")
Back to Archived Journals » International Journal of Interferon, Cytokine and Mediator Research » Volume 6
Inflammatory bowel disease: a focus on the involvement of dietary fats
Authors de Silva P, Korzenik J
Received 3 March 2014
Accepted for publication 10 May 2014
Published 22 July 2014 Volume 2014:6 Pages 19—25
DOI https://doi.org/10.2147/IJICMR.S35997
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Punyanganie S de Silva,1,2 Joshua R Korzenik1,2
1Department of Gastroenterology, Hepatology and Endoscopy, Brigham and Women's Hospital, 2Harvard Medical School, Boston, MA, USA
Abstract: Inflammatory bowel diseases (IBDs) are chronic immune-mediated diseases of the gastrointestinal tract well known to be associated with both genetic and environmental risk factors. Certain genotypes may develop clinical manifestations under particular environmental influences. There is increasing evidence to suggest that diet is a key environmental factor in IBD, and dietary fats in particular have been implicated in both pro- and anti-inflammatory roles. Previous epidemiological studies have highlighted variable results, most probably due to the complex pathways that may be mediated by fatty acid activity. We aim to review the available studies on dietary fats and IBD pathogenesis and explore mechanisms involved to identify potential opportunities for future research and therapies.
Keywords: inflammatory bowel disease, ulcerative colitis, Crohn's disease, lipids, n-3 polyunsaturated fatty acids, n-6 polyunsaturated fatty acids
Introduction
Inflammatory bowel diseases (IBDs) represent a spectrum of chronic inflammatory disorders of the intestine that is characterized by remitting and relapsing episodes. IBD is chiefly comprised of two main subtypes: ulcerative colitis (UC) and Crohn’s disease (CD). While UC is characterized predominantly by inflammation limited to the mucosa, in CD the inflammation is transmural, often resulting in stricturing or penetrating phenotypes.
The diseases were traditionally believed to be limited to the Western industrialized world, with high incidence and prevalence recorded in Western Europe and North America.1,2 However, in the last decade, a dramatic increase has been noted in countries that have recently adapted a more Westernized lifestyle, which includes dietary changes.3–6 In addition, there is an increasing number of first- and second-generation immigrants to countries with a historically high incidence and prevalence of IBD who have been diagnosed.7,8 This changing epidemiology suggests that environment, and particularly diet, may have a role to play in the etiopathogenesis.
While both diseases are distinct in terms of presentation and often response to treatment, it is believed that they share a common underlying pathogenic mechanism in that IBD is a dysregulated immune response to single or multiple environmental risk factors in a genetically susceptible host.9–11 It is also increasingly evident that the gut commensal microflora, genetic composition of the host, and the mucosal immune response may be altered through interaction with dietary components, thereby potentially increasing or reducing an individual’s risk of developing inflammation.
Overview of dietary fats
In the US, consumption of nutrient fat has increased by approximately 25% over the last 60 years.12 These dietary fats consist of saturated, trans, polyunsaturated, monounsaturated, and cholesterol forms. Saturated fats are commonly found in high-fat cheeses and cuts of meat, whole-fat milk, cream, and butter. Naturally occurring trans fat is found in the fatty parts of meat and dairy products. Artificial trans fat comes from foods that contain partially hydrogenated oil, and is formed when hydrogen is added to liquid oil, turning it into solid fat. Cholesterol is also derived from animal-based foods, such as meats, poultry, egg yolks, and whole milks. Olive oil and nuts are the commonest dietary source of monounsaturated fatty acids, and ω-6 (n-6) polyunsaturated fatty acids (PUFAs) are commonly derived from soybean, corn, and safflower oils along with red meats and pork. Sources rich in n-3 PUFAs include canola oil, walnuts, flaxseed, and fish, such as trout, herring, and salmon.13
Epidemiology of dietary fats in inflammatory bowel disease
In the past, studies have attempted to assess food groups and their role in IBD activity. The vast majority of these studies have been retrospective, and have not specifically assessed individual fatty acid intakes, thereby possibly giving rise to recall bias and confounding potential associations. Prospective studies that have investigated the effects of dietary factors in predicting the likelihood of clinical relapse are also limited, and results have been variable. Of the studies that have been carried out, there is evidence to suggest that lipids, and in particular PUFAs, may have a role to play in intestinal inflammation.
