure 4 Dectin-1 blockade reduced ICH-induced expression of pyroptosis-associated molecules. (A) Representative Western blot bands and protein quantitative analysis of GSDMD-N; n = 4. (B) Concentrations of IL-1β and IL-18 in the perihematomal zone according to ELISA; n = 5. Data are expressed as the mean ± SEM. *p < 0.05, ***p < 0.001 vs sham. #p < 0.05, ##p < 0.01 vs ICH + vehicle. |
Blockade of Dectin-1 Inhibited the Signal Axis of Dectin-1/SYK
Next, the mRNA and protein levels of Dectin-1 were assessed to determine whether the Dectin-1/SYK signal was activated following ICH. Double immunofluorescence staining showed that Dectin-1 co-localized with SYK, which suggested that an association exists between Dectin-1 and SYK (Figure 5A). The qRT-PCR results suggested that the transcription level of Dectin-1 in the ICH + vehicle group was significantly increased compared to that in the sham group, whereas laminarin treatment after ICH abrogated the up-regulation of Dectin-1 (ICH + laminarin vs ICH + vehicle: F = 136.772, p < 0.001 (Figure 5B). Moreover, the protein expression of Dectin-1 and phospho-SYK (p-syk) significantly increased following ICH, both of which were down-streamed by treatment with laminarin (ICH + laminarin vs ICH + vehicle—Dectin-1: F = 21.434, p = 0.001; p-syk: F = 84.656, p < 0.001) (Figure 5C and Figure 5D). These data showed that Dectin-1 signaling was activated after ICH and induced the recruitment and phosphorylation of SYK.
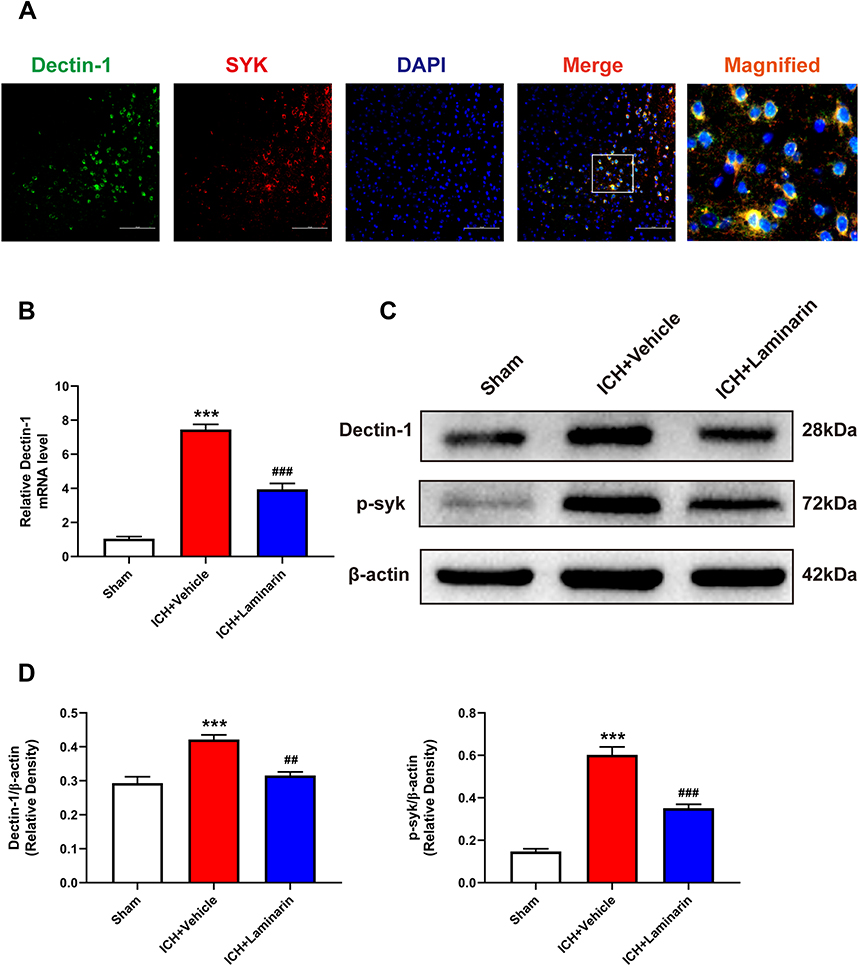 |
Figure 5 Laminarin inhibited the activation of the Dectin-1 signal after ICH. (A) Double immunofluorescence staining images of Dectin-1 co-located with SYK; n = 3. Scale bar = 100 μm. (B) Relative mRNA level of Dectin-1 in the perihematomal zone at 3 days following ICH; n = 6. (C and D) Protein bands and quantitative analysis of Dectin-1 and phosphorylation of SYK (p-syk); n = 4. Data are expressed as the mean ± SEM. ***p < 0.001 vs sham. ##p < 0.01, ###p < 0.001 vs ICH + vehicle. |
Dectin-1 Triggered NLRP3 Inflammasome Activation After ICH
We confirmed that Dectin-1 triggered pyroptosis after ICH. However, it is unclear whether Dectin-1 triggers pyroptosis by activating NLRP3 inflammasomes following ICH. To clarify this question, qRT-PCR and Western blot were conducted to determine the expression of NLRP3 inflammasomes and associated inflammatory cytokines (IL-1β, IL-18) after ICH. We found that the transcription levels of NLRP3, apoptosis-associated speck-like protein