")
Back to Journals » OncoTargets and Therapy » Volume 13
Individualized Management of Blood Concentration in Patients with Gastrointestinal Stromal Tumors
Received 2 September 2020
Accepted for publication 18 November 2020
Published 5 January 2021 Volume 2020:13 Pages 13345—13355
DOI https://doi.org/10.2147/OTT.S279998
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Federico Perche
Hao Xu,1 Qi Liu2
1Department of Gastrointestinal Surgery, The First Hospital of China Medical University, Shenyang, People’s Republic of China; 2Trauma Center, The First Hospital of China Medical University, Shenyang, People’s Republic of China
Correspondence: Qi Liu
Trauma Center, The First Hospital of China Medical University, Shenyang 110001, People’s Republic of China
Tel/Fax +86-24-83283321
Email [email protected]
Abstract: Gastrointestinal stromal tumor (GIST) is the most common mesenchymal tumor, and surgical resection is the first choice for the treatment of the disease, but since the advent of tyrosine kinase inhibitors (TKIs) such as imatinib (IM), the prognosis of the disease has undergone revolutionary changes. According to the current version of the guidelines, most GIST patients receive a fixed dose without taking into account their own individual differences, resulting in a wide difference in blood concentration, adverse reactions and prognosis. With more studies on the relationship between blood drug concentrations and prognosis, the concept of individualized therapy has been paid more attention by researchers. Therapeutic drug monitoring (TDM) has also been made available for the research field of GIST targeted therapy. How to reduce the incidence of drug resistance and adverse reactions in patients with GISTs has become the focus of the current research. This article reviews the common monitoring methods and timing of TKIs blood concentration, the reasonable range of blood drug concentration, the toxic or adverse effects caused by high blood drug concentration, some possible factors affecting blood drug concentration and recent research progress, in order to discuss and summarize the treatment strategy of individual blood drug concentration, improve the prognosis of patients and reduce the adverse effects as much as possible.
Keywords: GIST, therapeutic drug monitoring, TDM, imatinib, sunitinib, targeted therapy
Background and Introduction
GIST is the most common mesenchymal tumor, which can originate from any part of the gastrointestinal tract, even outside the gastrointestinal tract.1 Since the advent of TKIs represented by IM, revolutionary changes have taken place in the treatment of GIST, which has greatly improved the prognosis, especially those GISTs that cannot be resected or metastasized. At present, the most commonly used risk classification method for recurrence is M-NIH or AFIP classification,2 but more uncertain factors are still being confirmed, such as gastrointestinal bleeding and high Ki67 index may lead to poor prognosis of GIST.3–5 However, TKIs such as IM are often used at a fixed dose, resulting in differences in prognosis and toxicity among patients, which may be attributed to different blood concentrations among the patients. At present, oral TKIs, such as IM and sunitinib (SU) have high individual pharmacokinetic variability, but they are taken orally at a fixed dose, without considering the differences of individual factors such as body mass index (BMI), body surface area (BSA), etc. Because there are many factors that affect the fluctuation of blood drug concentration of TKIs, it is difficult