")
Back to Journals » Journal of Inflammation Research » Volume 15
Induction of Trained Immunity Protects Neonatal Mice Against Microbial Sepsis by Boosting Both the Inflammatory Response and Antimicrobial Activity
Authors Zhou H, Lu X, Huang J, Jordan P, Ma S, Xu L, Hu F, Gui H, Zhao H, Bai Z, Redmond HP, Wang JH, Wang J
Received 26 March 2022
Accepted for publication 21 June 2022
Published 7 July 2022 Volume 2022:15 Pages 3829—3845
DOI https://doi.org/10.2147/JIR.S363995
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Ning Quan
Huiting Zhou,1,* Xiaying Lu,2,3,* Jie Huang,1,* Patrick Jordan,2 Shurong Ma,1 Lingqi Xu,1 Fangjie Hu,1 Huan Gui,1 He Zhao,1 Zhenjiang Bai,1 H Paul Redmond,2 Jiang Huai Wang,2 Jian Wang1
1Institute of Pediatric Research, Children’s Hospital of Soochow University, Suzhou, People’s Republic of China; 2Department of Academic Surgery, University College Cork, Cork University Hospital, Cork, Ireland; 3Department of Physiology, Gannan Medical University, Ganzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jian Wang, Institute of Pediatric Research, Children’s Hospital of Soochow University, Suzhou, People’s Republic of China, Email [email protected] Jiang Huai Wang, Department of Academic Surgery, University College Cork, Cork University Hospital, Cork, Ireland, Email [email protected]
Background: Neonates are susceptible to a wide range of microbial infection and at a high risk to develop severe sepsis and septic shock. Emerged evidence has shown that induction of trained immunity triggers a much stronger inflammatory response in adult monocytes/macrophages, thereby conferring protection against microbial infection.
Methods: This study was carried out to examine whether trained immunity is inducible and exerts its protection against microbial sepsis in neonates.
Results: Induction of trained immunity by Bacillus Calmette-Guerin (BCG) plus bacterial lipoprotein (BLP) protected neonatal mice against cecal slurry peritonitis-induced polymicrobial sepsis, and this protection is associated with elevated circulating inflammatory cytokines, increased neutrophil recruitment, and accelerated bacterial clearance. In vitro stimulation of neonatal murine macrophages with BCG+BLP augmented both inflammatory response and antimicrobial activity. Notably, BCG+BLP stimulation resulted in epigenetic remodeling characterized by histone modifications with enhanced H3K4me3, H3K27Ac, and suppressed H3K9me3 at the promoters of the targeted inflammatory and antimicrobial genes. Critically, BCG+BLP stimulation led to a shift in cellular metabolism with increased glycolysis, which is the prerequisite for subsequent BCG+BLP-triggered epigenetic reprogramming and augmented inflammatory response and antimicrobial capacity.
Conclusion: These results illustrate that BCG+BLP induces trained immunity in neonates, thereby protecting against microbial infection by boosting both inflammatory and antimicrobial responses.
Keywords: trained immunity, inflammatory response, antimicrobial activity, epigenetic reprogramming, intracellular metabolic rewiring, neonatal sepsis
Introduction
Despite significant advances in critical care medicine and the best available supportive treatment, death associated with neonatal and infant sepsis has remained largely unchanged worldwide over the last several decades.1,2 Neonatal sepsis kills more than 1 million newborns each year and mortality rates associated with neonatal sepsis are substantially higher than those in children and adults.1,3 Microbial sepsis-related mortality rates in premature birth and very low-birth-weight infants are higher still, and the incidence could reach up to 50%.4 Even in full-term infants, their inefficient immune response to microbial pathogens not only predisposes but renders them more susceptible to infection.2,3 Moreover, neonates and infants who survive severe sepsis may undergo developmental and growth impairment, leading to long-term social and economic consequences.1–3
Neonates have well-described deficits in both innate and adaptive immunity, thereby making them more vulnerable to a wide range of microbial infection and at a high risk to develop severe sepsis and septic shock.2,4 Neonates experience very little exposure to antigens in utero, resulting in immaturity in their adaptive immunity during infancy.5,6 Accumulated evidence has revealed a number of deficiencies of adaptive immunity in neonates and infants for both cell- and antibody-mediated responses.2,6,7 Due to the immature state of adaptive immunity that favors tolerance and contributes little to host defense mechanism against microbial infection, neonates rely mainly on their innate immunity.2,8 However, in spite of a substantial dependence on innate immunity, neonates exhibit an insufficient innate response to microbial infection including reduced inflammatory cytokine production, diminished neutrophil antimicrobial activities, delayed dendritic cell maturation, and impaired NK cell activation.8,9 Our previous work found that neonate and infant polymorphonuclear neutrophils (PMNs) exhibited defective in vitro chemotaxis and in vivo recruitment, while their macrophages displayed delayed phagosome maturation and impaired bacterial killing.10 Furthermore, studies on transcriptomics have revealed that the early inflammatory response to microbial infection in neonates is less effective and vigorous in comparison with children and adults.2,11 Thus, neonates show defects in protective immune inflammatory responses to microbial infection, which may account for their reduced ability to clear infections and the increased risk in developing microbial sepsis.
The host immune response is traditionally divided into the innate and adaptive immune systems. The adaptive immune response is slower to develop but is highly specific due to antigen gene rearrangement, whereas the innate immune response reacts rapidly in a nonspecific and identical manner upon encountering a pathogen but lacks an ability to develop an immunological memory. However, emerged evidence that innate immune cells can exert adaptive characteristics and are able to build a nonspecific memory has challenged this concept.12 Notably, this phenomenon characterized by a newly built memory phenotype of innate immunity with an increased inflammatory response upon re-infection is also confirmed in mammals and humans, and a series of in vitro and in vivo experiments has demonstrated that this specific memory phenotype can be induced by several pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs).13–19 Ex vivo stimulation with β-glucan enhanced production of proinflammatory cytokines to a second stimulus in both mouse macrophages and human monocytes,13–15 whereas an in vivo challenge with β-glucan protected mice against subsequent Candida albicans (C. albicans) or Staphylococcus aureus (S. aureus) infection.13,16 Furthermore, stimulation with Bacillus Calmette-Guerin (BCG) led to a boosted inflammatory response with increased TNF-α and IL-6 release upon a secondary stimulus and protected against pulmonary tuberculosis and viral infection.17–19 Together, innate immune cells can adapt and build up a memory after a previous challenge and produce a much stronger immune response when encountering a second stimulation. This nonspecific immunological memory is termed as trained immunity or innate immune memory.12,20
Accumulated evidence has revealed that trained immunity is predominantly dependent on both epigenetic reprogramming and intracellular metabolic rewiring, which leads to a long-term memory phenotype of innate immunity.12,20 Several studies have shown that induction of trained immunity is fundamentally based on epigenetic remodeling, in particular histone modification with chromatin reconfiguration.13,14,20 Among histone modifications, H3K4me1, H3K4me3, and H3K27Ac denote positive marks associated with open or active chromatin, whereas H3K9me3 is a repressor mark associated with closed or inactive chromatin. Consequently, increased H3K4me1, H3K4me3, H3K27Ac, and/or suppressed H3K9me3 at the promoter of inflammatory cytokine genes are evident in β-glucan-, BCG- or C albicans-stimulated mouse macrophages and human monocytes with trained immunity.13,14,17,20 The shift in cellular metabolism is an essential driver in switching immune cell phenotype and activation, and acts as an important hallmark for induction of different inflammatory phenotypes in innate immunity. Notably, studies have shown that trained mouse macrophages and human monocytes display altered intracellular metabolic pathways with substantial increases in glycolysis, glutaminolysis, and cholesterol synthesis.15,17,21,22 Moreover, a shift of cellular metabolism in trained innate immune cells leads to accumulated metabolic intermediates and several metabolites including α-ketoglutarate, succinate, and fumarate have been found to participate in epigenetic remodeling by affecting histone modifications.20,22–24 Collectively, trained immunity is originated from two essential intracellular events, namely epigenetic reprogramming of gene transcription and shifts in intracellular metabolic pathways, ultimately resulting in protection against secondary infections.
In the present study, we examined whether trained immunity is inducible and exerts its protection against microbial sepsis in neonates. Here we report that BCG in combination with bacterial lipoprotein (BLP) induced trained immunity in neonatal mice, thereby conferring robust protection against cecal slurry peritonitis-induced polymicrobial sepsis. In vitro BCG+BLP boosted both the inflammatory response and antimicrobial capacity in neonatal murine macrophages with increased inflammatory cytokine and chemokine production and augmented bactericidal activity. Notably, BCG+BLP initiated epigenetic reprogramming with upregulated H3K4me3, H3K27Ac, and downregulated H3K9me3 at the promoters of the targeted genes. Importantly, BCG+BLP resulted in altered intracellular metabolic pathways with increased glycolysis, which contributes to subsequent BCG+BLP-triggered epigenetic remodeling and boosted both inflammatory and antimicrobial responses.
