")
Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Induction of mucosal immune responses and protection of cattle against direct-contact challenge by intranasal delivery with foot-and-mouth disease virus antigen mediated by nanoparticles
Authors Pan L, Zhang Z, Lv J, Zhou P, Hu W, Fang Y, Chen H, Liu X, Shao J, Zhao F, Ding Y, Lin T, Chang H, Zhang J, Zhang Y, Wang Y
Received 6 August 2014
Accepted for publication 11 October 2014
Published 2 December 2014 Volume 2014:9(1) Pages 5603—5618
DOI https://doi.org/10.2147/IJN.S72318
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Li Pan,1,2 Zhongwang Zhang,1,2 Jianliang Lv,1,2 Peng Zhou,1,2 Wenfa Hu,1,2 Yuzhen Fang,1,2 Haotai Chen,1,2 Xinsheng Liu,1,2 Junjun Shao,1,2 Furong Zhao,1,2 Yaozhong Ding,1,2 Tong Lin,1,2 Huiyun Chang,1,2 Jie Zhang,1,2 Yongguang Zhang,1,2 Yonglu Wang1,2
1State Key Laboratory of Veterinary Etiological Biology, National Foot-and-Mouth Disease Reference Laboratory, Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences (CAAS), Lanzhou, Gansu, People’s Republic of China; 2Jiangsu Co-innovation Center for Prevention and Control of Important Animal Infectious Diseases and Zoonoses, Yangzhou, People’s Republic of China
Abstract: The aim of this study was to enhance specific mucosal, systemic, and cell-mediated immunity and to induce earlier onset of protection against direct-contact challenge in cattle by intranasal delivery of a nanoparticle-based nasal vaccine against type A foot-and-mouth disease (FMD). In this study, two kinds of nanoparticle-based nasal vaccines against type A FMD were designed: (1) chitosan-coated poly(lactic-co-glycolic acid) (PLGA) loaded with plasmid DNA (Chi-PLGA-DNA) and (2) chitosan-trehalose and inactivated foot-and-mouth disease virus (FMDV) (Chi-Tre-Inactivated). Cattle were immunized by an intranasal route with nanoparticles and then challenged for 48 hours by direct contact with two infected donor cattle per pen. Donors were inoculated intradermally in the tongue 48 hours before challenge, with 0.2 mL cattle-passaged FMDV. Serological and mucosal antibody responses were evaluated, and virus excretion and the number of contact infections were quantified. FMDV-specific secretory immunoglobulin (Ig)A (sIgA) antibodies in nasal washes were initially detected at 4 days postvaccination (dpv) with two kinds of nanoparticles. The highest levels of sIgA expression were observed in nasal washes, at 10 dpv, from animals with Chi-PLGA-DNA nanoparticles, followed by animals immunized once by intranasal route with a double dose of Chi-Tre-Inactivated nanoparticles and animals immunized by intranasal route three times with Chi-Tre-Inactivated nanoparticles (P<0.05). FMDV-specific IgA antibodies in serum showed a similar pattern. All animals immunized by intranasal route developed low levels of detectable IgG in serum at 10 dpv. Following stimulation with FMDV, the highest levels of proliferation were observed in splenocytes harvested from Chi-PLGA-DNA-immunized animals, followed by proliferation of cells harvested from Chi-Tre-Inactivated nanoparticle-immunized animals (P<0.05). Higher protection rates were associated with the highest sIgA antibody responses induced in the Chi-PLGA-DNA nanoparticle-immunized group. Only one animal was clinically affected with mild signs after 7 days of contact challenge, after a delay of 2–3 days compared with the clinically affected negative-control group. Of the five animals directly challenged that were vaccinated by intranasal route with a double dose of Chi-Tre-Inactivated, four were clinically infected; however, the degree of severity of disease in this group was lower than in control cattle. The number of viral RNA copies in nasal swabs from the vaccinated, severely infected group was significantly higher than in swabs from the vaccinated, clinically protected group. These data suggested that intranasal delivery of Chi-PLGA-DNA nanoparticles resulted in higher levels of mucosal, systemic, and cell-mediated immunity than did of Chi-Tre-Inactivated nanoparticles. In conclusion, although intranasal delivery with FMDV antigen mediated by nanoparticles did not provide complete clinical protection, it reduced disease severity and virus excretion and delayed clinical symptoms. Chi-PLGA-DNA nanoparticle vaccines have potential as a nasal delivery system for vaccines.
Keywords: FMDV, nanoparticles, chitosan, trehalose, poly(lactic-co-glycolic acid), PLGA
Introduction
Foot-and-mouth disease (FMD) is one of the world’s most important infectious diseases of livestock.1 Currently available FMD vaccines are mainly based on inactivated viral antigens formulated with various proprietary adjuvants.2 Despite tremendous progress in FMD vaccine production in recent years, the use of conventional and chemically inactivated FMD virus (FMDV) vaccines is associated with a number of concerns and deficiencies:3–5 (1) large doses are required and induce only a short duration of immunity and a limited spectrum of antiviral immune responses; (2) unless highly purified, the vaccines do not allow differentiation between infected and vaccinated animals; (3) animals vaccinated with the current vaccines are generally protected against disease and induce serum immunoglobulin (Ig)G antibodies but have low stimulation of secretory IgA (sIgA) at respiratory mucosal sites and show sporadic CD8+ cytotoxic T lymphocytes; and (4) the current vaccines requires 7–14 days to induce protection.6 The time for protection to develop leaves an immunity gap, a window of susceptibility prior to induction of the adaptive immune response in which vaccinated animals are still susceptible to disease; (5) protection is short-lived, often requiring frequent revaccination, and manipulation of large amounts of virulent virus could result in viral dissemination; (6) vaccination does not prevent colonization of the upper respiratory tract and development of the carrier state.7,8 To address these issues and to induce an earlier onset of protection and longer duration of immunity, different vaccine administration routes, vaccine delivery systems, and alternative approaches to conventional vaccines are urgently needed.
Evidence suggests that mucosal immunity indicates good mucosal protection, efficiently preventing disease spread by destroying the causative agent directly at the respiratory or gastrointestinal mucosa, the sites that represent the main entrance for many pathogens.9,10 In contrast to other antibody isotypes, sIgA in the oronasal cavities, esophageal-pharyngeal fluids, and mucosal epithelium is relatively resistant to enzymatic degradation and prevents the attachment of bacteria and virus to the mucosa, preventing entry. This feature makes sIgA uniquely suitable for mucosal defense.11
Intranasal vaccination is superior to vaccination at other sites for eliciting protection against respiratory pathogens. Immune responses generated in nasal-associated lymphoid tissue provide long-term protection.12,13 The nasal cavity is one of the most promising administration sites because of its reduced enzymatic activity compared with the oral route. The nasal cavity has a high availability of immunoreactive sites because the nasal epithelium layer consists of specialized antigen-sampling microfold cells that overlay the nasal-associated lymphoid tissue, resulting in antigen uptake and subsequent mucosal and systemic immune induction.14,15 In addition to these advantages, the nasal route offers simpler and more cost-effective protocols for vaccination, with improved patient compliance.16 However, despite these encouraging characteristics, free antigens do not usually elicit protective responses following intranasal administration. The residence time of antigens in the nasal cavity is limited because of mucociliary clearance, which results in a very small dose reaching antigen-presenting cells in the subepithelial region. Moreover, the uptake of antigens through the nasal epithelium is restricted because of the large size of antigens and the tolerogenic nature of the mucosal epithelium, which can complicate a robust immune response through multiple mechanisms.17,18 For this reason, mucosal vaccine adjuvants and new delivery systems are necessary to improve the performance of existing and future antigens.