A large prospective dietary study of 191 patients with UC identified red and processed meat as a predictor of clinical relapse.14 It is well established that both these food groups contain large amounts of n-6 PUFAs, giving rise to a potential proinflammatory association. This is further supported by two case-control studies, from Israel and the Netherlands, which demonstrated statistically significant odds ratios of 6.515 and 5.116 for higher intakes of total PUFAs and the development of new-onset UC. The study from the Netherlands reported that the two highest tertiles of linoleic acid approximately doubled the risk, although this were not statistically significant.16 A prospective cohort study involving seven regions across Europe in the EPIC (European Prospective Investigation into Cancer and Nutrition) study17 reported that participants with the higher dietary intakes of linoleic acid (an n-6 PUFA and precursor to arachidonic acid), as measured by food-frequency questionnaires, had more than a doubling of the risk of developing UC (odds ratio 2.49, 95% confidence interval [CI] 1.23–5.07).17
Two subsequent prospective cohort studies of diet in the development of incident cases of UC also identified a positive association with total PUFA intake, and increased levels of the n-6 PUFAs linoleic acid and arachidonic acid with UC incidence increased by two- and fourfold, respectively.18,19 Furthermore, another study from this same European cohort revealed that an increased intake of dietary n-3 PUFAs, in particular eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) was associated with a reduced incidence of UC.20 A recent prospective cohort study by Chan et al also investigated the association between dietary fatty acid intake and CD in 229,702 participants recruited from nine European centers. Seventy-three incident cases of CD were identified during a 12-year follow-up period, and it was noted that there was a statistically significant reduced intake of DHA in cases compared to controls when adjusted for smoking, body mass index, oleic acid, EPA, linoleic acid, and total energy intake (P=0.04).21 However, these epidemiological studies were undertaken in older European populations with a male predominance, and did not specifically address disease activity after diagnosis or genetic susceptibility. Recent data from a large prospective cohort of younger North American women did not find cumulative energy-adjusted intake of total fat, saturated fats, unsaturated fats or n-6 and n-3 PUFAs to be associated with increased risk of CD or UC.22 However, within this same study, a greater intake of long-chain n-3 PUFAs was associated with a trend toward lower risk of UC (hazard ratio 0.72, 95% CI 0.51–1.01), and a high long-term intake of trans unsaturated fatty acids was associated with a trend toward an increased incidence of UC (hazard ratio 1.34, 95% CI 0.94–1.92).22 This variation in results may be explained by individual genetic variability, age, and other as-yet unidentified environmental factors leading to variable susceptibility to dietary risk factors. Case-control studies that included younger patients have also reported positive associations with PUFAs and linoleic acid, although these associations did not always reach statistical significance.15,16
Another point of note is that the majority of these epidemiological studies focused on UC and not CD. This may be partially due to lower numbers of CD patients in the cohorts studied, but in those that did include CD patients, the lack of association may also be partially explained by the fact that inflammation in CD is transmural and not confined to the mucosa as in UC, thereby giving rise to the hypothesis that dietary agents may have a lesser role to play in stimulating or suppressing nonmucosal inflammatory mechanisms.
Role of dietary fats in the pathophysiology of IBD
Dietary PUFAs and eicosanoids
There are many plausible biological mechanisms for how diet may be involved in the etiology of IBD. Dietary constituents are in direct contact with the intestinal wall of the colon. PUFAs are present in colonic cell membranes predominantly as arachidonic acid (AA, n-6 PUFA derived), EPA, and DHA (n-3 PUFA derived). Both n-3 and n-6 membrane phospholipids can be metabolized to eicosanoids, which include prostaglandins (PGs), thromboxanes, and leukotrienes (LTs). Those eicosanoids, derived from such n-6 PUFAs as PGE2 and leukotriene B4 (LTB4), are more proinflammatory than the n-3 PUFA-derived eicosanoids PGE3 and LTB5,23,24 and may subsequently induce or exacerbate UC. Their proinflammatory effects include neutrophil aggregation, chemotaxis, and release of lysosomal enzymes.24–28 Therefore, hypothetically, a high dietary intake of n-6 PUFAs and their incorporation into colonic cell membranes would lead to a source of proinflammatory molecules that may induce UC relapses. This is supported by finding increased levels of PGE2 and LTB4 in the mucosa of patients with UC when compared to healthy controls.27,28 There is also a positive correlation between these eicosanoids and the histological level of inflammation (r=0.89, P<0.05).29 Further supporting this hypothesis, first-line drugs used to treat UC, namely 5-aminosalicyclic acid compounds, suppress PGE2 levels in a dose-dependent manner.26
Dietary fats and resolvins
In addition to modifying inflammatory responses and influencing the biochemical composition of colonocyte membranes, PUFAs may alter the balance of a wide range of lipid signaling molecules and their effects on nuclear receptors.30 These molecules are involved in a complex network of pathways that regulate and resolve inflammation.31 Anti-inflammatory (“resolving”) molecules or resolvins include the lipoxins LXA4 and LXB4, which are formed from AA through multiple routes depending on tissue type, and aspirin-triggered lipoxin.32 It has been hypothesized that in IBD there is a decreased amount of lipoxins.33 It is thought that while high intakes of n-6 PUFAs seem to be well tolerated and may have beneficial effects in most people, there may be a subgroup of individuals who are unable to convert AA to lipoxins in adequate quantities, and as a result n-6 PUFA is directed toward proinflammatory PG synthesis.
