")
Back to Journals » Cancer Management and Research » Volume 12
Inhibition of CDK1 Reverses the Resistance of 5-Fu in Colorectal Cancer
Authors Zhu Y, Li K, Zhang J, Wang L, Sheng L, Yan L
Received 6 April 2020
Accepted for publication 19 August 2020
Published 4 November 2020 Volume 2020:12 Pages 11271—11283
DOI https://doi.org/10.2147/CMAR.S255895
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Seema Singh
Yiping Zhu,1 Kai Li,2 Jieling Zhang,2 Lu Wang,1 Lili Sheng,1 Liang Yan2
1Department of Oncology, The First Affiliated Hospital of Wannan Medical College, Wuhu, Anhui, People’s Republic of China; 2Provincial Key Laboratory of Biological Macro-molecules Research, Wannan Medical College, Wuhu, Anhui, People’s Republic of China
Correspondence: Lili Sheng; Liang Yan Email [email protected]; [email protected]
Introduction: Although the survival rate of colorectal cancer (CRC) patients can be improved by surgery, radiotherapy, and chemotherapy, the resistance to 5-fluorouracil (5-Fu) affects the effect of chemotherapy and the prognosis of patients. An increasing number of studies showed that 5-Fu resistance was the main reason for the failure of colorectal cancer treatment. The poor prognosis of colorectal cancer greatly harms people’s health. This study aimed to clarify the correlation between cyclin-dependent kinase 1 (CDK1) and 5-Fu-induced tumor resistance.
Materials and Methods: Cell proliferation and invasion experiments showed that down-regulation of CDK1 inhibited fluorouracil-resistant CRC cell proliferation. The expression level of CDK1 was detected in 5-Fu-resistant CRC cells in vitro. Tumor growth was inhibited by down-regulation of CDK1 in tumor xenograft mouse models.
Results: We found that CDK1 was highly expressed in tumor tissues, especially in fluorouracil-resistant tissues. We also confirmed that the differential expression of 5-Fu in tumor tissues was related to tumor site, lymph node metastasis and stage. CDK1 promoted migration, invasion and inhibited apoptosis in 5-Fu-resistant CRC cells. Down-regulation of CDK1 inhibited fluorouracil-resistant CRC cell proliferation and tumorigenesis in vivo.
Conclusion: High expression of CDK1 may lead to poor clinical prognosis, and inhibition of CDK1 enhances 5-Fu sensitivity in CRC. Our research suggested that CDK1 may be used to predict 5-Fu efficacy and as a therapeutic target for CRC.
Keywords: CDK1, colorectal cancer, 5-Fu, drug resistance
Background
Approximately 1,800,000 patients were diagnosed with colorectal cancer (CRC) in 2018, and some patients died of this disease.1 Although diagnostic methods and treatment have been greatly improved,2–4 for nearly 60% of colorectal cancer patients with distant metastases, the survival rate is very low.5 Clinical stages closely result in the outcomes of CRC patients. The 5-year survival rate for stage I patients exceeds 90%, while is less than 10% for stage IV patients.6 Surgery, radiotherapy, and chemotherapy are widely used in CRC. Radiotherapy is recommended as adjuvant/neoadjuvant treatment for the majority of rectal cancer patients,7 and radiotherapy combined with chemotherapy significantly reduces the risk of recurrence and metastasis.8 Adjuvant chemotherapy contained 5-fluorouracil (5-Fu) is currently recommended for patients with stage III CRC after surgery.9–11 Moreover, a combination of 5-Fu, irinotecan/oxaliplatin, and monoclonal antibodies significantly improved overall response rate (ORR), and prolonged survival time of advanced CRC patients.12–14
5-Fu is a common thymidylate synthase inhibitor, converting into 5-fluorouracil deoxynucleotide (5F-dUMP), inhibiting deoxythymidylate synthetase, preventing deoxy methylation of uridine (dUMP) into deoxythymidylate (dTMP), which affects DNA and RNA synthesis, and exerting an anti-tumor effect.15 However, nearly 50% of colorectal cancer patients showed no response to 5-Fu, and more and more studies reported that fluorouracil resistance led to failure of CRC treatment.16–18 But the mechanism is not clear. Therefore, biomarkers predicting the efficacy of fluorouracil are urgently needed in CRC treatment.
CDK1 (cyclin-dependent kinase 1) is a serine/threonine-like protein kinase. Activation of CDK1 can phosphorylate target proteins and produce corresponding physiological effects, such as regulating cell cycle and promoting cell proliferation.19 Multiple studies have reported that CDK1 is overexpressed in several malignancies.20,21 In CRC patients, the high nuclear/cytoplasmic ratio of CDK1 often predicted a poor prognosis.22 Moreover, CDK1 has been found to be closely associated with chemotherapy resistance in numerous tumors. Li et al23 found that silencing UBE2C in cisplatin-resistant ovarian cancer cells reversed its resistance to cisplatin via down-regulating the expression of CDK1. Shang et al24 also reported that LncRNA OR3A4 promoted drug resistance to cisplatin by upregulating CDK1 in non-small cell lung cancer (NSCLC). In CRC, BRAF mutation is a poor prognostic factor and often results in poor survival outcomes. A combination of CDK1 inhibitor and MEK/ERK inhibitor promoted apoptosis in BRAF mutant CRC cells, which suggested that CDK1 could mediate apoptosis resistance in colorectal cancer.25 Zhang26 showed that Wnt/β-catenin activation promoted the resistance of colorectal cancer to fluorouracil and oxaliplatin. However, the mechanism by which CDK1 promotes resistance remains to be elucidated. Our previous research revealed that there was a CeRNA regulatory relationship between LINC00365 and CDK1. CDK1 expression was positively correlated with Wnt/β-catenin activation.27,28 Therefore, we surmised that CDK1 might be associated with 5-Fu resistance in CRC.
Our study demonstrated that overexpression of CDK1 resulted in a poor prognosis in CRC patients. Besides, CDK1 up-regulation was also observed in fluorouracil-resistant CRC tissues. Then, we explored that silence of CDK1 in fluorouracil-resistant cells inhibited the proliferation, migration, and invasion, and reversed fluorouracil-resistance. CDK1 might be considered as a predictor for fluorouracil efficacy, and a therapeutic target for CRC.
Materials and Methods
Samples of CRC Patients
From April 2012 to September 2014, we collected tumor and paracancerous tissues of 80 CRC patients who were admitted to the First Affiliated Hospital of Wannan Medical College. All these 80 cases underwent surgery. Meanwhile, we also collected tumor tissues from 76 metastatic CRC (mCRC) patients. None of the samples were exposed to radiotherapy or chemotherapy. All the fresh tissues were stored in the −80°C refrigerator until RNA extraction.All specimens were confirmed by two independent pathologists. Tumors were staged according to the seventh edition of cancer staging criteria of the American Joint Committee on Cancer (AJCC). Clinicopathological characteristics of 80 patients undergoing surgery were collected, and the correlation between CDK1 and clinical case characteristics was analyzed. All 76 mCRC patients received first-line chemotherapy containing 5-Fu. Tumor response to chemotherapy was confirmed by CT every two cycles and evaluated according to RECIST. Among the 76 patients, 43 cases achieved partial response (PR) and complete response (CR), 33 cases achieved stable disease (SD) and progression disease (PD). This study complied with the Helsinki Declaration. We obtained permission from Yijishan Hospital Ethics Committee (approval number 2018–34) and got informed consent from all patients.