containing CARD (ASC), caspase-1, IL-1β, and IL-18 in the ICH + vehicle group were significantly elevated compared to those in the sham group, which were importantly inhibited by the administration of laminarin (ICH + laminarin vs ICH + vehicle—NLRP3: F = 33.776, p = 0.029; ASC: F = 40.545, p = 0.013; caspase-1: F = 46.174, p < 0.001; IL-1β: F = 26.392, p < 0.001; IL-18: F = 12.658, p = 0.004) (Figure 6A). In addition, the protein levels of NLRP3, ASC, cleaved caspase-1, proIL-1β, IL-1β, and IL-18 were significantly increased in the ICH + vehicle group compared to those in the sham group; however, the increments of these proteins were all markedly inhibited in ICH mice treated with laminarin (ICH + laminarin vs ICH + vehicle—NLRP3: F = 23.316, p = 0.021; ASC: F = 16.044, p = 0.002; cleaved caspase-1: F = 34.555, p < 0.001; proIL-1β: F = 8.520, p = 0.017; IL-1β: F = 37.601, p < 0.001; IL-18: F = 42.144, p < 0.001) (Figure 6B–E). These results revealed that Dectin-1 was involved in orchestrating neuroinflammation triggered by NLRP3 inflammasomes after ICH.
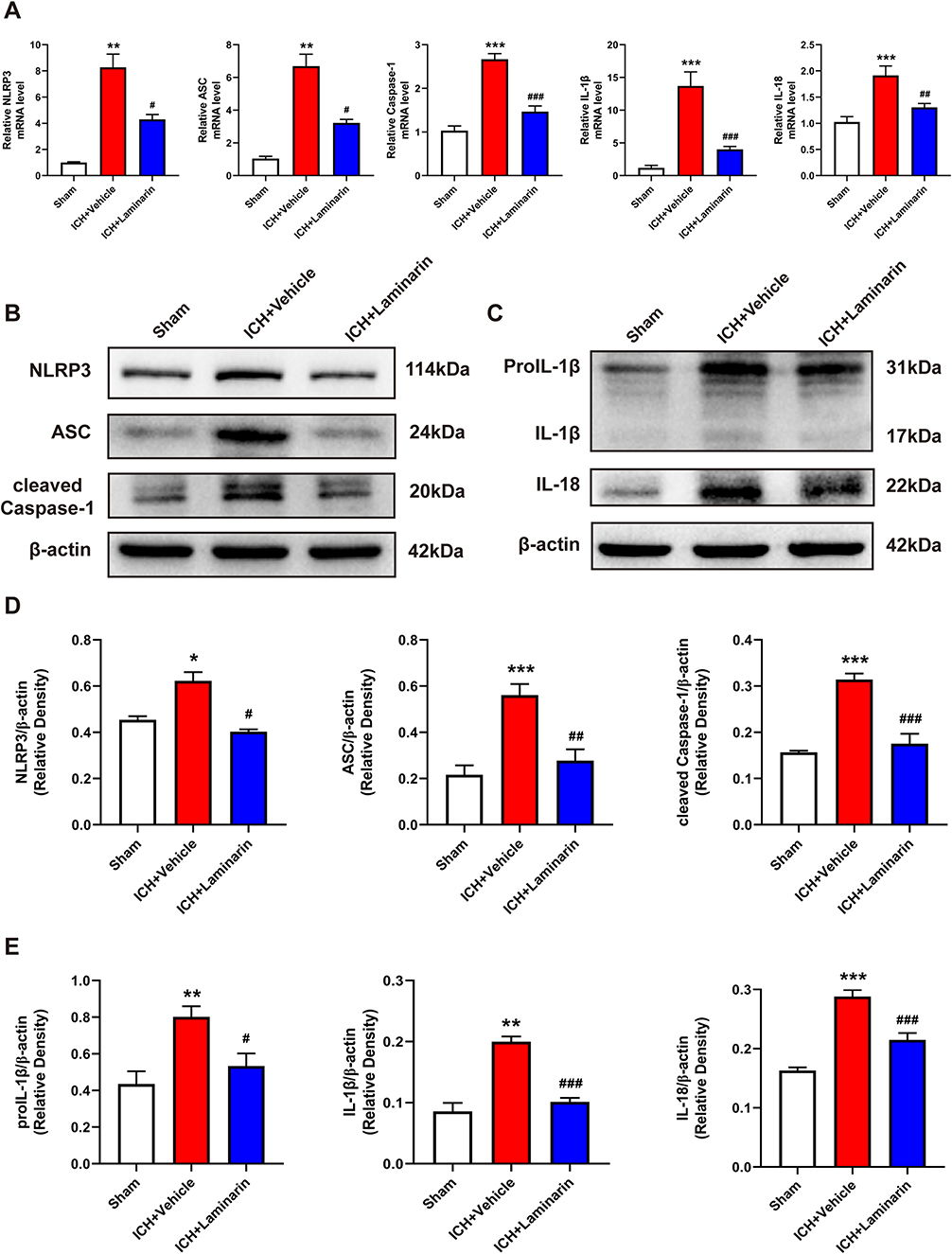 |
Figure 6 Dectin-1 blockade downregulated the expression of NLRP3 inflammasomes and associated inflammatory cytokines. (A) Transcription levels of NLRP3, ASC, caspase-1, IL-1β, and IL-18 at 3 days following ICH; n = 6. (B–D) Protein bands and protein quantitative analysis of NLRP3, ASC, and cleaved caspase-1. (C–E) Protein bands and protein quantitative analysis of IL-1β and IL-18; n = 4. Data are expressed as the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 vs sham. #p < 0.05, ##p < 0.01, ###p < 0.001 vs ICH + vehicle. |
Inhibition of Dectin-1 Attenuated LPS-Stimulated Dectin-1 Signaling Activation and Microglial Pyroptosis in vitro
In an in vitro experiment, BV2 microglia cells were stimulated with LPS, and laminarin was administered for intervention. To determine whether laminarin exhibited cytotoxicity toward BV2 microglia, a CCK-8 assay was performed at 24 h after treatment with different concentrations of laminarin (0, 10, 100, 300, 500, and 1000 μg/mL). The results showed that treatment with these concentrations of laminarin did not exhibit cytotoxic effects on BV2 microglia cells (Supplemental Figure S1). Therefore, based on previous studies,22,23 BV2 microglia cells were treated with laminarin (300 μg/mL). Then, Western blot was performed to assess the expression of the Dectin-1 signal axis, NLRP3 inflammasomes, and associated inflammatory cytokines. In