Among possible delivery systems, nanocarriers prepared with biodegradable and mucoadhesive substances are promising because of their capacity to protect encapsulated bioactive macromolecules, such as peptides, proteins, or nucleic acid-based antigens.19 This encapsulation increases antigen residence time and controls the release of antigen over a prolonged time.20 This increased time raises the chance of uptake by the epithelium,21 and so nanocarriers have been widely investigated for their biological potential.22,23 Based on these points, designing optimized vaccine nanocarriers is a promising strategy for nasal mucosal vaccination.
Previous study has shown that FMDV 3C proteinase processes the viral structural protein precursor, P1-2A, into the capsid proteins, VP0, VP3, and VP1, and the nonstructural peptide, 2A. These proteins then self-assemble to form empty icosahedral capsid particles that contain 60 copies of each protein.24 Immunological study has identified linear and conformational sites that are present on both empty capsids and virions, and antiserum raised against either form has the same serological specificity.25 Thus, the structural protein precursor, P1-2A, and the 3C protease of FMDV are desirable immune antigens for new vaccine development.
Plasmid DNA can be easily prepared in large scale, repeatedly administered, and is highly stable compared with proteins and other biological polymers.26 Plasmid DNA vaccines have become an attractive and potentially effective strategy for generating antigen-specific immune responses. Since DNA plasmid used to stimulate mucosal immunity can be easily degraded by DNases present at mucosal surfaces, DNA plasmids were adsorbed onto chitosan-coated poly(lactic-co-glycolic acid) (PLGA) particles and were shown to be protected against enzymatic degradation.27 Investigation of the immunogenicity of antigens encapsulated in PLGA nanoparticles showed that serum IgG and mucosal sIgA levels in samples from saliva and nasal fluids were significantly higher than levels elicited by naked plasmid, in mice administered chitosan-coated PLGA nanoparticles.28–31 We reported previously substantial significant cellular and humoral immune responses elicited by cationic PLGA nanoparticles loaded with FMDV DNA vaccine. We demonstrated that intranasal delivery of nanoparticles loaded with FMDV DNA vaccine formulations encoding interleukin 6 (IL-6) as a molecular adjuvant enhanced protective immunity against FMDV.32
Other reports obtained potent nasal mucosal and systemic immune responses using intranasal delivery of whole, inactivated influenza vaccine powder formulations. Powder-formulated vaccine elicited a significant serum antibody response in rats that was at least as strong as the response from liquid vaccine administered by intranasal route or by intramuscular injection.33,34 In these reports, a mouse model was used to assess vaccine efficacy mediated by nanoparticle delivery by intranasal delivery, against influenza virus, parainfluenza, hepatitis B, and Streptococcus pneumoniae pathogens.
Knowledge about the protective efficacy of vaccines in a suitable large animal model is limited. Here, we studied the cattle vaccine potency of two kinds of nanoparticle delivery system: chitosan-coated PLGA nanoparticles loaded with plasmid DNA (Chi-PLGA-DNA); and chitosan-trehalose nanoparticles loaded with inactivated FMDV (Chi-Tre-Inactivated). We investigated the two intranasal delivery systems in reinforcing the mucosal immune response and in protecting cattle against direct-contact challenge at the primary portal for virus entry. The aim of this study was to evaluate the ability of these two intranasal delivery systems to induce early onset protection, and specific mucosal, systemic, and cell-mediated immunity; and to determine possible suitable antigen carrier systems for nasal administration.
Materials and methods
Materials
D-(+)-trehalose and chitosan were from Sigma-Aldrich Corp (St Louis, MO, USA). PLGA (50 kDa) was from Shandong Institute of Medical Instruments (Jinan, Shandong, People’s Republic of China). Inactivated FMD vaccine was provided by Lanzhou Veterinary Research Institute, Chinese Academy of Agriculture (Beijing, People’s Republic of China). Fluorescein isothiocyanate (FITC)-conjugated sheep anti-rabbit IgG, horseradish-peroxidase (HRP)-conjugated rabbit anti-guinea pig IgG, and HRP-conjugated sheep anti-bovine IgA were from Bioward. FITC-labeled anti-bovine CD4, phycoerythrin (PE)-labeled anti-bovine CD8, and isotype controls were from BD Biosciences Pharmingen (San Diego, CA, USA). 4-(2-hydroxyethy1)-1-piperazine-ethanesulfonic acid (HEPES) (15 mM), 10% (vol/vol) heat-inactivated fetal bovine serum, and Roswell Park Memorial Institute (RPMI)-1640 media were from Gibco® (Life Technologies, Carlsbad, CA, USA) or Sigma-Aldrich Corp.
Experimental animals
Cattle were obtained from an FMDV-free region and checked for absence of FMDV-specific antibodies, by liquid-phase blocking enzyme-linked immunosorbent assay (LB-ELISA), before immunization. During the experimental period, animals were kept in biosafety level 3A facilities, according to biosecurity and animal welfare regulations. Experiments conformed to local (Institutional Animals Use and Care Committee of Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences [CAAS]) guidelines on the ethical use of animals.
Preparation of the Chi-PLGA-DNA nanoparticles
Two pairs of specific primers were used to amplify P12A and 3C genes from pGEM-P12A and pGEM-3C plasmids maintained in our laboratory (Table 1). P12A was digested using BamH I and EcoR I, and 3C was digested with EcoR I and Xbal I, and fragments were subcloned into the corresponding sites of pcDNA3.1(+) (Invitrogen®; Life Technologies) using T4 DNA ligase to generate recombinant plasmids pcDNA3.1/P12A3C. The sequence accuracy of recombinant plasmids was authenticated by specific polymerase chain reaction (PCR), double digestion, and DNA sequencing.
 | Table 1 Primers used to construct plasmids pcDNA3.1/P12A3C |
To confirm the expression of structural proteins, purified pcDNA3.1/P12A3C was transfected into BHK-21 cells using Lipofectamine™ 2000 (Invitrogen®; Life Technologies) according to the manufacturer’s instructions. After 48-hour transfection, cells were harvested and analyzed for expression of FMDV P12A3C by indirect immunofluorescence test. Cell monolayers were cultured on cover slips and fixed in cold acetone (−20°C for 30 minutes). Samples were incubated with rabbit anti-FMDV serum (1:1,000 at 37°C for 30 minutes) in a humidified chamber and stained with FITC-conjugated goat anti-rabbit IgG (1:200) for 1 hour at 37°C and then, observed microscopically. FMDV capsids were observed by transmission electron microscopy (TEM) in BHK-21 cells transfected with pcDNA3.1 or P12A3C.
The Chi-PLGA-DNA nanoparticles were prepared using an emulsion-diffusion-evaporation method as previously described,32,35 except that a microball mill (Pulverisette® 7; Fritsch GmbH, Idar-Oberstein, Germany,) was used to reduce particle size. Nanoparticles were stored at 4°C.