Patients with UC are known to have reduced LXA4 synthesis, and blockage of LXA synthesis in animal models has also been shown to induce UC.34 This association between LXA4 and UC was further demonstrated in a study by Vong et al,35 where colonic mucosal biopsies were collected from 20 healthy volunteers with or without a prior history of UC, individuals with UC experiencing active disease (n=8), and UC patients in medically induced remission (n=16). It was noted that in individuals in remission, mucosal expression of LXA4 was significantly elevated by nearly threefold compared to the other groups, further supporting LXA4’s anti-inflammatory role.
Resolution of inflammation is also known to be mediated by the n-3-derived eicosanoids resolvin E and resolvin D, which are derived from EPA and DHA, respectively.30,36–39 A recent study of healthy volunteers demonstrated that following ingestion of 1 g of EPA, sera collected 3 hours later had elevated levels of 18-hydroxyeicosapentaenoate, which is a precursor to resolvins.37 The n-3 PUFA-derived mediator resolvin E1 has also shown important positive effects on dextran sulfate sodium and 2,4,6-trinitrobenzene sulfonic acid-induced colitis, reducing mortality and colon damage38 in addition to reducing polymorphonuclear infiltration and production of the proinflammatory cytokines TNFα, IL-12p40, and IL-1β in colonic tissue.39
Animal models have also demonstrated that resolvin D1 and D2 are effective in preventing UC, mainly secondary to their ability to inhibit polymorphonuclear infiltration, downregulating NF-κB, and reducing proinflammatory cytokines, chemokines, and some adhesion molecules.40 Mouse models have also demonstrated that n-3 PUFAs reduce cytokine- and adhesion-molecule production, enhance specialized pro-resolving mediator production, and decrease leukocyte– endothelial cell adhesive interactions.37
Genetic regulation of fatty acid desaturases and dietary fat intake
The enzymes fatty acid desaturase (FADS)-1 and FADS2 are the rate-limiting enzymes in the synthesis of AA, EPA, and DHA from their dietary precursors linoleic acid and α-linolenic acid41 (Figure 1). The mammalian δ6-desaturase coded by FADS2 (HSA11q12–q13.1) catalyzes the first and rate-limiting step for the biosynthesis of long-chain PUFAs.42,43
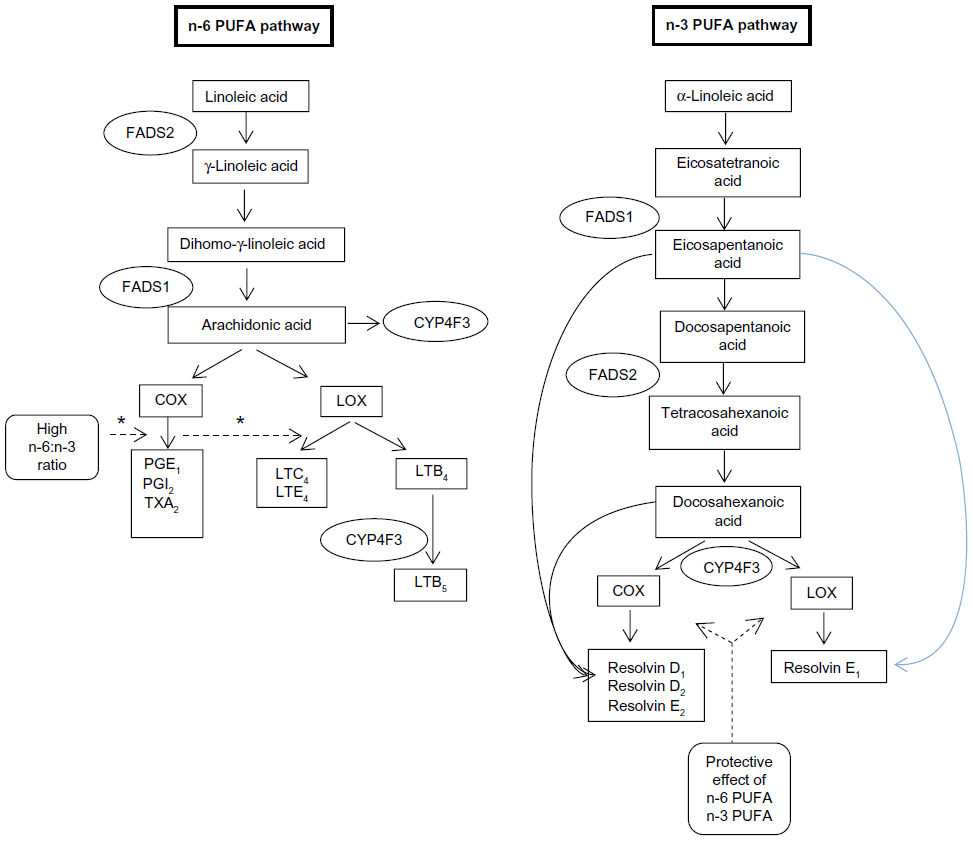 | Figure 1 Schematic diagram of key n-3 and n-6 PUFA-metabolism pathways associated with inflammation in inflammatory bowel disease. |
A recent large-scale study of 2,066 participants from the European Prospective Investigation into Cancer and Nutrition – Potsdam study investigating the association between the rs174546 genotype single-nucleotide peptide block on chromosome 11 and dietary fatty acid intake, as measured through food-frequency questionnaires and serum, identified a strong correlation between this single-nucleotide peptide when coding for δ6-desaturase and fatty acid intake.44 δ6-Desaturase is the key rate-limiting enzyme that converts EPA to DHA, which in turn is a precursor to resolvin D1 and D2. This association between FADS genes and IBD was explored in a cohort of pediatric CD patients.45 It was noted in