agreement with the in vivo results, the expression of Dectin-1, p-syk, NLRP3, ASC, procaspase-1, proIL-1β, and pyroptosis-related molecules (cleaved caspase-1, IL-1β, and GSDMD-N) was significantly increased in the lysates of LPS-stimulated BV2 microglia compared to the control cells, all of which were reversed with laminarin treatment (LPS + laminarin vs LPS—Dectin-1: F = 11.889, p = 0.002; p-syk: F = 45.720, p < 0.001; NLRP3: F = 10.012, p = 0.022; ASC: F = 12.620, p = 0.004; procaspase-1: F = 44.723, p = 0.045; proIL-1β: F = 99.214, p < 0.001; cleaved caspase-1: F = 12.051, p = 0.006; IL-1β: F = 25.108, p = 0.020; GSDMD-N: F = 19.592, p = 0.030) (Figure 7A and Figure 7B). According to these results, we confirmed that Dectin-1 activation could amplify neuroinflammation and microglia pyroptosis by activating the NLRP3 inflammasome.
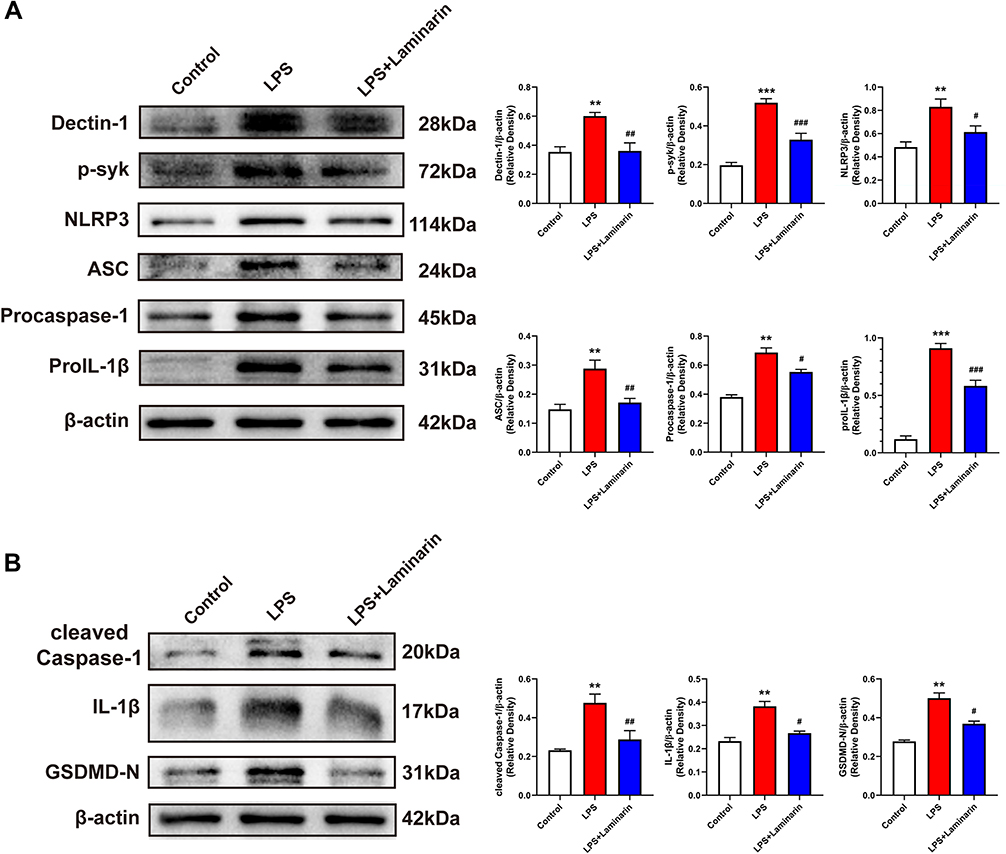 |
Figure 7 Inhibition of Dectin-1 attenuated LPS-stimulated Dectin-1 signaling and microglial pyroptosis in vitro. (A) Western blot bands and protein quantitative analysis of Dectin-1, p-syk, NLRP3, ASC, procaspase-1, and proIL-1β in BV2 microglia lysates. (B) Western blot bands and protein quantitative analysis of pyroptosis-associated proteins, including cleaved caspase-1, IL-1β, and GSDMD-N, in BV2 microglia lysates; n = 4. Data are expressed as the mean ± SEM. **p < 0.01, ***p < 0.001 vs control. #p < 0.05, ##p < 0.01, ###p < 0.001 vs LPS. |
Discussion
In this study, we uncovered an important role for Dectin-1 in triggering neuroinflammatory injury after ICH. First, we observed that the protein levels of Dectin-1 significantly increased, and the Dectin-1 receptor co-localized with microglia following ICH. In addition, inhibition of Dectin-1 by laminarin reduced ICH-induced neurological impairment and brain edema. Moreover, suppression of Dectin-1 inhibited microglial pyroptosis, activation of the NLRP3 inflammasome, and the production of IL-1β and IL-18. Together, we conclude that the inhibition of Dectin-1 pathways eases neuroinflammatory injury by alleviating NLRP3 inflammasome-mediated pyroptosis following ICH and may be beneficial in the clinic.