to use a fixed dose of TKI to make all GIST patients reach a reasonable blood concentration range. Besides, there may be differences in tolerance to the same dose of TKIs because of ethnic or weight differences in Europe, America, or Asia. For example, in patients with drug resistance or KIT exon 9 mutations, the recommended dose for Chinese patients is 600 mg, instead of 800 mg, as recommended in the NCCN guidelines because the tolerance of Chinese patients to high doses of IM is significantly lower than the patients in Europe and the United States.6,7 A study shows that the Cmin of IM in Chinese GIST patients are higher than that in Europe at the same dose.8
For patients with GISTs treated with TKIs, due to the influence of economic, adverse effects and other factors, the number of patients terminating treatment is gradually increasing, and patient compliance plays an important role in the treatment.9 Patients with poor compliance may cause fluctuations in blood concentrations and even develop drug resistance. Some studies have confirmed that the overall survival (OS) of patients with high blood concentration of IM is longer than that of patients with low blood concentration.10 However, continuous high blood concentration will bring unnecessary adverse effects and toxicity, so the monitoring of TKI blood concentration can not be ignored.11
Current research shows that the concept of individualized therapy has attracted more attention. In the past, TDM was often used to detect the blood concentration of antibiotics, immunosuppressants and antiepileptic drugs, but for various reasons, TDM is not common in GISTs.12 However, TDM is a very effective individualized therapy and will be beneficial to the prognosis of patients. Recent studies have shown that TDM can improve the safety and efficacy of a variety of oral targeted anticancer drugs.13 TDM can not only provide dose guidance during treatment but also reduce the total cost of clinical treatment.14 Although few prospective clinical studies have confirmed the safety and efficacy of TDM in TKIs such as IM and SU. However, a large number of retrospective studies have shown that TDM is indispensable for the treatment of GISTs. Therefore, this article reviews the range of blood drug concentration of GIST-related TKIs, the time and method of measuring blood drug concentration, and the factors that may affect the blood drug concentration, in order to explore and summarize the treatment strategy of individual blood drug concentration in GIST patients, so as to make each GIST patient reach the standard of “personal tailor”, so as to improve the prognosis of patients and reduce the adverse effects of patients.
Opportunity for Monitoring the Blood Concentration of TKIs
IM is metabolized mainly by CYP3A4 and CYP3A5 in liver and small intestine,15 and the blood concentration is affected by many factors. However, at present, the clinical application of TDM is still not popular, but routine blood concentration monitoring is very helpful for clinical treatment. At present, high-performance liquid chromatography-tandem mass spectrometry (HPLC-tandem MS/MS) is widely used to monitor the blood concentration of IM.
