")
Back to Journals » Infection and Drug Resistance » Volume 8
Influence of host resistance on viral adaptation: hepatitis C virus as a case study
Authors Plauzolles A, Lucas M, Gaudieri S
Received 27 November 2014
Accepted for publication 18 December 2014
Published 7 April 2015 Volume 2015:8 Pages 63—74
DOI https://doi.org/10.2147/IDR.S49891
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Suresh Antony
Anne Plauzolles,1 Michaela Lucas,2,3 Silvana Gaudieri4
1Centre for Forensic Science, 2School of Medicine and Pharmacology, Harry Perkins Institute, 3School of Pathology and Laboratory Medicine, 4School of Anatomy, Physiology and Human Biology, University of Western Australia, Perth, WA, Australia
Abstract: Genetic and cellular studies have shown that the host's innate and adaptive immune responses are an important correlate of viral infection outcome. The features of the host's immune response (host resistance) reflect the coevolution between hosts and pathogens that has occurred over millennia, and that has also resulted in a number of strategies developed by viruses to improve fitness and survival within the host (viral adaptation). In this review, we discuss viral adaptation to host immune pressure via protein–protein interactions and sequence-specific mutations. Specifically, we will present the “state of play” on viral escape mutations to host T-cell responses in the context of the hepatitis C virus, and their influence on infection outcome.
Keywords: hepatitis C virus, viral adaptation, immune escape, adaptive immune response
Hepatitis C virus: an overview
Hepatitis C affects 3% of the human population, with an estimated 170 million people worldwide infected with the hepatitis C virus (HCV).1–3 Hepatitis C constitutes a global health problem, given the progression to cirrhosis (20%–35%) in a significant, proportion of individuals with chronic HCV infection, and it is a significant risk factor for hepatocellular carcinoma (increased rate of 3%–6% per year) and liver transplantation.4,5 The impact of HCV infection on the population is further exacerbated by the often asymptomatic nature of the disease for years after infection, such that many individuals infected with HCV are unaware of their condition and unknowingly transmit the virus, leading to a “silent epidemic”.6
HCV is a blood-borne virus. Before the discovery of HCV by Choo et al in 1989 and the subsequent implementation of efficient blood-screening methods, blood transfusion was one of the main routes of HCV transmission.7 In developed countries, transmission via mucosal exposure to infected blood or blood products is commonly due to unsafe drug-injecting practices (intravenous drug use [IDU]), with 60%–80% of newly identified cases attributed to IDU,8–10 while in developing countries transmission occurs via contaminated medical equipment/contaminated blood and IDU.11,12 Transmission via sexual activity is less common,13,14 as is mother-to-child transmission.15,16
HCV is a positive strand-enveloped ribonucleic acid (RNA) virus of approximately 9,600 nucleotides in length, belonging to the Hepacivirus genus of the Flaviviridae family. The single-stranded RNA molecule consists of a large open reading frame that encodes a polyprotein precursor of approximately 3,000 residues, which is processed during and after translation into three structural proteins (core protein and the envelope proteins E1 and E2), a small viroporin protein p7, and six nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B).
The NS5B protein is an RNA-dependent RNA polymerase that lacks 3′-5′ exonuclease proofreading activity, resulting in a high rate of mutation that has been estimated to be around 1.2×10−4 substitutions per nucleotide per cell infection (greater than the estimated mutation rate for HIV at 2.4×10−5).17,18 Evolutionary forces, such as random genetic drift and selection, affect the frequency of these mutations in the circulating HCV strains. These changes may affect the virus at different levels, including replicative fitness, pathogenicity, immune escape, and response to viral treatment.19
Hepatitis C virus: diverse RNA virus
Due to a high mutation rate, many different strains of HCV exist worldwide. Based on genetic heterogeneity, seven major genotypes and more than 70 subtypes have been described.20,21 These genotypes differ in their amino acid sequences by more than 20%, while their subtypes differ from each other by 10%–20% (Table 1). However, for the most part, the natural history of the infection is similar for genotypes and subtypes, with the exception of HCV genotype 3, which presents a higher prevalence of steatosis in comparison to other genotypes.22 Importantly, genotype is a significant predictor of therapy outcome with the immunomodulatory IFNα/ribavirin (RBV)-based therapy;6 potentially reflecting different IFN-responsiveness.
The HCV infecting a host circulates in the blood and liver as a heterogeneous population. This population consists of a pool of closely related viral strains referred to as quasispecies (Table 1).
 | Table 1 Genomic heterogeneity of hepatitis C virus (HCV) |
The observed diversity across the HCV genome is not uniform, and is mainly due to differences in functional and structural constraints for the different proteins. Therefore, some regions that code for important proteins for the virus’s life cycle present a restricted number of changes. In contrast, the envelope region presents a high degree of amino acid variation, especially in the N-terminus of the E2 protein, designated as hypervariable regions 1 and 2 (HVR1 and HVR2). The amino acid sequence within these HVRs can vary up to 80% between HCV genotypes.23 The envelope region is a known immunogenic region for neutralizing antibodies, and it is likely that the immune pressure exerted on this region is the main driving force of the observed envelope-sequence variation resulting in the emergence of escape mutants.24,25–27
Factors that influence HCV-infection outcome
Following infection with HCV, about 25%–30% of individuals can spontaneously resolve infection, but in the majority of cases the infection persists to establish chronic infection. The interplay of several viral and host factors largely influences HCV-infection outcome. From the host’s perspective, age, sex, liver function, and the immune response are all important components that influence infection outcome. A strong and sustained HCV-specific T-cell response is associated with spontaneous clearance; however cells, and molecules in the innate arm of the immune response have also been implicated in determining outcome following HCV infection.
Host-resistance factors that influence HCV-infection outcome: immune response
The host’s immune response is an important correlate of infection outcome, and several studies have examined variations in candidate genes involved in the immune system and HCV-infection outcome (Table 2).28 These association studies, either candidate-gene studies or genome-wide association studies, underline the crucial role played by the host’s immune responses in the control of HCV infection.