Dectin-1, expressed mainly on myeloid cells, is a C-type lectin receptor with an extracellular carbohydrate-recognition domain, a stalk and transmembrane region, and an intracellular cytoplasmic tail that contains an immune-receptor tyrosine-based activation motif (ITAM).35,36 Initial research has indicated that the activation of Dectin-1 contributes to the fungal immunity inflammatory response.37,38 However, recent research has indicated that Dectin-1 may also contribute to sterile inflammation. For example, Dectin-1 increases after myocardial IR injury and aggravates myocardial injury by releasing inflammatory cytokines, whereas Dectin-1 blockade can ease inflammation and myocardial injury.21,26 In ischemic stroke mice, activation of Dectin-1/SYK signaling can trigger neuroinflammation and microglia activation, and aggravate neurological damage.23 In ICH-induced mice, suppression of Dectin-1 can regulate microglial polarization to alleviate neuroinflammation.22 In contrast, Dectin-1 limits neuroinflammation in experimental autoimmune encephalomyelitis mice by inducing oncostatin M expression via a Card9-independent pathway.39 Activation of Dectin-1 can trigger neuroinflammation to facilitate central nervous system axon regeneration.19 Therefore, Dectin-1 may play different roles in different pathological diseases. Although it has been shown that the inhibition of Dectin-1 can regulate microglial polarization following ICH, the mechanism by which Dectin-1 functions in the context of neuroinflammation remains incompletely understood. Therefore, it is important to explore the precise regulatory mechanism of Dectin-1 in ICH-induced neuroinflammation.
In the present study, we found that the protein levels of Dectin-1 in the hemorrhagic cerebral hemispheres increased with a time-dependent trend following ICH, significantly increasing at 12 h after surgery, reaching a peak at 3 days, and maintaining high expression at 7 days. Double immunofluorescence staining indicated that Dectin-1 is positively expressed on microglia but not co-localized with astrocytes and neurons following ICH. There is increasing evidence to suggest that receptors co-localized with microglia, highly expressed after ICH, may regulate neuroinflammation.7,40,41 In the in vitro experiments, we found that protein levels of Dectin-1 increased in the LPS-stimulated BV2 microglia cells. As a pattern recognition receptor, Dectin-1 can recognize pathogen-associated molecular patterns (PAMPs) in microorganisms.42 In addition, LPS, which belongs to PAMPs, can induce neuroinflammation in microglia. Thus, LPS can be recognized by Dectin-1, resulting in activation and an increasing expression of Dectin-1. Taken together, these findings suggest that Dectin-1 may increase to contribute to neuroinflammation in the acute phase after ICH.