It is generally believed that the pharmacokinetics of IM or SU is rarely affected by clinical-related factors. Moreover, the pharmacokinetics of GISTs and Chronic Myelocytic Leukemia (CML) patients were similar. The blood concentration of IM reached the peak after 1.8–4.0 hours (Cmax),16 but it will take 6–12 hours for SU to reach the Cmax.17 However, the important monitoring index that can reflect the therapeutic effect is based on the steady-state blood trough concentration (Cmin) level of IM or SU, and Cmin is relatively stable and easier to be monitored. Demetri et al found that18 the GIST patients who used IM and obtained steady-state Cmin were divided into quartiles, and the Cmin was higher than the lowest quartile of 1100ng/mL, their disease was effectively controlled. This is also a commonly used reference threshold, although the optimal blood concentration range of IM and SU is still controversial, as shown in Table 1.
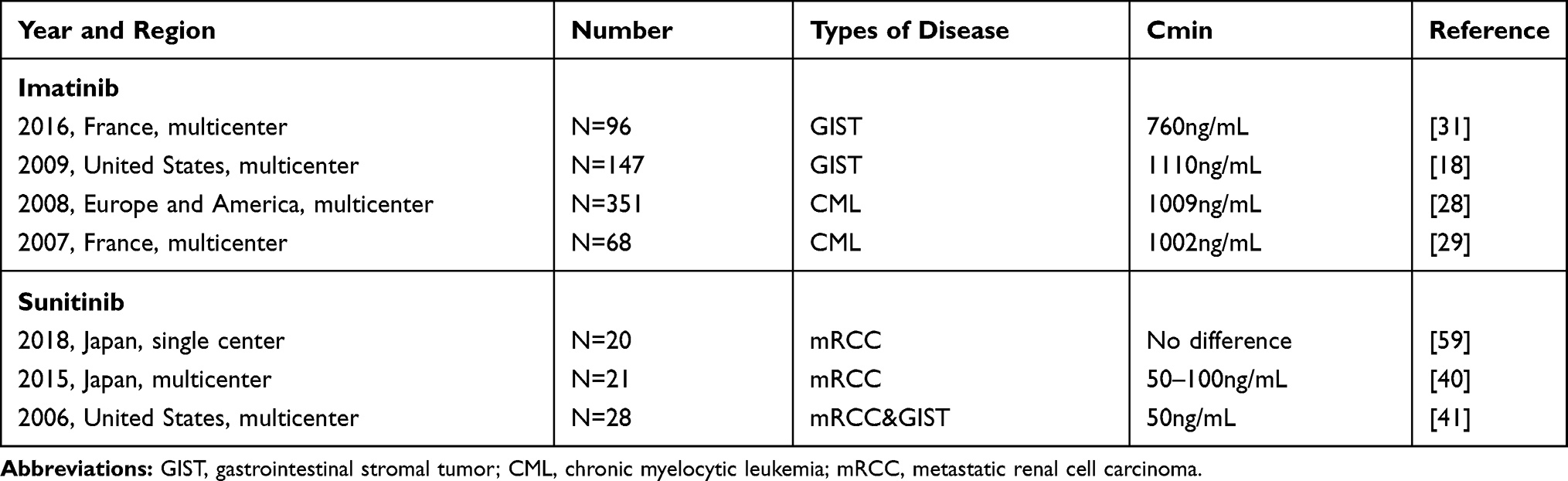 |
Table 1 Clinical Trials of Blood Concentration of Imatinib and Sunitinib |
Pharmacokinetic studies of IM in GISTs, CMLs and healthy volunteers showed that its bioavailability was 98%,15 while the bioavailability of SU was relatively low, about 50%.19 Recent studies have found that one month after the first administration, the bioavailability of IM decreased by about 17%, and three months after the first administration, the bioavailability of the IM gradually decreased by about 30%, resulting in a gradual decrease in blood concentration.20 A retrospective pharmacokinetic analysis also confirmed that the clearance rate of IM increased over time.21 Therefore, it is not recommended to measure the blood concentration within one month. It is recommended that the time to measure the blood concentration is to use IM regularly for 1–3 months, and start to monitor the blood concentration once every 3–6 months. After reaching a stable state, the monitoring period can be appropriately prolonged.
Because the half-life of IM is about 18–20 hours, in the case of oral administration once a day, the time to measure the blood concentration is 24 hours ±3 hours after administration,15,22 while the half-life of SU is longer, about 40–60 hours.23 If we infer from the Cmin method of calculating IM, the Cmin of SU should appear 24 hours ±7 hours after taking the medicine.24 However, due to the limitations of various conditions, it is difficult for some patients to collect samples in an appropriate time range for blood concentration monitoring, resulting in inaccurate results.
The Optimal Range of Blood Concentration of TKIs
The Optimal Range of Blood Concentration of IM
The bioavailability of IM after oral administration is more than 90%, which is widely metabolized and cleared in the liver, and 68% of the IM is eventually excreted through feces.15 Although IM selectively inhibits the phosphorylation of vascular endothelial growth factor R2 and platelet-derived growth factor receptor β in a time-and dose-dependent manner, but it is mainly dose-dependent. The in vitro study of GISTs showed that IM inhibited PDGFR and C-KIT in a concentration-dependent manner. The half inhibitory concentration (IC50) of IM on PDGFR and C-KIT was 49.4 ng/mL. When the blood concentration of IM was 493.6 ng/mL, c-Kit was completely inhibited,25,26 while when the blood concentration was above 50ng/mL, it had a strong inhibitory effect on the above two targets.27
Although there have been many large sample studies on the blood concentration of IM in CML patients, many of the studies in GIST patients are retrospective analysis and lack of prospective studies with a higher level of evidence. For CMLs, when the blood concentration is more than 1000 ng/mL, a better clinical treatment result will be obtained,28,29 At present, clinical treatment also takes 1000ng/mL as the threshold standard to monitor whether the blood concentration of CML patients is in the most appropriate range.