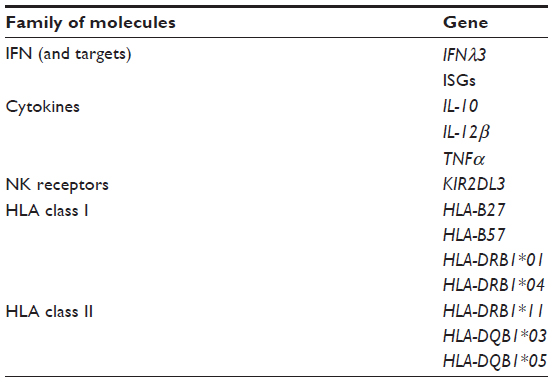 | Table 2 Genetic variants of genes involved in the host immune response associated with HCV infection outcomea |
Non-pathogen-specific immunity
The host’s innate immune response constitutes the first line of defense against an infection. This primary response relies on two strategies: “microbial-nonself” and “missing-self” identification. The missing-self strategy is based on the recognition of molecules expressed exclusively on normal healthy cells of the host.29 These molecules are not produced by microorganisms, and their expression can be lost when the host’s cell is infected with a pathogen. Following viral infection, the absence of such molecules thus constitutes a missing self that will then be identified by natural killer (NK) cells (through their receptors). On the other hand, microbial nonself identification is the first pattern-recognition strategy. This process is based on the recognition of molecular structures that are not produced by the host, but are common within microorganisms.30,31 Such viruses as HCV produce viral pathogen-associated molecular patterns (PAMPs). These PAMPs act as a “molecular signature” of the infecting microbial organism, and are able to initiate a cascade of events leading to innate intracellular immunity. Cells recognize these PAMPs via pattern-recognition receptors, such as Toll-like receptors (TLRs); an evolutionarily conserved gene family.22 In the case of HCV infection, the virus engages TLR3 and RIG-1, which are receptors that specifically recognize double-stranded RNA (dsRNA),32 a product that is produced by most viruses during their life cycle. This recognition subsequently results in the activation of signaling pathways (or “proinflammatory” signaling pathways), leading to the production of antimicrobial effectors and proinflammatory and antiviral cytokines (eg, IFN, TNF, IL-1), as well as gene products that prime the adaptive immune responses.32 Accordingly, candidate-gene studies have shown that genetic variations in the TLR genes RIG-I and TNF, and several interleukins in the innate immune-response pathway have been associated with HCV-infection outcome.28
HCV RNA triggers expression of IFN type I (α/β) and III (λ), which play a role in the defense against viral infections, due to their antiviral properties inducing a state of viral resistance that inhibits viral replication via several IFN-stimulated genes (ISGs).32,33,34–37 These ISGs include double-stranded PKR, and Mx protein.22,38,39 PKR is an intracellular sensor of viral infection that is activated in the presence of dsRNA produced by many viruses, including HCV. The activation of PKR results in the block of viral protein synthesis via eIF2α phosphorylation.22,40,41 Similarly to PKR, OAS is a family of IFN-inducible enzymes that are activated by dsRNA and result in the activation of RNAse L, which degrades viral RNA.42,43 Finally, Mx proteins belong to the class of dynamin-like large guanosine triphosphatases, whose expression is regulated by type I and type III IFNs.44 Their activation blocks viral transport inside the cell.22
In 2009, several genome-wide association studies identified single-nucleotide polymorphisms (SNPs) present near the IL28B gene (rs12979860 and rs8099917), encoding type III IFNλ3, that were associated with treatment response to pegylated (peg)-IFNα/RBV therapy45–48 and spontaneous clearance48,49 in HCV-infected individuals. Subsequently, Thompson et al observed the rates of SVR among different ethnic groups (ie, Caucasian, African-American, and Hispanic populations), and correlated these values with carriage of the “good” CC genotype for the SNP rs12979860.50 Similarly, IFNλ4, a recently discovered additional member of the IFNλ family, shows similar antiviral activity to IFNλ3, as well as a strong genetic association with spontaneous and treatment-induced HCV clearance.51 At present, the IFNλ genotype is the strongest host pre-IFNα-based treatment predictor.52 It is still unclear how variants of these genes affect HCV-infection outcome, but recent evidence suggests that variations in the promoter and other untranslated regions of the gene are associated with differential binding of transcription factors and transcript stability.53,54
It is now well established that NK cells constitute a crucial role in the fight against viral infections as part of the innate immune system. Target cells, such as virally infected cells, typically have low or no expression of human leukocyte antigen (HLA) class I molecules on their surface (to indicate “normal self” to the immune system), and are recognized and lysed by NK cells in a process referred to as missing-self recognition.55 Downregulation of HLA class I molecules on virally infected cells is thought to be an immune evasion strategy utilized by viruses against cytotoxic CD8+ T cells, which are reliant on the recognition of viral peptides in complex with HLA class I, and are therefore susceptible to missing-self recognition by NK cells. Furthermore, it has recently been shown in viral infections that NK cells may modulate the host’s adaptive immune response by directly deleting activated CD4+ and CD8+ T cells.56,57
The cytotoxic capacity of NK cells is regulated via signals derived from a complex array of inhibitory and activating receptors. NK cell receptors, such as KIRs and CD94/NKG2A, allow NK cells to sample cells for the presence of HLA class I molecules (ligands) that are typically expressed on healthy cells.58,59 While the NKG2 family is relatively conserved through evolution, KIRs on the contrary are diverse and polymorphic.60 Due to the diversity of KIRs (including a total of 14 genes), they are more likely to generate interindividual variation in immune responses to pathogens than the less polymorphic NKG2 family.
KIRs are located within the leukocyte-receptor complex on chromosome 19 (19q13.4), encoding for inhibitory and activating receptors that recognize the HLA class I molecules HLA-A, -B, and -C.61,62 The presence of particular combinations of HLA Class I alleles and KIRs within a host will determine the activation threshold of NK cells and modulate NK cell-mediated responses. The KIR–ligand interaction is highly variable, and this evolution is driven by selective pressure from exposure to pathogens.63–66 Furthermore, KIRs are expressed on NK cells in a stochastic manner, increasing the complexity of the receptor–ligand interaction.
Due to linkage disequilibrium, KIR haplotypes are defined by a combination of specific genes present at the centromeric and telomeric ends of the complex. The KIR A haplotype is relatively fixed in terms of gene content, while the B haplotype is characterized by variable KIR gene numbers and comprises more activating KIR genes. Differences in infection outcome have been associated with different genes within a specific KIR haplotype, including in the study by Dring et al,67 which found that the B haplotype-associated genes KIR2DS3 and KIR2DL5 appear to be more frequent in chronic HCV-infected subjects in comparison to resolvers.
Genetic association studies examining single KIR genes with a particular phenotype can be limiting, given the coordinated interaction between multiple KIRs and the hierarchy of interactions with their HLA class I ligands. Accordingly, studies that examine KIR haplotypes and/or KIR–HLA epistatic interactions can help clarify the overall influence of KIR on infection outcome. One such study by Martin et al showed that when single KIR genes were correlated with human immunodeficiency virus (HIV)-infection outcome, KIR3DS1 was associated with faster progression to disease, but this effect was reversed in the presence of a subset of HLA-Bw4 alleles with isoleucine at position 80.68 Khakoo et al then showed, in the context of HCV infection, that homozygosity for the NK cell-inhibitory receptor KIR2DL3 when in the presence of its ligand, HLA-C1, results in a higher probability of infection resolution.69 This combined genotype results in relatively weak inhibition of NK cells, and thus likely protects against HCV by rendering NK cells more easily activated than in other subjects. While both HIV and HCV studies have suggested that a diminished inhibitory response can be associated with good outcome following infection, human papillomavirus-induced cervical cancer appears more likely, with HLA–KIR interactions that favor NK-cell activation.70 Therefore, the role of NK cells in contributing to such viral infections as hepatitis C and disease progression (particularly to hepatocellular carcinoma) requires further investigation.
Pathogen-specific immunity
Activation of the innate immune system is typically followed by stimulation of adaptive immune responses to viral pathogens. The antigen-specific nature of the adaptive immune response is due to rearrangements of genes (and additional somatic mutations) within T cells and B cells that encode for specific antigen receptors (T-cell receptors and immunoglobulin, respectively).22 Clearance of HCV seems to require a rapid and effective adaptive immune response targeting multiple antigenic targets of the virus polyprotein.22,71 Following priming of naïve T cells and B cells with viral antigens, memory cells are formed with the aim of preventing reinfection due to a stronger and higher avidity response upon reexposure. However, in the case of HCV, reinfection can be observed, particularly with a different strain of HCV, most likely due to the genetic heterogeneity observed for HCV genotypes and subtypes.