Brain edema is a risk factor resulting in poor neurological prognosis of ICH.5 Laminarin, an inhibitor of Dectin-1, has been reported as an important biodegradable polysaccharide without toxicity.43 We found that 450 mg/kg of laminarin effectively reduced neurological damage and brain edema following ICH. Lee T-K found that 50 mg/kg of laminarin attenuated brain injury following transient forebrain cerebral ischemic,29 while Ye X-C found that 300 mg/kg of laminarin improved neurological functions after ischemic stroke.23 Therefore, laminarin may have different metabolic efficiencies in different diseases. It is meaningful to explore the pharmacokinetics of laminarin in these diseases, and we are preparing another study to explore the pharmacokinetics of laminarin in ICH mice.
Pyroptosis, an inflammatory type of programmed cell death, results in extracellular spilling of proinflammatory cytokines.44 GSDMD-N, the pyroptosis executor, releases inflammasome-associated cytokines (IL-1β and IL-18) by forming cytotoxic pores on the plasma membrane.9,10 Therefore, we evaluated the expression of GSDMD-N, IL-1β, and IL-18, and investigated whether Dectin-1 was involved in pyroptosis following ICH. We found that GSDMD-N was upregulated in ICH mice and in LPS-stimulated BV2 microglia. In addition, the concentrations of IL-1β and IL-18 were significantly increased in the perihematomal area following ICH. However, in both in vivo and in vitro experiments, inhibition of Dectin-1 by laminarin effectively attenuated the increase in IL-1β and GSDMD-N, indicating that Dectin-1 contributed to microglial pyroptosis following ICH.
We next investigated the potential mechanism by which Dectin-1 triggers microglial pyroptosis. The NLRP3 inflammasome consists of three components (NLRP3, ASC, and procaspase-1).14,45 Once the NLRP3 inflammasome complex is activated, procaspase-1 is induced into activated caspase-1, which promotes the formation of IL-1β and IL-18, and cleaves GSDMD to GSDMD-N resulting in cell pyroptosis.10,14 It is known that SYK phosphorylation can activate the NLRP3 inflammasome,46 and activated Dectin-1 can induce the recruitment and phosphorylation of SYK.47 Thus, we assumed that Dectin-1 might induce microglial pyroptosis by activating the NLRP3 inflammasome after ICH. Interestingly, we found that the Dectin-1 receptor inhibitor laminarin downregulated the expression of Dectin-1 and reversed the phosphorylation of SYK both in vivo and in vitro, suggesting that laminarin could markedly inhibit Dectin-1 signal activation. Furthermore, we first found that inhibition of Dectin-1 decreased the expression of NLRP3 inflammasome components and GSDMD-N after ICH. These data showed that Dectin-1 induces microglial pyroptosis by activating the NLRP3 inflammasome. Based on these results, we discovered for the first time that there is a link between Dectin-1 and NLPR3 inflammasome. We found that the inhibition of Dectin-1 alleviated neuroinflammation by attenuating NLRP3 inflammasome-mediated pyroptosis after ICH, which provided a novel insight into the function and role of Dectin-1 in regulating neuroinflammation injury after ICH.
This study has several limitations that warrant discussion. First, it is known that Dectin-1 promotes inflammation, apoptosis, and axon regeneration.19,23,48 As a result, there are concerns regarding the inflammatory effects of Dectin-1; therefore, it is meaningful to elucidate other effects of Dectin-1 in the nervous system. Second, we did not conduct transmission electron microscopy to observe the morphological changes of microglial pyroptosis in this research, although this will be used in our future studies.
Conclusions
Our study is the first to indicate that the inhibition of Dectin-1 alleviated neuroinflammation by attenuating NLRP3 inflammasome-mediated pyroptosis after ICH (Figure 8). Dectin-1 blockade might be a potential method for treating patients with ICH.
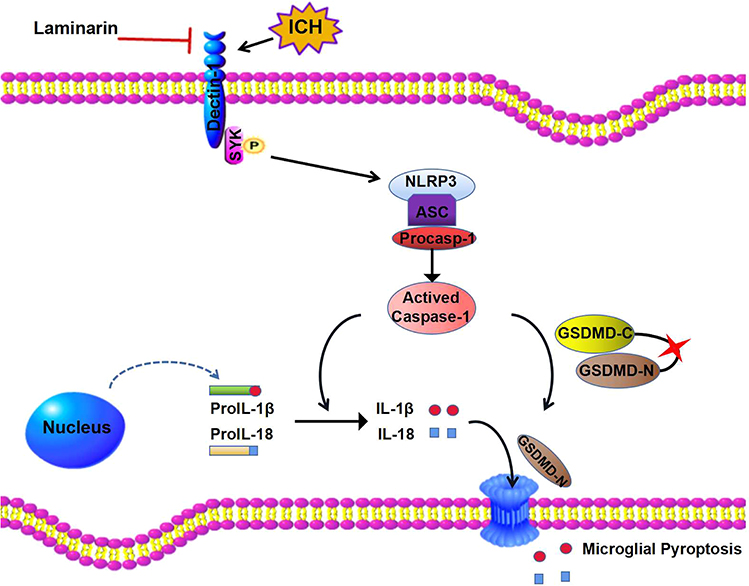 |
Figure 8 Diagram of Dectin-1-mediated microglial pyroptosis after ICH. The level of Dectin-1 is upregulated following ICH injury. Dectin-1 initiates SYK phosphorylation and mobilizes the NLRP3/caspase-1 downstream pathway to generate inflammatory mediators. In addition, the activated caspase-1 induced by Dectin-1 initiation cleaves GSDMD to release an N-terminal fragment, which forms pores on microglia, resulting in the extracellular release of inflammatory factors such as IL-1β and IL-18. Inhibiting Dectin-1 by laminarin alleviates inflammatory responses, resulting in easing of intracerebral hemorrhage. |
Data Availability
All of the data used in the study are available from the corresponding author upon reasonable request.