Materials and Methods
Reagents, Antibodies, Bacteria
The following are the reagents, antibodies, and bacteria used in the experiment: BLP (EMC Microcollections, Tubingen, Germany), BCG (Chengdu Institute Biological Products Co. Ltd, Chengdu, China), UNC1999 (Sigma-Aldrich, St. Louis, MO), C646 (Sigma-Aldrich), 2-deoxy-D-glucose (2-DG) (Sigma-Aldrich), anti-NF-κB p65 antibody (Santa Cruz Biotechnology, Santa Cruz, CA), anti-H3K4me3 antibody (Active Motif, Carlsbad, CA), anti-H3K27Ac antibody (Diagenode, Denville, NJ), anti-H3K9me3 antibody (Active Motif), S. aureus (American Type Culture Collection, Manassas, VA), and Salmonella typhimurium (S. typhimurium) (American Type Culture Collection).
Mice, Polymicrobial Sepsis
Pyrogen-free, 5- to 7-day-old neonatal C57BL/6 mice were obtained from JOINN Laboratories (Suzhou, China). All animal experiments were approved by the Soochow University Institutional Animal Care and Use Committee and complied with the Animal Welfare Act. All procedures were performed according to its guidelines and regulations. Neonatal mice were injected i.p. with PBS as the control, BCG alone (250 µg/g body weight), BLP alone (5 µg/g body weight), or BCG plus BLP (250 µg + 5 µg/g body weight) and rested for 3 days to induce trained immunity in vivo. These mice were further subjected to cecal slurry peritonitis-induced mid-grade or high-grade polymicrobial sepsis as previously described.10,25 Briefly, cecal contents of adult C57BL/6 mice were suspended in 5% dextrose solution (Sigma-Aldrich) with a final concentration of 80 mg/mL. The cecal slurry was briefly vortexed before injection to create a homogenous suspension and was used within 2 h of preparation. Neonatal mice received an i.p. injection of the cecal content suspension at 0.85 mg/g body weight or 1.25 mg/g body weight to induce a mid-grade or high-grade polymicrobial sepsis model with an overall mortality rate at approximately 35% or 85%. Survival was monitored for at least 7 days.
Assessment of Bacteria Loads in the Blood and Visceral Organs
Bacterial counts in the blood, liver, spleen, and lungs were assessed as previously described.26,27 Briefly, neonatal mice were culled at different time points after cecal slurry peritonitis-induced polymicrobial sepsis. The heparinized blood samples and homogenized visceral organs were diluted in sterile water containing 0.5% Triton X-100 (Sigma-Aldrich) and incubated on brain heart infusion agar (BD Biosciences, San Jose, CA) for 24 h at 37°C to determine bacterial CFU.
Assessment of Peritoneal PMN and Macrophage Subpopulations
Peritoneal lavage was harvested before and after cecal slurry peritonitis-induced polymicrobial sepsis. Peritoneal exudate cells were incubated with FITC, PE, PerCP, or APC-conjugated anti-CD11b (BD Pharmingen, San Diego, CA), anti-CD11c (BioLegend, San Diego, CA), anti-F4/80 (BD Pharmingen), and anti-Gr1 (BioLegend) monoclonal antibodies. Subpopulations of PMNs (CD11b+F4/80−Gr1hi) and macrophages (CD11b+F4/80+CD11clo) in the peritoneal cavity were analyzed by flow cytometry (BD Biosciences).
Measurement of Serum Cytokines
Heparinized whole-blood samples were collected at different time points after cecal slurry peritonitis-induced polymicrobial sepsis. Serum TNF-α and IL-6 were measured by ELISA (R&D Systems, Minneapolis, MN) according to the manufacturer’s instruction.
Isolation and Training of Murine Macrophages
Neonatal murine peritoneal macrophages were collected, pooled, and incubated in 96- or 24-well plates (Falcon, Lincoln Park, NJ) for 90 min to remove non-adherent cells as previously described.28,29 Neonatal murine bone marrow-derived macrophages (BMMs) were isolated from the femurs and cultured in DMEM containing 20% heat-inactivated FCS, 100 units/mL penicillin, 100 μg/mL streptomycin sulfate, and 10 ng/mL recombinant mouse macrophage-CSF (R&D Systems) for 7 days as previously described.28,29 Isolated neonatal murine peritoneal macrophages or BMMs were stimulated with BCG (5 µg/mL), BLP (100 ng/mL), or BCG plus BLP (5 µg/mL + 100 ng/mL) for 24 h, washed with PBS, and rested in fresh complete culture medium for additional 3 days to induce trained immunity in vitro. These macrophages were further infected with S. aureus or S. typhimurium to determine inflammatory cytokine and chemokine release, bactericidal activity, epigenetic reprogramming, and shifts in cellular metabolism. Neonatal murine peritoneal macrophages or BMMs were also treated with the histone methyltransferase inhibitor UNC1999 (200 nM), histone acetyltransferase inhibitor C646 (100 nM), and 2-DG (1 mM) 1 h before incubation with PBS or stimulation with BCG plus BLP.
Measurement of Inflammatory Cytokines and Chemokines
Isolated neonatal murine peritoneal macrophages were seeded in 96-well plates (Falcon) and stimulated with BCG, BLP, or BCG plus BLP for 24 h. After rested for additional 3 days as described above, these macrophages were further challenged with heat-killed S. aureus or S. typhimurium (bacteria:macrophage = 50:1) for 18 h. Concentrations of TNF-α, IL-6, IL-12p70, and CXCL2 in the supernatants were measured by ELISA (R&D Systems).
Assessment of Bacterial Phagocytosis and Killing
Bacterial phagocytosis and intracellular bacterial killing were determined as previously described.30,31 Briefly, isolated neonatal murine peritoneal macrophages were stimulated with BCG, BLP, or BCG plus BLP for 24 h, rested for additional 3 days as described above, and further challenged with heat-killed, FITC-labeled S. aureus and S. typhimurium (bacteria:macrophage = 20:1) for 60 min to assess bacterial phagocytosis or live S. aureus and S. typhimurium (bacteria:macrophage = 20:1) for 2 h in the presence or absence of cytochalasin B (5 µg/mL) (Sigma-Aldrich) to assess bacterial killing. Bacterial phagocytosis was analyzed by flow cytometry (BD Biosciences) after the external fluorescence of the bound, but non-ingested, bacteria was quenched with 0.025% crystal violet (Sigma-Aldrich). Intracellular bacterial killing was calculated based on the total and extracellular bacterial killings, and both total and extracellular bacterial killings were determined by incubation of serial 10-fold dilutions of macrophage lysates on tryptone soy agar (Merck) plates at 37°C for 24 h.
Determination of Phagosome Luminal pH
Phagosomal pH was determined as previously described.27,32 Briefly, heat-killed S. aureus and S. typhimurium were labeled with both a pH-sensitive fluorescent probe, carboxyfluorescein-SE (5 µg/mL) (Molecular Probes, Eugene, OR) and a pH-insensitive fluorescent probe, carboxytetramethylrhodamine-SE (10 µg/mL) (Molecular Probes). After being stimulated with BCG, BLP, or BCG plus BLP for 24 h and rested for additional 3 days as described above, isolated neonatal murine peritoneal macrophages were incubated with the labeled S. aureus and S. typhimurium (bacteria:macrophage = 20:1) for different time points. The fluorescein- and rhodamine-generated mean fluorescence intensity (MFI) on fluorescent channel 1 and 2 was analyzed by flow cytometry (BD Biosciences). Phagosome luminal pH was determined based on the MFI ratio generated by fluorescein and rhodamine.
Measurement of Phagosome Maturation in a Cell-Free Organelle System
After being stimulated with BCG, BLP, or BCG plus BLP for 24 h and rested for additional 3 days as described above, isolated neonatal murine peritoneal macrophages were labeled with 20 µM PKH26 (Sigma-Aldrich), a red fluorescent cell membrane linker, for subsequent phagosome recognition as previously described.27,33 PKH26-labeled macrophages were incubated with heat-killed bacteria (bacteria:macrophage = 20:1) for different time points and lysed to isolate the phagosome. After being permeabilized with 0.2% saponin (Sigma-Aldrich), isolated phagosomes were stained with FITC-conjugated anti-LAMP-1 monoclonal antibody (Abcam, Cambridge, MA) that specifically recognizes late endosomes/lysosomes. Phagosome maturation, as represent by LAMP-1-generated green fluorescence on the red fluorescent phagosomes that have phagocytosed bacteria, was analyzed by flow cytometry (BD Biosciences).
Chromatin Immunoprecipitation (ChIP)
Isolated neonatal murine BMMs were stimulated with BCG, BLP, or BCG plus BLP for 24 h and rested for additional 3 days as described above. Cells were then washed with PBS, fixed with 1% formaldehyde, and sonicated to obtain 200–700 bp DNA fragments. ChIP was performed by incubation of the obtained DNA fragments with antibodies against NF-κB p65, H3K4me3, H3K27Ac, and H3K9me3 at 4°C overnight. The chromatin/antibody complex was mixed with protein A/G magnetic beads (Active Motif) and rotated for 2 h. Protein–DNA complexes were eluted from the beads, and further incubated with 200 mM NaCl and treated with proteinase K to reverse cross-links and to digest proteins. Immunoprecipitated DNA and non-immunoprecipitated DNA (input control) were amplified with gene-specific primers by quantitative PCR (qPCR).