Studies have shown that a vigorous, broad, and sustained CD4+ and CD8+ T-cell response directed against HCV is generally observed within an individual experiencing a self-limited course of HCV infection.72–81 Sequential liver biopsies performed on infected chimpanzees revealed that viral clearance coincided with the accumulation of HCV-specific CD4+ and CD8+ T cells in the liver at 8–14 weeks after exposure to HCV.78,81 In addition, studies have shown a decrease in viremia that temporarily correlates with the emergence of cellular immunity within an individual mounting a brief HCV-specific T-cell response followed by a viral rebound after the loss of these T-cell responses.73,77 Finally, T cell-depletion studies in chimpanzees have confirmed the crucial role of cellular immune responses (CD4+ and CD8+ T cells) in the control of viral infection.81,82 On the contrary, chronic HCV infection is associated with an impaired or stunted immunological profile.72–75,80,83–91
Impairment of cellular immune responses is a recurrent factor among chronic HCV-infected individuals. It is assumed that progression to chronicity among infected individuals is due to either weak or stunted T-cell responses developed by the host and/or immune escape (a strategy developed by the virus; discussed in Sequence-specific viral adaptations).
For both CD4+ and CD8+ T cells, the T-cell receptor is responsible for recognizing antigens bound to HLA molecules. This peptide–HLA complex is formed, processed, and presented on the surface of an antigen-presenting cell (APC).22 The HLA molecules are highly polymorphic, and can be separated into two classes. HLA class I molecules (HLA-A, -B, and -C) are expressed on the surface of all nucleated cells, and are recognized by “cytotoxic” CD8+ T cells,92 while HLA class II molecules (HLA-DR, -DQ) present on the surface of professional APCs (eg, dendritic cells, macrophages, B cells) and present antigens that are recognized by CD4+ T cells. The HLA repertoire of the host determines the set of viral peptides or targets presented to the T cells. Not surprisingly, a number of HLA class I and II alleles have been associated with HCV-infection outcome.28 Only a few of these associations are replicated, due to lack of power (partly as a result of the high heterogeneity of the HLA loci), unknown infecting virus, and the linkage disequilibrium between HLA class I (and to a lesser extent with the more distant class II loci-forming haplotypes, similar to what is observed for the KIR loci), which complicates the ability to identify associations between specific HLA alleles and infection outcome.
Although these host-“resistance” molecules are effective against pathogens, HCV (and other viruses) has developed several strategies to combat the host’s immune response. These strategies involve protein–protein interactions and specific mutations within the virus to escape recognition by the immune system.
Viral adaptation: relevance to infection outcome
Protein–protein interactions
To counteract the actions of the host’s immune response, HCV has developed several mechanisms to subvert the endogenous IFN response, including blocking the induction of IFNs, interference with signals triggered by IFNs, or the inhibition of ISGs.39 As such, several studies have demonstrated that HCV NS3/4A affects the induction of IFNβ, as well as subsequent ISGs,93,94 by interfering with the activation of IRF-3,95 by disrupting RIG-I signaling94,96–98 and inhibiting TLR3 signaling,93,99 which are all essential steps of the IFN response. In parallel, NS5A and the envelope protein E2 are also assumed to block activity of PKR by binding to PKR and inhibiting formation of the PKR dimer.100–102 Additionally, the HCV core protein has been found to interfere with the Jak-STAT pathway that is involved in the expression of numerous ISGs.103–107 Overall, HCV uses numerous mechanisms to disrupt the IFN system, which can affect the action of endogenous IFN to control the HCV infection and the efficacy of IFNα-based treatment.
HCV has also been shown to affect HLA class I and II expression on the surface of APCs. The proteins core108 and NS3109 have been shown to affect the function of the proteasome complex110 and potentially HLA class I presentation. The core protein has also been implicated in reducing HLA class I expression via the transporter TAP1,111 although in this case the change in HLA class I expression on target cells did not appear to affect CD8+ T-cell recognition, and may instead have been related to NK-cell cytotoxicity. Downregulation of HLA class I molecules has important consequences in relation to CD8+ T-cell immunity, but also NK-cell cytotoxicity via missing self, particularly for HLA-B and -C. However, it is still unclear which of the HLA class I loci are affected, as many of these studies have used pan-HLA class I antibodies. For HIV, the virus can downregulate specific HLA class I loci, but retain others as a strategy to subvert both T-cell and NK cell-mediated immune responses.112
The HCV proteins core and NS5A are also thought to be involved in the downregulation of HLA class II expression on APCs.113 Again, this is an important mechanism of viral escape from the immune system, as CD4+ T cells that engage HLA class II antigen presentation are critical in HCV-infection outcome, given that lack of CD4+ T-cell help and the loss of HCV-specific CD4+ T-cell responses following the acute phase of infection is strongly associated with viral persistence.114,115
Sequence-specific viral adaptations
The viral population in an infected subject consists of closely related but nonidentical genomes, referred to as viral quasispecies. The presence of such viral diversity is mainly due to the error-prone RNA-dependent RNA polymerase that does not have a 3′-5′ exonuclease proofreading activity. Random genetic drift and selection pressures on the virus can change the composition of the quasispecies over time. The host immune responses as well as antiviral drugs have been identified as the main selection pressures shaping viral quasispecies.
In the context of the host’s T-cell immune response, this selection pressure can drive the evolution of HCV populations during the course of infection within a single host and circulating within the human population.116 As the host’s T cells see HCV in the context of the HLA molecules present on the surface of APCs, these viral mutations or viral adaptations are specific for HLA alleles. In population-based genetic studies that sampled the HLA type and viral sequence of a large number of virus-infected individuals, HLA-associated viral polymorphisms marked true in vivo targets of the host’s T-cell immune response,117,118 as was originally shown for HIV.119 Such studies on viral adaptation at the population level have tended to examine chronic HCV-infected subjects, given the difficulty in accessing acute HCV-infection cohorts and also the ability to detect HLA “footprints” that are fixed (or within the major circulating viruses using bulk Sanger-based sequencing approaches) in the individual. However, there are data to suggest that bottlenecks in HCV infection occur during the acute phase of infection and emerging viral strains are likely to exhibit viral adaptations to the host’s HCV-specific immune response.120 Accordingly, analysis of viral evolution during the acute phase of infection is important to better understand host–virus interplay and its effect on infection outcome.