Ethics Approval and Consent to Participate
All animal studies were approved by the Ethics Committee of Zhujiang Hospital of Southern Medical University.
Acknowledgments
The authors wish to thank all of the staff at the Laboratory Animal Center of Zhujiang Hospital of Southern Medical University.
Author Contributions
This work was conducted with the cooperation of the authors. Qinghua Wang conceived and jointly designed the study with Zhiquan Ding, Haitao Sun, and Run Zhang; Zhiquan Ding, Zhenzhong Zhong, and Jun Wang conducted this study and wrote the manuscript; Run Zhang, Jinlian Shao, Yulong Li, Guiwei Wu, Huiru Tu, and Wen Yuan conducted data acquisition and data analysis; Zhiquan Ding, Zhenzhong Zhong, Jun Wang, Run Zhang, Jinlian Shao, Yulong Li, Guiwei Wu, Huiru Tu, Wen Yuan, Qinghua Wang, and Haitao Sun revised the manuscript. All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (81701200); the Natural Science Fund of Guangdong Province (2020A1515010038); the Presidential Foundation of Zhujiang Hospital of Southern Medical University (2021yzjj2020qn15); and the Presidential Foundation of Zhujiang Hospital of Southern Medical University (yzjj2018rc03).
Disclosure
The authors declare no conflicts of interest in this study.
References
1. Nadeau CA, Dietrich K, Wilkinson CM, et al. Prolonged blood-brain barrier injury occurs after experimental intracerebral hemorrhage and is not acutely associated with additional bleeding. Transl Stroke Res. 2019;10(3):287–297. doi:10.1007/s12975-018-0636-9
2. Bai Q, Xue M, Yong VW. Microglia and macrophage phenotypes in intracerebral haemorrhage injury: therapeutic opportunities. Brain. 2020;143(5):1297–1314. doi:10.1093/brain/awz393
3. Zhu H, Wang Z, Yu J, et al. Role and mechanisms of cytokines in the secondary brain injury after intracerebral hemorrhage. Prog Neurobiol. 2019;178:101610. doi:10.1016/j.pneurobio.2019.03.003
4. Chen S, Peng J, Sherchan P, et al. TREM2 activation attenuates neuroinflammation and neuronal apoptosis via PI3K/Akt pathway after intracerebral hemorrhage in mice. J Neuroinflammation. 2020;17(1):168. doi:10.1186/s12974-020-01853-x
5. Chen S, Yang Q, Chen G, Zhang JH. An update on inflammation in the acute phase of intracerebral hemorrhage. Transl Stroke Res. 2015;6(1):4–8. doi:10.1007/s12975-014-0384-4
6. Zhou Y, Wang Y, Wang J, Anne Stetler R, Yang QW. Inflammation in intracerebral hemorrhage: from mechanisms to clinical translation. Prog Neurobiol. 2014;115:25–44. doi:10.1016/j.pneurobio.2013.11.003
7. Chen S, Zhao L, Sherchan P, et al. Activation of melanocortin receptor 4 with RO27-3225 attenuates neuroinflammation through AMPK/JNK/p38 MAPK pathway after intracerebral hemorrhage in mice. J Neuroinflammation. 2018;15(1). doi:10.1186/s12974-018-1140-6
8. Li Q, Cao Y, Dang C, et al. Inhibition of double‐strand DNA‐sensing cGAS ameliorates brain injury after ischemic stroke. EMBO Mol Med. 2020;12(4). doi:10.15252/emmm.201911002
9. Xu P, Hong Y, Xie Y, et al. TREM-1 exacerbates neuroinflammatory injury via NLRP3 inflammasome-mediated pyroptosis in experimental subarachnoid hemorrhage. Transl Stroke Res. 2020;12(4):643–659. doi:10.1007/s12975-020-00840-x
10. Xu P, Zhang X, Liu Q, et al. Microglial TREM-1 receptor mediates neuroinflammatory injury via interaction with SYK in experimental ischemic stroke. Cell Death Dis. 2019;10:555. doi:10.1038/s41419-019-1777-9
11. Li Y, Song W, Tong Y, et al. Isoliquiritin ameliorates depression by suppressing NLRP3-mediated pyroptosis via miRNA-27a/SYK/NF-κB axis. J Neuroinflammation. 2021;18(1). doi:10.1186/s12974-020-02040-8
12. Chang Y, Zhu J, Wang D, et al. NLRP3 inflammasome-mediated microglial pyroptosis is critically involved in the development of post-cardiac arrest brain injury. J Neuroinflammation. 2020;17(1). doi:10.1186/s12974-020-01879-1
13. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16(7):407–420. doi:10.1038/nri.2016.58
14. Luo Y, Reis C, Chen S. NLRP3 inflammasome in the pathophysiology of hemorrhagic stroke: a review. Curr Neuropharmacol. 2019;17(7):582–589. doi:10.2174/1570159x17666181227170053
15. Wang S, Yuan YH, Chen NH, Wang HB. The mechanisms of NLRP3 inflammasome/pyroptosis activation and their role in Parkinson’s disease. Int Immunopharmacol. 2019;67:458–464. doi:10.1016/j.intimp.2018.12.019
16. Xiao L, Zheng H, Li J, Wang Q, Sun H. Neuroinflammation Mediated by NLRP3 inflammasome after intracerebral hemorrhage and potential therapeutic targets. Mol Neurobiol. 2020;57(12):5130–5149. doi:10.1007/s12035-020-02082-2
17. Yuan D, Guan S, Wang Z, et al. HIF-1α aggravated traumatic brain injury by NLRP3 inflammasome-mediated pyroptosis and activation of microglia. J Chem Neuroanat. 2021;116:101994. doi:10.1016/j.jchemneu.2021.101994
18. Ran Y, Su W, Gao F, et al. Curcumin ameliorates white matter injury after ischemic stroke by inhibiting microglia/macrophage pyroptosis through NF-κB suppression and NLRP3 inflammasome inhibition. Oxid Med Cell Longev. 2021;2021:1–25. doi:10.1155/2021/1552127
19. Baldwin KT, Carbajal KS, Segal BM, Giger RJ. Neuroinflammation triggered by β-glucan/dectin-1 signaling enables CNS axon regeneration. Proc Natl Acad Sci. 2015;112(8):2581–2586. doi:10.1073/pnas.1423221112
20. Cao M, Wu Z, Lou Q, et al. Dectin-1-induced RIPK1 and RIPK3 activation protects host against Candida albicans infection. Cell Death Differ. 2019;26(12):2622–2636. doi:10.1038/s41418-019-0323-8
21. Fan Q, Tao R, Zhang H, et al. Dectin-1 contributes to myocardial ischemia/reperfusion injury by regulating macrophage polarization and neutrophil infiltration. Circulation. 2019;139(5):663–678. doi:10.1161/circulationaha.118.036044
22. Fu X, Zeng H, Zhao J, et al. Inhibition of Dectin-1 ameliorates neuroinflammation by regulating microglia/macrophage phenotype after intracerebral hemorrhage in mice. Transl Stroke Res. 2021;12(6):1018–1034. doi:10.1007/s12975-021-00889-2
23. Ye XC, Hao Q, Ma WJ, et al. Dectin-1/Syk signaling triggers neuroinflammation after ischemic stroke in mice. J Neuroinflammation. 2020;17(1). doi:10.1186/s12974-019-1693-z
24. Wagener M, Hoving JC, Ndlovu H, Marakalala MJ. Dectin-1-Syk-CARD9 Signaling Pathway in TB Immunity. Front Immunol. 2018;9. doi:10.3389/fimmu.2018.00225
25. Peng Y, Chen Y, Ma J, et al. Role and mechanism of the Dectin‑1‑mediated Syk/NF‑κB signaling pathway in Talaromyces marneffei infection. Exp Ther Med. 2021;23(1). doi:10.3892/etm.2021.11007
26. Li X, Bian Y, Pang P, et al. Inhibition of Dectin-1 in mice ameliorates cardiac remodeling by suppressing NF-κB/NLRP3 signaling after myocardial infarction. Int Immunopharmacol. 2020;80:106116. doi:10.1016/j.intimp.2019.106116
27. Ng ACK, Yao M, Cheng SY, et al. Protracted morphological changes in the corticospinal tract within the cervical spinal cord after intracerebral hemorrhage in the right striatum of mice. Front Neurosci. 2020;14. doi:10.3389/fnins.2020.00506
28. Xiao L, Zheng H, Li J, et al. Targeting NLRP3 inflammasome modulates gut microbiota, attenuates corticospinal tract injury and ameliorates neurobehavioral deficits after intracerebral hemorrhage in mice. Biomed Pharmacother. 2022;149:112797. doi:10.1016/j.biopha.2022.112797
29. Lee TK, Ahn JH, Park CW, et al. Pre-treatment with laminarin protects hippocampal CA1 pyramidal neurons and attenuates reactive gliosis following transient forebrain ischemia in gerbils. Mar Drugs. 2020;18(1):52. doi:10.3390/md18010052
30. Fourrier C, Remus-Borel J, Greenhalgh AD, et al. Docosahexaenoic acid-containing choline phospholipid modulates LPS-induced neuroinflammation in vivo and in microglia in vitro. J Neuroinflammation. 2017;14(1). doi:10.1186/s12974-017-0939-x