Measurement of Glucose, Lactate, OCR, ECAR, and Metabolites
Isolated neonatal murine peritoneal macrophages were stimulated with BCG, BLP, or BCG plus BLP for 24 h and rested for additional 3 days as described above, and further challenged with heat-killed bacteria (bacteria:macrophage = 50:1) for different time points. Concentrations of lactate, glucose, glutamate, and acetyl-CoA were assessed using the fluorometric quantification assay kit (BioVision, Milpitas, CA), glutamate colorimetric assay kit (Abcam), and acetyl-coenzyme fluorometric assay kit (Sigma-Aldrich), respectively. Extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) were measured as previously described.17,34 Briefly, isolated neonatal murine macrophages were stimulated with BCG, BLP, or BCG plus BLP as described above. After rested in fresh culture medium for 3 days, macrophages plated onto overnight-calibrated cartridges were incubated in RPMI 1640 medium supplemented with glutamine (0.6 mM), glucose (5 mM), and pyruvate (1 mM), pH 7.4) at 37°C for 1 h. ECAR and OCR were measured using the glycolysis stress test kit (Seahorse Bioscience, Chicopee, MA) and cell mito stress kit (Seahorse Bioscience), respectively.
Statistical Analyses
A log rank test was used for survival analysis, the analysis of variance (ANOVA) or Mann–Whitney U-test were used for all others, and all results expressed as the mean ± standard deviation (SD). GraphPad Prism 8 (GraphPad Software, La Jolla, CA) was used to perform the statistical analysis, and p<0.05 was considered statistically significant.
Results
Induction of Trained Immunity in vivo by BCG+BLP Protects Neonatal Mice Against Microbial Sepsis
To ascertain whether BCG+BLP-induced trained immunity confers in vivo protection against microbial sepsis-associated lethality, neonatal mice received PBS as the control or BCG alone, BLP alone, and BCG plus BLP to induce trained immunity, and these mice were then rested for 3 days and further challenged with cecal slurry peritonitis-induced mid-grade or high-grade polymicrobial sepsis. Treatment with BCG alone and BLP alone enhanced the survival in neonatal mice challenged with mid-grade sepsis from 35% seen in PBS-treated mice to 50% and 61%, respectively, but did not reach the statistically significant differences, whereas treatment with BCG+BLP afforded maximal protection with an overall survival at 89% (p=0.0011) (Figure 1A). Furthermore, treatment with BCG+BLP rescued 45% neonatal mice from high-grade sepsis compared with a 6% survival rate in PBS-treated mice (p=0.002); by contrast, neither BCG alone nor BLP alone was able to provide any survival benefit (Figure 1B). We next examined whether BCG+BLP-induced trained immunity in vivo boosts both inflammatory and antimicrobial responses as represented by systemic proinflammatory cytokine release, bacterial clearance in the circulation and visceral organs, and PMN recruitment in the infected site. Treatment with BCG+BLP resulted in substantially increased serum levels of proinflammatory cytokines TNF-α and IL-6 upon septic challenge compared to PBS-, BCG-, and BLP-treated mice (p<0.05, p<0.01) (Figure 1C). Importantly, treatment with BCG+BLP significantly diminished bacterial loads in the circulation and visceral organs including the liver, spleen, and lungs compared to PBS-, BCG-, and BLP-treated mice (p<0.05) (Figure 1D), indicating an accelerated bacterial clearance. By contrast, treatment with BCG alone only slightly enhanced serum TNF-α and IL-6 but had no effect on bacterial clearance, whereas BLP alone partially reduced bacterial counts but did not increase circulating proinflammatory cytokines (Figure 1C and D). Recruitment of circulating PMNs to the infectious site during microbial infection is a critical step for host to eradicate the invaded microbial pathogens from the body, and successful clearance of microbial infection relies predominantly on a fast PMN influx into the infected site.35,36 Markedly increased PMN subpopulation, but not the macrophage subpopulation, in the peritoneal cavity, the infected site was observed in BCG+BLP-treated mice at 6 and 12 h after septic challenge compared to PBS-, BCG-, and BLP-treated mice (p<0.01) (Figure 1E). Collectively, these results indicate that BCG+BLP-induced trained immunity protects neonatal mice against microbial sepsis via augmentations in both inflammatory and antimicrobial responses.
 |
Figure 1 Trained immunity induced by BCG+BLP protects neonatal mice against microbial sepsis. Neonatal mice were injected with BCG, BLP, or BCG+BLP and rested for 3 days to induce trained immunity, and then challenged with cecal slurry peritonitis-induced polymicrobial sepsis. (A and B) Substantially improved survival in BCG+BLP-treated mice subjected to either mid-grade sepsis (p=0.0011) (A) or high-grade sepsis (p=0.002) (B) (n=18 per group) compared to PBS-treated mice (n=17). (C) Data shown are the results of serum TNF-α and IL-6 levels at 0, 2, and 6 h post septic challenge. (D) Bacterial counts in the blood, liver, spleen, and lungs assessed at 12 and 24 h post septic challenge. (E) Data shown are subpopulations (%) of PMNs (CD11b+F4/80−Gr1hi) and macrophages (CD11b+F4/80+CD11clo) in the peritoneal lavage collected at 0, 6, and 12 h post septic challenge. Data in (C–E) are mean ± SD (4–5 mice per time point) and representative of three independent experiments. *p<0.05, **p<0.01 versus PBS-treated mice; ≠p<0.05, ≠≠p<0.01 versus BCG- or BLP-treated mice. |
Induction of Trained Immunity in vitro by BCG+BLP Boosts Both the Inflammatory Response and Antimicrobial Capacity
By functioning as the main participants in innate immunity-triggered inflammatory responses, both inflammatory cytokines and chemokines are critically involved in the host defense against microbial infection.37,38 We first assessed whether stimulation of neonatal murine macrophages with BCG+BLP triggers augmented inflammatory cytokine and chemokine production. Stimulation with BCG+BLP resulted in a robust release of proinflammatory cytokines TNF-α, IL-6, and IL12-p70 as well as chemokine CXCL2 from neonatal macrophages upon S. aureus or S. typhimurium challenge compared to PBS-, BCG-, and BLP-stimulated macrophages (p<0.01), while stimulation with BCG alone, but not BLP alone, to a less extent also increased TNF-α, IL-6, IL12-p70, and CXCL2 release compared to PBS-stimulated macrophages (p<0.05, p<0.01) (Figure 2A). Consistent with an increased inflammatory cytokine and chemokine release, stimulation of neonatal murine macrophages with BCG+BLP substantially enhanced NF-κB p65 nuclear transactivation at the promoters of TNF-α, IL-6, IL12-p70, and CXCL2 upon S. aureus or S. typhimurium challenge compared to PBS-, BCG-, and BLP-stimulated macrophages (p<0.01) (Figure 2B). A slightly increased NF-κB p65 recruitment at the promoters of TNF-α, IL-6, IL12-p70, and CXCL2 was also seen in BCG-stimulated macrophages (p<0.05) (Figure 2B).