Mutational escape can impact the presentation process and subsequently alter or impair the immune responses initiated by HLA molecules. These mutations provide a short-term benefit for the virus, in order to cope with immune responses. Nevertheless, absence of reversion after transmission can be observed for some mutant viruses, indicating minimal “fitness cost”.121 As a consequence, such escape mutants may spread through the population, regardless of HLA type, and ultimately the “wild-type” strain circulating in a population becomes the adapted form.119
Studies on single-source outbreaks in Germany and Ireland overcame some of the issues of studying viral adaptation, as the source strain was known and all exposed individuals could be assessed irrespective of the clinical nature of the acute infection (which is often asymptomatic in HCV infection). In both cases, the adaptation potential of the source virus was likely to have influenced the specific HLA alleles associated with infection outcome.122,123 For example, in the study of Irish women who were infected through the administration of anti-D immunoglobulin contaminated with an HCV genotype 1b strain, HLA-B8 was associated with poor outcome, and sequencing the source strain revealed existing viral adaptation within an immunodominant NS3 HLA-B8-restricted T-cell epitope,122,124 which is likely to have been disadvantageous for new hosts expressing HLA-B8. Similarly, in the same cohort, Fitzmaurice et al125 found a novel T-cell epitope restricted by HLA-A3 in the NS3 protease (TVYHGAGTK position 1080–1088), which was targeted by 60% of women in this cohort and associated with a strong “HLA footprint”, in that women who carried HLA-A3 and became chronically infected with HCV tended to exhibit a mutation within this target (K1088R). The HLA-A3 allele was found to be protective, which could be explained by the requirement of a second substitution (T1087A) to offset the significant loss of fitness associated with the K1088R change that was also found in HLA-A3 chronic HCV-infected women in this cohort. In the cohort of German women who had received HCV-contaminated anti-D immunoglobulin, spontaneous HCV clearance was associated with HLA-B-27,123 and the source sequence showed no evidence of viral adaptation in an immunodominant HLA-B27 T-cell epitope. Mutations in this epitope are commonly observed in chronic HCV-infected subjects with HLA-B27. Similarly, preservation of targeted epitopes for the HLA-B57 allele is also associated with viral clearance.126
But what is the contribution of viral adaptation to HLA-specific T-cell responses to overall viral diversity? HIV and simian immunodeficiency virus studies have demonstrated the role of CD8+ T-cell selective pressure in shaping viral evolution, showing that more than 50% of mutations developed after acute infection are associated with CD8+ T-cell responses,127 including the reversion of CD8+ T-cell escape mutations, likely due to viral fitness cost.128–130 Similar observations were made with HCV studies;124,131 however, these viral adaptations were observed to a lesser extent, as shown by Kuntzen et al,132 with up to 11% of mutations occurring within regions targeted by detectable CD8+ T-cell responses (in nonenvelope regions). Interestingly, of the de novo mutations observed in the Kuntzen et al study, 19% of these changes were reversions to wild type, with the majority (80%) of these reversions occurring upon transmission or during the acute stage of the infection. We have also previously shown less extent of HCV variation during the acute phase of infection compared to HIV and a high number of synonymous changes relative to nonsynonymous changes observed between longitudinal sequences in subjects, reflecting greater constraint along the HCV genome than for HIV.133 These HCV studies suggest that reversion is likely to happen upon transmission if immune pressure is no longer present, as escape mutations are generally associated with a fitness cost and overall HCV may not be as flexible as HIV. It should be noted that the number of known T-cell epitopes for the two viruses is heavily biased toward HIV, and this may account for some of the discrepancy reported in the proportion of viral adaptations of total viral diversity observed during the natural course of early infection.
More recently, there has been some evidence to suggest that NK cells can directly influence viral evolution. Alter et al134 looked at NK cell-mediated immune pressure on HIV evolution via KIR-associated amino acid polymorphisms along the HIV-1 sequence of chronic HIV-infected individuals. Their study revealed polymorphisms associated with the presence of inhibitory receptor KIR2DL2 in Vpu and Env, showing that these polymorphisms allow binding of KIR2DL2 upon HLA-C presentation, whereas the HIV wild-type sequences do not. These KIR-associated polymorphisms can modulate the interaction of KIR+ NK cells with HIV-1-infected CD4+ T cells, thus demonstrating that the viral variants presenting these “KIR footprints” seem to impact on NK-cell recognition and/or antiviral activity and constitute another mechanism by which HIV can escape NK cell-mediated immunity, a sequence-specific manner akin to what has been observed for viral immune escape from T cells.
Today, the impact of NK cells on HCV infection has only been investigated with respect to the presence of a specific KIR and outcome of infection. However, given the numerous features shared by HIV and HCV, specifically their high mutation rates and genetic association studies showing an association between infection outcome and specific KIRs and NK-cell activation, HCV evolution could also be subject to NK cell-mediated immune pressure. At present, such an analysis has not been done with HCV-infected subjects, and further research is necessary to understand the role and impact of NK cells on HCV-infection outcome.
Overlap between selection pressures: relevance to treatment outcome
In addition to immune selective pressure, antiviral treatment is another factor that can influence within-host viral diversity. The use of IFN-based treatment could be taken as an indicator of IFN-based immune pressure on the virus. However, data on the impact of IFN-based treatment on viral diversity are limited and contradictory.135–138 No specific resistance mutations have been associated with impairment of IFN therapy, which can be explained by the indirect action of IFNs on viral infections.139 Rather than specific mutations, there is evidence to suggest that sequence heterogeneity within areas of the HCV genome may affect IFN-based treatment outcome. For example, Enomoto et al140,141 demonstrated in Japanese subjects infected with HCV genotype 1b strains that greater sequence heterogeneity within the IFN sensitivity-determining region of the NS5A protein was associated with IFN-treatment outcome. However, overall there are limited data to suggest specific viral variants are selected by IFN-based therapy (and likely IFN responses in general).
Unlike IFN, the new direct-acting antiviral (DAA) drugs used for hepatitis C do not enhance existing immune responses, but directly target the action of viral proteins involved in the virus’s life cycle. First-generation inhibitors of NS3/4a serine protease, telaprevir, and boceprevir, in conjunction with peg-IFNα/RBV, were approved for clinical use over 12 months ago, and more recently the second-generation NS3/4a protease inhibitor simeprevir and the NS5B inhibitor sofosbuvir have been approved for clinical use in combination with peg-IFNα/RBV. These new DAA drugs have a >90% efficacy rate against HCV genotype 1. However, HCV is a rapidly evolving virus with a high mutation rate, and the rapid emergence of drug-resistance mutations can jeopardize treatment outcome for these drugs that directly target the virus (unlike IFN-based therapies).142–144 Furthermore, reports have shown evidence of drug-resistant variants in treatment-naïve subjects, but typically at low frequencies.145
Of interest are those sites along the viral genome that are likely to be under two selection pressures: immune response and antiviral treatment. HIV studies have previously shown the influence of immunological host factors on the emergence of drug-resistance variants, demonstrating the increased risk for the subject to develop mutations at sites at which the two selective forces intersect. John et al146 examined HIV sequences and HLA data from a cohort of 487 HIV-1-infected individuals, and identified an increased incidence of drug-resistance mutations among individuals that expressed an HLA that targeted the same region. In another cohort of 94 HIV-1-positive subjects, Mueller et al147 demonstrated that several drug resistance-associated mutations along the protease acted as CD8+ T-cell escape mutants. Few HCV studies have shown similar results, given the recent introduction of the new DAAs. However, Salloum et al looked at substitutions R155K and A156T, which constitute key positions for resistance to DAAs, such as telaprevir and boceprevir, and the overlap with the HLA-A68-restricted epitope (HAVGIFRAAV) from position 1175–1184.148 They investigated the impact of these protease inhibitor-resistant mutants at the replication level, as well their influence on the HCV-specific antiviral CD8+ T-cell response. Their replication assay showed a low and intermediate fitness cost associated with the R155K and A156T changes, respectively, and that these variants enabled immune escape from HLA-A68-restricted CD8+ T cells. HLA-A68 is a common allele in Native American populations, but less so in Caucasian and Asian populations (http://www.allelefrequencies.net). We also analyzed HCV sequences from over 400 subjects infected with HCV genotypes 1a, 1b, and 3a, and evaluated the prevalence of drug-resistance mutations within sites along the HCV NS3 protease and NS5B polymerase genes for which drug and immune selective pressures are likely to intersect. We identified sites within immunodominant T-cell epitopes in which host immune pressure may affect the frequency of drug-resistant variations in subjects undergoing specific DAA treatment and the site and variation of interest is a known immune escape mutation that is often offset with surrounding compensatory mutations.149
In parallel to DAAs, agents targeting host proteins or nucleic acids are being developed. These compounds target cellular proteins and microRNAs that are essential for the viral life cycle, and thus inhibit the ability of HCV to use host-cell components for continued infection. These pharmaceutical agents include cyclophilin inhibitors, which aim to disrupt the interaction of NS5a and cyclophilin A,150 and an antagonist of microRNA 122 that inhibits its binding to the 5′ untranslated region of the HCV genome, an essential step for HCV replication.151,152 These agents could be an important addition to DAAs in the treatment of HCV, as they have a high genetic barrier to resistance, with no cross-resistance with DAAs and present pan-HCV genotypic activity.153
Conclusion
The host’s immune response is an important correlate of infection outcome. Accordingly, coevolution between host and pathogen has resulted in a number of strategies for both organisms to improve fitness. In the context of human pathogens, such as HCV, the life cycle and mutation rate between host and pathogen are different, and reflect the strategies undertaken by each to improve survival. For the host, the innate and adaptive immune responses combine to combat the pathogen using nonspecific (but nonself) and specific means to rid the pathogen or at least minimize damage. The evolution of these molecules, such as IFN, TLRs, KIR, and HLA, has resulted in multicopy, polymorphic systems that can identify and combat a large number of pathogens. However, such pathogens as HCV can subvert a number of the host’s immune mechanisms via protein–protein interactions, such as downregulation of HLA molecules on the surface of the APC and interfering with the IFN signaling pathway and via a rapid mutation rate. Viral adaptation via sequence-specific changes is associated with T-cell and B-cell escape. In the case of T cells, these viral adaptations are recognizable at the population level and within a single infected individual.