31. Nam HY, Nam JH, Yoon G, et al. Ibrutinib suppresses LPS-induced neuroinflammatory responses in BV2 microglial cells and wild-type mice. J Neuroinflammation. 2018;15(1). doi:10.1186/s12974-018-1308-0
32. Dang R, Yang M, Cui C, et al. Activation of angiotensin‐converting enzyme 2/angiotensin (1–7)/mas receptor axis triggers autophagy and suppresses microglia proinflammatory polarization via forkhead box class O1 signaling. Aging Cell. 2021;20(10). doi:10.1111/acel.13480
33. Ren H, Kong Y, Liu Z, et al. Selective NLRP3 (Pyrin Domain–Containing Protein 3) inflammasome inhibitor reduces brain injury after intracerebral hemorrhage. Stroke. 2018;49(1):184–192. doi:10.1161/strokeaha.117.018904
34. Zeng J, Chen Y, Ding R, et al. Isoliquiritigenin alleviates early brain injury after experimental intracerebral hemorrhage via suppressing ROS- and/or NF-κB-mediated NLRP3 inflammasome activation by promoting Nrf2 antioxidant pathway. J Neuroinflammation. 2017;14(1). doi:10.1186/s12974-017-0895-5
35. Brown GD, Willment JA, Whitehead L. C-type lectins in immunity and homeostasis. Nat Rev Immunol. 2018;18(6):374–389. doi:10.1038/s41577-018-0004-8
36. Tian J, Ma J, Ma K, et al. β-Glucan enhances antitumor immune responses by regulating differentiation and function of monocytic myeloid-derived suppressor cells. Eur J Immunol. 2013;43(5):1220–1230. doi:10.1002/eji.201242841
37. Lilly LM, Gessner MA, Dunaway CW, et al. The β-glucan receptor dectin-1 promotes lung immunopathology during fungal allergy via IL-22. J Immunol Res. 2012;189(7):3653–3660. doi:10.4049/jimmunol.1201797
38. Quintin J, Saeed S, Martens Joost HA, et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe. 2012;12(2):223–232. doi:10.1016/j.chom.2012.06.006
39. Deerhake ME, Danzaki K, Inoue M, et al. Dectin-1 limits autoimmune neuroinflammation and promotes myeloid cell-astrocyte crosstalk via Card9-independent expression of Oncostatin M. Immunity. 2021;54(3):484–498.e8. doi:10.1016/j.immuni.2021.01.004
40. Zhang Y, Chen Y, Wu J, et al. Activation of Dopamine D2 receptor suppresses neuroinflammation through αB-crystalline by inhibition of NF-κB nuclear translocation in experimental ICH mice model. Stroke. 2015;46(9):2637–2646. doi:10.1161/strokeaha.115.009792
41. Li M, Li Z, Ren H, et al. Colony stimulating factor 1 receptor inhibition eliminates microglia and attenuates brain injury after intracerebral hemorrhage. J Cereb Blood Flow Metab. 2016;37(7):2383–2395. doi:10.1177/0271678x16666551
42. Li M, Zhang R, Li J, Li J. The role of C-type lectin receptor signaling in the intestinal microbiota-inflammation-cancer axis. Front Immunol. 2022;13. doi:10.3389/fimmu.2022.894445
43. Costa AMS, Rodrigues JMM, Pérez-Madrigal MM, Dove AP, Mano JF. Modular functionalization of laminarin to create value-added naturally derived macromolecules. J Am Chem Soc. 2020;142(46):19689–19697. doi:10.1021/jacs.0c09489
44. Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci. 2017;42(4):245–254. doi:10.1016/j.tibs.2016.10.004
45. Yang Y, Wang H, Kouadir M, Song H, Shi F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019;10(2). doi:10.1038/s41419-019-1413-8
46. Patel D, Gaikwad S, Challagundla N, Nivsarkar M, Agrawal-Rajput R. Spleen tyrosine kinase inhibition ameliorates airway inflammation through modulation of NLRP3 inflammosome and Th17/Treg axis. Int Immunopharmacol. 2018;54:375–384. doi:10.1016/j.intimp.2017.11.026
47. Geijtenbeek TBH, Gringhuis SI. C-type lectin receptors in the control of T helper cell differentiation. Nat Rev Immunol. 2016;16(7):433–448. doi:10.1038/nri.2016.55
48. Zongyun C, Bixia L, Fadian D, et al. The role of dectin-1-mediated m1 macrophage polarization in cerebral ischemia-reperfusion injury. Emerg Med Int. 2021;2021:1–7. doi:10.1155/2021/6697271
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.