Recent retrospective studies have shown that steady-state Cmin of IM can predict the prognosis of GISTs and CMLs,18,28 so it is important to include the blood concentration monitoring of IM in the guidelines and guide the postoperative management of GIST patients. The B222 study shows that for advanced GIST patients, if the blood concentration is less than 1100mg/mL, the clinical benefit is limited.18 Therefore, 1100ng/mL is often used as an effective threshold to monitor the blood concentration of GIST patients after surgery or neoadjuvant therapy. In a retrospective study,30 the Cmin of 75% of the GIST patients was lower than the efficacy threshold of 1100 ng/mL. But the group of patients were given pharmacokinetic guidance to increase the IM dose, and 63% of the patients were successful. However, PFS was similar in patients with increased dose and those who did not increase the dose of IM. This may be due to the insufficient sample size and the less number of advanced patients. In a French multicenter retrospective study in 2016,31 it was concluded that a stable blood concentration higher than 760ng/mL would achieve better therapeutic results. Therefore, the reasonable range of optimal plasma concentration of IM is still controversial. As for GISTs with KIT exon 9 mutation, metastasis or recurrence, increase the dose to 800mg to make a higher blood concentration, and closely monitor the blood concentration, which can also prolong the relapse free survival (RFS),32 but it is accompanied by the emergence of more adverse effects.
The Optimal Range of Blood Concentration of SU
SU inhibits many tyrosine kinase receptor families, including PDGFR, VEGFR, and C-KIT. It is currently used to treat recurrent or metastatic renal cell carcinoma (mRCC) and IM-resistant, unresectable or advanced GISTs. Of course, SU is currently in a number of clinical trials of many solid tumors, which may be used in more solid tumors. It is well known that the current recommended dose of SU is 50mg/d, for 4 weeks on and 2 weeks off. There is also another option that is taken for 2 weeks and stopped for 1 week, which is to reduce the occurrence of toxic effects.33 Recent studies have shown that the continuous use of 37.5 mg has also achieved good results and is more easily tolerated by patients than the previous two options.34
The absorption of SU is slow after oral administration, it usually takes 6–12 hours to reach the Cmax, and the bioavailability of SU is low, about 50%.19,35 Due to the long elimination half-life of SU, its stable concentration generally appears 10–14 days after the first administration.36 The elimination half-life of N-desethyl-sunitinib, it’s main active metabolite, is longer. More than 60% of the dose of SU was excreted in feces, and only a small portion was found in urine.37 On the other hand, the permeation of SU in cells was not affected by active transport. The transporter can also prevent SU from passing through the blood-brain barrier.
Some recent studies have shown that when the blood concentration of SU is reduced to 50 ng/mL, it is necessary to increase the dose, otherwise it may lead to the decrease of RFS and even increase drug resistance.38,39 But a Japanese study shows that patients with blood concentrations higher than 100ng/mL may have a worse clinical outcome because these patients suffer from severe toxicity.40 Therefore, it is safe, reasonable and evidence-based to consider that the blood concentration of patients should be controlled between 50–100ng/mL. The recommended blood concentration is 60–80mg/mL. Of course, this needs to be confirmed by more studies.41 At present, the blood concentration of SU is recommended to be 50–100ng/mL in clinical treatment. At the same time, for unnecessary adverse effects, the blood concentration of SU should not be higher than that of 100ng/mL.12 A new study shows that switching to SU for drug-resistant GISTs, can achieve a longer progression-free survival than simply increasing the dose of IM.42
As for other new TKIs, such as the third-line drug regorafenib, and the fourth-line drug avapritinib (BLU-285), ripretinib (DCC-2618), which has just been approved by the FDA of the United States, the optimal range of their blood concentration needs further observation and study, through dose optimization and adjustment, so as to maximize the benefit of the GIST patients.