Preparation of the Chi-Tre-Inactivated nanoparticles
Whole inactivated FMDV, strain AF/72, was from Lanzhou Veterinary Research Institute, Chinese Academy of Agriculture. FMDV was concentrated and purified following a standard sucrose density gradient centrifugation method36 and resuspended in NET buffer (0.1 M NaCl, 0.004 M ethylenediaminetetraacetic acid [EDTA], 0.05 M tris(hydroxymethyl)aminomethane [Tris]; pH 8.0). One mL antigen suspension (5 mg/mL) was stirred and placed in vials for freeze-drying using a Kinetics Flexi-Dry™ system. Vials were frozen at −20°C and placed on the manifold at −55°C, with chamber pressure 5×10−3 mm Hg, for 48 hours. Trehalose was freeze-dried using the lyophilized FMDV antigen method, for 48 hours.
Powder was made by mixing inactivated FMDV with trehalose at 1:100 ratio antigen to trehalose, then lyophilized and milled, using a microball mill (Pulverisette 7), in a dry box at <15% relative humidity to reduce particle size for small quantities of powder.37 Vibrational milling was for 15 minutes with milling speeds of 450 rpm, repeated three times at intervals of 3 minutes per grinding cycle. Milled powders were sieved with 300, 125, and 45 μm sieves. Powders were sieved for 2 hours in tap mode, and particles in the 45 to 125 μm range were collected. Chitosan was milled using a cryomilling technique at 1,600 rpm for 20 minutes. The resulting powder was sieved as described above. The final formulation contained 500 μg FMDV-inactivated antigen blended in 50 mg trehalose and 1.4 mg chitosan. To minimize humidity effects, the powder blend was placed in vials for freeze-drying and vibrated by milling for 30 minutes to achieve uniformity.
Particle size measurement by scanning electron microscopy
Double-sided adhesive tape was placed on an aluminum specimen holder, and a small amount of powder was added. Particles were sprinkled with approximately 10–20 nm gold powder, sputtered using a sputter coater device K650X (Emitech, Hailsham, UK), and analyzed with a JEOL scanning electron microscope (JSM-5600LV; Jeol, Tokyo, Japan). Measurement of mean size diameter of nanoparticles was based on a dynamic light scattering technique.
Immunization and challenge protocol
Immunization and challenge were performed on 24 cattle (Table 2). Two groups received a booster dose at 4 and 7 days postvaccination (dpv). The number of cattle and vaccine regimens are summarized in Table 2. The treatments received by the groups were: 1) Group 1 (Gr-1) was immunized by intramuscular route with 0.2 mL FMD inactivated vaccine and used as a control; 2) Group 2 (Gr-2) was immunized by intranasal route 3 times with Chi-Tre-Inactivated nanoparticles on days 1, 4 and 7; 3) Group 3 (Gr-3) was immunized by intranasal route with a double dose for one time of Chi-Tre-Inactivated nanoparticles; 4) Group 4 (Gr-4) was immunized by intranasal route 3 times with Chi-PLGA-DNA nanoparticles on days 1, 4 and 7; 5) Group 5 (Gr-5) was administered with 150 mg nanoparticles of chitosan-PLGA and trehalose by intranasal route as a negative control.
 | Table 2 Summary of cattle treatments |
Sample collection
Serum and nasal swabs were collected at 3, 5, 7, and 10 dpv for antibody detection. Additionally, after challenge, saliva, nasal, and oropharyngeal fluid samples were collected for virus detection. Oropharyngeal fluid samples were collected at 3, 5, 7, 9, 11, 13, and 15 days postchallenge (dpc) for analysis of viral RNA copies by real-time reverse-transcription (RT)-PCR. Immediately after collection, 0.2 mL oropharyngeal fluid was mixed with 0.3 mL lysis buffer (F. Hoffman-La Roche Ltd) and stored at −70°C.
Assessment of humoral responses
Quantification of FMDV-specific IgA antibodies
The presence of FMDV-specific sIgA in saliva and nasal washes and IgA in sera of immunized cattle was detected by capture ELISA, with some modifications.38,39 Briefly, 96-well microtiter plates were coated with rabbit anti-FMDV antibody and stored at 4°C overnight. After washing, inactivated FMDV antigen (100 mL/well) was added and incubated for 1 hour at 37°C. Wells were washed and incubated for 1 hour at 37°C with twofold serial dilutions of samples of serum, saliva, or nasal secretions, starting with a 1:4 dilution. Secondary antibody HRP-conjugated sheep anti-bovine IgA was diluted 1:1,000 and added into wells, and incubated at 37°C for 1 hour. Plates were washed and incubated with substrate (10 mg o-phenylenediamine + 20 mL 0.015% hydrogen peroxide in phosphate/citrate buffer) for 10 minutes at room temperature. Color development was stopped with 0.5 M of H2SO4 after 15 minutes. Optical density of the ELISA plates was read at 492 nm.
Quantification of FMDV-specific IgG antibodies
FMDV-specific IgG antibodies were detected by LB-ELISA kits (provided by Lanzhou Veterinary Research Institute Chinese Academy of Agriculture) according to the supplier’s instructions. Cattle serum samples collected from unvaccinated and vaccinated animals were tested in parallel using inactivated virus antigen. A twofold dilution of test sera was generated from 1:8 to 1:1,024, and 75 μL diluted test serum was mixed with 75 μL antigen in deep-well plates and incubated overnight at 4°C. Then, 100 μL of mixture was transferred in duplicate to microplates precoated with rabbit anti-FMDV capture serum. The plates were washed and incubated with guinea pig anti-FMDV serum and incubated at 37°C for one hour. After washing, HRP-conjugated rabbit anti-guinea pig IgG was added at 1:2,000 and incubated at 37°C for one hour. The plates were washed and incubated with substrate (10 mg o-phenylenediamine + 20 mL 0.015% hydrogen peroxide in phosphate/citrate buffer) for 10 minutes at room temperature. Color development was stopped with 0.5 M of H2SO4 after 15 minutes. Optical density of the ELISA plates was read at 492 nm.