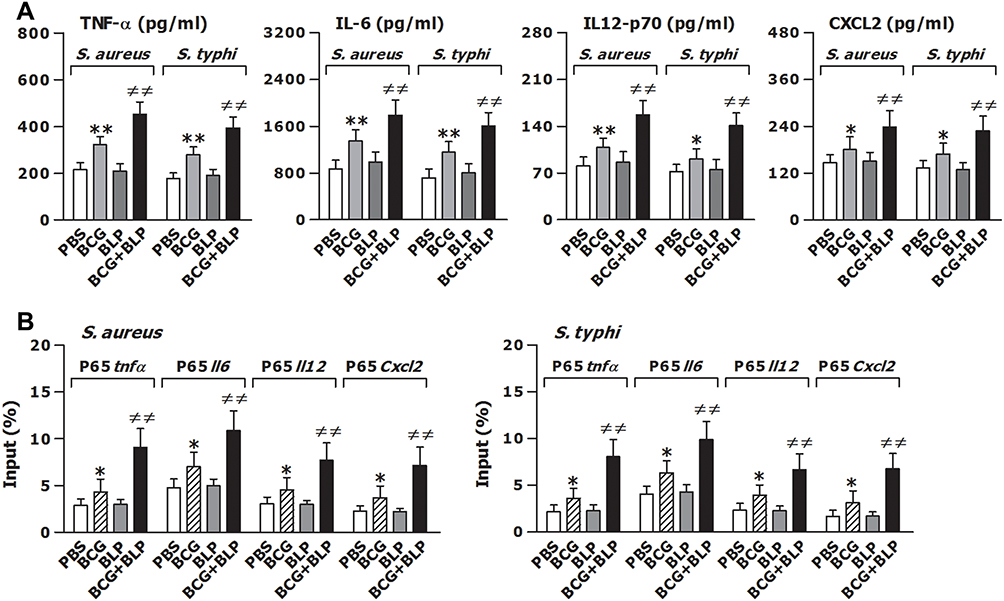 |
Figure 2 Training innate immunity by BCG+BLP enhances nuclear transactivation of NF-κB p65 and boosts inflammatory cytokine and chemokine release. Isolated neonatal murine peritoneal macrophages or BMMs were stimulated with BCG, BLP, or BCG+BLP for 24 h, rested for additional 3 days, and further challenged with heat-killed bacteria. (A) TNF-α, IL-6, IL-12p70, and CXCL2 in the supernatants were measured 18 h post bacterial challenge. (B) The binding of p65 to TNF-α, IL-6, IL-12p70, and CXCL2 promoters were measured 1 h post bacterial challenge. Data are mean ± SD from four to six separate experiments. *p<0.05, **p<0.01 versus PBS-incubated macrophages; ≠≠p<0.01 versus BCG- or BLP-stimulated macrophages. |
Host innate immunity-associated antimicrobial response is triggered by the pattern-recognition receptor (PRR)-associated detection of microbial pathogens and subsequently, the invaded pathogens are ingested by the professional phagocytes and killed through a process of phagosome/lysosome fusion.39,40 We first assessed whether stimulation of neonatal murine macrophages with BCG+BLP potentiates innate phagocyte-associated bactericidal activity. Stimulation with BCG+BLP significantly enhanced phagocytosis of S. aureus and S. typhimurium (Figure 3A) with substantially increased intracellular killing of the engulfed both bacteria (Figure 3B) in neonatal macrophages compared to PBS-, BCG-, and BLP-stimulated macrophages (p<0.01), indicating an augmentation in bactericidal capability. After engulfment of bacteria, phagosome maturation of innate phagocytes is a critical event for the killing and destruction of the ingested bacteria, thus playing an important role in host defense against bacterial infection.39–41 We further assessed whether stimulation of neonatal murine macrophages with BCG+BLP accelerates phagosome maturation by assessing both phagosomal acidification and phagolysosome fusion. A significant acceleration in phagosomal acidification after engulfment of S. aureus or S. typhimurium was observed in BCG+BLP-stimulated neonatal macrophages compared to PBS-, BCG-, and BLP-stimulated macrophages (p<0.01) (Figure 3C). Neonatal macrophages stimulated by BCG+BLP also displayed substantially enhanced phagolysosome fusion after engulfment of S. aureus or S. typhimurium compared to PBS-, BCG-, and BLP-stimulated macrophages (p<0.05, p<0.01) (Figure 3D). Stimulation with BLP alone enhanced antimicrobial capacity in neonatal macrophages with slightly increased bactericidal activity and accelerated phagosome maturation (p<0.05) (Figure 3).
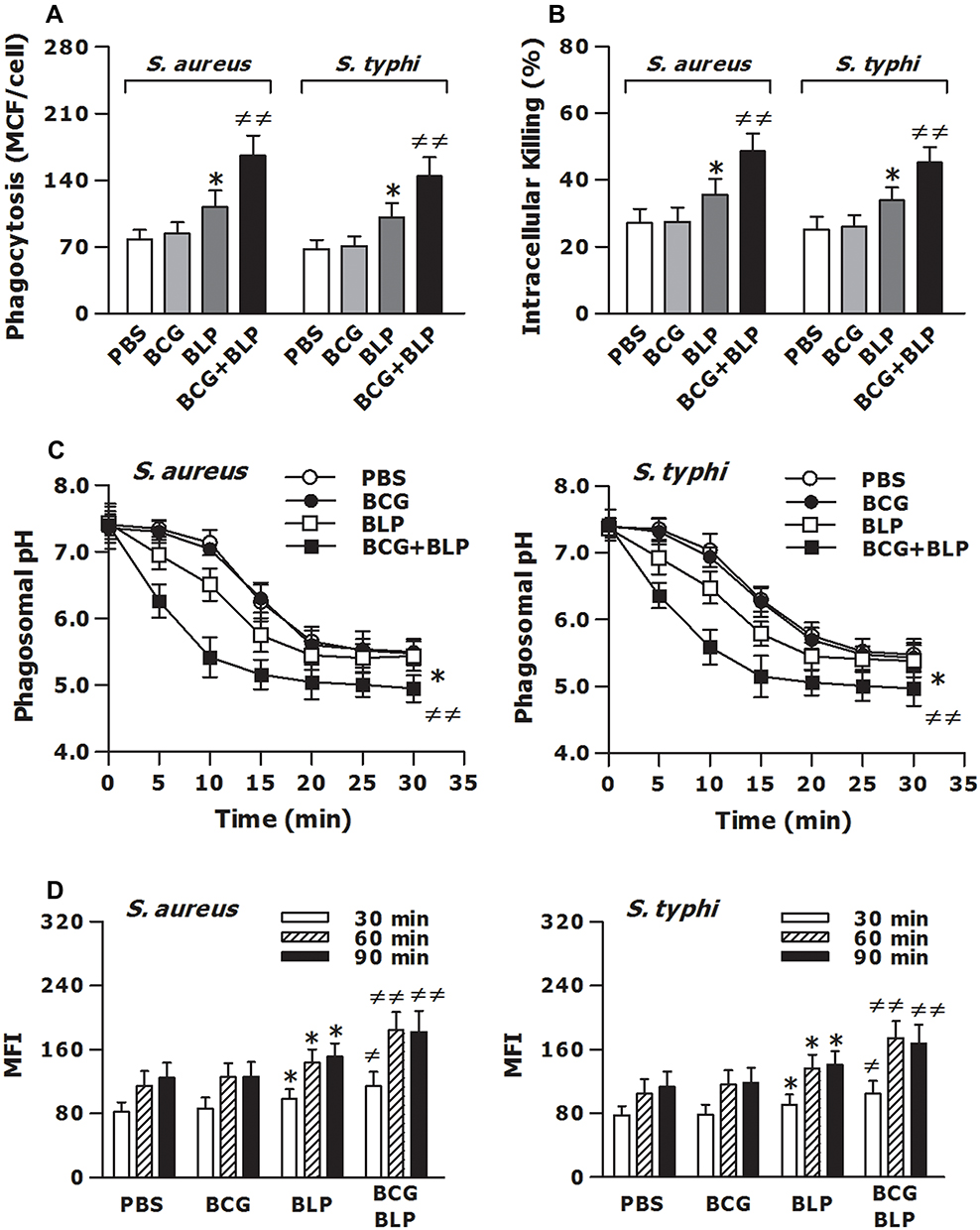 |
Figure 3 Training innate immunity by BCG+BLP accelerates phagosome maturation and augments phagocyte-associated bactericidal activity. Isolated neonatal murine peritoneal macrophages were stimulated with BCG, BLP, or BCG+BLP for 24 h and rested for additional 3 days. (A) After being incubated with heat-killed, FITC-conjugated S. aureus or S. typhi for 60 min, bacterial phagocytosis was assessed in neonatal macrophages. (B) After being challenged with live S. aureus or S. typhi for 2 h, intracellular killing of the ingested bacteria was assessed in neonatal macrophages. (C) After being incubated with fluorescent probe-labeled S. aureus or S. typhi for different time points, phagosomal pH was assessed in neonatal macrophages. (D) After being incubated with heat-killed S. aureus or S. typhi for 30, 60, and 90 min, phagolysosome fusion was assessed in neonatal macrophages. Data are mean ± SD from five to six separate experiments in duplicate or triplicate. *p<0.05 versus PBS-incubated macrophages; ≠p<0.05, ≠≠p<0.01 versus BCG- or BLP-stimulated macrophages. |
Histone Modification-Associated Epigenetic Reprogramming is Responsible for BCG+BLP-Boosted Both Inflammatory and Antimicrobial Responses
It has been shown that induction of trained immunity is primarily dependent on epigenetic remodeling and specifically, histone modification with chromatin reconfiguration is the central process for trained immunity.13,14,20 We first asked whether stimulation of neonatal murine macrophages with BCG+BLP induces epigenetic reprogramming characterized by histone modifications at the promoters of the targeted genes. We selected TNF-α and IL-6 as the inflammatory response genes, and Acp5 and Camp as the antimicrobial effector genes. Stimulation of neonatal macrophages with BCG+BLP strongly upregulated H3K4me3 and H3K27Ac, two positive marks associated with transcriptionally active chromatin, not only at the promoters of TNF-α and IL-6 (Figure 4A and C) but also at the promoters of Acp5 and Camp (Figure 4B and D) (p<0.01 versus PBS-, BCG-, and BLP-stimulated macrophages). Consistently, stimulation with BCG+BLP markedly downregulated H3K9me3, a repressor mark associated with inactive chromatin, at the promoters of TNF-α and IL-6 (Figure 4E) as well as at the promoters of Acp5 and Camp (Figure 4F) in neonatal macrophages (p<0.01 versus PBS-, BCG-, and BLP-stimulated macrophages). By contrast, BCG alone-induced upregulation of H3K4me3 and H3K27Ac and downregulation of H3K9me3 were only occurred at the promoters of TNF-α and IL-6, while BLP alone-triggered alterations in histone modifications were only observed at the promoters of Acp5 and Camp (p<0.05, p<0.01) (Figure 4).