Cell Culture and Establishment of Drug-Resistant Cell Lines
Human CRC cells (SW620, SW480, HCT116, HT-29) and human colorectal mucosal epithelial cells-NCM460 were purchased from Shanghai Cell Research Institute. We obtained HT-29/5-Fu and SW480/5-Fu by gradually increasing 5-Fu concentration to treat HT-29 and SW480 cells in 6 months. Cells were cultured at 37°C with 5% CO2.
qRT-PCR
Total RNA was extracted from the tissues with Trizol (Invitrogen). RNA was reverse-transcribed to cDNA with Reverse Transcription Kit (Ribo, China). qRT-PCR was performed on the ABI7500 PCR operating system. Sequences of primers are listed as follows: GAPDH Forward: GAACGGGAAGCTCACTGG, Reverse: GCCTGCTTCACCACCTTCT; CDK1 Forward: GGTTCCTAGTACTGCAATTCG, Reverse: TTTGCCAGAAATTCGTTTGG. Relative CDK1 expression was normalized using equation 2–ΔΔCT. GAPDH served as a reference.
Transfections
Small interfering RNA targeting CDK1 and its negative control were synthesized by Ribo, and its sequences are listed below: siRNA1: GGAACTTCGTCATCCAAAT, siRNA2: GTACTGCAATTCGGGAAAT, siRNA3: GGTTATATCTCATCTTTGA. Lipo3000 was used for cell transfection according to the protocol. Cells transfected with siRNA were collected for further experiments. Then, the knockdown efficiency of CDK1 was detected by RT-PCR.
Establishment of Stable Transfected Cell Lines
siRNA3 targeting CDK1 was selected for further study. Based on its sequence, GenePharma (Shanghai, China) synthesized the short hairpin RNA (shRNA) and packaged it into a recombinant lentivirus. Simultaneously, a negative control was synthesized with an empty vector. Infected with shRNA for 48 hours, cells were treated with puromycin (Genomeditech, China) at different concentrations (SW480: 1.0 μg/mL; HT-29:0.6 μg/mL). Puromycin concentrations were stably maintained for 6 weeks to establish stable transfected cell lines.
Methyl Thiazolyl Tetrazolium (MTT) Assay
Cells were cultured in 96-well plate, 180 µl of culture medium was added into each well. Thereafter, 20 μL of MTT solution was added into each well at 24, 48, 72, and 96 h. After incubation for 4 h, we added 150 μL of dimethyl sulfoxide into each well. Ten minutes later, the samples were detected at 490nm wavelength.
Flow Cytometry
In 6-well plates, floating and adherent cells were harvested and centrifuged. On the ice, the cells were stained with Annexin V/propidium iodide (LiankeBio, China). Next, cell apoptosis detection was performed on flow cytometry (BD, USA).
Colony Formation Assay
In 12-well plates, 500 cells were cultured each well, and the medium was changed every 5 days until visible colonies formed. Then, colonies were fixed and stained with giemsa. The images were captured with a fluorescence microscope and analyzed with Image J software.
Wound Healing Assay
We scraped cells with a 20µl pipette tip when they were confluent. Floating cells were washed out. Then, cells were incubated with serum-free medium. Images were captured at 0, 24, and 48 h with a fluorescence microscope. The pictures were processed with Image J software to analyze the ability of cell migration.
Transwell Assay
Matrigel (Matrigel: RPMI-1640, 1:9) was used to treat the transwell chamber. Then, cells were resuspended to a suspension with a density of 105/mL, and 100 μL suspension was added into the upper chamber. On the next day, cells which passed through the matrix gel were fixed and stained. Images were captured with a fluorescence microscope.
Western Blotting
Radioimmunoprecipitation assay buffer (Beyotine, China) was used to extract protein; 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis was used for protein separation. Subsequently, we transferred proteins to polyvinylidene fluoride (PVDF) membrane. After blocking with BSA, the PVDF membrane was incubated with a specific antibody. The Enhanced Chemiluminescence System (Invitrogen, CA) was used to detect protein bands with specific antibodies. Anti-CDK1 (1:1000, Cat.ab18, Abcam, UK) and anti-β-actin (1:1000, Cat.ab8226, Abcam, UK) were primary antibodies. β-actin was considered as an endogenous control. Image J software was used to analyze the quantity of protein.
Tumor Xenograft Mouse Models
Experimental Animal Center of Nanjing Medical University provided the 4–5-week-old BALB/c nude mice. The animal experiments were approved by the Yijishan Hospital Ethics Committee and followed the guidelines published by the Yijishan Hospital Ethics Committee. . Each nude mouse was inoculated subcutaneously with 2×106 cells. From day 8 to day14, the mice were injected intraperitoneally with 50 mg/kg of 5-Fu every other day. We measured tumor size every three days, sacrificed mice, and stripped tumors on the 15th day. The CDK1 expression level was examined with WB and RT-PCR. Immunohistochemical assay was used to detect the expression of Ki-67.
Immunohistochemistry
The transplanted tumors were fixed, embedded, and cut into sections at 4 µm thickness. The samples were baked for 30 minutes, and dewaxed to remove endogenous catalase. Sections and anti-Ki-67 (1:1000, Cat.ab92742, Abcam, UK) were incubated together at 4°C overnight. The second day, sections and secondary HRP-conjugated antibody were co-incubated for 30 minutes. Two independent pathologists analyzed the data.
TUNE Assay
Transplanted tumor tissue sections were dewaxed, and treated successively with ethanol and proteinase K. After fixing and blocking, sections were incubated in a 50 μL reaction mixture of terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL). We captured images with a fluorescence microscope.
Statistical Analysis
SPSS 17.0 and GraphPad Prism 7.0 were used to analyze all data. Student’s t-test was used to determine the difference between the two groups. Survival curves were analyzed using Log-rank test. We used univariate and multivariate analyses to analyze the prognostic factor. P<0.05 was considered significantly different. All experiments were repeated in triplicate.
Results
CDK1 Overexpression in CRC Tissue Resulted in Poor Outcome
We detected CDK1 in tumor tissues and paired paracancerous tissues. Higher CDK1 expression was observed in tumor tissues than their paired paracancerous tissues (Figure 1A and B). Correlations between the CDK1 and the clinicopathological characteristics are shown in Table 1. Tumor location, numbers of lymph node metastasis and tumor stage led to different expressions of CDK1 (Figure 1C–E). High-CDK1-expression CRC patients had a poor clinical outcome (Figure 1F). Univariate and multivariate analysis revealed that tumor location, number of lymph node metastases, tumor stage, and CDK1 expression were poor prognosis independent predictors (Table 2). We also found that CDK1 expression in 33 fluorouracil-resistant tissues was significantly increased compared with 43 fluorouracil-sensitive tissue (Figure 1G and H).