T lymphocyte proliferation assay
T lymphocyte proliferation activity was evaluated by carboxyfluorescein diacetate succinimidyl ester (CFSE), originally developed by Lyons and Parish.40 Peripheral blood mononuclear cells (PBMCs) were isolated from heparinized blood using Ficoll-Hypaque density gradients (Axis-Shield Diagnostics Ltd, Dundee, UK) for 20 minutes at room temperature. PBMCs were washed twice with phosphate-buffered saline (PBS) and resuspended in RPMI-1640 culture medium without fetal bovine serum. CFSE was dissolved to 5 mM in dimethyl sulfoxide (DMSO) and stored in aliquots at −20°C. PBMCs (1.0–2.0×107 cells/mL in RPMI-1640) were stained with 5 μM CFSE in the same buffer (15 minutes, 37°C, in darkness) with occasional stirring. Staining was stopped by adding culture medium and washing three times with a 10× volume of PBS containing 3% fetal bovine serum. Centrifugation was carried out at 250 g/min for ten minutes. After discarding the supernatant, sediment was resuspended in culture medium (2×106 cells/mL). CFSE-labeled cells were cultured in duplicate under sterile conditions and stimulated with inactivated FMDV, at 37°C in a 5% CO2 environment for 60 hours, with 10 μg/mL concanavalin A (ConA) as the polyclonal stimulator for the positive control. Negative control cultures were treated with 200 μL RPMI-1640. At the end of incubations, cells were washed twice with PBS and analyzed by FACScan flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
Flow cytometric analysis of CD4+ and CD8+ T lymphocytes subsets
A total of 100 μL heparinized blood was collected from immunized cattle at 10 dpv and incubated with anti-CD4-FITC and anti-CD8-PE or with the corresponding isotype controls (anti-CD8-PE and anti-IgG2a-PE, or anti-CD4-FITC and anti-IgG2a-FITC) for 30 minutes at room temperature. Added to the cells was 2 mL 1× FACS red blood cell lysis buffer (BD Biosciences), and then, cells were incubated for 15 minutes at room temperature. Centrifugation was carried out at 300 g/min for 5 minutes. After discarding the supernatant, pellets were washed three times with 2 mL PBS, to thoroughly resuspend cells. Centrifugation was carried out at 300 g/min for 5 minutes, discarding the supernatant. Cells were resuspended in 0.3 mL PBS, and fluorescence profiles analyzed on a FACScan flow cytometer (BD Biosciences) by System II software (Beckman Coulter Inc, Brea, CA, USA).
FMDV RNA detection by real-time RT-PCR
Quantification of FMDV RNA copies in saliva, nasal washes, and oropharyngeal fluids used a dilution series of an RNA standard (RNA transcribed in vitro from a plasmid clone containing a fragment of the FMDV 3D region run in parallel with each plate of RT-PCR assays) as previously described.41,42 An automated nucleic acid robotic work station (BioRobot® MDx; Qiagen, Venlo, the Netherlands) was used to generate nucleic acid templates from samples for real-time RT-PCR. Nucleic acid extraction was by a customized automated protocol using reagents from a QIAamp® All Nucleic Acid MDx kit (Qiagen). Total nucleic acids were eluted to a final volume of 50 μL. Real-time RT-PCR reactions were prepared manually by adding 4 μL template to 21 μL real-time RT-PCR reaction mix containing 12.5 μL of 2× one step RT-PCR buffer, 0.5 μL of Prime Script RT Enzyme Mix, 0.5 μL TakaRa Ex Taq enzyme HS (5 u/μL), 0.5 μL each of forward and reverse primer (10 μM), 0.3 μL dual-labeled TaqMan probe, and 6.2 μL nuclease-free dH2O, to a final volume of 25 μL. Samples and standards were run in triplicate in an Mx3005P Multiplex Quantitative PCR System, at 42°C for 50 minutes and 95°C for 10 seconds, followed by 40 cycles of 95°C for 5 seconds and 60°C for 30 seconds. Samples with cycle threshold values <33 were considered positive. Primers and probe for the FMDV 3D region and standard plasmid were kindly provided by Dr Huifang Bao.
Statistical analysis
Data are presented as mean ± standard deviation (SD). Statistically significant differences were tested using one-way analysis of variance (ANOVA) test. Statistical differences are noted as P<0.05 or ns = no significant difference (P>0.05). Data analysis was performed using GraphPad Prism 5.00 (GraphPad Software Inc., La Jolla, CA, USA).
Results
Construction and plasmid characterization
The recombinant plasmid pcDNA3.1/P12A3C, carrying complementary (c)DNA encoding the capsid polypeptide P12A and the 3C protease of FMDV, was constructed and confirmed by PCR (Figure 1A), enzyme digestion (Figure 1B), and sequencing analysis. Cells transfected with pcDNA3.1/P12A3C showed fluorescence compared with negative controls (Figure 2A and B). TEM revealed that cells transfected with plasmid pcDNA3.1/P12A3C had detectable empty capsid structures, but cells transfected with pcDNA3.1 did not (Figure 2C and D).
 | Figure 1 Construction of the recombinant plasmid pcDNA3.1/P12A3C. PCR (A), enzyme digestion (B). |
 | Figure 2 Detection of FMDV structural protein and FMDV capsid. |
Characterization of microparticle properties
Particle characterization by scanning electron microscopy is shown in Figure 3. After freeze-drying, average diameters were about 500 nm for Chi-PLGA-DNA nanoparticles (Figure 3A) and 3–20 μm for Chi-Tre-Inactivated virus nanoparticles (Figure 3B).
 | Figure 3 Scanning electron micrographs of nanoparticle morphology. |
Humoral immune responses
Substantial antibody responses corresponding to sIgA production in nasal washes and IgA in serum were observed in groups immunized through an intranasal route (Figure 4A and B), except for in the animals of Gr-5. FMDV-specific sIgA antibodies in nasal washes were initially detected at 4 dpv by an intranasal route with the two kinds of nanoparticles. The highest levels of sIgA expression were observed in nasal washes at 10 dpv, from animals of Gr-4, followed by animals of Gr-3 and animals of Gr-2 (P<0.05). No anti-FMDV sIgA antibodies were detected in nasal washes from the animals of of Gr-5 or Gr-1. The dynamic patterns of the IgA responses in serum samples were similar to responses from nasal wash samples, but the IgA responses in serum from animals of Gr-1 were higher than the animals of Gr-5 (P<0.05), although vaccination resulted in low IgA responses in serum (data not shown).
 | Figure 4 Humoral response induced in cattle, to FMDV antigen mediated by nanoparticles. |
To examine systemic immune responses elicited using nanoparticles through an intranasal route, specific IgG antibodies were analyzed by LB-ELISA for serum samples collected at 7 and 10 dpv. Serum IgG (Figure 4C) showed that nasal administration of nanoparticles elicited a low but detectable FMDV-specific systemic IgG antibody response at 10 dpv compared with administration of FMD inactivated vaccine through an intramuscular route. A significant IgG response was observed in animals (Gr-1) immunized through an intramuscular route with FMD inactivated vaccine (P<0.05). In addition, we observed low levels of IgG titers following nasal delivery of nanoparticle-encapsulated FMDV inactivated vaccine until 14 dpv (unpublished results). These results suggested that the intramuscular route might be superior to the nasal route for inducing systemic immune responses.
T lymphocyte proliferation activity by CFSE
Following stimulation with FMDV, the highest levels of T lymphocyte proliferation were observed in splenocytes harvested from animals immunized with Chi-PLGA DNA (3×IN) (Figure 5), followed by proliferation of cells harvested from animals immunized with inactivated vaccine (IM) or Chi-Tre-Inactivated vaccine (3×IN) (P<0.05). Low levels of proliferation were observed in animals immunized with Chi-Tre-Inactivated vaccine (1×IN). Proliferation was significantly higher in cells harvested from animals in all vaccine groups compared with the negative-control group (P<0.05).