Toxic or Adverse Effects Caused by High Blood Concentration of TKIs
Toxic or Adverse Effects of High Dose of IM
Regular and quantitative use of TKIs is an important factor to ensure the effect of treatment, and reducing the adverse effects caused by TKIs is the premise of better compliance. The therapeutic efficacy and drug adverse effects of IM vary greatly among GISTs,43 and about 98% of patients have grade 1 or 2 adverse reactions during treatment.44 A retrospective study showed that when the blood concentration of IM was more than 3200μg/L, the adverse effects increased significantly.45 The currently recommended blood concentration monitoring flow chart is shown in Figure 1. About 50% of the GIST patients will have different adverse effects after taking TKIs, about 30% of the patients will stop taking TKIs because of serious adverse effects,46 and 25% of the patients will have relatively serious adverse effects after taking TKIs. It may be necessary to suspend the treatment of IM if necessary, and these adverse effects are related to the blood concentration.44 However, it has not been confirmed whether there is a linear relationship between the blood concentration of IM and the adverse effects.47 However, the current study found that neutropenia was not affected by the dose of IM,48,49 but the study also found that increasing the dose to 800mg/d increased the risk of rash by 46.6%.49 In these GIST patients, nausea and vomiting, diarrhea, anemia and skin toxicity are also dose-related or dose-dependent.50 Another study shows that patients with higher blood concentrations are more likely to develop thrombocytopenia,51 and that patients with blood concentrations of higher than 1451.6 ng/mL of IM were more likely to develop myelosuppression.52
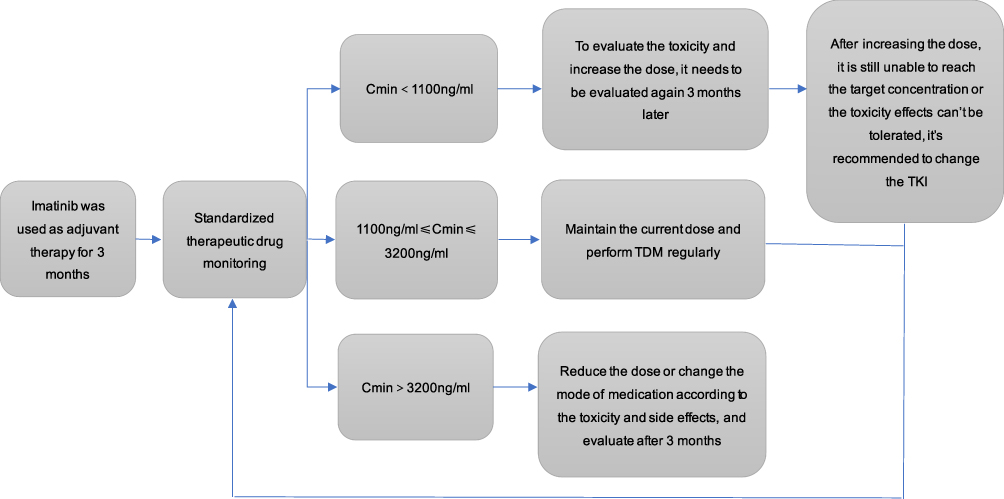 |
Figure 1 Recommended blood drug monitoring flow chart. |
A study found that higher protein levels in the body can significantly reduce the concentrations of free IM and N-desmethyl-imatinib, thus affecting their pharmacological effects. This study also found that the adverse effects of IM were related to the blood concentration of N-desmethyl-imatinib. If the total concentration of IM or free IM concentration reached the threshold of IM or N-desmethyl-imatinib, patients were more likely to have adverse effects.53
Toxic or Adverse Effects of High Dose of SU
Similar to IM, most patients are tolerable with SU. The main adverse effects are anemia, skin discoloration, fatigue, gastrointestinal reactions and stomatitis. Some common toxic or adverse effects and clinical treatment principles of TKIs are listed in Table 2. Rare adverse reactions in a very small number of patients include renal failure, heart failure, pulmonary embolism, gastrointestinal perforation or bleeding.54 Due to the thyroid toxicity of SU, special care should be taken when patients need to use both SU and thyroxine.55 The incidence of asymptomatic proteinuria in patients receiving SU was 21–63%, and the incidence of severe proteinuria was 6.5%, in a dose- and time-dependent manner.56 The consensus in China suggests that reducing the dose of SU from 50 mg/d to 37.5 mg/d, can reduce the degree of neutropenia or thrombocytopenia.57