In this review, we have focused on viral adaptation to HCV-specific T-cell responses, but clearly antibodies directed against specific HCV antigens generated from B cells also influence viral diversity, particularly in the envelope proteins. Importantly, knowledge of viral adaptation to the different arms of the host’s immune response will be useful in the design of a preventative vaccine for both HIV and HCV, as well as in identifying new treatment strategies.
Disclosure
The authors report no conflicts of interest in this work.
References
Alter HJ, Seeff LB. Recovery, persistence, and sequelae in hepatitis C virus infection: a perspective on long-term outcome. Semin Liver Dis. 2000;20(1):17–35. | |
Chisari FV. Unscrambling hepatitis C virus-host interactions. Nature. 2005;436(7053):930–932. | |
Tan SL. Hepatitis C Viruses: Genomes and Molecular Biology. Abingdon, UK: Taylor and Francis; 2006. | |
Lok AS, Seeff LB, Morgan TR, et al. Incidence of hepatocellular carcinoma and associated risk factors in hepatitis C-related advanced liver disease. Gastroenterology. 2009;136(1):138–148. | |
Seeff LB. The history of the “natural history” of hepatitis C (1968–2009). Liver Int. 2009;29 Suppl 1:89–99. | |
Fabry S, Narasimhan RA. 100 Questions and Answers About Hepatitis C: A Lahey Clinic Guide. Sudbury (MA): Jones and Bartlett; 2006. | |
Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244(4902):359–362. | |
Nelson PK, Mathers BM, Cowie B, et al. Global epidemiology of hepatitis B and hepatitis C in people who inject drugs: results of systematic reviews. Lancet. 2011;378(9791):571–583. | |
Grebely J, Dore GJ. What is killing people with hepatitis C virus infection? Semin Liver Dis. 2011;31(4):331–339. | |
Grebely J, Dore GJ. Can hepatitis C virus infection be eradicated in people who inject drugs? Antiviral Res. 2014;104:62–72. | |
Lavanchy D. Evolving epidemiology of hepatitis C virus. Clin Microbiol Infect. 2011;17(2):107–115. | |
Thursz M, Fontanet A. HCV transmission in industrialized countries and resource-constrained areas. Nat Rev Gastroenterol Hepatol. 2014;11(1):28–35. | |
Grady BP, Prins M, van der Loeff MS. The sexual transmission rate of HCV among heterosexual couples. Hepatology. 2013;58(5):1865–1866. | |
Terrault NA, Dodge JL, Murphy EL, et al. Sexual transmission of hepatitis C virus among monogamous heterosexual couples: the HCV partners study. Hepatology. 2013;57(3):881–889. | |
Gentile I, Zappulo E, Buonomo AR, Borgia G. Prevention of mother-to-child transmission of hepatitis B virus and hepatitis C virus. Expert Rev Anti Infect Ther. 2014;12(7):775–782. | |
Murakami J, Nagata I, Iitsuka T, et al. Risk factors for mother-to-child transmission of hepatitis C virus: maternal high viral load and fetal exposure in the birth canal. Hepatol Res. 2012;42(7):648–657. | |
Sanjuan R, Nebot MR, Chirico N, Mansky LM, Belshaw R. Viral mutation rates. J Virol. 2010;84(19):9733–9748. | |
Diamond MS. Evasion of innate and adaptive immunity by flaviviruses. Immunol Cell Biol. 2003;81(3):196–206. | |
Fishman SL, Branch AD. The quasispecies nature and biological implications of the hepatitis C virus. Infect Genet Evol. 2009;9(6):1158–1167. | |
Simmonds P. Genetic diversity and evolution of hepatitis C virus – 15 years on. J Gen Virol. 2004;85(Pt 11):3173–3188. | |
Smith DB, Bukh J, Kuiken C, et al. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment web resource. Hepatology. 2014;59(1):318–327. | |
Jirillo E. Hepatitis C Virus Disease: Immunobiology and Clinical Applications. Heidelberg: Springer; 2007. | |
Weiner AJ, Christopherson C, Hall JE, et al. Sequence variation in hepatitis C viral isolates. J Hepatol. 1991;13 Suppl 4:S6–S14. | |
Forns X, Purcell RH, Bukh J. Quasispecies in viral persistence and pathogenesis of hepatitis C virus. Trends Microbiol. 1999;7(10):402–410. | |
Farci P, Shimoda A, Wong D, et al. Prevention of hepatitis C virus infection in chimpanzees by hyperimmune serum against the hypervariable region 1 of the envelope 2 protein. Proc Natl Acad Sci U S A. 1996;93(26):15394–15399. | |
Zibert A, Kraas W, Meisel H, Jung G, Roggendorf M. Epitope mapping of antibodies directed against hypervariable region 1 in acute self-limiting and chronic infections due to hepatitis C virus. J Virol. 1997;71(5):4123–4127. | |
Dowd KA, Netski DM, Wang XH, Cox AL, Ray SC. Selection pressure from neutralizing antibodies drives sequence evolution during acute infection with hepatitis C virus. Gastroenterology. 2009; 136(7):2377–2386. | |
Rauch A, Gaudieri S, Thio C, Bochud PY. Host genetic determinants of spontaneous hepatitis C clearance. Pharmacogenomics. 2009; 10(11):1819–1837. | |
Kärre K. How to recognize a foreign submarine. Immunol Rev. 1997; 155(1):5–9. | |
Janeway CA Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol. 1989;54 Pt 1:1–13. | |
Janeway CA Jr. The immune system evolved to discriminate infectious nonself from noninfectious self. Immunol Today. 1992;13(1):11–16. | |
Gale M Jr, Foy EM. Evasion of intracellular host defence by hepatitis C virus. Nature. 2005;436(7053):939–945. | |
Thimme R, Lohmann V, Weber F. A target on the move: innate and adaptive immune escape strategies of hepatitis C virus. Antiviral Res. 2006;69(3):129–141. | |
Su AI, Pezacki JP, Wodicka L, et al. Genomic analysis of the host response to hepatitis C virus infection. Proc Natl Acad Sci U S A. 2002;99(24):15669–15674. | |
Chen L, Borozan I, Feld J, et al. Hepatic gene expression discriminates responders and nonresponders in treatment of chronic hepatitis C viral infection. Gastroenterology. 2005;128(5):1437–1444. | |
Feld JJ, Nanda S, Huang Y, et al. Hepatic gene expression during treatment with peginterferon and ribavirin: identifying molecular pathways for treatment response. Hepatology. 2007;46(5):1548–1563. | |
Sarasin-Filipowicz M, Oakeley EJ, Duong FH, et al. Interferon signaling and treatment outcome in chronic hepatitis C. Proc Natl Acad Sci U S A. 2008;105(19):7034–7039. | |
Janeway CA Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. | |
Haller O, Weber F. Pathogenic viruses: smart manipulators of the interferon system. Curr Top Microbiol Immunol. 2007;316:315–334. | |
Williams BR. PKR; a sentinel kinase for cellular stress. Oncogene. 1999;18(45):6112–6120. | |
Sadler AJ, Williams BR. Interferon-inducible antiviral effectors. Nat Rev Immunol. 2008;8(7):559–568. | |