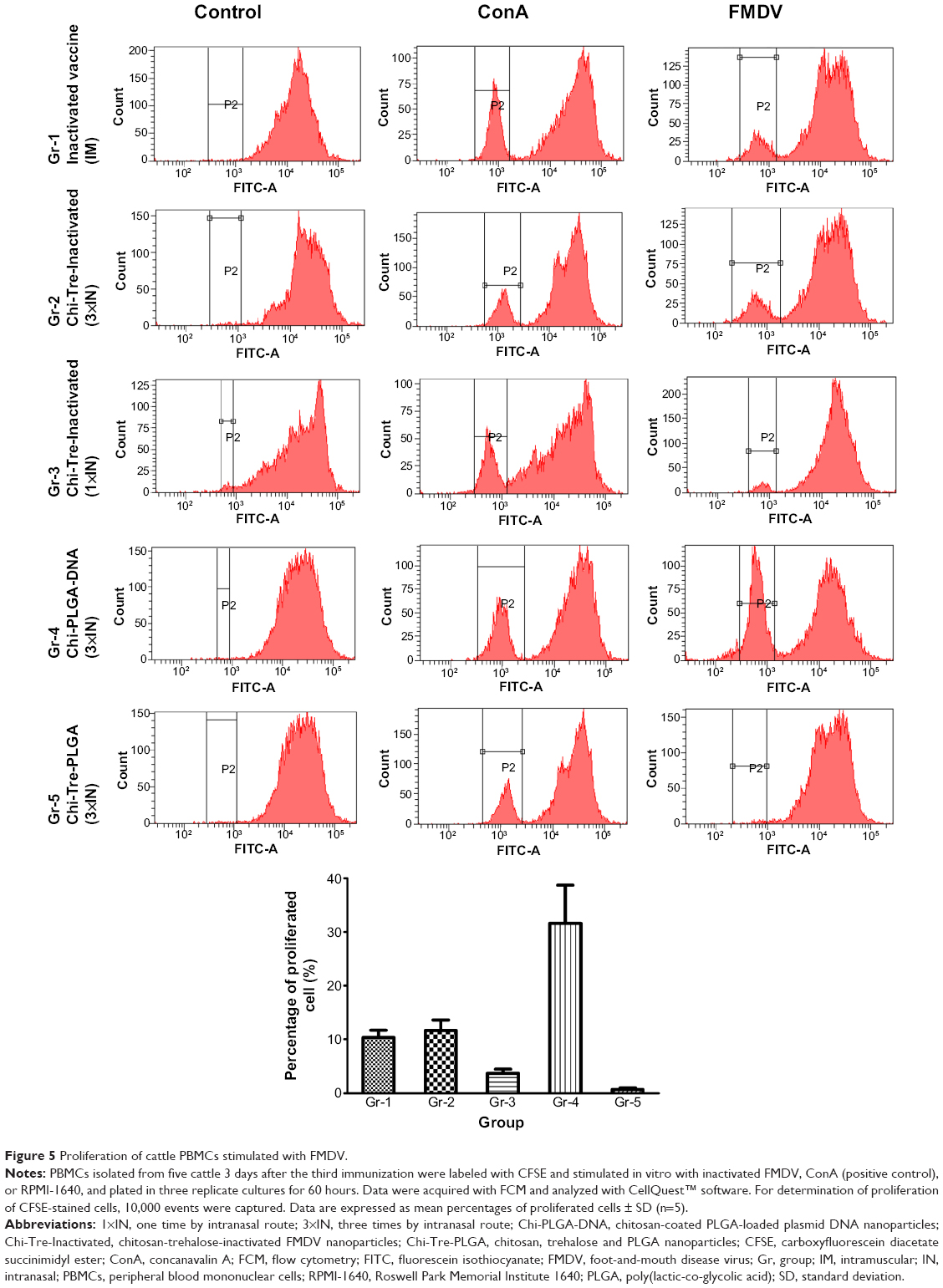 | Figure 5 Proliferation of cattle PBMCs stimulated with FMDV. |
Analysis of CD4+ and CD8+ T lymphocytes levels
Percentages of CD4+CD8− T cells increased significantly in the group of animals immunized with Chi-PLGA-DNA (3×IN), followed by the group with Chi-Tre-Inactivated (3×IN). The lowest increase was seen in the group given Chi-Tre-Inactivated vaccine (1×IN) or inactivated vaccine (IM) compared with the negative control (P<0.05) (Figure 6). Percentages of CD4−CD8+ T cells from the cattle immunized with Chi-PLGA-DNA (3×IN) or Chi-Tre-Inactivated (3×IN) increased significantly, followed by the group given inactivated vaccine (IM). The group given Chi-Tre-Inactivated vaccine (1×IN) had the lowest increase compared with the negative control (P<0.05). The CD4−CD8+ percentages of all cattle in the immunized group increased significantly compared with the negative control (P<0.05).
 | Figure 6 Flow cytometry for percentages of lymphocyte subpopulations between experimental and control groups. |
Assessment of protective activity
At 10 dpv, all animals were challenged for 48 hours with direct contact with two infected donor cattle per pen that had been inoculated intradermally at 48 hours prechallenge with 0.2 mL of cattle-passaged FMDV AF/72. Six needle-challenged donor cattle in three groups developed fever and severe clinical disease, with lesions on all four feet, within 1–2 days postinfection. The control animals (Gr-5) started exhibiting fever and the first vesicles appeared between 3–5 dpc. They showed lameness and developed severe vesicular lesions on the buccal mucosa and on all four feet by 7 dpc. In contrast, none of the animals vaccinated by the intramuscular route with inactivated vaccine (Gr-1) showed clinical signs of FMD during the course of the experiment and were fully protected up to 15 dpc. For animals vaccinated by intranasal route with nanoparticles of Chi-PLGA-DNA (Gr-4), only one (number 95) was clinically affected with mild signs (a lesion on the dental pad) at 9 dpc; this was a delay of 2–3 days compared with the clinically affected control group (Gr-5). We observed that three of four vaccinated animals in Gr-4 did not develop fever and were completely protected from clinical disease. Among animals vaccinated by the intranasal route three times with Chi-Tre-Inactivated nanoparticles (Gr-2), two of three vaccinated animals did not show clinical signs of FMD and were completely protected from direct challenge; only one animal (number 39) in this group showed a single lesion and increased rectal temperature. These symptoms were first apparent at 7 dpv, and this animal was considered protected since the disease did not spread systemically, as shown by the absence of viremia and lesions on the feet. Animals 52, 59, and 92 died from illnesses unrelated to FMDV. In contrast, four of five cattle vaccinated by the intranasal route with a single injection of Chi-Tre-Inactivated nanoparticles (Gr-3) were clinically infected; however, the degree of severity of disease was lower than in the control cattle (Table 3).
 | Table 3 Protective responses of experimental cattle, against FMDV in the direct-contact challenge |
Detection of RNA copies in nasal, saliva and oropharyngeal fluid
No viral RNA was detected from either nasal or oropharyngeal fluid from vaccinated, clinically protected cattle. At 5 dpc, the nasal swabs of severely infected cattle (numbers 13, 18, 38, 66, and 32) had a significantly higher viral RNA copy numbers than did the swabs from vaccinated cattle with mild clinical signs (numbers 95 and 65) (Figure 7).
 | Figure 7 FMDV RNA copy number detected by real-time RT-PCR from nasal samples collected from noses of immunized cattle. |
Although a higher copy number of RNA was detected in the nasal swabs of control animals than of the vaccinated, infected cattle, no significant differences (P>0.05) in RNA copy number were found at 1 and 3 dpc (Figure 7). However, in nasal swab or oropharyngeal fluid samples collected from vaccinated, clinically infectious animals, viral RNA was detected up to 9 dpc, irrespective of vaccination status, whereas viral RNA was detected for longer. Viral RNA copy number increased in all infected cattle from 3 to 9 dpc, except for in a vaccinated animal with mild clinical signs (number 95) for which RNA copy number was decreased (Figure 7). The viral RNA copy number was detected by real-time PCR in infected cattle and was lower in saliva samples than in nasal and oropharyngeal fluid (data not shown).