this study that there was an interaction between the FADS2 single-nucleotide polymorphism rs11230815 and dietary n-3 PUFAs (P=0.042). When the dietary ratio of n-6 PUFA:n-3 PUFA was considered, significant interactions (P<0.05) were noted in the CYP4F3 and FADS2 genotypes rs1290617, rs1290620, rs11230815, rs17831757, rs968567, and rs174627. This suggests that there is an association between the dietary ratio of these fatty acids and CD according to the CYP4F3 and FADS2 genotypes.
Dietary fats and the PPAR receptors
Another potential mechanism for PUFAs in the etiology of IBD involves their effects on nuclear receptors, in particular peroxisome proliferator-activated receptors (PPARs). These act as transcription factors controlling the expression of genes related to a wide range of key functions, such as cell differentiation, lipid metabolism, and inflammation.46 PUFAs and the arachidonic acid metabolite PGJ2 act as ligands for PPARα and PPARγ,47,48 and cyclooxygenase expression has been shown to be decreased through PPARγ signaling.49 This association is emphasized in studies highlighting the complex relationship between PUFAs and cyclooxygenase expression.50,51 These pathways are complex however, and studies have demonstrated that linoleic acid metabolites are ligands for PPARδ, which in turn upregulates PPARγ activity.52 The 5-aminosalicyclic acid drugs are also known to bind PPARγ.53 The role of these PPARs in IBD is not fully elucidated, but these may transpire to be targets for future advanced therapies.51,52 NF-κB expression is also downregulated by PPARγ activity, and has been shown to reduce experimentally induced UC.46,54 Furthermore, PPAR agonists can counteract the increased expression of proinflammatory cytokines induced through Toll-like receptor signaling.55 Further studies are needed to better understand these complex associations.
Dietary fats, the microbiome, and IBD
The human gut contains a vast number of microorganisms known collectively as the “gut microbiota”. A further potential mechanism for dietary lipids in the pathogenesis of IBD may involve its alteration of the normal intestinal microbiota. Variations in the gut microbiota have been noted in IBD with increases in proinflammatory Bacteroides with long-term diets, particularly protein and animal fat.56,57 While there have been several studies that have demonstrated how alterations in dietary fiber can influence the microbiome,58,59 a high intake of PUFAs may also modify the gut microbiota, because approximately 2% of PUFAs consumed arrive intact in the colon, and thus can hypothetically alter the survival of bacteria.60
Clinical significance and future prospects
As described, there are several potential mechanisms for how dietary lipids may be involved in the etiology of IBD, but further experimental investigation is required. Providing data in support of dietary fatty acid risk factors would strengthen the evidence for causality and provide a further basis for future potential dietary interventions as treatment in IBD. It would also help us to further understand the specific potential mechanisms by which diet may influence activity in UC and CD.