Kumar M, Carmichael GG. Antisense RNA: function and fate of duplex RNA in cells of higher eukaryotes. Microbiol Mol Biol Rev. 1998; 62(4):1415–1434. | |
Samuel CE. Antiviral actions of interferons. Clin Microbiol Rev. 2001;14(4):778–809. | |
Holzinger D, Jorns C, Stertz S, et al. Induction of MxA gene expression by influenza A virus requires type I or type III interferon signaling. J Virol. 2007;81(14):7776–7785. | |
Tanaka Y, Nishida N, Sugiyama M, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet. 2009;41(10):1105–1109. | |
Suppiah V, Moldovan M, Ahlenstiel G, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet. 2009;41(10):1100–1104. | |
Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461(7262):399–401. | |
Rauch A, Kutalik Z, Descombes P, et al. Genetic variation in IL28B is associated with chronic hepatitis C and treatment failure: a genome-wide association study. Gastroenterology. 2010;138(4):1338–1345, 1345. e1–e7. | |
Thomas DL, Thio CL, Martin MP, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature. 2009;461(7265):798–801. | |
Thompson AJ, Muir AJ, Sulkowski MS, et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology. 2010;139(1):120–129.e18. | |
Prokunina-Olsson L, Muchmore B, Tang W, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet. 2013; 45(2):164–171. | |
Lange CM, Zeuzem S. IL28B single nucleotide polymorphisms in the treatment of hepatitis C. J Hepatol. 2011;55(3):692–701. | |
McFarland AP, Horner SM, Jarret A, et al. The favorable IFNL3 genotype escapes mRNA decay mediated by AU-rich elements and hepatitis C virus-induced microRNAs. Nat Immunol. 2014;15(1):72–79. | |
Chinnaswamy S, Chatterjee S, Boopathi R, Mukherjee S, Bhattacharjee S, Kundu TK. A single nucleotide polymorphism associated with hepatitis C virus infections located in the distal region of the IL28B promoter influences NF-κB-mediated gene transcription. PloS One. 2013;8(10):e75495. | |
Kärre K, Ljunggren HG, Piontek G, Kiessling R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature. 1986;319(6055):675–678. | |
Nielsen N, Ødum N, Ursø B, Lanier LL, Spee P. Cytotoxicity of CD56(bright) NK cells towards autologous activated CD4+ T cells is mediated through NKG2D, LFA-1 and TRAIL and dampened via CD94/NKG2A. PloS One. 2012;7(2):e31959. | |
Peppa D, Gill US, Reynolds G, et al. Up-regulation of a death receptor renders antiviral T cells susceptible to NK cell-mediated deletion. J Exp Med. 2013;210(1):99–114. | |
Fields BN, Knipe DM, Howley PM. Fields Virology. Vol 1. Philadelphia: Lippincott Williams & Wilkins; 2007. | |
Paul WE. Fundamental Immunology. Philadelphia: Lippincott Williams and Wilkins; 2008. | |
Jamil KM, Khakoo SI. KIR/HLA interactions and pathogen immunity. J Biomed Biotechnol. 2011;2011:298348. | |
Wilson MJ, Torkar M, Haude A, et al. Plasticity in the organization and sequences of human KIR/ILT gene families. Proc Natl Acad Sci U S A. 2000;97(9):4778–4783. | |
Trowsdale J. Genetic and functional relationships between MHC and NK receptor genes. Immunity. 2001;15(3):363–374. | |
Parham P, Abi-Rached L, Matevosyan L, et al. Primate-specific regulation of natural killer cells. J Med Primatol. 2010;39(4):194–212. | |
Hershberger KL, Shyam R, Miura A, Letvin NL. Diversity of the killer cell Ig-like receptors of rhesus monkeys. J Immunol. 2001; 166(7):4380–4390. | |
Khakoo SI, Rajalingam R, Shum BP, et al. Rapid evolution of NK cell receptor systems demonstrated by comparison of chimpanzees and humans. Immunity. 2000;12(6):687–698. | |
Uhrberg M, Valiante NM, Shum BP, et al. Human diversity in killer cell inhibitory receptor genes. Immunity. 1997;7(6):753–763. | |
Dring MM, Morrison MH, McSharry BP, et al. Innate immune genes synergize to predict increased risk of chronic disease in hepatitis C virus infection. Proc Natl Acad Sci U S A. 2011;108(14):5736–5741. | |
Martin MP, Gao X, Lee JH, et al. Epistatic interaction between KIR3DS1 and HLA-B delays the progression to AIDS. Nat Genet. 2002;31(4):429–434. | |
Khakoo SI, Thio CL, Martin MP, et al. HLA and NK cell inhibitory receptor genes in resolving hepatitis C virus infection. Science. 2004;305(5685):872–874. | |
Rajagopalan S, Long EO. Understanding how combinations of HLA and KIR genes influence disease. J Exp Med. 2005;201(7):1025–1029. | |
Gershwin E, Vierling JM, Manns MP. Liver Immunology: Principles and Practice. New York: Humana; 2007. | |
Diepolder HM, Zachoval R, Hoffmann RM, et al. Possible mechanism involving T-lymphocyte response to non-structural protein 3 in viral clearance in acute hepatitis C virus infection. Lancet. 1995; 346(8981):1006–1007. | |
Gerlach JT, Diepolder HM, Jung MC, et al. Recurrence of hepatitis C virus after loss of virus-specific CD4(+) T-cell response in acute hepatitis C. Gastroenterology. 1999;117(4):933–941. | |
Missale G, Bertoni R, Lamonaca V, et al. Different clinical behaviors of acute hepatitis C virus infection are associated with different vigor of the anti-viral cell-mediated immune response. J Clin Invest. 1996;98(3):706–714. | |
Lechner F, Wong DK, Dunbar PR, et al. Analysis of successful immune responses in persons infected with hepatitis C virus. J Exp Med. 2000;191(9):1499–1512. | |
Takaki A, Wiese M, Maertens G, et al. Cellular immune responses persist and humoral responses decrease two decades after recovery from a single-source outbreak of hepatitis C. Nat Med. 2000;6(5):578–582. | |
Thimme R, Oldach D, Chang KM, Steiger C, Ray SC, Chisari FV. Determinants of viral clearance and persistence during acute hepatitis C virus infection. J Exp Med. 2001;194(10):1395–1406. | |
Thimme R, Bukh J, Spangenberg HC, et al. Viral and immunological determinants of hepatitis C virus clearance, persistence, and disease. Proc Natl Acad Sci U S A. 2002;99(24):15661–15668. | |
Day CL, Lauer GM, Robbins GK, et al. Broad specificity of virus-specific CD4+ T-helper-cell responses in resolved hepatitis C virus infection. J Virol. 2002;76(24):12584–12595. | |
Gruner NH, Gerlach TJ, Jung MC, et al. Association of hepatitis C virus-specific CD8+ T cells with viral clearance in acute hepatitis C. J Infect Dis. 2000;181(5):1528–1536. | |
Cooper S, Erickson AL, Adams EJ, et al. Analysis of a successful immune response against hepatitis C virus. Immunity. 1999;10(4):439–449. | |
Dalod M, Dupuis M, Deschemin JC, et al. Weak anti-HIV CD8(+) T-cell effector activity in HIV primary infection. J Clin Invest. 1999; 104(10):1431–1439. | |