Discussion
Mucosal immunization with polymer-based vaccines by a nasal route is a substitute for conventional immunization that results in a major immunological defense: induction of sIgA that prevents the attachment of bacteria and viruses to the mucosa, preventing entry.11 Efforts are currently underway to produce FMDV vaccines that generate considerable sIgA and maintain high serum IgG titers through mucosal immunization. Our previous study32 demonstrated that substantial cellular and humoral immune responses are elicited by a vaccine of cationic PLGA nanoparticles loaded with FMDV DNA. An intranasal route immunization strategy with this formulation could be developed as an FMDV mucosal vaccine for use in animals following additional testing in protection-challenge experiments and using an animal model that can test the efficacy in this type of challenge.32 However, how immunization with cationic PLGA nanoparticles loaded with FMDV DNA induces protective immunity after challenge in cattle is not known. Since cattle are the most economically important livestock susceptible to FMD,43 demonstrating the feasibility of intranasal delivery of an antigen mediated by nanoparticles in this species is necessary. Our unpublished data showed that cattle immunized with a dry powder nasal vaccine formulation of inactivated FMDV and a mucoadhesive compound developed an FMDV-specific sIgA response in nasal fluid and substantial lymphocyte proliferation. This study investigated these two delivery systems for reinforcing the mucosal immune response, systemic and cell immunity, and protection of cattle against direct-contact challenge at the primary portal for virus entry. We aimed to determine a suitable possible antigen carrier system for nasal administration.
Early evidence that the respiratory tract43 was the natural route of infection implicated the nasal cavity44 or lungs45 as the sites of primary replication of FMDV in cattle. Detailed description of previremic stages after experimental aerosol infection in cattle indicated that the nasopharynx is the most important site of primary infection, that infection initiates in the nasopharynx and is promptly followed by pulmonary infection, and that the onset of viremia is coincident with increased viral load in the lungs and decreased virus in the nasopharynx.46,47 Blocking the primary site of FMDV infection is critical to substantial decreased shedding, transmission, and severity of clinical signs in early FMD epidemics. Induction of FMDV-specific sIgA prevents attachment of viruses to the mucosa, preventing entry, and making blocking the primary site of FMDV infection suitable for mucosal defense. We determined the onset of the local anti-FMDV antibody response in nasal swabs, saliva, and in serum samples collected at days 1, 4, 7, and 10. Specific sIgA antibodies were initially detected at 4 dpv in nasal washes of cattle immunized through an intranasal route but not detected in cattle immunized through an intramuscular route with FMD inactivated vaccine. This result suggested that an intranasal route was superior to an intramuscular route for eliciting mucosal antibodies. All animals vaccinated through an intranasal route had low but detectable FMDV-specific neutralizing antibody responses on the day of challenge and a boost in titer after challenge (data not shown). Titers were significantly lower than neutralizing antibody titers (P<0.05) in the Gr-1 group immunized by an intramuscular route. However, after direct-contact challenge (10 dpv), three of four vaccinated animals in Gr-4 directly exposed to actively infected animals for a 2-day challenge period did not develop fever, were completely protected from clinical disease, and had enhanced nasal mucosal immune responses. The two cattle that were completely protected against contact challenge had the highest induced IgA responses after vaccination. Vaccination with cationic Chi-PLGA-DNA through the intranasal route resulted in better clinical protection of the inoculated cattle, and fewer contacted cattle became infected. In cattle, protection against challenge at 3 weeks after vaccination is correlated with induced virus-neutralizing antibodies caused by vaccination.48 However, Golde et al49 demonstrated that a commercially available, standard-dose vaccine formulation fully protected cattle against direct challenge with virus in as few as 7 days, with partial protection in 4 days. Further, animals challenged on day 4 had delayed and less severe disease and reduced virus in plasma and nasopharyngeal secretions. Our results demonstrated that intranasal delivery to cattle of FMDV antigen mediated by nanoparticles protected against clinical disease after vaccination against FMD as soon as 10 dpv. The reason for the difference in the start and duration of isotype-specific IgA and IgG responses and protection between our result and other studies might be attributed to the more severe challenge regime in our experiments.
To design the challenge regime, we used a 48-hour, direct-contact challenge to simulate an encounter with FMDV. For this, extrapolations were made to estimate challenges in natural conditions. In this study, cohabitation for more than 48 hours was adopted following our preliminary experiments on direct-contact challenge in cattle. Eight control cattle were housed with two infected donor cattle inoculated intradermally in the tongue at 48 hours before challenge with 0.2 mL cattle-passaged FMDV AF/72. After 6, 12, 18, and 24 hours of direct-contact challenge, donors were separated and killed. Of eight control cattle, only two groups (group 18 hours and group 24 hours) were clinically affected within 3–5 days after contact challenge, a delay of 2–3 days compared with the two donors, which showed clinical disease within 24–48 hours after inoculation (unpublished data). Thus, cohabitation for more than 24 hours was suitable for direct-contact challenge of cattle. In our severe challenge model, intranasal delivery with FMDV antigen mediated by nanoparticles did not fully protect cattle against clinical disease; however, it reduced disease severity and virus excretion, and delayed clinical symptoms compared with negative-control animals. Thus, this mucosal vaccine would be likely to reduce the amount of released virus shortly after vaccination and in a field situation, reduce the risk of viral transmission to other herds or vaccinated animals. Even better protection might be observed after less severe FMDV challenge, eg, following indirect exposure to virus in the field when animal movement restrictions are in place.50,51
Empty viral capsids contain the entire repertoire of immunogenic sites of intact virus, are antigenically similar to infectious virus, and are as immunogenic as virions in animals.52,53 Using recombinant DNA technology we produced an FMD construct, pcDNA3.1/P12A3C, that included the coding region for the viral structural protein precursor P1-2A and 3C proteinase, the nonstructural protein required for capsid precursor processing and assembly. However, the construct lacked the coding regions for most other nonstructural proteins.54,55 TEM showed that cells transfected with plasmid pcDNA3.1/P12A3C had detectable empty capsid structures that contained all or most epitopes and induced both humoral and cell-mediated immune responses. We detected isotype-specific sIgA in nasal mucosa samples, and IgA and IgG responses in serum after vaccination of cattle at different time points. The isotype-specific sIgA responses in nasal secretion were initially detected at 4 dpv by an intranasal route, with cationic PLGA nanoparticles loaded with pcDNA3.1/P12A3C vaccine. Responses increased at 7 dpv and 10 dpv. Grubman et al56 reported that the most rapid and long-lasting protection against FMDV challenge in swine was achieved by vaccination with antiviral interferon (IFN)a. Grubman et al found that swine inoculated with the type 1 IFN antiviral agent induced immediate protection, within 1 day, and lasted for 3–5 days. Consequently, to induce an earlier onset of protection and longer immunity duration, the combination of nanoparticle-based nasal vaccines and type 1 IFN could contribute to protection against FMDV infection. This strategy would reduce viral replication and shedding, decreasing the spread of disease and reducing the number of animals that have to be destroyed during an outbreak. Therefore, the induction of mucosal immunity by intranasal immunization requires further research.