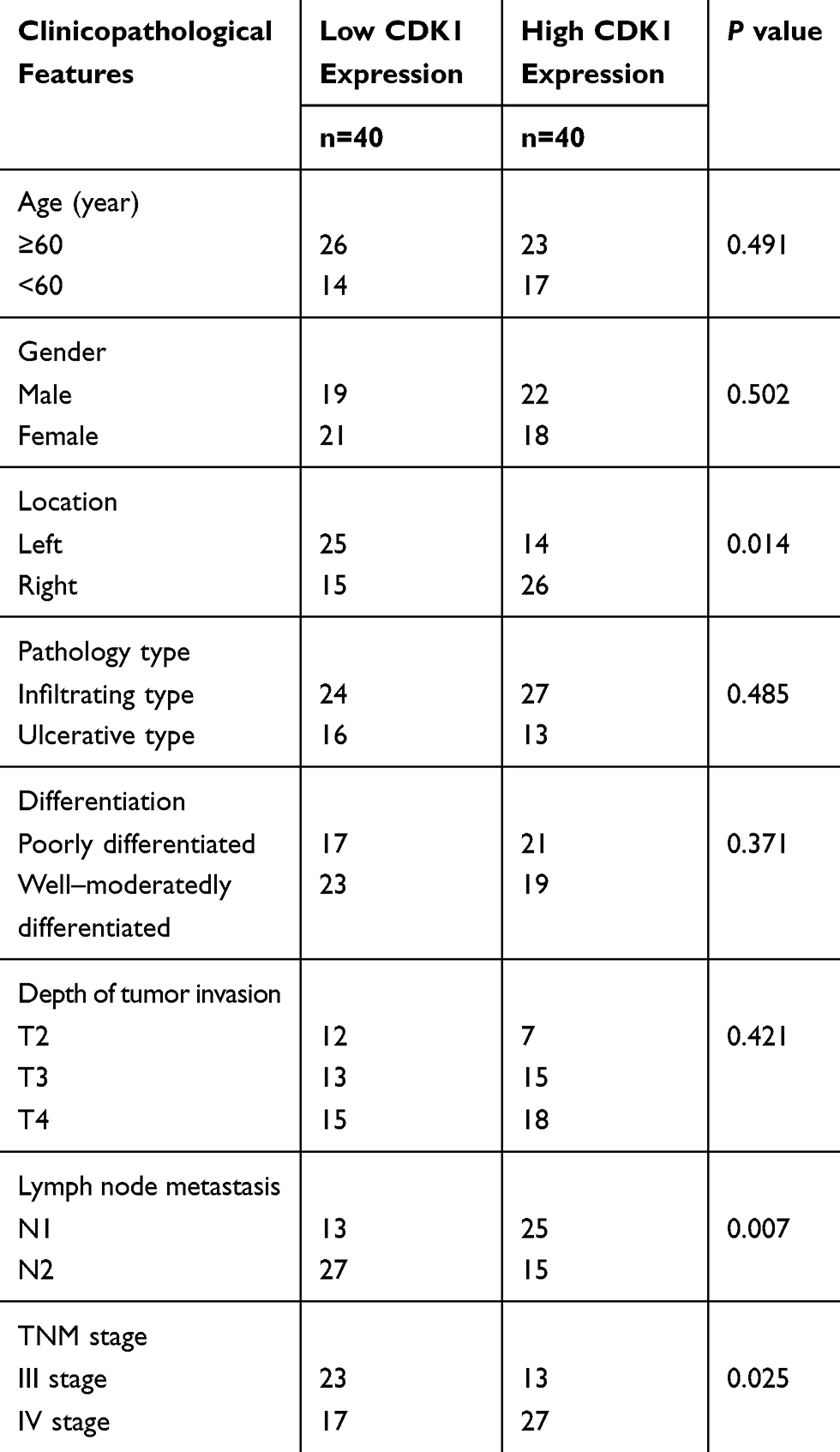 |
Table 1 Clinicopathologic Features of 80 Patients with Colorectal Cancer |
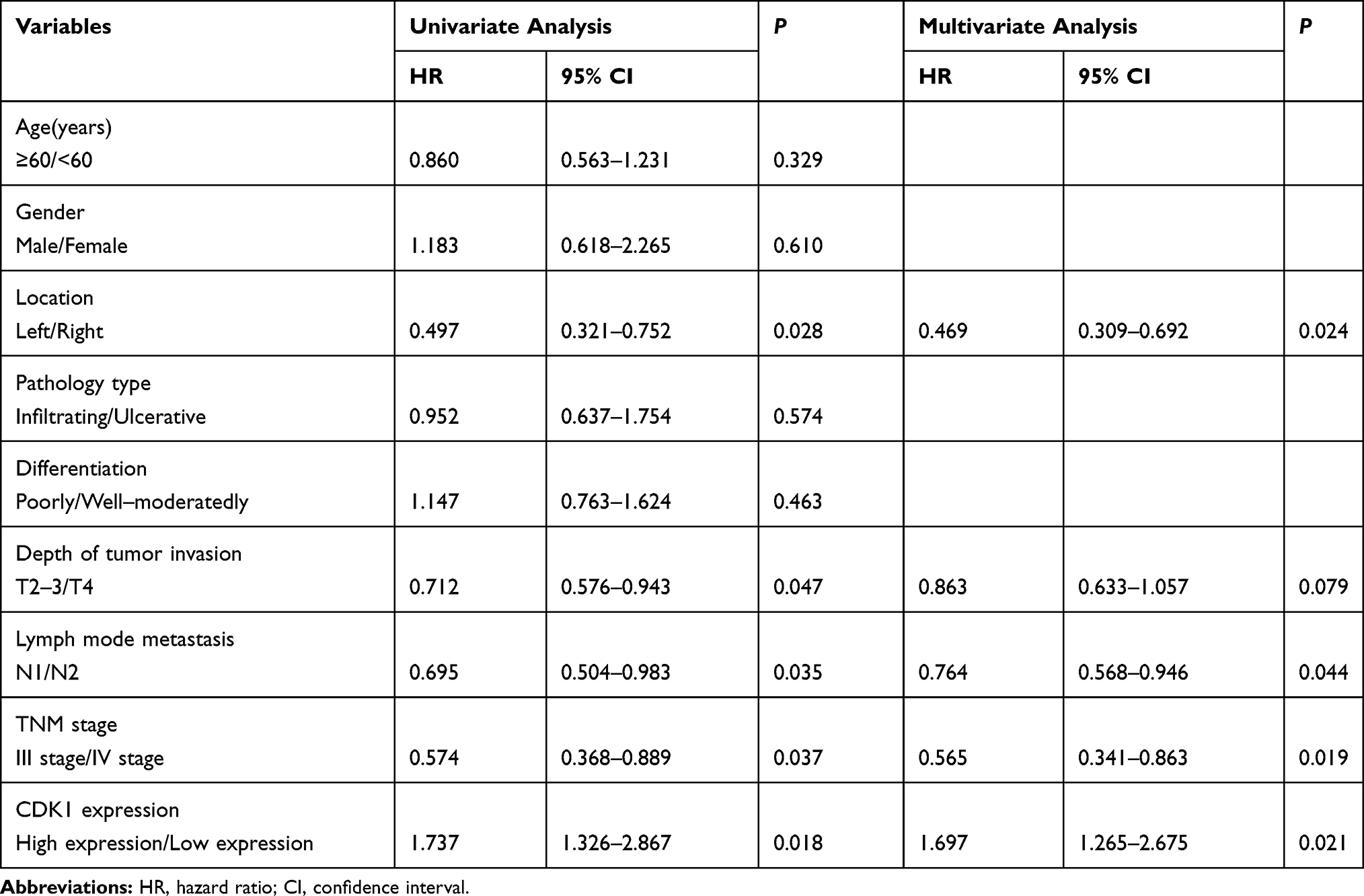 |
Table 2 Cox’s Proportional Hazard Model Analysis of Prognostic in Patients with CRC |
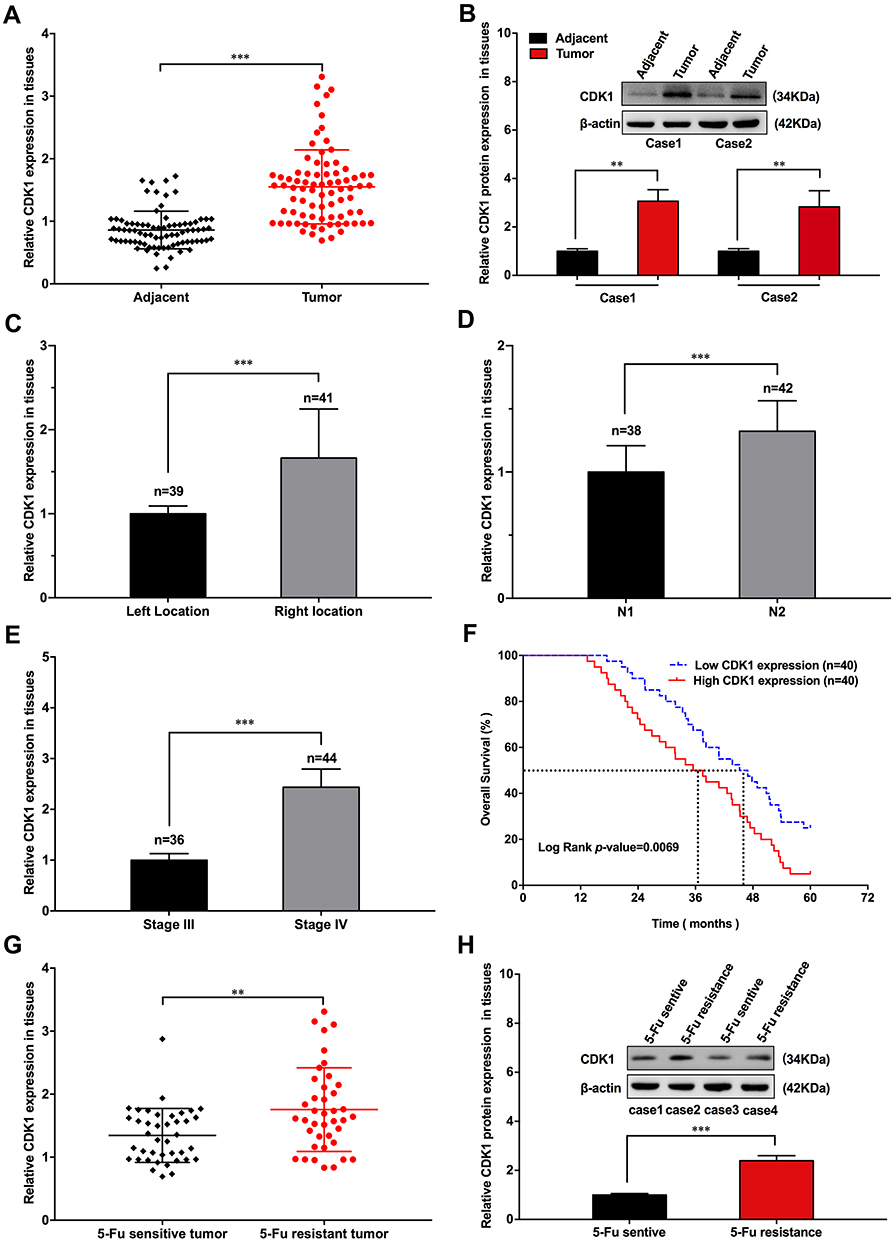 |
Figure 1 CDK1 overexpression in CRC tissue resulting in poor outcome. |
CDK1 Up-Regulation in Fluorouracil-Resistant CRC Cell
CDK1 was up-regulated in CRC cells, and significant differences were observed in HT-29 and SW480 (Figure 2A). Therefore, we selected these two cell lines for further research. We observed that the 50% inhibitory concentration of HT-29 was 44.60 ± 1.65μM, while in HT-29/5-Fu, it was 12.30 ± 0.48 μM, which was significantly different (Figure 2B and C). A similar phenomenon was also observed in SW480 and SW480/5-Fu cells (27.97 ± 1.05 μM VS 5.60 ± 0.23 μM) (Figure 2D and E). As shown in Figure 2F, we also found that CDK1 expression significantly increased in fluorouracil-resistant cells. These studies suggested that CDK1 might participate in the drug resistance and serve as a preditor for chemotherapy response in CRC.
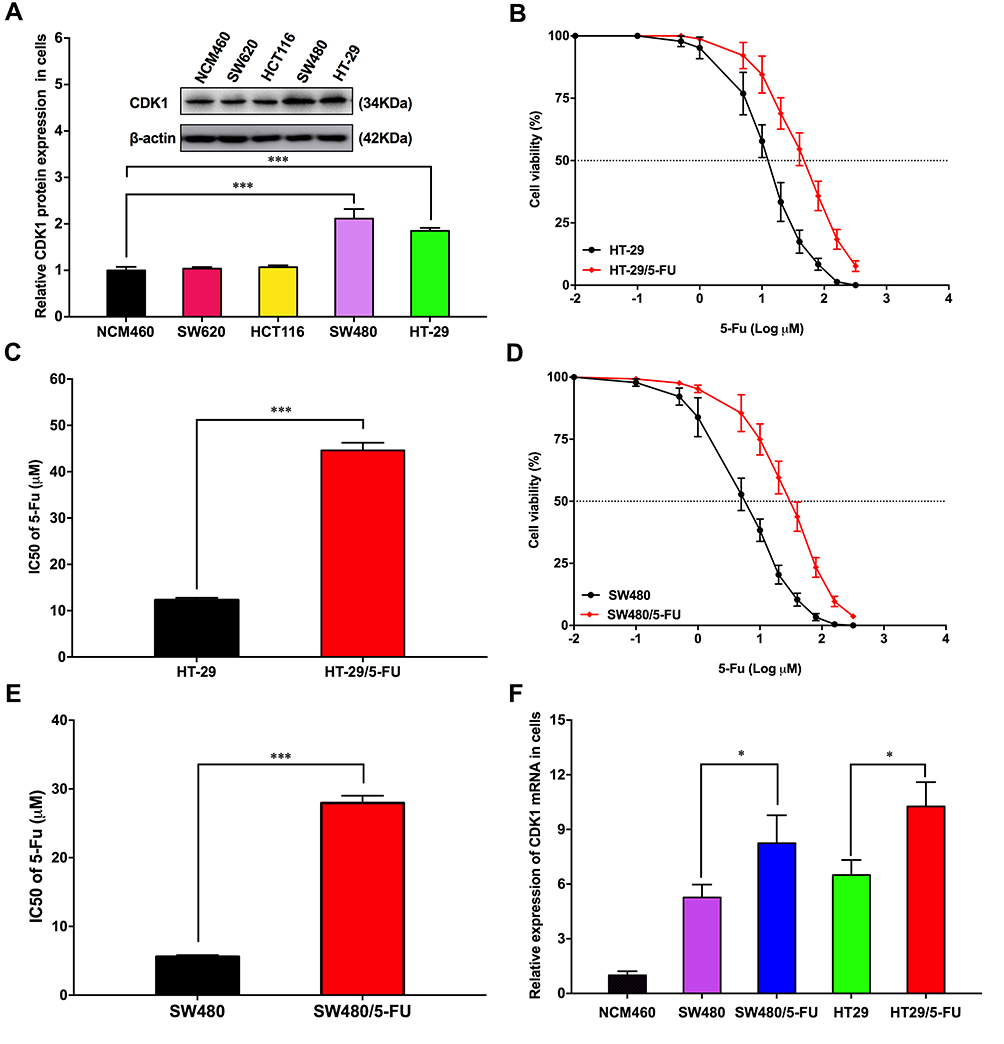 |
Figure 2 CDK1 up-regulation in fluorouracil-resistant CRC cell. |
Down-Regulation of CDK1 Inhibited Fluorouracil-Resistant CRC Cells Proliferation