Battegay M, Fikes J, Di Bisceglie AM, et al. Patients with chronic hepatitis C have circulating cytotoxic T cells which recognize hepatitis C virus-encoded peptides binding to HLA-A2.1 molecules. J Virol. 1995;69(4):2462–2470. | |
Cerny A, McHutchison JG, Pasquinelli C, et al. Cytotoxic T lymphocyte response to hepatitis C virus-derived peptides containing the HLA A2.1 binding motif. J Clin Invest. 1995;95(2):521–530. | |
Chang KM, Thimme R, Melpolder JJ, et al. Differential CD4(+) and CD8(+) T-cell responsiveness in hepatitis C virus infection. Hepatology. 2001;33(1):267–276. | |
Ferrari C, Valli A, Galati L, et al. T-cell response to structural and nonstructural hepatitis C virus antigens in persistent and self-limited hepatitis C virus infections. Hepatology. 1994;19(2):286–295. | |
Koziel MJ, Dudley D, Afdhal N, et al. HLA class I-restricted cytotoxic T lymphocytes specific for hepatitis C virus. Identification of multiple epitopes and characterization of patterns of cytokine release. J Clin Invest. 1995;96(5):2311–2321. | |
Wong DK, Dudley DD, Dohrenwend PB, et al. Detection of diverse hepatitis C virus (HCV)-specific cytotoxic T lymphocytes in peripheral blood of infected persons by screening for responses to all translated proteins of HCV. J Virol. 2001;75(3):1229–1235. | |
Lauer GM, Ouchi K, Chung RT, et al. Comprehensive analysis of CD8(+)-T-cell responses against hepatitis C virus reveals multiple unpredicted specificities. J Virol. 2002;76(12):6104–6113. | |
Koziel MJ, Dudley D, Afdhal N, et al. Hepatitis C virus (HCV)-specific cytotoxic T lymphocytes recognize epitopes in the core and envelope proteins of HCV. J Virol. 1993;67(12):7522–7532. | |
Rehermann B, Chang KM, McHutchinson J, et al. Differential cytotoxic T-lymphocyte responsiveness to the hepatitis B and C viruses in chronically infected patients. J Virol. 1996;70(10):7092–7102. | |
Neumann-Haefelin C, Blum HE, Chisari FV, Thimme R. T cell response in hepatitis C virus infection. J Clin Virol. 2005;32(2):75–85. | |
Li K, Foy E, Ferreon JC, et al. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci U S A. 2005;102(8):2992–2997. | |
Foy E, Li K, Sumpter R Jr, et al. Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc Natl Acad Sci U S A. 2005;102(8):2986–2991. | |
Foy E, Li K, Wang C, et al. Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science. 2003; 300(5622):1145–1148. | |
Vilasco M, Larrea E, Vitour D, et al. The protein kinase IKKµ can inhibit HCV expression independently of IFN and its own expression is downregulated in HCV-infected livers. Hepatology. 2006;44(6):1635–1647. | |
Johnson CL, Owen DM, Gale M Jr. Functional and therapeutic analysis of hepatitis C virus NS3.4A protease control of antiviral immune defense. J Biol Chem. 2007;282(14):10792–10803. | |
Binder M, Kochs G, Bartenschlager R, Lohmann V. Hepatitis C virus escape from the interferon regulatory factor 3 pathway by a passive and active evasion strategy. Hepatology. 2007;46(5):1365–1374. | |
Meylan E, Curran J, Hofmann K, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437(7062):1167–1172. | |
Gale M Jr, Blakely CM, Kwieciszewski B, et al. Control of PKR protein kinase by hepatitis C virus nonstructural 5A protein: molecular mechanisms of kinase regulation. Mol Cell Biol. 1998;18(9):5208–5218. | |
Taylor DR, Shi ST, Romano PR, Barber GN, Lai MM. Inhibition of the interferon-inducible protein kinase PKR by HCV E2 protein. Science. 1999;285(5424):107–110. | |
Gale MJ Jr, Korth MJ, Tang NM, et al. Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology. 1997; 230(2):217–227. | |
Bode JG, Ludwig S, Ehrhardt C, et al. IFN-alpha antagonistic activity of HCV core protein involves induction of suppressor of cytokine signaling-3. FASEB J. 2003;17(3):488–490. | |
Melén K, Fagerlund R, Nyqvist M, Keskinen P, Julkunen I. Expression of hepatitis C virus core protein inhibits interferon-induced nuclear import of STATs. J Med Virol. 2004;73(4):536–547. | |
Lin W, Kim SS, Yeung E, et al. Hepatitis C virus core protein blocks interferon signaling by interaction with the STAT1 SH2 domain. J Virol. 2006;80(18):9226–9235. | |
Kawamura H, Govindarajan S, Aswad F, et al. HCV core expression in hepatocytes protects against autoimmune liver injury and promotes liver regeneration in mice. Hepatology. 2006;44(4):936–944. | |
Lin W, Choe WH, Hiasa Y, et al. Hepatitis C virus expression suppresses interferon signaling by degrading STAT1. Gastroenterology. 2005;128(4):1034–1041. | |
Miyamoto H, Moriishi K, Moriya K, et al. Involvement of the PA28γ-dependent pathway in insulin resistance induced by hepatitis C virus core protein. J Virol. 2007;81(4):1727–1735. | |
Khu YL, Tan YJ, Lim SG, Hong W, Goh PY. Hepatitis C virus non-structural protein NS3 interacts with LMP7, a component of the immunoproteasome, and affects its proteasome activity. Biochem J. 2004;384(Pt 2):401–409. | |
Osna NA. Hepatitis C virus and ethanol alter antigen presentation in liver cells. World J Gastroenterol. 2009;15(10):1201–1208. | |
Herzer K, Falk CS, Encke J, et al. Upregulation of major histocompatibility complex class I on liver cells by hepatitis C virus core protein via p53 and TAP1 impairs natural killer cell cytotoxicity. J Virol. 2003;77(15):8299–8309. | |
Cohen GB, Gandhi RT, Davis DM, et al. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity. 1999; 10(6):661–671. | |
Kim H, Mazumdar B, Bose SK, et al. Hepatitis C virus-mediated inhibition of cathepsin S increases invariant-chain expression on hepatocyte surface. J Virol. 2012;86(18):9919–9928. | |
Schulze Zur Wiesch J, Ciuffreda D, Lewis-Ximenez L, et al. Broadly directed virus-specific CD4+ T cell responses are primed during acute hepatitis C infection, but rapidly disappear from human blood with viral persistence. J Exp Med. 2012;209(1):61–75. | |
Lucas M, Ulsenheimer A, Pfafferot K, et al. Tracking virus-specific CD4+ T cells during and after acute hepatitis C virus infection. PloS One. 2007;2(7):e649. | |
Klenerman P, McMichael A. AIDS/HIV. Finding footprints among the trees. Science. 2007;315(5818):1505–1507. | |
Gaudieri S, Rauch A, Park LP, et al. Evidence of viral adaptation to HLA class I-restricted immune pressure in chronic hepatitis C virus infection. J Virol. 2006;80(22):11094–11104. | |
Rauch A, James I, Pfafferott K, et al. Divergent adaptation of hepatitis C virus genotypes 1 and 3 to human leukocyte antigen-restricted immune pressure. Hepatology. 2009;50(4):1017–1029. | |