In the Western diet, there is a predominance of n-6 PUFAs. In a recent study conducted in the UK and the Netherlands, a ratio of n-6:n-3 PUFA intake of 8:1 was noted in patients treated in gastroenterology clinics,61 while a ratio of 2:1–4:1, which is closer to that of prehistoric diets, has been recommended.62,63
The genetic loci associated with CD or UC can be broadly categorized into those involving abnormalities in innate immune response, mucosal barrier function, adaptive immunity, or immune regulatory response. These pathways are probably influenced by such environmental factors as dietary lipid agents. Therefore, it is likely that depending on the presence or absence of CD- or UC-risk alleles, response to dietary lipids may differ. Investigating the role of different polymorphisms in IBD may provide further insights; those that have already been identified are only able to explain a small percentage of cases,64 while there may be as-yet unidentified others that directly interact with dietary lipids.
Further evaluation of the role of lipid-mediated receptors in relation to dietary fatty acid intake would be useful in order to further understand the exact mechanisms by which these dietary agents affect IBD activity, which in turn could potentially lead to future dietary therapies for IBDs. Although interest in dietary therapies for IBD date back to the early stages of its management in the era of modern medicine,65 progress to date has been poor. This is primarily due to the complexities surrounding diet and the variability of nutrient intake in humans, variations in dosage, treatment length, confounding medication, or translating evidence from the laboratory to the clinic. However, it is increasingly evident that the environment and particularly diet have a significant role to play in IBD pathogenesis.
The strongest evidence for associations between diet and IBD pathogenesis comes from interventional studies. Studies have shown that clinical remission and mucosal healing can be achieved in CD by a switch from a normal diet to a formula-defined enteral feed, but the mechanisms for this response are not well understood. Potential mechanisms include changes in gut-microbiota type or quantity, improved nutritional status, reduced allergenicity of gut contents, avoidance of food additives, or provision of an anti-inflammatory factor such as TGFβ.66,67
While initial results for supplementation of n-3 PUFA in CD were encouraging, this was not consistently reproduced. A further meta-analysis of six published trials shows a small benefit for maintenance supplementation (relative risk of relapse 0.77, 95% CI 0.61–0.98), but the studies were significantly heterogeneous (consistency index I2=58.4%, P for heterogeneity =0.03), and two large studies showed no benefit.68 However, it is to be noted that some of the negative studies may have had variable bioactivity of n-3 PUFAs due to the different preparations used. The same meta-analysis also assessed dietary n-3 PUFA trials in UC. This was limited to three trials, and no significant benefit (relative risk of relapse 1.02, 95% CI 0.51–2.03) was noted.
More recently, an n-3 PUFA food-exchange table was used to assess changes in disease activity in individuals with IBD who underwent a dietary intervention to increase n-3:n-6 PUFA ratio levels. It was noted that the n-3:n-6 PUFA ratio was significantly higher in those patients who did not relapse during follow-up compared to those who did experience a flare.69
Dietary interventions may be beneficial in IBD. However, there is now an opportunity to identify novel approaches to preventing and reversing/resolving chronic inflammatory disease based upon the downstream metabolic products of EPA and DHA and their mechanisms of action or the interaction of these n-3 PUFAs with free fatty acid receptors.
Conclusion
In summary, current epidemiological and translational studies suggest that dietary lipids have a role in IBD pathogenesis. Further studies to understand these complex lipid-mediated mechanisms may open the window to effective therapeutic or complementary nutrition-modulation strategies for patients with UC or CD.
Disclosure
The authors report no conflicts of interest in this work.