Three siRNAs for silencing CDK1 were transfected into HT-29 and SW480 cells. All siRNAs significantly reduced CDK1 expression compared with negative control, and the silencing ability of siRNA3 was highest (Figure 3A–D). CDK1 protein significantly increased in drug-resistant cells. When transfected with siRNA3, CDK1 expression was significantly attenuated in drug-resistant cells, with no difference compared to that in HT-29 and SW480 (Figure 3E and F). Subsequently, we explored the effect of CDK1 on IC50 values in drug-resistant cells, and found that with CDK1 knockdown, the IC50 was significantly reduced (44.60±1.65 μM vs 19.33±0.69 μM; 27.97±1.05 μM vs 8.57±0.28 μM). This suggested that inhibition of CDK1 expression reversed 5-Fu resistance in CRC (Figure 4A and B). MTT assay revealed that the proliferation ability of drug-resistant cells was significantly enhanced. However, with the silence of CDK1, the proliferation ability was significantly weakened, and no significant difference was observed between fluorouracil-resistant cells and fluorouracil-sensitive cells (Figure 4C and D). Exposed with 5-Fu (IC50), the colony-forming ability of drug-resistant cells was significantly increased compared with fluorouracil-sensitive cells. While CDK1 was knocked down, the colony-forming ability was significantly suppressed in drug-resistant HT-29 and SW480 (Figure 4E and F). These results indicated that CDK1 promoted the proliferation in CRC and contributed to 5-Fu-resistance.