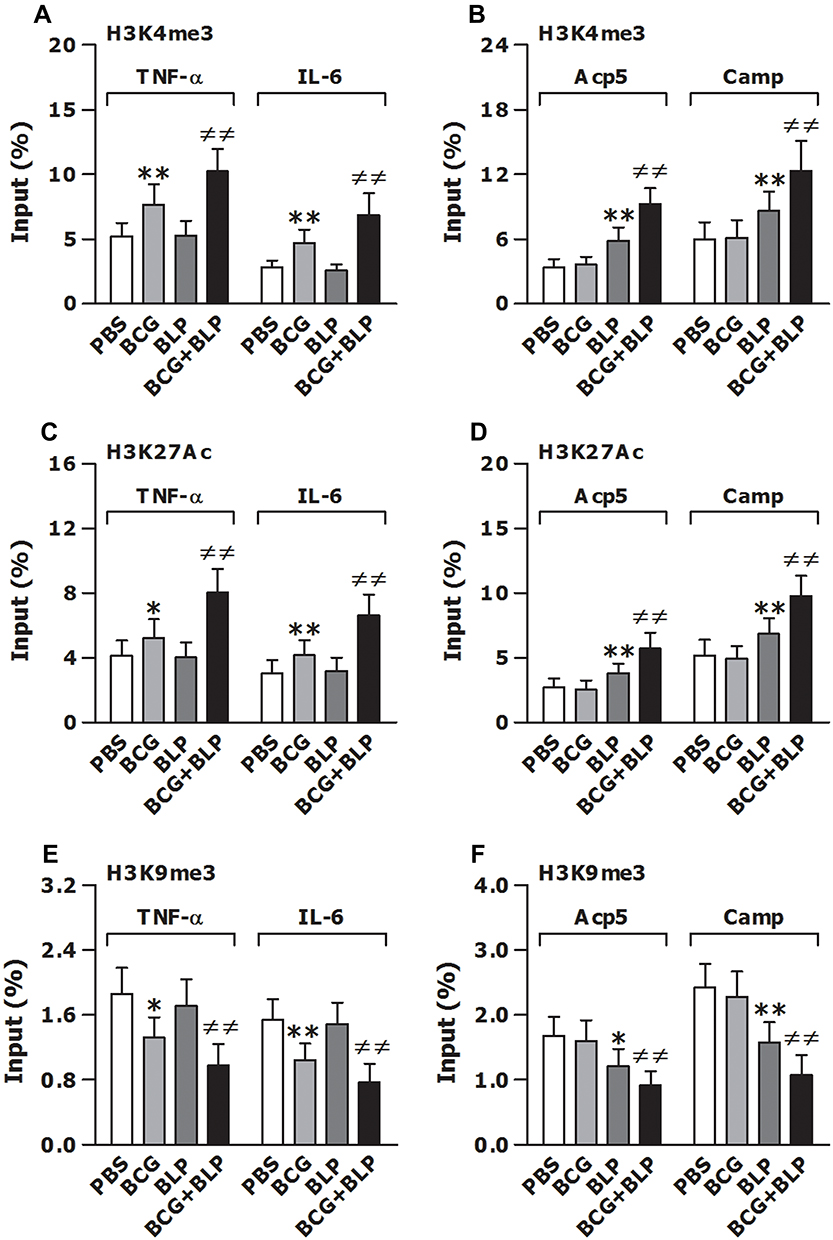 |
Figure 4 Training innate immunity by BCG+BLP induces epigenetic reprogramming with histone modifications at the promoters of TNF-α, IL-6, Acp5, and Camp. Isolated neonatal murine BMMs were stimulated with BCG, BLP, or BCG+BLP for 24 h and rested for additional 3 days. H3K4me3 (A and B), H3K27Ac (C and D), and H3K9me3 (E and F) at the promoters of inflammatory response genes TNF-α and IL-6 (A, C, E) and antimicrobial effector genes Acp5 and Camp (B, D, F) were assessed in neonatal macrophages. Data are mean ± SD from five separate experiments in duplicate. *p<0.05, **p<0.01 versus PBS-incubated macrophages; ≠≠p<0.01 versus BCG- or BLP-stimulated macrophages. |
We further asked whether histone modification-associated epigenetic reprogramming is responsible for BCG+BLP-boosted both inflammatory and antimicrobial responses. To test this, we examined whether blockage of histone modifications with their specific inhibitors in neonatal murine macrophages abrogates BCG+BLP-induced epigenetic signatures of histone modifications, namely upregulated H3K4me3, H3K27Ac, and downregulated H3K9me3, thereby eradicating BCG+BLP-augmented inflammatory response and bactericidal activity. Pretreatment of neonatal macrophages with UNC1999, a histone methyltransferase inhibitor, or C646, an inhibitor of histone acetyltransferases, strongly attenuated BCG+BLP-triggered upregulation of H3K4me3 or H3K27Ac at the promoters of TNF-α, IL-6, Acp5, and Camp (p<0.01 versus DMSO-pretreated and BCG+BLP-stimulated macrophages) (Figure 5A). Remarkably, pretreatment with a combination of UNC1999 and C646 effectively prevented BCG+BLP-boosted TNF-α, IL-6, IL12-p70, and CXCL2 release from neonatal macrophages (p<0.01 versus DMSO-pretreated and BCG+BLP-stimulated macrophages) (Figure 5B). Moreover, pretreatment with UNC1999 plus C646 substantially abrogated BCG+BLP-augmented bactericidal activity as evidenced by markedly diminished bacterial phagocytosis and intracellular killing, and BCG+BLP-accelerated phagosome maturation as represented by significantly delayed phagosomal acidification and phagolysosome fusion (p<0.01 versus DMSO-pretreated and BCG+BLP-stimulated macrophages) (Figure 5C).
 |
Figure 5 Blockage of histone modifications abrogates BCG+BLP-boosted inflammatory response and bactericidal activity. Isolated neonatal murine peritoneal macrophages or BMMs were pretreated with DMSO, UNC1999 (UNC), C646, or UNC1999+C646 (Inhibitors) for 1 h before training of innate immunity. (A) H3K4me3 and H3K27Ac at the promoters of TNF-α, IL-6, Acp5, and Camp were assessed in neonatal macrophages before bacterial challenge. (B) TNF-α, IL-6, IL-12p70, and CXCL2 in the supernatants were measured 18 h post bacterial challenge. (C) Bacterial phagocytosis and intracellular killing were assessed in neonatal macrophages 60 min and 2 h after being incubated with FITC-conjugated or live S. aureus. Phagosomal pH and phagolysosome fusion were assessed in neonatal macrophages after being incubated with fluorescent probe-labeled or heat-killed S. aureus. Data are mean ± SD from five to six separate experiments in duplicate or triplicate. **p<0.01 versus DMSO-pretreated, PBS-incubated macrophages; ≠p<0.05, ≠≠p<0.01 versus DMSO-pretreated, BCG+BLP-stimulated macrophages. |
Shifts in Intracellular Metabolic Pathways Contribute to BCG+BLP-Triggered Epigenetic Remodeling and Boosted Both Inflammatory and Antimicrobial Responses
We first assessed whether stimulation of neonatal murine macrophages with BCG+BLP induces a metabolic phenotype characterized by altered glucose metabolism, glutaminolysis, and cholesterol synthesis. We selected glucose consumption, lactate secretion, ECAR, and OCR to reflect glucose metabolism, and glutamate and acetyl-CoA to reflect glutaminolysis and cholesterol synthesis, respectively. Stimulation with BCG+BLP resulted in substantial elevations in glucose consumption (Figure 6A) and lactate secretion (Figure 6B) in neonatal macrophages upon S. aureus or S. typhimurium challenge compared to PBS-, BCG-, and BLP-stimulated macrophages (p<0.01), indicating an increased glycolysis. In addition, stimulation with BCG+BLP significantly increased ECAR (Figure 6C) while did not affect OCR (Figure 6D) in neonatal macrophages (p<0.01 versus PBS-, BCG-, and BLP-stimulated macrophages), suggesting that glucose metabolism changes from oxidative phosphorylation to glycolysis. By contrast, stimulation of neonatal macrophages with either BCG alone or BLP alone only slightly increased glucose consumption and lactate concentrations (Figure 6A and B) (p<0.05 versus PBS-stimulated macrophages) but had no effect on ECAR (Figure 6C and D). Moreover, strong accumulations in both glutamate (Figure 6E) and acetyl-CoA (Figure 6F) upon S. aureus or S. typhimurium challenge were observed in BCG+BLP-stimulated neonatal macrophages (p<0.01 versus PBS-, BCG-, and BLP-stimulated macrophages), whereas stimulation with BCG alone or BLP alone did not affect the levels of glutamate and acetyl-CoA (Figure 6E and F). Together, these results indicate that BCG+BLP-induced trained immunity alters intracellular metabolic pathways with increased glycolysis.