We also showed that vaccination with nanoparticles of Chi-Tre-Inactivated through an intranasal route induced systemic, mucosal sIgA responses and partial protection in cattle. To our knowledge, this is the first time that sIgA responses following nasal vaccination with FMDV nanoparticles of whole inactivated virus have been reported, although induction of systemic and mucosal immune responses by nasal vaccination with whole inactivated influenza virus has been reported in rats.34,57 Our results indicated that animals in Gr-2 immunized three times by intranasal route with Chi-Tre-Inactivated nanoparticles on days 1, 4, and 7 had IgA responses that were slightly higher than the group immunized once with two intranasal doses of Chi-Tre-Inactivated nanoparticles (Gr-3) on 7 dpv and 10 dpv. Induction of IgA responses after vaccination and possible protection against challenge with FMDV requires further research.
In conclusion, we showed that although intranasal vaccination with FMDV antigen mediated by nanoparticles did not provide complete clinical or virological protection, it reduced virus excretion significantly, decreased disease severity, and delayed clinical symptoms. Moreover, IgA response after vaccination possibly protected against infection. The results of this study further showed that Chi-PLGA-DNA nanoparticles containing chitosan-coated PLGA-loaded FMDV DNA have potential as a nasal delivery system for vaccines.
Acknowledgments
This work was supported by grants from the Chinese “863” National Programs for High Technology Research and Development (grant number 2011AA10A211), the European Commission’s Seventh Framework Programme (grant number 226556), the International Science and Technology Cooperation Program of Gansu Province (grant number 1011WCGA160), the National Pig Industrial System (CARS-36-068), and the Special Fund for Agro-scientific Research in the Public Interest (201203039). We would wish to thank Dr Junwu Ma (Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Science), for providing the inactivated FMDV antigen, and Dr Huifang Bao (Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Science), for assistance in performing real-time RT-PCR assays.
Disclosure
The authors report no conflicts of interest in this work.
References
Bachrach HL. Foot-and-mouth disease and its antigens. In: Atassi MZ, Bachrach HL, editors. Immunobiology of Proteins and Peptides-III. New York, NY: Plenum Press; 1985:27–46. | ||
Rodriguez LL, Grubman MJ. Foot and mouth disease virus vaccines. Vaccine. 2009;27 Suppl 4:D90–D94. | ||
Cox SJ, Parida S, Voyce C, et al. Further evaluation of higher potency vaccines for early protection of cattle against FMDV direct contact challenge. Vaccine. 2007;25(44):7687–7695. | ||
Grubman MJ, Baxt B. Foot and mouth disease. Clin Microbiol Rev. 2004;17(2):465–493. | ||
Segundo FD, Weiss M, Pérez-Martín E, Dias CC, Grubman MJ, Santos Tde L. Inoculation of swine with foot-and-mouth disease SAP-mutant virus induces early protection against disease. J Virol. 2012;86(3):1316–1327. | ||
Grubman MJ. Development of novel strategies to control foot-and-mouth disease: marker vaccines and antivirals. Biologicals. 2005;33(4):227–234. | ||
Cunliffe HR. Observations on the duration of immunity in cattle after experimental infection with foot-and-mouth disease virus. Cornell Vet. 1964;54:501–510. | ||
Doel TR. Optimisation of the immune response to foot-and-mouth disease vaccines. Vaccine. 1999;17(13–14):1767–1771. | ||
Zanvit P, Havlícková M, Tácner J, et al. Protective and cross-protective mucosal immunization of mice by influenza virus type A with bacterial adjuvant. Immunol Lett. 2008;115(2):144–152. | ||
Neutra MR. M cells in antigen sampling in mucosal tissues. Curr Top Microbiol Immunol. 1999;236:17–32. | ||
Krishnakumar D, Kalaiyarasi D, Bose JC, Jaganathan KS. Evaluation of mucoadhesive nanoparticle based nasal vaccine. Journal of Pharmaceutical Investigation. 2012;42:315–326. | ||
Singleton R, Etchart N, Hou S, Hyland L. Inability to evoke a long-lasting protective immune response to respiratory syncytial virus infection in mice correlates with ineffective nasal antibody responses. J Virol. 2003;77(21):11303–11311. | ||
Liang B, Hyland L, Hou S. Nasal-associated lymphoid tissue is a site of long-term virus-specific antibody production following respiratory virus infection of mice. J Virol. 2001;75(11):5416–5420. | ||
Soane RJ, Frier M, Perkins AC, Jones NS, Davis SS, Illum L. Evaluation of the clearance characteristics of bioadhesive systems in humans. Int J Pharm. 1999;178(1):55–65. | ||
Ugwoke MI, Agu RU, Verbeke N, Kinget R. Nasal mucoadhesive drug delivery: background, applications, trends and future perspectives. Adv Drug Deliv Rev. 2005;57(11):1640–1665. | ||
Csaba N, Garcia-Fuentes M, Alonso MJ. Nanoparticles for nasal vaccination. Adv Drug Deliv Rev. 2009;61(2):140–157. | ||
Zinkernagel RM. Localization dose and time of antigens determine immune reactivity. Semin Immunol. 2000;12(3):163–171; discussion 257–344. | ||
Stano A, Nembrini C, Swartz MA, Hubbell JA, Simeoni E. Nanoparticle size influences the magnitude and quality of mucosal immune responses after intranasal immunization. Vaccine. 2012;30(52):7541–7546. | ||
Pérez C, Castellanos IJ, Costantino HR, Al-Azzam W, Griebenow K. Recent trends in stabilizing protein structure upon encapsulation and release from bioerodible polymers. J Pharm Pharmacol. 2002;54(3):301–313. | ||
Jaganathan KS, Vyas SP. Strong systemic and mucosal immune responses to surface-modified PLGA microspheres containing recombinant hepatitis B antigen administered intranasally. Vaccine. 2006;24(19):4201–4211. | ||
Clark MA, Jepson MA, Hirst BH. Exploiting M cells for drug and vaccine delivery. Adv Drug Deliv Rev. 2001;50(1–2):81–106. | ||
Reddy ST, van der Vlies AJ, Simeoni E, et al. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat Biotechnol. 2007;25(10):1159–1164. | ||
Hu Y, Litwin T, Nagaraja AR, et al. Cytosolic delivery of membrane-impermeable molecules in dendritic cells using pH-responsive core-shell nanoparticles. Nano Lett. 2007;7(10):3056–3064. | ||
Knipe T, Rieder E, Baxt B, Ward G, Mason PW. Characterization of synthetic foot-and-mouth disease virus provirions separates acid-mediated disassembly from infectivity. J Virol. 1997;71(4):2851–2856. | ||
Doel TR, Chong WK. Comparative immunogenicity of 146S, 75S and 12S particles of foot-and-mouth disease virus. Arch Virol. 1982;73(2):185–191. | ||
Wang G, Pan L, Zhang Y. Approaches to improved targeting of DNA vaccines. Hum Vaccin. 2011;7(12):1271–1281. | ||