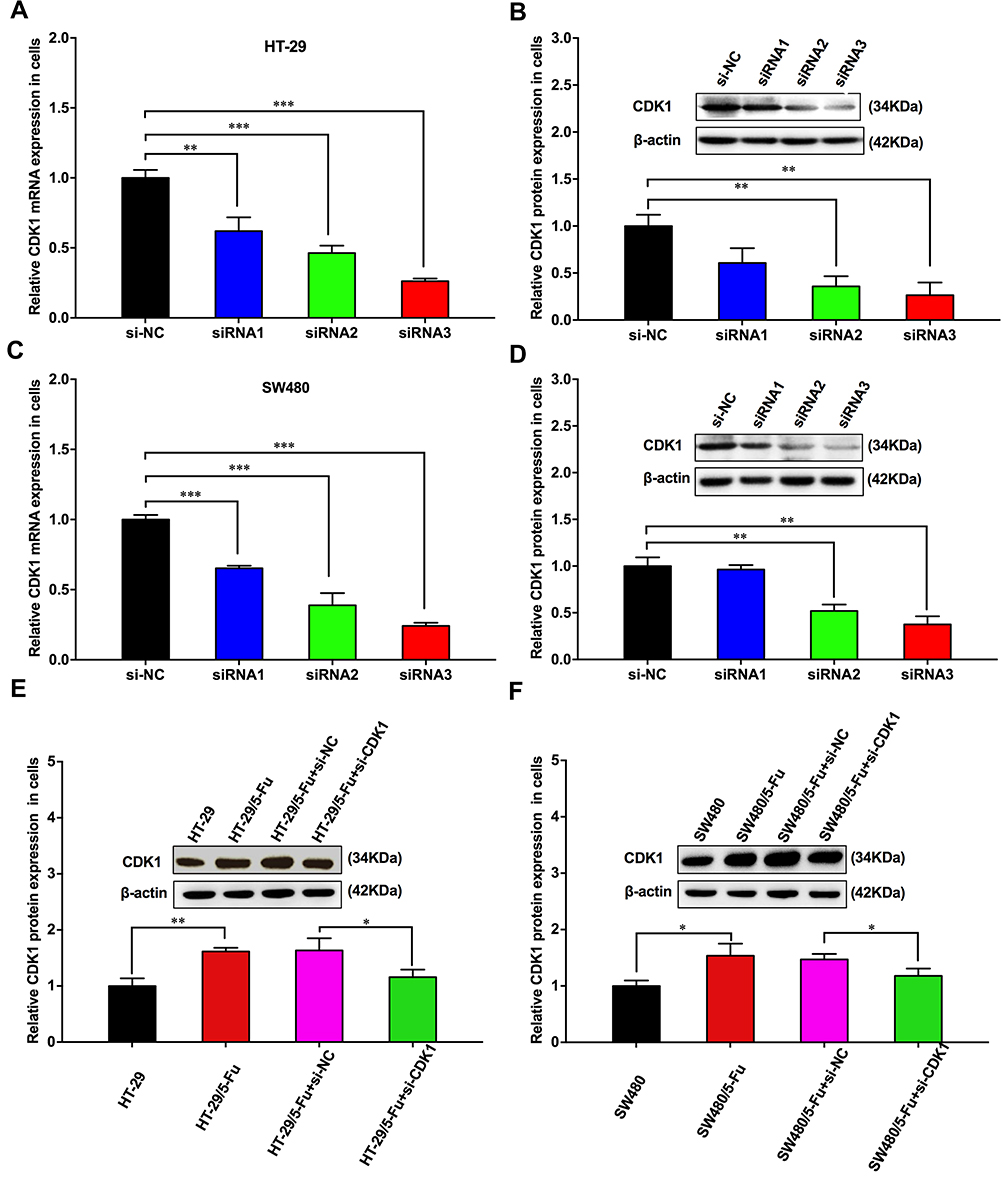 |
Figure 3 CDK1 was knocked down by using siRNA. |
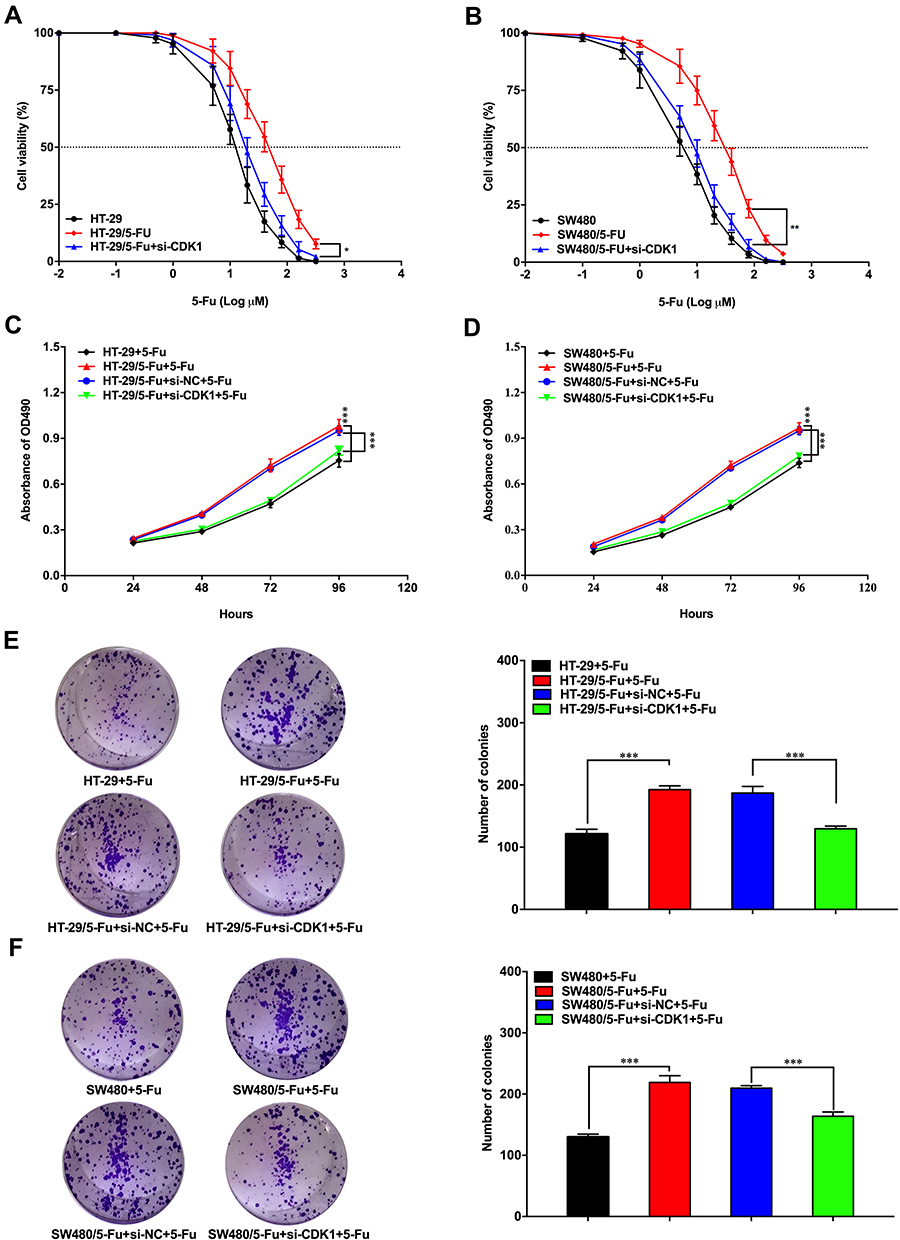 |
Figure 4 Down-regulation of CDK1 inhibited proliferation of fluorouracil-resistant CRC cells. |
CDK1 Promoted Migration, Invasion and Inhibited Apoptosis in 5-Fu- Resistant CRC Cells
In HT-29/5-Fu and SW480/5-Fu cells, the migration ability was significantly enhanced compared with HT-29 and SW480, and CDK1 knockdown significantly inhibited the migration ability (Figure 5A and B). Exposed with 5-Fu (IC50), less invasive cells were observed in fluorouracil-sensitive groups compared with fluorouracil-resistant groups. With the silence of CDK1, the invasive ability of fluorouracil-resistant cells was significantly reduced compared with control groups (Figure 5C and D). Flow cytometry showed that treated with 5-Fu (IC50), the apoptosis rate of HT-29 was 49.42±2.62% and SW480 was 49.80±2.14%. While in fluorouracil-resistant groups, the apoptosis rate decreased significantly, which was 25.92±1.66% (HT-29/5-Fu) and 29.82±1.60% (SW480/5-Fu). We silenced CDK1 in fluorouracil-resistant cells and observed apoptosis rate increased significantly (46.41 ± 3.02% vs 30.1 ± 1.43%, P<0.01; 