References
Molodecky NA, Soon IS, Rabi DM, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142:46–54. e42. | |
Shivananda S, Lennard-Jones J, Logan R, et al. Incidence of inflammatory bowel disease across Europe: is there a difference between north and south? Results of the European Collaborative Study on Inflammatory Bowel Disease (EC-IBD). Gut. 1996;39:690–697. | |
Ng SC, Tang W, Ching JY, et al. Incidence and phenotype of inflammatory bowel disease based on results from the Asia-Pacific Crohn’s and colitis epidemiology study. Gastroenterology. 2013;145:158–165. e2. | |
Benchimol EI, Fortinsky KJ, Gozdyra P, Van den Heuvel M, Van Limbergen J, Griffiths AM. Epidemiology of pediatric inflammatory bowel disease: a systematic review of international trends. Inflamm Bowel Dis. 2011;17:423–439. | |
Loftus EV. Clinical epidemiology of inflammatory bowel disease: incidence, prevalence, and environmental influences. Gastroenterology. 2004;126:1504–1517. | |
Bernstein CN, Shanahan F. Disorders of a modern lifestyle: reconciling the epidemiology of inflammatory bowel diseases. Gut. 2008;57:1185–1191. | |
Li X, Sundquist J, Hemminki K, Sundquist K. Risk of inflammatory bowel disease in first- and second-generation immigrants in Sweden: a nationwide follow-up study. Inflamm Bowel Dis. 2011;17:1784–1791. | |
Tsironi E, Feakins RM, Probert CS, Rampton DS, Phil D. Incidence of inflammatory bowel disease is rising and abdominal tuberculosis is falling in Bangladeshis in East London, United Kingdom. Am J Gastroenterol. 2004;99:1749–1755. Erratum in: Am J Gastroenterol. 2005;100:255. | |
Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med. 2009;361:2066–2078. | |
Sartor RB. Genetics and environmental interactions shape the intestinal microbiome to promote inflammatory bowel disease versus mucosal homeostasis. Gastroenterology. 2010;139:1816–1819. | |
Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. | |
Risek RL, Friend B, Page L. Fat in today’s food supply – level of use and sources. J Am Oil Chem Soc. 1974;51:244–254. | |
Centers for Disease Control and Prevention. Dietary fat. 2012. Available from: http://www.cdc.gov/nutrition/everyone/basics/fat. Accessed June 4, 2014. | |
Jowett SL, Seal CJ, Pearce MS, et al. Influence of dietary factors on the clinical course of ulcerative colitis: a prospective cohort study. Gut. 2004;53:1479–1484. | |
Reif S, Klein I, Lubin F, Farbstein M, Hallak A, Gilat T. Pre-illness dietary factors in inflammatory bowel disease. Gut. 1997;40:754–760. | |
Geerling BJ, Dagnelie PC, Badart-Smook A, Russel MG, Stockbrügger RW, Brummer RJ. Diet as a risk factor in the development of ulcerative colitis. Am J Gastroenterol. 2000;95:1008–1013. | |
IBD in EPIC Study Investigators, Tjonneland A, Overvad K, et al. Linoleic acid, a dietary n-6 polyunsaturated fatty acid, and the aetiology of ulcerative colitis: a nested case-control study within a European prospective cohort study. Gut. 2009;58:1606–1611. | |
Hart AR, Luben R, Olsen A, et al. Diet in the aetiology of ulcerative colitis: a European prospective cohort study. Digestion. 2008;77: 57–64. | |
de Silva PS, Olsen A, Christensen J, et al. An association between dietary arachidonic acid, measured in adipose tissue, and ulcerative colitis. Gastroenterology. 2010;139:1912–1917. | |
John S, Luben R, Shrestha SS, Welch A, Khaw KT, Hart AR. Dietary n-3 polyunsaturated fatty acids and the aetiology of ulcerative colitis: a UK prospective cohort study. Eur J Gastroenterol Hepatol. 2010;22:602–606. | |
Chan SS, Luben R, Olsen A, et al. Association between high dietary intake of the n-3 polyunsaturated fatty acid docosahexaenoic acid and reduced risk of Crohn’s disease. Aliment Pharmacol Ther. 2014;39:834–842. | |
Ananthakrishnan AN, Khalili H, Konijeti GG, et al. Long-term intake of dietary fat and risk of ulcerative colitis and Crohn’s disease. Gut. 2014;63:776–784. | |
Lee TH, Menica-Huerta JM, Shih C, Corey EJ, Lewis RA, Austen KF. Characterization and biological properties of 5,12-dihydroxy derivatives of eicosapentaenoic acid, including leukotriene B5 and the double lipoxygenase product. J Biol Chem. 1984;259:2383–2389. | |