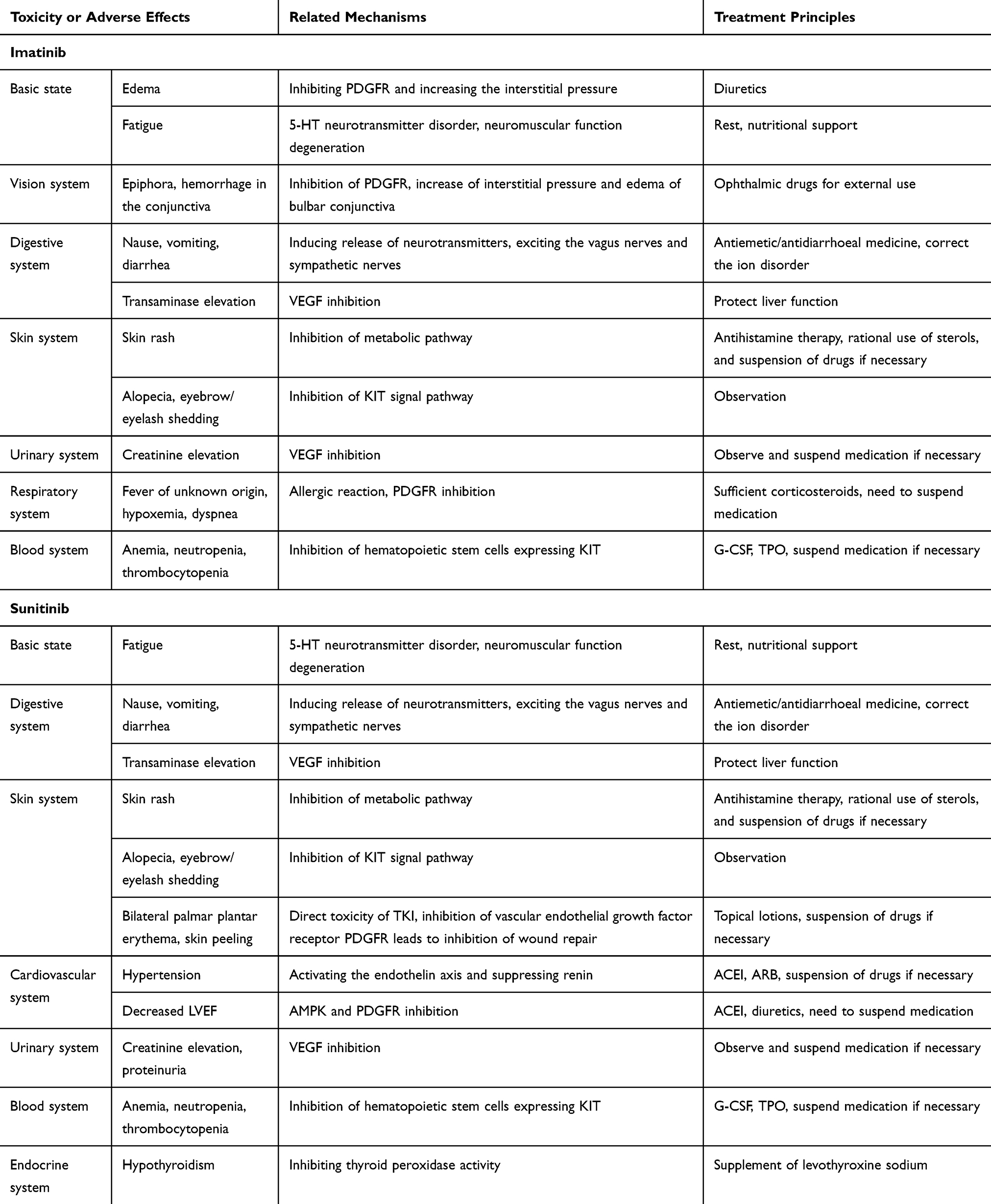 |
Table 2 Some Toxicity or Adverse Effects of TKIs and Clinical Treatment Principles |
In many solid tumors, increasing the dose will get better RFS and OS. A study in patients with mRCC or GIST confirmed that increasing the dose of SU can improve RFS, OS, and have a higher chance of anti-tumor response, but at the same time, it also increases the risk of adverse effects.58
A single center study of mRCC in Japan shows that whether the blood concentration of SU is higher than that of 50ng/mL has no significant difference on OS, but the toxicity should be considered.59 Patients with a concentration of higher than 100 ng/mL had a higher incidence of grade 3 or more toxicity. If patients have serious adverse effects caused by excessive blood concentration of SU, Cmin of SU should be monitored regularly and adjusted appropriately.8
Factors Affecting the Plasma Concentration of TKIs
Drug-Related Factors
The pharmacokinetic differences between different patients, which may lead to sub-or supratherapeutic exposures, and affect the outcome of treatment.14 IM and SU are similar in that they metabolize mainly CYP3A4/5 produced by the liver, so the combination of IM and CYP3A4/5 inhibitors may inhibit the metabolism of IM. For example, ketoconazole can significantly increase the exposure of IM. On the contrary, when used with CYP3A4/5 inducers such as phenytoin sodium, carbamazepine, rifampicin, dexamethasone, etc., IM exposure was reduced. Macrolide antibiotics, azole antifungal agents and HIV protease inhibitors such as indinavir and ritonavir can also lead to a significant decrease in SU blood concentration.12,60 Whether these drugs can be used to increase or reduce blood concentration purposefully and safely requires more research. At present, the combined use of these drugs should pay attention to the fluctuation of blood concentration. Some calcium channel blockers can inhibit CYP3A4, and increase the blood concentration of SU,61 so calcium channel antagonists are not recommended in GIST patients. In addition, although the dose of IM exposure decreased in some patients after subtotal gastrectomy, no significant correlation was found between the use of proton pump inhibitors and imatinib exposure.24
Metabolic Related Factors
IM is mainly metabolized by cytochrome P450 system in the liver, and effluxed by ATP Binding Cassette Subfamily B Member 1 (ABCB1, P-glycoprotein) and2 (ABCG2, BCRP), etc.). IM is mainly metabolized by CYP3A4 into its active metabolite called N-desmethyl metabolite (NDI). At present, the research on individualized treatment of IM is mainly focused on the genetic polymorphism of the drug metabolic enzymes.62 The blood concentration of people over 60 years old is relatively stable because of their slow metabolic level. Diet has no effect on the speed or degree of bioavailability, but it has an effect on blood drug concentration. The excretion of IM is mainly excreted through feces, while SU is excreted through feces and urine.23 Recent studies have shown that some pharmacokinetics models describe the positive correlation between IM clearance and body weight.21 At the same dose of IM, the Cmin of male patients is lower than that of female patients. The