47.39 ± 1.12% vs 27.49 ± 2.52%, P<0.01). The siRNA-CDK1 group and fluorouracil-sensitive cells had similar apoptosis rates, with no statistical difference (Figure 5E and F). Silencing of CDK1 promoted apoptosis and reversed the resistance of CRC to 5-Fu.
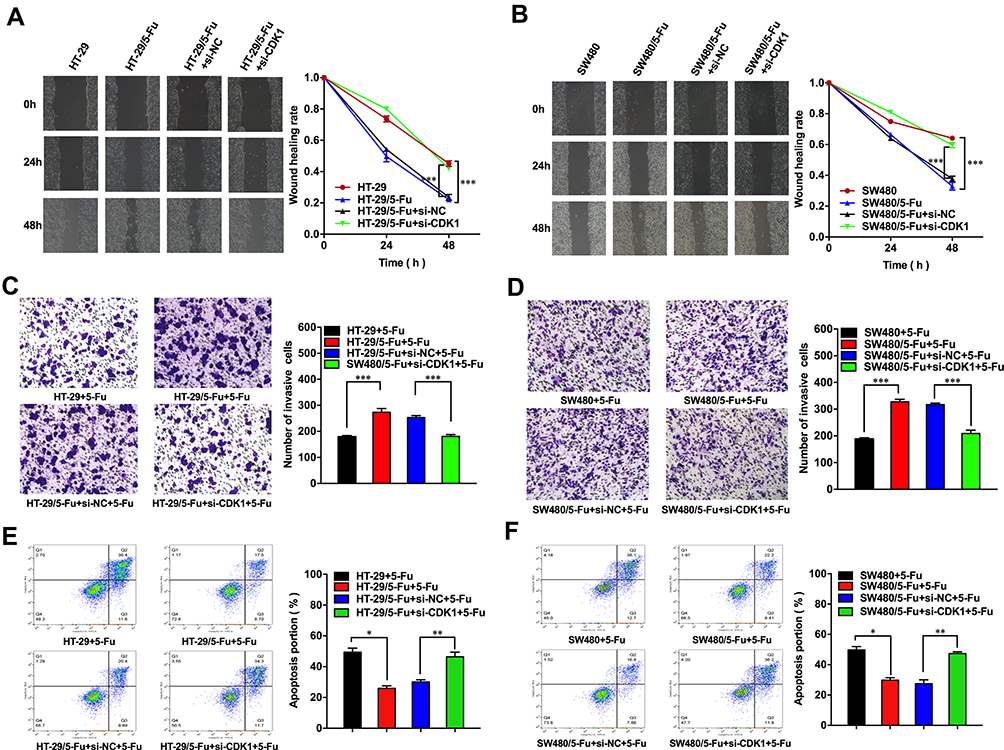 |
Figure 5 Down-regulation of CDK1 inhibited migration, invasion and promoted apoptosis in fluorouracil-resistant CRC cells. |
CDK1 Down-Regulation Inhibited CRC Tumorigenesis in vivo
Infected with sh-CDK1 or sh-NC, HT-29/5-Fu cells were subcutaneously inoculated into the armpit of BALB/c mice. From the eighth day, 50 mg/kg of 5-Fu was intraperitoneally injected into each nude mouse every two days. Simultaneously, we recorded tumor size every three days. On the 15th day, mice were sacrificed and the transplanted tumor tissue was stripped (Figure 6A). In the sh-CDK1 group, the growth rate of subcutaneous transplanted tumors was significantly lower, and CDK1 expression was significantly reduced (Figure 6B–D). More Tunel-positive cells and lower-intensity Ki-67 staining were also observed in the sh-CDK1 group (Figure 6E and F). These results demonstrated that the down-regulation of CDK inhibited tumor growth in vivo.