Goldman DW, Pickett WC, Goetzl EJ. Human neutrophil chemotactic and degranulating activities of leukotriene B5 (LTB5) derived from eicosapentaenoic acid. Biochem Biophys Res Comm. 1983;117:282–288. | |
Rampton D, Collins C. Review article: thromboxanes in inflammatory bowel disease – pathogenic and therapeutic implications. Aliment Pharmacol Ther. 1993;7:357–367. | |
Sharon P, Ligumsky M, Rachmilewitz D, Zor U. Role of prostaglandins in ulcerative colitis. Enhanced production during active disease and inhibition by sulfasalazine. Gastroenterology. 1978;75:638–640. | |
Heller A1, Koch T, Schmeck J, van Ackern K. Lipid mediators in inflammatory disorders. Drugs. 1998;55:487–496. | |
Sharon P, Stenson WF. Enhanced synthesis of leukotriene B4 by colonic mucosa in inflammatory bowel disease. Gastroenterology. 1984;86:453–460. | |
Nishida T, Miwa H, Shigematsu A, Yamamoto M, Iida M, Fujishima M. Increased arachidonic acid composition of phospholipids in colonic mucosa from patients with active ulcerative colitis. Gut. 1987;28:1002–1007. | |
Calder PC. Fatty acids and immune function: relevance to inflammatory bowel diseases. Rev Immunol. 2009;28:506–534. | |
Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349–361. | |
Serhan CN. Lipoxins and aspirin-triggered 15-epi-lipoxins are the first lipid mediators of endogenous anti-inflammation and resolution. Prostaglandins Leukot Essent Fatty Acids. 2005;73:141–162. | |
Gewirtz AT, Neish AS, Madara JL. Mechanisms of active intestinal inflammation and potential down-regulation via lipoxins. Adv Exp Med Biol. 2002;507:229–236. | |
Mangino MJ, Brounts L, Harms B, Heise C. Lipoxin biosynthesis in inflammatory bowel disease. Prostaglandins Other Lipid Mediat. 2006;79:84–92. | |
Vong L, Ferraz JG, Dufton N, et al. Up-regulation of annexin-A1 and lipoxin A(4) in individuals with ulcerative colitis may promote mucosal homeostasis. PLoS One. 2012;7:e39244. | |
Mas E, Croft KD, Zahra P, Barden A, Mori TA. Resolvins D1, D2, and other mediators of self-limited resolution of inflammation in human blood following n-3 fatty acid supplementation. Clin Chem. 2012;58:1476–1484. | |
Oh SF, Pillai PS, Recchiuti A, Yang R, Serhan CN. Pro-resolving actions and stereoselective biosynthesis of 18S E-series resolvins in human leukocytes and murine inflammation. J Clin Invest. 2011;121:569–581. | |
Arita M, Yoshida M, Hong S, et al. Resolvin E1, an endogenous lipid mediator derived from omega-3 eicosapentaenoic acid, protects against 2,4,6-trinitrobenzene sulfonic acid-induced colitis. Proc Natl Acad Sci U S A. 2005;102:7671–7676. | |
Ishida T, Yoshida M, Arita M, et al. Resolvin E1, an endogenous lipid mediator derived from eicosapentaenoic acid, prevents dextran sulfate sodium-induced colitis. Inflamm Bowel Dis. 2010;16:87–95. | |
Bento AF, Claudino RF, Dutra RC, Marcon R, Calixto JB. Omega-3 fatty acid-derived mediators 17(R)-hydroxy docosahexaenoic acid, aspirin-triggered resolvin D1 and resolvin D2 prevent experimental colitis in mice. J Immunol. 2011;187:1957–1969. | |
Nakamura MT, Nara TY. Structure, function, and dietary regulation of δ6, δ5, and δ9 desaturases. Annu Rev Nutr. 2004;24:345–376. | |
Cho HP, Nakamura MT, Clarke SD. Cloning, expression, and nutritional regulation of the mammalian delta-6 desaturase. J Biol Chem. 1999;274:471–477. | |
Cho HP, Nakamura MT, Clarke SD. Cloning, expression, and fatty acid regulation of the human delta-5 desaturase. J Biol Chem. 1999;274:37335–37339. | |
Zietemann V, Kröger J, Enzenbach C, et al. Genetic variation of the FADS1 FADS2 gene cluster and n-6 PUFA composition in erythrocyte membranes in the European Prospective Investigation into Cancer and Nutrition-Potsdam study. Br J Nutr. 2010;104:1748–1759. | |
Costea I, Mack DR, Lemaitre RN, et al. Interactions between the dietary polyunsaturated fatty acid ratio and genetic factors determine susceptibility to pediatric Crohn’s disease. Gastroenterology. 2014;146:929–931. | |
Celinski K, Dworzanski T, Korolczuk A, et al. Activated and inactivated PPARs-γ modulate experimentally induced colitis in rats. Med Sci Monit. 2011;17:BR116–BR124. | |