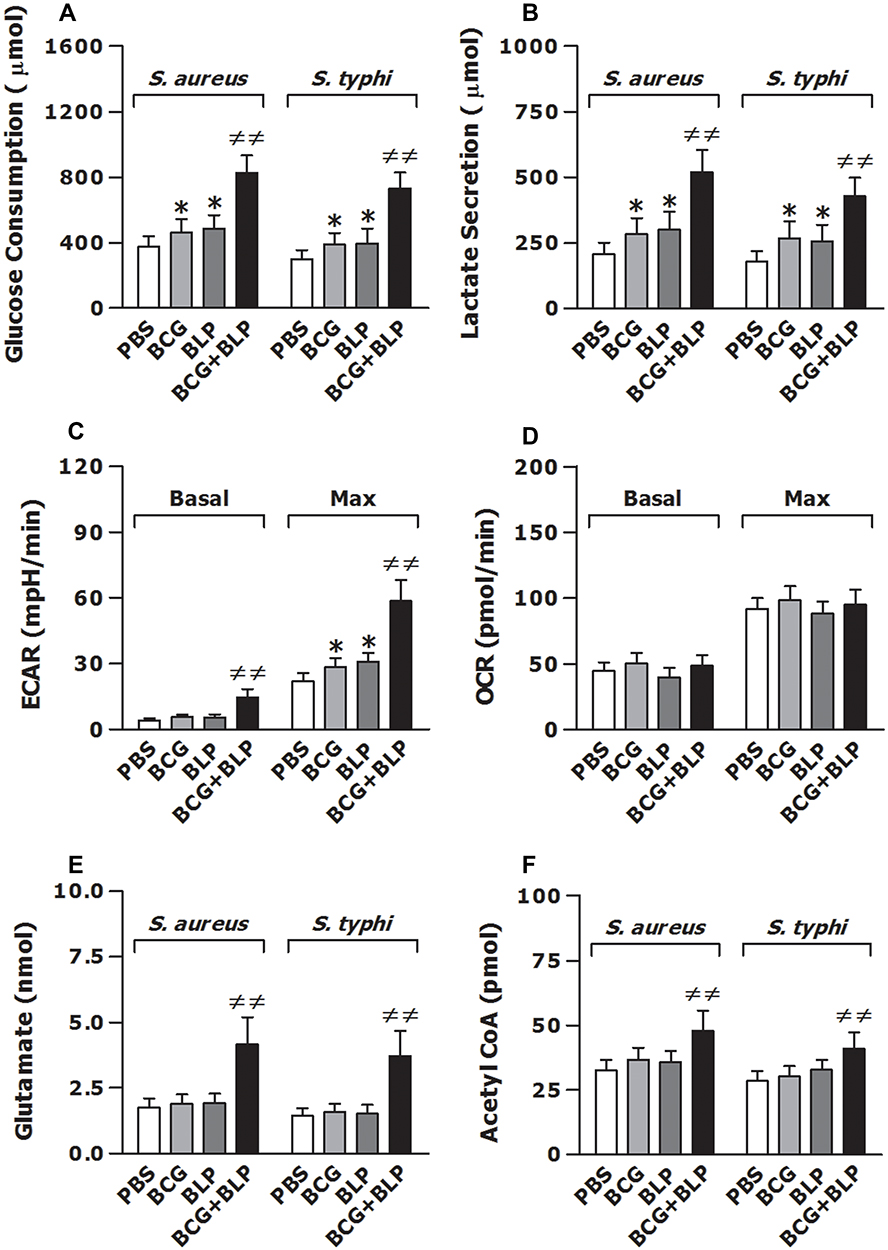 |
Figure 6 Training innate immunity by BCG+BLP leads to altered intracellular metabolic pathways. Isolated neonatal murine peritoneal macrophages were stimulated with BCG, BLP, or BCG+BLP for 24 h and rested for additional 3 days. After being incubated with heat-killed S. aureus or S. typhi for 6 h, glucose consumption (A) and lactate secretion (B) were assessed in neonatal macrophages. Basal and max extracellular acidification rate (ECAR) (C) and oxygen consumption rate (OCR) (D) were measured in neonatal macrophages before bacterial challenge. Glutamate (E) and acetyl-CoA (F) were assessed in neonatal macrophages after being incubated with heat-killed S. aureus or S. typhi for 6 h. Data are mean ± SD from four to five separate experiments in duplicate or triplicate. *p<0.05 versus PBS-incubated macrophages; ≠≠p<0.01 versus BCG- or BLP-stimulated macrophages. |
We next asked whether a shift in intracellular metabolic pathways is the prerequisite for BCG+BLP-induced trained immunity and examined whether inhibition of glycolysis disrupts BCG+BLP-triggered epigenetic reprogramming and -augmented both inflammatory and antimicrobial responses. Pretreatment with 2-DG efficiently interrupted BCG+BLP-initiated upregulation of H3K4me3 (Figure 7A) and downregulation of H3K9me3 (Figure 7B) at the promoters of TNF-α, IL-6, Acp5, and Camp (p<0.01 versus culture medium-pretreated and BCG+BLP-stimulated macrophages). Importantly, pretreatment with 2-DG strongly precluded BCG+BLP-boosted inflammatory response and bactericidal activity, as evidenced by significantly attenuated TNF-α, IL-6, IL12-p70, and CXCL2 release upon S. aureus challenge (Figure 7C) and substantially reduced phagocytosis (Figure 7D) and intracellular killing (Figure 7E) of S. aureus and S. typhimurium (p<0.01 versus culture medium-pretreated and BCG+BLP-stimulated macrophages). Collectively, these results indicate that stimulation with BCG+BLP leads to switches in cellular metabolism with increased glycolysis, which is the prerequisite for subsequent BCG+BLP-triggered epigenetic reprogramming and boosted both inflammatory and antimicrobial responses.
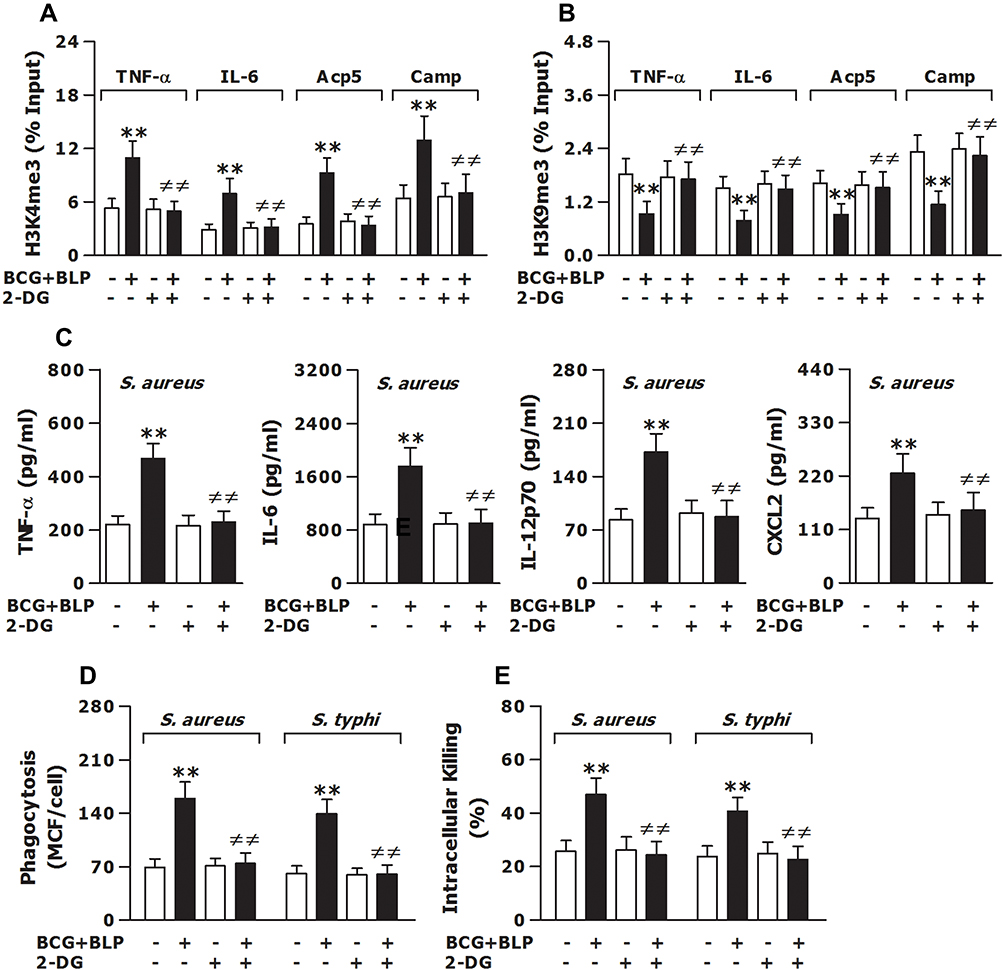 |
Figure 7 Inhibition of glycolysis prevents BCG+BLP-induced trained immunity with disrupted epigenetic reprogramming and abrogated augmentations in both inflammatory and antimicrobial responses. Isolated neonatal murine peritoneal macrophages or BMMs were pretreated with culture medium (CM) or 2-DG for 1 h before training of innate immunity. (A and B) H3K4me3 and H3K9me3 at the promoters of TNF-α, IL-6, Acp5, and Camp were assessed in neonatal macrophages before bacterial challenge. (C) TNF-α, IL-6, IL-12p70, and CXCL2 in the supernatants were measured 18 h post bacterial challenge. (D and E) Bacterial phagocytosis and intracellular killing were assessed in neonatal macrophages 60 min and 2 h after being incubated with FITC-conjugated or live S. aureus or S. typhi. Data are mean ± SD from five to six separate experiments in duplicate or triplicate. **p<0.01 versus CM-pretreated, PBS-incubated macrophages; ≠≠p<0.01 versus CM-pretreated, BCG+BLP-stimulated macrophages. |
Discussion
In this study, we show that stimulation with BCG+BLP induces trained immunity in neonatal murine macrophages, characterized by boosted both the inflammatory response and antimicrobial capacity with enhanced inflammatory cytokine and chemokine production and augmented bactericidal activity. Notably, this in vitro trained immunity is predominantly dependent on BCG+BLP-triggered histone modification-associated epigenetic reprogramming and shifts in cellular metabolism with increased glycolysis. Critically, BCG+BLP-induced trained immunity confers robust protection against cecal slurry peritonitis-induced polymicrobial sepsis in neonatal mice.