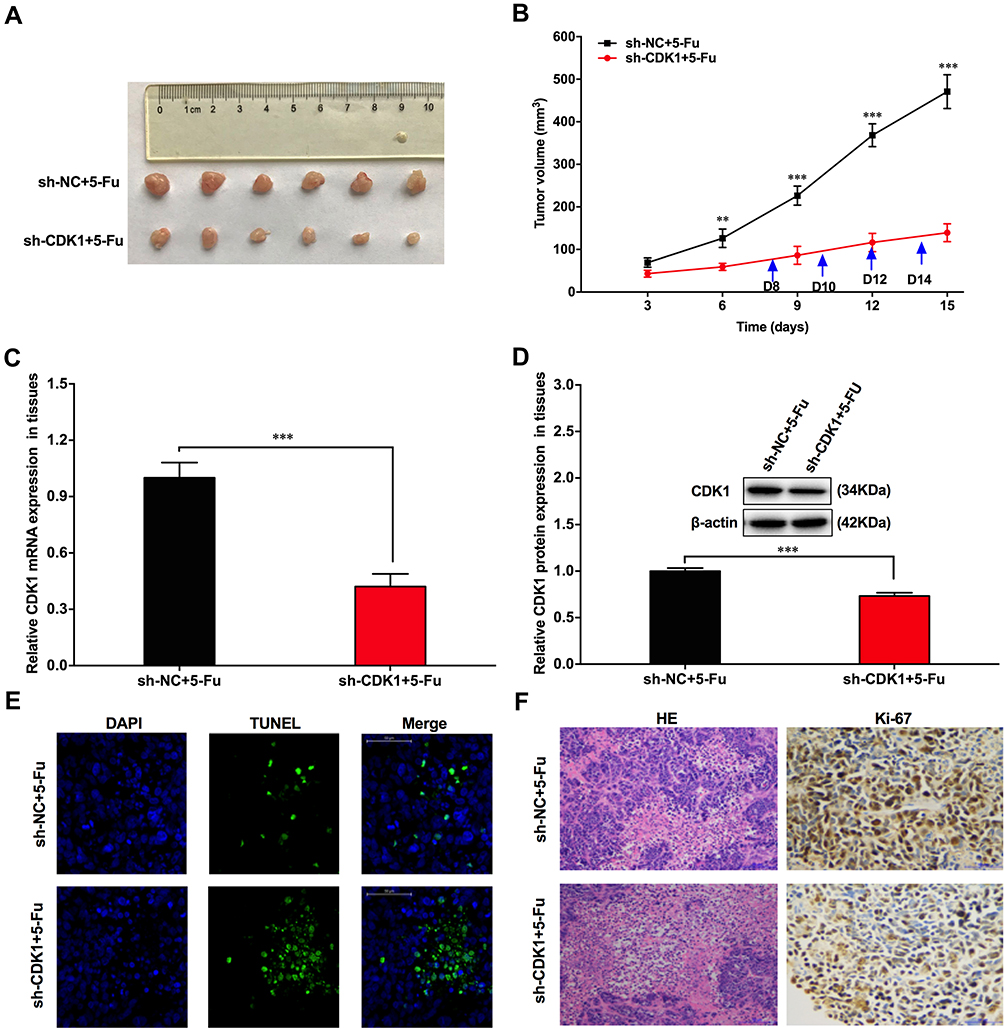 |
Figure 6 CDK1 down-regulation inhibited CRC tumorigenesis in vivo. |
Discussion
CDK1 was abnormally expressed in several tumors.29 Phosphorylated CDK1 was highly expressed and related to poor outcome in cholangiocarcinoma patients with immunohistochemical analysis. Roscovitine, a CDK inhibitor, could significantly decrease the expression of p-CDK1. With p-CDK1 down-regulation, the ability of proliferation and invasion was significantly inhibited, and the cell cycle was stagnated.30 CDK1 phosphorylated TFCP2L1 at Thr177, thereby affecting the cell cycle progression, pluripotency, and differentiation in embryonic stem cells. Activation of the CDK1-TFCP2L1 pathway stimulated cell proliferation, self-renewal and invasion in human aggressive bladder cancer.31 Deng32 reported that the up-regulation of CDK1 inhibited the G2/M phase block and promoted cervical cancer resistance to radiotherapy. CDK1 was also found to be stably activated by CDK1-KCTD12 interaction in BRAF-mutated colon cancer models resistant to Vemurafenib. When the CDK1-KCTD12 interaction was disrupted, the activation of CDK1 was inhibited, which reversed the resistance of Vemurafenib in CRC.33
CDK1, a cell cycle checkpoint regulator, facilitated cell viability via mediating anti-apoptotic factors survivin34,35 and Mcl-1.36 Shapiro et al have proposed that CDK1 inhibitors might serve as the potential therapeutic target for tumors.37 In KRAS-mutated colorectal and pancreatic cancer cells, CDK1 inhibitors had been shown to reduce the proportion of S-phase cells in KRAS-mutant cells and regulate Rb (the main control point of G1/S). CDK1 could be a synthetic lethal therapeutic target for KRAS mutations.38 Another study showed that a combination of CDK1 inhibitors and sorafenib could overcome the resistance of sorafenib in hepatocellular carcinoma by down-regulating CDK1 and inactivating downstream PDK1 and β-catenin.39 Hansel reported that Cdc2/Cdk1 was differentially up-regulated in adenocarcinoma compared with normal esophageal mucosal epithelium. Treated with CDC2/CDK1 inhibitor, the proliferation of esophageal adenocarcinoma cells showed a dose-dependent reduction.40
In the present study, we observed higher expression of CDK1 in tumor tissues, especially in fluorouracil-resistant tissues and cells. In fluorouracil-resistant cells, the ability of proliferation, migration, and invasion was significantly enhanced, and the apoptosis significantly reduced. We also demonstrated that inhibition of CDK1 expression could suppress CRC progression in vivo and in vitro. CDK1 was closely related to the clinicopathological features. We revealed that the silencing of CDK1 reversed the resistance of 5-Fu in CRC. This indicated that CDK1 might be a predictor of 5-Fu resistance and targeting CDK1 might be a novel strategy in CRC. However, our study did not detect the CDK1 downstream key protein level, when the CDK1 was knocked down, the mechanisms of CDK1 leading to drug resistance should be further explored.
Funding
Funding of “Peak” Training Program for Scientific Research of Yijishan Hospital, Wannan Medical College (GF2019G11) supported this study.
Disclosure
The authors declare no competing interests.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi:10.3322/caac.21492
2. Amd W, Eth F, Church TR, et al. Colorectal cancer screening for average-risk adults: 2018 guideline update from the American cancer society. CA Cancer J Clin. 2018;68:250–281.
3. Corley DA, Jensen CD, Quinn VP, et al. Association between time to colonoscopy after a positive fecal test result and risk of colorectal cancer and cancer stage at diagnosis. JAMA. 2017;317:1631–1641. doi:10.1001/jama.2017.3634
4. Li L, Ma BB. Colorectal cancer in Chinese patients: current and emerging treatment options. Onco Targets Ther. 2014;7:1817–1828. doi:10.2147/OTT.S48409
5. Schmidt-Kittler O, Ragg T, Daskalakis A, et al. From latent disseminated cells to overt metastasis: genetic analysis of systemic breast cancer progression. Proc Natl Acad Sci U S A. 2003;100:7737–7742. doi:10.1073/pnas.1331931100
6. Dienstmann R, Salazar R, Tabernero J. Personalizing colon cancer adjuvant therapy: selecting optimal treatments for individual patients. J Clin Oncol. 2015;33:1787–1796.
7. Saltz LB, Minsky B. Adjuvant therapy of cancers of the colon and rectum. Surg Clin North Am. 2002;82:1035–1058. doi:10.1016/S0039-6109(02)00041-5
8. Robinson E, Bartal A, Cohen Y, Haim N, Mohilever J, Mekori T. Combined adjuvant therapy of radically operated colorectal cancer patients. Cancer Chemother Pharmacol. 1982;8:35–40. doi:10.1007/BF00292869
9. Kerr DJ, Gray R, McConkey C, Barnwell J. Adjuvant chemotherapy with 5-fluorouracil, L-folinic acid and levamisole for patients with colorectal cancer: non-randomised comparison of weekly versus four-weekly schedules–less pain, same gain. QUASAR colorectal cancer study group. Ann Oncol. 2000;11:947–955. doi:10.1023/A:1008303229469
10. Shigeta K, Ishii Y, Hasegawa H, Okabayashi K, Kitagawa Y. Evaluation of 5-fluorouracil metabolic enzymes as predictors of response to adjuvant chemotherapy outcomes in patients with stage II/III colorectal cancer: a decision-curve analysis. World J Surg. 2014;38:3248–3256. doi:10.1007/s00268-014-2738-1
11. Tajima Y, Shimada Y, Kameyama H, et al. Association between poorly differentiated clusters and efficacy of 5-fluorouracil-based adjuvant chemotherapy in stage III colorectal cancer. Jpn J Clin Oncol. 2017;47:313–320. doi:10.1093/jjco/hyw209
12. Innocenti F, Ou FS, Qu X, et al. Mutational analysis of patients with colorectal cancer in CALGB/SWOG 80405 identifies new roles of microsatellite instability and tumor mutational burden for patient outcome. J Clin Oncol. 2019;37:1217–1227. doi:10.1200/JCO.18.01798
13. Modest DP, Martens UM, Riera-Knorrenschild J, et al. FOLFOXIRI plus panitumumab as first-line treatment of RAS wild-type metastatic colorectal cancer: the randomized, open-label, phase II VOLFI Study (AIO KRK0109). J Clin Oncol. 2019;37:3401–3411.