Moore CB, John M, James IR, Christiansen FT, Witt CS, Mallal SA. Evidence of HIV-1 adaptation to HLA-restricted immune responses at a population level. Science. 2002;296(5572):1439–1443. | |
Bull RA, Luciani F, McElroy K, et al. Sequential bottlenecks drive viral evolution in early acute hepatitis C virus infection. PLoS Pathog. 2011;7(9):e1002243. | |
Leslie A, Kavanagh D, Honeyborne I, et al. Transmission and accumulation of CTL escape variants drive negative associations between HIV polymorphisms and HLA. J Exp Med. 2005;201(6):891–902. | |
Merani S, Petrovic D, James I, et al. Effect of immune pressure on hepatitis C virus evolution: insights from a single-source outbreak. Hepatology. 2011;53(2):396–405. | |
Neumann-Haefelin C, McKiernan S, Ward S, et al. Dominant influence of an HLA-B27 restricted CD8+ T cell response in mediating HCV clearance and evolution. Hepatology. 2006;43(3):563–572. | |
Ray SC, Fanning L, Wang XH, Netski DM, Kenny-Walsh E, Thomas DL. Divergent and convergent evolution after a common-source outbreak of hepatitis C virus. J Exp Med. 2005;201(11):1753–1759. | |
Fitzmaurice K, Petrovic D, Ramamurthy N, et al. Molecular footprints reveal the impact of the protective HLA-A*03 allele in hepatitis C virus infection. Gut. 2011;60(11):1563–1571. | |
Kim AY, Kuntzen T, Timm J, et al. Spontaneous control of HCV is associated with expression of HLA-B 57 and preservation of targeted epitopes. Gastroenterology. 2011;140(2):686–696. e1. | |
Allen TM, Altfeld M, Geer SC, et al. Selective escape from CD8+ T-cell responses represents a major driving force of human immunodeficiency virus type 1 (HIV-1) sequence diversity and reveals constraints on HIV-1 evolution. J Virol. 2005;79(21):13239–13249. | |
Friedrich TC, Dodds EJ, Yant LJ, et al. Reversion of CTL escape-variant immunodeficiency viruses in vivo. Nat Med. 2004;10(3):275–281. | |
Herbeck JT, Nickle DC, Learn GH, et al. Human immunodeficiency virus type 1 env evolves toward ancestral states upon transmission to a new host. J Virol. 2006;80(4):1637–1644. | |
Leslie AJ, Pfafferott KJ, Chetty P, et al. HIV evolution: CTL escape mutation and reversion after transmission. Nat Med. 2004; 10(3):282–289. | |
Cox AL, Mosbruger T, Mao Q, et al. Cellular immune selection with hepatitis C virus persistence in humans. J Exp Med. 2005;201(11):1741–1752. | |
Kuntzen T, Timm J, Berical A, et al. Viral sequence evolution in acute hepatitis C virus infection. J Virol. 2007;81(21):11658–11668. | |
Pfafferott K, Gaudieri S, Ulsenheimer A, et al. Constrained pattern of viral evolution in acute and early HCV infection limits viral plasticity. PloS One. 2011;6(2):e16797. | |
Alter G, Heckerman D, Schneidewind A, et al. HIV-1 adaptation to NK-cell-mediated immune pressure. Nature. 2011;476(7358):96–100. | |
Martell M, Esteban JI, Quer J, et al. Hepatitis C virus (HCV) circulates as a population of different but closely related genomes: quasispecies nature of HCV genome distribution. J Virol. 1992;66(5):3225–3229. | |
Farci P, Bukh J, Purcell RH. The quasispecies of hepatitis C virus and the host immune response. Springer Semin Immunopathol. 1997;19(1):5–26. | |
Manzin A, Solforosi L, Clementi M. Dynamics of viral quasispecies in hepatitis C virus infection. Res Virol. 1997;148(2):171–176. | |
Farci P, Shimoda A, Coiana A, et al. The outcome of acute hepatitis C predicted by the evolution of the viral quasispecies. Science. 2000;288(5464):339–344. | |
Wohnsland A, Hofmann WP, Sarrazin C. Viral determinants of resistance to treatment in patients with hepatitis C. Clin Microbiol Rev. 2007;20(1):23–38. | |
Enomoto N, Sakuma I, Asahina Y, et al. Comparison of full-length sequences of interferon-sensitive and resistant hepatitis C virus 1b. Sensitivity to interferon is conferred by amino acid substitutions in the NS5A region. J Clin Invest. 1995;96(1):224–230. | |
Enomoto N, Sakuma I, Asahina Y, et al. Mutations in the nonstructural protein 5A gene and response to interferon in patients with chronic hepatitis C virus 1b infection. N Engl J Med. 1996;334(2):77–81. | |
Lin C, Gates CA, Rao BG, et al. In vitro studies of cross-resistance mutations against two hepatitis C virus serine protease inhibitors, VX-950 and BILN 2061. J Biol Chem. 2005;280(44):36784–36791. | |
Lin C, Lin K, Luong YP, et al. In vitro resistance studies of hepatitis C virus serine protease inhibitors, VX-950 and BILN 2061: structural analysis indicates different resistance mechanisms. J Biol Chem. 2004;279(17):17508–17514. | |
Sarrazin C, Kieffer TL, Bartels D, et al. Dynamic hepatitis C virus genotypic and phenotypic changes in patients treated with the protease inhibitor telaprevir. Gastroenterology. 2007;132(5):1767–1777. | |
Applegate TL, Gaudieri S, Plauzolles A, et al. Naturally occurring dominant drug resistance mutations occur infrequently in the setting of recently acquired hepatitis C. Antivir Ther. Epub August 8, 2014. | |
John M, Moore CB, James IR, Mallal SA. Interactive selective pressures of HLA-restricted immune responses and antiretroviral drugs on HIV-1. Antivir Ther. 2005;10(4):551–555. | |
Mueller SM, Schaetz B, Eismann K, et al. Dual selection pressure by drugs and HLA class I-restricted immune responses on human immunodeficiency virus type 1 protease. J Virol. 2007;81(6):2887–2898. | |
Salloum S, Kluge SF, Kim AY, Roggendorf M, Timm J. The resistance mutation R155K in the NS3/4A protease of hepatitis C virus also leads the virus to escape from HLA-A*68-restricted CD8 T cells. Antiviral Res. 2010;87(2):272–275. | |
Gaudieri S, Rauch A, Pfafferott K, et al. Hepatitis C virus drug resistance and immune-driven adaptations: relevance to new antiviral therapy. Hepatology. 2009;49(4):1069–1082. | |
Schaefer EA, Chung RT. Anti-hepatitis C virus drugs in development. Gastroenterology. 2012;142(6):1340–1350. e1. | |
Janssen HL, Reesink HW, Lawitz EJ, et al. Treatment of HCV infection by targeting microRNA. N Engl J Med. 2013;368(18):1685–1694. | |
Lanford RE, Hildebrandt-Eriksen ES, Petri A, et al. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science. 2010;327(5962):198–201. | |
Wendt A, Bourlière M. An update on the treatment of genotype-1 chronic hepatitis C infection: lessons from recent clinical trials. Ther Adv Infect Dis. 2013;1(6):191–208. | |
Hepatitis C Virus (HCV) Database Project [database on the Internet]. Los Alamos National Security. Available from: http://hcv.lanl.gov/content/index. Accessed January 8, 2015. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.