possible reason is that the BMI, BSA, or metabolic level of male patients is higher than that of female patients.63 SU is similar to IM, and the clearance rate of SU decreases with the decrease of body weight and BSA.64,65 Women have lower apparent clearance than men, but even if there is such a difference, there is no evidence that women’s doses need to be adjusted.66 With the increase of age, the clearance rate of SU decreased slightly by about 0.7%. At the same time, the clearance rate of SU in females was lower than that in males.67 The blood concentration of SU fluctuates slightly between different ages, but a study shows that the exposure dose of mRCC patients over the age of 70 is significantly lower than that of young patients.68
There is no difference in the metabolism of IM between patients with normal or abnormal liver function, but it is recommended that the maximum dose of mild liver dysfunction is 500mg/d,69 and a small range of GIST liver metastasis will not affect the metabolism of IM. About 50% of the patients in the outpatient follow-up are unable to reach effective drug concentrations, and there is a risk of treatment failure or drug resistance, while about 5% of the patients’ blood concentrations are higher than 2000ng/mL, and their AST, ALT levels are all elevated, which may be related to liver metabolic disorders.24 There was no significant difference in SU exposure between patients with Child-Pugh A or B, but more severe liver injury was not included in the study.70
Studies have found that patients with renal failure have higher blood concentration, which may be hypothesized that patients with renal failure will lead to a decrease in cytochrome P450 activity, affecting the clearance of IM from the liver, and then increase the blood concentration.71 Another hypothesis is that patients with renal failure have higher levels of urotoxin, which may inhibit liver metabolism of IM.72 Similarly, SU dose exposure in patients with abnormal renal function was similar to that in patients with normal renal function. However, among GIST patients who needed hemodialysis, SU exposure decreased to 47% of those with normal renal function.12 And a study suggests that the dose of SU does not need to be adjusted in patients undergoing hemodialysis.73 When to use the drug had no effect on the blood concentration and treatment outcome. But a study shows that patients who take SU in the morning seem to have fewer adverse effects.74
Other Related Factors
For gastric GISTs, surgical resection of part or all of the stomach has a certain impact on the absorption of TKIs. But interestingly, there is a study that shows that the blood concentration of patients with partial or total gastrectomy is significantly lower than that of patients without gastrectomy, but it has little effect after small intestinal resection.31,75 The absorption of IM is affected by gastrectomy, but the gastric prognosis is better than non-gastric, which indicates that the prognosis of GIST is due to the mixture of many factors, and the blood concentration is only one of them. Therefore, after regular oral administration of TKIs after partial or total gastrectomy, the blood concentration should be measured routinely.