Accumulated evidence has shown that stimulation with a number of PAMPs and DAMPs including β-glucan and BCG induces trained immunity in adult murine macrophages and human monocytes/macrophages characterized by a boosted inflammatory response upon a second stimulation.13–15,17,42 Moreover, induction of trained immunity in vivo by β-glucan or BCG protects against subsequent bacterial and viral infections in adult mice and humans.13,16,19 Our previous work has shown that pretreatment of adult murine macrophages and human monocytes with BLP, a TLR2 agonist, results in diminished TNF-α and IL-6 release, but an augmented antimicrobial capacity in response to a secondary bacterial challenge.30,41,43 Importantly, in vivo pretreatment with BLP confers protection against both lethal bacterial infection and severe polymicrobial sepsis in adult mice.26,30 This BLP-afforded protection is dependent on BLP-initiated reprogramming in innate phagocytes characterized by a blunted inflammatory cytokine production and simultaneously, an augmented antimicrobial capability with enhanced bacterial ingestion and intracellular killing.26,30,44 In this study, we first examined whether BCG alone, BLP alone, or BCG+BLP is capable of inducing trained immunity in neonatal murine macrophages and mice, and further examined whether training innate immunity leads to boosted both the inflammatory response and antimicrobial activity, thereby conferring protection against microbial sepsis-associated lethality in neonatal mice. Stimulation of neonatal murine macrophages with BCG+BLP resulted in not only a robust release of TNF-α, IL-6, IL12-p70, and CXCL12 upon S. aureus or S. typhimurium challenge but also substantially enhanced bacterial ingestion and killing with accelerated phagosome maturation, indicating augmentations in both inflammatory response and bactericidal capability. By contrast, stimulation with BCG alone only induced an enhanced inflammatory response with moderate increases in inflammatory cytokine and chemokine release similar to the previously reported,17,19,42 but failed to augment innate phagocyte-associated bactericidal activity, whereas stimulation with BLP alone resulted in an opposite effect ie, an augmented antimicrobial capability with increased bacterial phagocytosis and killing as previously described,41,43 but not the inflammatory response. In vivo neither BCG alone nor BLP alone afforded any protection against cecal slurry peritonitis-induced polymicrobial sepsis in neonatal mice. Remarkably, BCG+BLP protected neonatal mice against both mid-grade and high-grade polymicrobial sepsis with substantially improved survival, which is closely associated with the increased circulating TNF-α and IL-6, enhanced PMN recruitment, and accelerated bacterial clearance. These results demonstrate that BCG+BLP, but not BCG alone or BLP alone, is capable of inducing trained immunity in neonatal murine macrophages and mice characterized by boosted both inflammatory and antimicrobial responses, thereby conferring robust protection against microbial sepsis.
Epigenetic remodeling and shifts in cellular metabolism are two intracellular events essential for the induction, regulation, and maintenance of trained immunity.12,20,24 It has been shown that induction of trained immunity is fundamentally dependent on epigenetic reprogramming of gene transcription and specifically, several histone modifications including H3K4me1, H3K4me3, H3K4Ac, H3K27Ac, and H3K9me3 at the promoters of the targeted innate immune genes play a central role in the formation of trained immunity.20,45 Several studies have reported that β-glucan-, BCG- or C albicans-induced trained immunity in mouse macrophages and human monocytes is associated with increased H3K4me1, H3K4me3, H3K27Ac, three positive marks associated with transcriptionally active chromatin, and/or suppressed H3K9me3, a repressor mark associated with inactive chromatin, at the promoters of inflammatory cytokine genes.13,14,17 On the other hand, induction of trained immunity leads to profound alterations in intracellular metabolic pathways including glycolysis, oxidative phosphorylation, and lipid and amino acid metabolism, thereby potentiating the response capacity of innate phagocytes to a second stimulus.12,20,45 Several studies have shown that there is substantially altered cellular metabolism in trained mouse macrophages and human monocytes characterized by significantly increased glycolysis, glutaminolysis, and cholesterol synthesis.15,17,21,22 Importantly, metabolites generated and/or accumulated from the altered intracellular metabolic pathways during the process of training innate immunity participate in the establishment of trained immunity.20,22–24 In the present study, we first examined whether training innate immunity with BCG+BLP triggers histone modification-associated epigenetic remodeling at the promoters of the targeted genes in neonatal macrophages. We found that stimulation of neonatal murine macrophages with BCG+BLP resulted in strong upregulation of H3K4me3, H3K27Ac and downregulation of H3K9me3 at the promoters of both the inflammatory response and antimicrobial effector genes including TNF-α, IL-6, Acp5, and Camp. Remarkably, blockage of histone methylation and acetylation with their specific inhibitors in neonatal murine macrophages effectively prevented BCG+BLP-boosted TNF-α, IL-6, IL12-p70, and CXCL2 release, and abrogated BCG+BLP-augmented bactericidal activity with significantly attenuated bacterial phagocytosis and intracellular killing, indicating that histone modification-associated epigenetic reprogramming of innate immune gene transcription is the underlying molecular event responsible for BCG+BLP-boosted both inflammatory and antimicrobial responses. Consistent with the previous finding,17,41–43 the enhanced H3K4me3, H3K27Ac and suppressed H3K9me3 induced by BCG alone were only occurred at the promoters of TNF-α and IL-6, whereas the altered histone modifications triggered by BLP alone were only observed at the promoters of Acp5 and Camp. We next determined whether training innate immunity with BCG+BLP switches intracellular metabolic pathways and induces a metabolic phenotype in neonatal macrophages. Stimulation of neonatal murine macrophages with BCG+BLP resulted in a substantially enhanced ECAR together with significant increases in glucose consumption and lactate secretion, indicating that glucose metabolism shifts from oxidative phosphorylation to glycolysis. Critically, inhibition of glycolysis with 2-DG interrupted BCG+BLP-triggered upregulation of H3K4me3 and downregulation of H3K9me3 at the promoters of both the inflammatory response and antimicrobial effector genes, and precluded BCG+BLP-augmented inflammatory response and bactericidal activity with significantly attenuated TNF-α, IL-6, IL12-p70, and CXCL2 release and substantially reduced bacterial phagocytosis and intracellular killing, indicating that the shift in intracellular metabolic pathways such as increased glycolysis during the process of training innate immunity contributes primarily to BCG+BLP-triggered epigenetic remodeling and boosted both inflammatory and antimicrobial responses.
Taken together, in the present study we show that BCG+BLP, but not BCG alone and BLP alone, induces trained immunity in neonatal murine macrophages characterized by boosted both the inflammatory response and antimicrobial activity. Notably, induction of trained immunity in neonatal macrophages depends primarily on BCG+BLP-initiated histone modification-associated epigenetic reprogramming and shifts in cellular metabolism. Critically, BCG+BLP-induced trained immunity confers robust protection against cecal slurry peritonitis-induced polymicrobial sepsis-associated lethality in neonatal mice.
Acknowledgments
This work was supported by National Natural Science Foundation of China (81871594, 82172132, 31670853, and 81420108022), Natural Science Foundation of Jiangsu Province (BK20190053, BRA2018393, and H2019002), Suzhou Program of Gusu Medical Talent (GSWS2019015 and GSWS2020043), Science and Technology Foundation of Suzhou (SYS2018067), and Pediatric Precise Surgical Clinical Medical Center of Suzhou (SZZXJ201505).
Disclosure
The authors have no conflicts of interest.