Bordelon H, Biris AS, Sabliov CM, Monroe WT. Characterization of plasmid DNA location within chitosan/PLGA/pDNA nanoparticle complexes designed for gene delivery. J Nanomater [serial on the Internet]. 2011;2011:952060 [about 9 pages]. Available from: http://dx.doi.org/10.1155/2011/952060. Accessed October 19, 2014. | ||
Vila A, Sánchez A, Pérez C, Alonso MJ. PLA-PEG nanospheres: new carriers for transmucosal delivery of proteins and plasmid DNA. Polym Adv Technol. 2002;13(10–12):851–858. | ||
Nafee N, Taetz S, Schneider M, Schaefer UF, Lehr CM. Chitosan-coated PLGA nanoparticles for DNA/RNA delivery: effect of the formulation parameters on complexation and transfection of antisense oligonucleotides. Nanomedicine. 2007;3(3):173–183. | ||
Slütter B, Bal S, Keijzer C, et al. Nasal vaccination with N-trimethyl chitosan and PLGA based nanoparticles: nanoparticle characteristics determine quality and strength of the antibody response in mice against the encapsulated antigen. Vaccine. 2010;28(38):6282–6291. | ||
Xu J, Dai W, Wang Z, Chen B, Li Z, Fan X. Intranasal vaccination with chitosan-DNA nanoparticles expressing pneumococcal surface antigen a protects mice against nasopharyngeal colonization by Streptococcus pneumoniae. Clin Vaccine Immunol. 2011;18(1):75–81. | ||
Wang G, Pan L, Zhang Y, et al. Intranasal delivery of cationic PLGA nano/microparticles-loaded FMDV DNA vaccine encoding IL-6 elicited protective immunity against FMDV challenge. PLoS One. 2011;6(11):e27605. | ||
Garmise RJ, Staats HF, Hickey AJ. Novel dry powder preparations of whole inactivated influenza virus for nasal vaccination. AAPS Pharm Sci Tech. 2007;8(4):E81. | ||
Huang J, Garmise RJ, Crowder TM, et al. A novel dry powder influenza vaccine and intranasal delivery technology: induction of systemic and mucosal immune responses in rats. Vaccine. 2004;23(6):794–801. | ||
Ravi Kumar MN, Bakowsky U, Lehr CM. Preparation and characterization of cationic PLGA nanospheres as DNA carriers. Biomaterials. 2004;25(10):1771–1777. | ||
Rweyemamu MM, Black L, Boge A, Thorne AC, Terry GM. The relationship between the 140S antigen dose in aqueous foot-and-mouth disease vaccines and the serum antibody response of cattle. J Biol Stand. 1984;12(1):111–120. | ||
Garmise RJ, Mar K, Crowder TM, et al. Formulation of a dry powder influenza vaccine for nasal delivery. AAPS Pharm Sci Tech. 2006;7(1):E19. | ||
Hamblin C, Barnett IT, Hedger RS. A new enzyme-linked immunosorbent assay (ELISA) for the detection of antibodies against foot-and-mouth disease virus. I. Development and method of ELISA. J Immunol Methods. 1986;93(1):115–121. | ||
Parida S, Anderson J, Cox SJ, Barnett PV, Paton DJ. Secretory IgA as an indicator of oro-pharyngeal foot-and-mouth disease virus replication and as a tool for post vaccination surveillance. Vaccine. 2006;24(8):1107–1116. | ||
Lyons AB, Parish CR. Determination of lymphocyte division by flow cytometry. J Immunol Methods. 1994;171(1):131–137. | ||
Reid SM, Ebert K, Bachanek-Bankowska K, et al. Performance of real-time reverse transcription polymerase chain reaction for the detection of foot-and-mouth disease virus during field outbreaks in the United Kingdom in 2007. J Vet Diagn Invest. 2009;21(3):321–330. | ||
Bao HF, Li D, Guo JH, et al. A highly sensitive and specific multiplex RT-PCR to detect foot-and-mouth disease virus in tissue and food samples. Arch Virol. 2008;153(1):205–209. | ||
Alexandersen S, Mowat N. Foot-and-mouth disease: host range and pathogenesis. Curr Top Microbiol Immunol. 2005;288:9–42. | ||
Korn G. Experimentelle untersuchungen zum virusnachweis im inkubationsstadium der maulund klauenseuche und zu ihrer pathogneses. Arch Exp Veterinarmed. 1957;11:637–649. German. | ||
Sutmoller P, McVicar JW. Pathogenesis of foot-and-mouth disease: the lung as an additional portal of entry of the virus. J Hyg (Lond). 1976;77(2):235–243. | ||
Arzt J, Pacheco JM, Rodriguez LL. The early pathogenesis of foot-and-mouth disease in cattle after aerosol inoculation. Identification of the nasopharynx as the primary site of infection. Vet Pathol. 2010;47(6):1048–1063. | ||
Pacheco JM, Arzt J, Rodriguez LL. Early events in the pathogenesis of foot-and-mouth disease in cattle after controlled aerosol exposure. Vet J. 2010;183(1):46–53. | ||
Pay TW, Hingley PJ. Correlation of 140S antigen dose with the serum neutralizing antibody response and the level of protection induced in cattle by foot-and-mouth disease vaccines. Vaccine. 1987;5(1):60–64. | ||
Golde WT, Pacheco JM, Duque H, et al. Vaccination against foot-and-mouth disease virus confers complete clinical protection in 7 days and partial protection in 4 days: Use in emergency outbreak response. Vaccine. 2005;23(50):5775–5782. | ||
Salt JS, Barnett PV, Dani P, Williams L. Emergency vaccination of pigs against foot-and-mouth disease: protection against disease and reduction in contact transmission. Vaccine. 1998;16(7):746–754. | ||
Barnett PV, Cox SJ, Aggarwal N, Gerber H, McCullough KC. Further studies on the early protective responses of pigs following immunisation with high potency foot and mouth disease vaccine. Vaccine. 2002;20(25–26):3197–3208. | ||
Lewis SA, Morgan DO, Grubman MJ. Expression, processing, and assembly of foot-and-mouth disease virus capsid structures in heterologous systems: induction of a neutralizing antibody response in guinea pigs. J Virol. 1991;65(12):6572–6580. | ||
Rowlands DJ, Sangar DV, Brown F. A comparative chemical and serological study of the full and empty particles of foot-and mouth disease virus. J Gen Virol. 1975;26(3):227–238. | ||
Chinsangaram J, Beard C, Mason PW, Zellner MK, Ward G, Grubman MJ. Antibody response in mice inoculated with DNA expressing foot-and-mouth disease virus capsid proteins. J Virol. 1998;72(5):4454–4457. | ||
Mayr GA, Chinsangaram J, Grubman MJ. Development of replication-defective adenovirus serotype 5 containing the capsid and 3C protease coding regions of foot-and-mouth disease virus as a vaccine candidate. Virology. 1999;263(2):496–506. | ||
Grubman MJ, Lewis SA, Morgan DO. Protection of swine against foot-and-mouth disease with viral capsid proteins expressed in heterologous systems. Vaccine. 1993;11(8):825–829. | ||
Moon HJ, Lee JS, Talactac MR, et al. Mucosal immunization with recombinant influenza hemagglutinin protein and poly gamma-glutamate/chitosan nanoparticles induces protection against highly pathogenic influenza A virus. Vet Microbiol. 2012;160(3–4):277–289. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.