14. Oki E, Kato T, Bando H, et al. A multicenter clinical phase ii study of FOLFOXIRI plus bevacizumab as first-line therapy in patients with metastatic colorectal cancer: QUATTRO study. Clin Colorectal Cancer. 2018;17:147–155. doi:10.1016/j.clcc.2018.01.011
15. Joag MG, Sise A, Murillo JC, et al. Topical 5-fluorouracil 1% as primary treatment for ocular surface squamous neoplasia. Ophthalmology. 2016;123:1442–1448. doi:10.1016/j.ophtha.2016.02.034
16. Bian Z, Jin L, Zhang J, et al. LncRNA-UCA1 enhances cell proliferation and 5-fluorouracil resistance in colorectal cancer by inhibiting miR-204-5p. Sci Rep. 2016;6:23892. doi:10.1038/srep23892
17. Wang M, Hu H, Wang Y, et al. Long non-coding RNA TUG1 mediates 5-fluorouracil resistance by acting as a ceRNA of miR-197-3p in colorectal cancer. J Cancer. 2019;10:4603–4613. doi:10.7150/jca.32065
18. Zhang LH, Li LH, Zhang PF, Cai YF, Hua D. LINC00957 acted as prognostic marker was associated with fluorouracil resistance in human colorectal cancer. Front Oncol. 2019;9:776. doi:10.3389/fonc.2019.00776
19. Huang WW, Ko SW, Tsai HY, et al. Cantharidin induces G2/M phase arrest and apoptosis in human colorectal cancer colo 205 cells through inhibition of CDK1 activity and caspase-dependent signaling pathways. Int J Oncol. 2011;38:1067–1073. doi:10.3892/ijo.2011.922
20. Xi Q, Huang M, Wang Y, et al. The expression of CDK1 is associated with proliferation and can be a prognostic factor in epithelial ovarian cancer. Tumour Biol. 2015;36:4939–4948. doi:10.1007/s13277-015-3141-8
21. Zheng HP, Huang ZG, He RQ, et al. Integrated assessment of CDK1 upregulation in thyroid cancer. Am J Transl Res. 2019;11:7233–7254.
22. Sung WW, Lin YM, Wu PR, et al. High nuclear/cytoplasmic ratio of Cdk1 expression predicts poor prognosis in colorectal cancer patients. BMC Cancer. 2014;14:951. doi:10.1186/1471-2407-14-951
23. Li J, Zhi X, Shen X, et al. Depletion of UBE2C reduces ovarian cancer malignancy and reverses cisplatin resistance via downregulating CDK1. Biochem Biophys Res Commun. 2020;523:434–440.
24. Shang J, Xu YD, Zhang YY, Li M. Long noncoding RNA OR3A4 promotes cisplatin resistance of non-small cell lung cancer by upregulating CDK1. Eur Rev Med Pharmacol Sci. 2019;23:4220–4225. doi:10.26355/eurrev_201905_17926
25. Zhang P, Kawakami H, Liu W, et al. Targeting CDK1 and MEK/ERK overcomes apoptotic resistance in BRAF-mutant human colorectal cancer. Mol Cancer Res. 2018;16:378–389. doi:10.1158/1541-7786.MCR-17-0404
26. Zhang K, Li M, Huang H, et al. Dishevelled1-3 contribute to multidrug resistance in colorectal cancer via activating Wnt/β-catenin signaling. Oncotarget. 2017;8:115803–115816. doi:10.18632/oncotarget.23253
27. Zhu Y, Bian Y, Zhang Q, et al. Construction and analysis of dysregulated lncRNA-associated ceRNA network in colorectal cancer. J Cell Biochem. 2019;120(6):9250–9263. doi:10.1002/jcb.28201
28. Zhu Y, Bian Y, Zhang Q, et al. LINC00365 promotes colorectal cancer cell progression through the Wnt/β-catenin signaling pathway. J Cell Biochem. 2020;121:1260–1272. doi:10.1002/jcb.29359
29. Piao J, Zhu L, Sun J, et al. High expression of CDK1 and BUB1 predicts poor prognosis of pancreatic ductal adenocarcinoma. Gene. 2019;701:15–22. doi:10.1016/j.gene.2019.02.081
30. Yamamura M, Sato Y, Takahashi K, Sasaki M, Harada K. The cyclin‑dependent kinase pathway involving CDK1 is a potential therapeutic target for cholangiocarcinoma. Oncol Rep. 2020;43:306–317. doi:10.3892/or.2019.7405
31. Heo J, Noh BJ, Lee S, et al. Phosphorylation of TFCP2L1 by CDK1 is required for stem cell pluripotency and bladder carcinogenesis. EMBO Mol Med. 2020;12:e10880. doi:10.15252/emmm.201910880
32. Deng YR, Chen XJ, Chen W, et al. Sp1 contributes to radioresistance of cervical cancer through targeting G2/M cell cycle checkpoint CDK1. Cancer Manag Res. 2019;11:5835–5844. doi:10.2147/CMAR.S200907
33. Yang J, Xu WW, Hong P, et al. Adefovir dipivoxil sensitizes colon cancer cells to vemurafenib by disrupting the KCTD12-CDK1 interaction. Cancer Lett. 2019;451:79–91. doi:10.1016/j.canlet.2019.02.050
34. O’Connor DS, Grossman D, Plescia J, et al. Regulation of apoptosis at cell division by p34cdc2 phosphorylation of survivin. Proc Natl Acad Sci U S A. 2000;97:13103–13107. doi:10.1073/pnas.240390697
35. O’Connor DS, Wall NR, Porter AC, Altieri DC. A p34(cdc2) survival checkpoint in cancer. Cancer Cell. 2002;2:43–54. doi:10.1016/S1535-6108(02)00084-3
36. Rossi AG, Sawatzky DA, Walker A, et al. Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat Med. 2006;12:1056–1064. doi:10.1038/nm1468
37. Shapiro GI. Cyclin-dependent kinase pathways as targets for cancer treatment. J Clin Oncol. 2006;24:1770–1783. doi:10.1200/JCO.2005.03.7689
38. Costa-Cabral S, Brough R, Konde A, et al. CDK1 is a synthetic lethal target for KRAS mutant tumours. PLoS One. 2016;11:e0149099. doi:10.1371/journal.pone.0149099
39. Wu CX, Wang XQ, Chok SH, et al. Blocking CDK1/PDK1/β-Catenin signaling by CDK1 inhibitor RO3306 increased the efficacy of sorafenib treatment by targeting cancer stem cells in a preclinical model of hepatocellular carcinoma. Theranostics. 2018;8:3737–3750. doi:10.7150/thno.25487
40. Hansel DE, Dhara S, Huang RC, et al. CDC2/CDK1 expression in esophageal adenocarcinoma and precursor lesions serves as a diagnostic and cancer progression marker and potential novel drug target. Am J Surg Pathol. 2005;29:390–399. doi:10.1097/00000478-200503000-00014
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.