References
1. Liu L, Oza S, Hogan D, et al. Global, regional, and national causes of child mortality in 2000–13, with projections to inform post-2015 priorities: an updated systematic analysis. Lancet. 2015;385:430–440. doi:10.1016/S0140-6736(14)61698-6
2. Raymond SL, Stortz JA, Mira JC, Larson SD, Wynn JL, Moldawer LL. Immunological defects in neonatal sepsis and potential therapeutic approaches. Front Pediatr. 2017;5:14. doi:10.3389/fped.2017.00014
3. Shane AL, Sánchez PJ, Stoll BJ. Neonatal sepsis. Lancet. 2017;390:1770–1780. doi:10.1016/S0140-6736(17)31002-4
4. Wynn JL. Defining neonatal sepsis. Curr Opin Pediatr. 2016;28:135–140. doi:10.1097/MOP.0000000000000315
5. PrabhuDas M, Adkins B, Gans H, et al. Challenges in infant immunity: implications for responses to infection and vaccines. Nat Immunol. 2011;12:189–194. doi:10.1038/ni0311-189
6. Zaghouani H, Hoeman CM, Adkins B. Neonatal immunity: faulty T-helpers and the shortcomings of dendritic cells. Trends Immunol. 2009;30:585–591. doi:10.1016/j.it.2009.09.002
7. Kollmann TR, Kampmann B, Mazmanian SK, Marchant A, Levy F. Protecting the newborn and young infant from infectious diseases: lessons from immune ontogeny. Immunity. 2017;46:350–363. doi:10.1016/j.immuni.2017.03.009
8. Wynn JL, Scumpia PO, Winfield RD, et al. Defective innate immunity predisposes murine neonates to poor sepsis outcome but is reversed by TLR agonists. Blood. 2008;112:1750–1758. doi:10.1182/blood-2008-01-130500
9. Kollmann TR, Crabtree J, Rein-Weston A, et al. Neonatal innate TLR-mediated responses are distinct from those of adults. J Immunol. 2009;183:7150–7160. doi:10.4049/jimmunol.0901481
10. Zhang Q, Coveney AP, Yu S, et al. Inefficient antimicrobial functions of innate phagocytes render infant mice more susceptible to bacterial infection. Eur J Immunol. 2013;43:1322–1332. doi:10.1002/eji.201243077
11. Kan B, Razzaghian HR, Lavoie PM. An immunological perspective on neonatal sepsis. Trends Mol Med. 2016;22:290–302. doi:10.1016/j.molmed.2016.02.001
12. Netea MG, Joosten LA, Latz E, et al. Trained immunity: a program of innate immune memory in health and disease. Science. 2016;352:aaf1098. doi:10.1126/science.aaf1098
13. Quintin J, Saeed S, Martens JH, et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe. 2012;12:223–232. doi:10.1016/j.chom.2012.06.006
14. Saeed S, Quintin J, Kerstens HH, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. 2014;345:1251086. doi:10.1126/science.1251086
15. Bekkering S, Arts RJW, Novakovic B, et al. Metabolic induction of trained immunity through the mevalonate pathway. Cell. 2018;172:135–146. doi:10.1016/j.cell.2017.11.025
16. Cheng SC, Scicluna BP, Arts RJ, et al. Broad defects in the energy metabolism of leukocytes underlie immunoparalysis in sepsis. Nat Immunol. 2016;17:406–413. doi:10.1038/ni.3398
17. Arts RJW, Carvalho A, La Rocca C, et al. Immunometabolic pathways in BCG-induced trained immunity. Cell Rep. 2016;17:2562–2571. doi:10.1016/j.celrep.2016.11.011
18. Kaufmann E, Sanz J, Dunn JL, et al. BCG educates hematopoietic stem cells to generate protective innate immunity against tuberculosis. Cell. 2018;172:176–190. doi:10.1016/j.cell.2017.12.031
19. Arts RJW, Moorlag S, Novakovic B, et al. BCG vaccination protects against experimental viral infection in humans through the induction of cytokines associated with trained immunity. Cell Host Microbe. 2018;23:89–100. doi:10.1016/j.chom.2017.12.010
20. Fanucchi S, Domínguez-Andrés J, Joosten LAB, Netea MG, Mhlanga MM. The intersection of epigenetics and metabolism in trained immunity. Immunity. 2021;54:32–43. doi:10.1016/j.immuni.2020.10.011
21. Cheng SC, Quintin J, Cramer RA, et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. 2014;345:1250684. doi:10.1126/science.1250684
22. Arts RJ, Novakovic B, Ter Horst R, et al. Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab. 2016;24:807–819. doi:10.1016/j.cmet.2016.10.008
23. Penkov S, Mitroulis I, Hajishengallis G, Chavakis T. Immunometabolic crosstalk: an ancestral principle of trained immunity? Trends Immunol. 2019;40:1–11. doi:10.1016/j.it.2018.11.002
24. Bekkering S, Domínguez-Andrés J, Joosten LAB, Riksen NP, Netea MG. Trained immunity: reprogramming innate immunity in health and disease. Annu Rev Immunol. 2021;39:667–693. doi:10.1146/annurev-immunol-102119-073855
25. Wynn JL, Scumpia PO, Delano MJ, et al. Increased mortality and altered immunity in neonatal sepsis produced by generalized peritonitis. Shock. 2007;28:675–683. doi:10.1097/shk.0b013e3180556d09
26. O’Brien GC, Wang JH, Redmond HP. Bacterial lipoprotein induces resistance to Gram-negative sepsis in TLR4-deficient mice via enhanced bacterial clearance. J Immunol. 2005;174:1020–1026. doi:10.4049/jimmunol.174.2.1020
27. Buckley JM, Liu JH, Li CH, et al. Increased susceptibility of ST2-deficient mice to polymicrobial sepsis is associated with an impaired bactericidal function. J Immunol. 2011;187:4293–4299. doi:10.4049/jimmunol.1003872
28. Liu J, Buckley JM, Redmond HP, Wang JH. ST2 negatively regulates TLR2 signaling, but is not required for bacterial lipoprotein-induced tolerance. J Immunol. 2010;184:5802–5808. doi:10.4049/jimmunol.0904127
29. Coveney AP, Wang W, Kelly J, et al. Myeloid-related protein 8 induces self-tolerance and cross-tolerance to bacterial infection via TLR4- and TLR2-mediated signal pathways. Sci Rep. 2015;5:13694. doi:10.1038/srep13694
30. Wang JH, Doyle M, Manning BJ, et al. Cutting edge: bacterial lipoprotein induces endotoxin-independent tolerance to septic shock. J Immunol. 2003;170:14–18. doi:10.4049/jimmunol.170.1.14
31. Zhou HT, Coveney AP, Wu M, et al. Activation of both TLR and NOD signaling synergizes to confer host innate immunity-mediated protection against microbial infection. Front Immunol. 2019;9:3082. doi:10.3389/fimmu.2018.03082
32. Savina A, Jancic C, Hugues S, et al. NOX2 controls phagosomal pH to regulate antigen processing during cross presentation by dendritic cells. Cell. 2006;126:205–218. doi:10.1016/j.cell.2006.05.035
33. Hmama Z, Sendide K, Talal A, Garcia R, Dobos K, Reiner NE. Quantitative analysis of phagolysosome fusion in intact cells: inhibition by mycobacterial lipoarabinomannan and rescue by an 1alpha, 25-dihydroxyvitamin D3 phosphoinositide 3-kinase pathway. J Cell Sci. 2004;117:2131–2140. doi:10.1242/jcs.01072
34. Keating ST, Groh L, Thiem K, et al. Rewiring of glucose metabolism defines trained immunity induced by oxidized low-density lipoprotein. J Mol Med. 2020;98:819–831. doi:10.1007/s00109-020-01915-w
35. Chen W, Zhao S, Ita M, et al. An early neutrophil recruitment into the infectious site is critical for bacterial lipoprotein tolerance-afforded protection against microbial sepsis. J Immunol. 2020;204:408–417. doi:10.4049/jimmunol.1801602
36. Alves-Filho JC, Sonego F, Souto FO, et al. Interleukin-33 attenuates sepsis by enhancing neutrophil influx to the site of infection. Nat Med. 2010;16:708–712. doi:10.1038/nm.2156
37. Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–826. doi:10.1038/nature06246
38. Ishii KJ, Koyama S, Nakagawa A, Coban C, Akira S. Host innate immune receptors and beyond: making sense of microbial infections. Cell Host Microbe. 2008;3:352–363. doi:10.1016/j.chom.2008.05.003
39. Blander JM, Medzhitov R. Regulation of phagosome maturation by signals from toll-like receptors. Science. 2004;304:1014–1018. doi:10.1126/science.1096158
40. Kinchen JM, Ravichandran KS. Phagosome maturation: going through the acid test. Nat Rev Mol Cell Biol. 2008;9:781–795. doi:10.1038/nrm2515
41. Liu J, Xiang J, Li X, et al. NF-κB activation is critical for bacterial lipoprotein tolerance-enhanced bactericidal activity in macrophages during microbial infection. Sci Rep. 2017;7:40418. doi:10.1038/srep40418
42. Bekkering S, Blok BA, Joosten LA, Riksen NP, van Crevel R, Netea MG. In vitro experimental model of trained innate immunity in human primary monocytes. Clin Vaccine Immunol. 2016;23:926–933. doi:10.1128/CVI.00349-16
43. Wang JH, Doyle M, Manning BJ, Wu QD, Blankson S, Redmond HP. Induction of bacterial lipoprotein tolerance is associated with suppression of toll-like receptor 2 expression. J Biol Chem. 2002;277:36068–36075. doi:10.1074/jbc.M205584200
44. Buckley JM, Wang JH, Redmond HP. Cellular reprogramming by gram-positive bacterial components: a review. J Leukoc Biol. 2006;80:731–741. doi:10.1189/jlb.0506312
45. Domínguez-Andrés J, Joosten LA, Netea MG. Induction of innate immune memory: the role of cellular metabolism. Curr Opin Immunol. 2019;56:10–16.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.