Kliewer SA, Sundseth SS, Jones SA, et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma. Proc Natl Acad Sci U S A. 1997;94:4318–4323. | |
Xu HE, Lambert MH, Montana VG, et al. Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol Cell. 1999;3:397–403. | |
Yang WL, Frucht H. Activation of the PPAR pathway induces apoptosis and COX-2 inhibition in HT-29 human colon cancer cells. Carcinogenesis. 2001;22:1379–1383. | |
Chêne G, Dubourdeau M, Balard P, et al. n-3 and n-6 Polyunsaturated fatty acids induce the expression of COX-2 via PPARgamma activation in human keratinocyte HaCaT cells. Biochim Biophys Acta. 2007;1771:576–589. | |
Coyne GS, Kenny DA, Childs S, Sreenan JM, Waters SM. Dietary n-3 polyunsaturated fatty acids alter the expression of genes involved in prostaglandin biosynthesis in the bovine uterus. Theriogenology. 2008;70:772–782. | |
Zuo X, Wu Y, Morris JS, et al. Oxidative metabolism of linoleic acid modulates PPAR-beta/delta suppression of PPAR-gamma activity. Oncogene. 2006;25:1225–1241. | |
Rousseaux C, Lefebvre B, Dubuquoy L, et al. Intestinal antiinflammatory effect of 5-aminosalicylic acid is dependent on peroxisome proliferator-activated receptor-gamma. J Exp Med. 2005;201:1205–1215. | |
Desreumaux P, Dubuquoy L, Nutten S, et al. Attenuation of colon inflammation through activators of the retinoid X receptor (RXR)/peroxisome proliferator-activated receptor gamma (PPARγ) heterodimer. A basis for new therapeutic strategies. J Exp Med. 2001;193:827–838. | |
Zhao W, Wang L, Zhang M, et al. Peroxisome proliferator-activated receptor gamma negatively regulates IFN-beta production in Toll-like receptor (TLR) 3- and TLR4-stimulated macrophages by preventing interferon regulatory factor 3 binding to the IFN-beta promoter. J Biol Chem. 2011;286:5519–5528. | |
Noor SO, Ridgway K, Scovell L, et al. Ulcerative colitis and irritable bowel patients exhibit distinct abnormalities of the gut microbiota. BMC Gastroenterol. 2010;10:134. | |
Tlaskalová-Hogenová H, Stěpánková R, Kozáková H, et al. The role of gut microbiota (commensal bacteria) and the mucosal barrier in the pathogenesis of inflammatory and autoimmune diseases and cancer: contribution of germ-free and gnotobiotic animal models of human diseases. Cell Mol Immunol. 2011;8:110–120. | |
Thibault R, Blachier F, Darcy-Vrillon B, et al. Butyrate utilization by the colonic mucosa in inflammatory bowel diseases: a transport deficiency. Inflamm Bowel Dis. 2010;16:684–695. | |
Steed H, Macfarlane GT, Macfarlane S. Prebiotics, synbiotics and inflammatory bowel disease. Mol Nutr Food Res. 2008;52:898–905. | |
Knapp HR, Melly MA. Bactericidal effects of polyunsaturated fatty acids. J Infect Dis. 1986;154:84–94. | |
Pot GK, Majsak-Newman G, Geelen A, et al. Fish consumption and markers of colorectal cancer risk: a multicenter randomized controlled trial. Am J Clin Nutr. 2009;90:354–361. | |
Kuipers RS, Luxwolda MF, Dijck-Brouwer DA, et al. Estimated macronutrient and fatty acid intakes from an East African Paleolithic diet. Br J Nutr. 2010;104:1666–1687. | |
Uchiyama K, Nakamura M, Odahara S, et al. N-3 polyunsaturated fatty acid diet therapy for patients with inflammatory bowel disease. Inflamm Bowel Dis. 2010;16:1696–1707. | |
Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. | |
Wright R, Truelove SC. A controlled therapeutic trial of various diets in ulcerative colitis. Br Med J. 1965;2:138–141. | |
Fell JM. Control of systemic and local inflammation with transforming growth factor beta containing formulas. J Parenter Enteral Nutr. 2005;29:S126–S128. | |
Richman E, Rhodes JM. Review article: evidence-based dietary advice for patients with inflammatory bowel disease. Aliment Pharmacol Ther. 2013;38(10):1156–1171. | |
Turner D, Shah PS, Steinhart AH, Zlotkin S, Griffiths AM. Maintenance of remission in inflammatory bowel disease using omega-3 fatty acids (fish oil): a systematic review and meta-analyses. Inflamm Bowel Dis. 2011;17:336–345. | |
Uchiyama K, Nakamura M, Odahara S, et al. N-3 polyunsaturated fatty acid diet therapy for patients with inflammatory bowel disease. Inflamm Bowel Dis. 2010;16:1696–1707. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.