The blood concentration of IM varies greatly,76 and there are ethnic differences. At present, the concept of blood concentration monitoring has not been fully popularized, especially in developing countries. In addition, due to a variety of reasons, affected the compliance of patients. If patients are willing to receive regular monitoring of IM blood concentration, it represents that the compliance of these patients will be better. A recent study showed that only about 65% of patients who took IM were tested for blood concentrations.9,77 Of course, patient compliance is an important factor leading to greater differences in RFS and OS, whether in CML or GIST patients.78
Due to the decline of physiological function, elderly patients will have different changes in the distribution, absorption and metabolism of TKIs, coupled with poor tolerance to TKIs, which makes the treatment of the elderly GIST patients more complicated.79 However, A study have shown that there is no significant difference in blood concentration between the elderly and non-elderly patients with the same dose of IM.68 A preclinical experiment has shown that hypoproteinemia may lead to a decrease in the total concentration of SU and N-desethyl-sunitinib as well as increasing the distribution.37 The fluctuation of blood concentration in GIST patients with is related to race, lifestyle and other factors, and this uncertainty needs to be confirmed by more real-world studies.
Mutation analysis of KIT and PDGFRA is the most important for optimal care of GISTs. KIT/PDGFRA wild-type GIST and PDGFRA D842V do not respond to IM or other TKIs, but PDGFRA D842V most may respond to avapritinib.80 SDH-deficient also had no response to IM and limited response to SU or regorafenib. NF1 mutation also have little response to TKIs. As for KIT exon 9 mutation, although this type responds to IM, it usually needs to be increased to 800mg/d. Due to drug resistance or lack of large sample clinical studies, the range of blood drug concentration in these special mutant types of GISTs is still controversial, so individualized treatment is needed.
Conclusions
The current targeted therapy strategy for GISTs is to use a fixed dose of TKI, without considering other factors. But at the same time, there are some voices that support individualized treatment doses to reduce unnecessary toxicity or adverse effects, make patients more tolerable, and gain more clinical benefits.24,39
Of course, the concentration of the TKIs will not be the only factor affecting the prognosis. The plan of individualized treatment is not only based on the pathological parameters of the tumor, or the gene type of the mutation, but also needs to be considered in an all-round way to achieve “Personal Tailor”. The dose of TKI is adjusted by pharmacokinetics and TDM. When this method fails, replacing TKI is also an option.
Disclosure
The authors report no conflicts of interest for this work and declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. Rammohan A, Sathyanesan J, Rajendran K, et al. A gist of gastrointestinal stromal tumors- review. World J Gastrointest Oncol. 2013;5(6):102–112.
2. Joensuu H, Martin-Broto J, Nishida T, Reichardt P, Schoffski P, Maki RG. Follow-up strategies for patients with gastrointestinal stromal tumour treated with or without adjuvant imatinib after surgery. Eur J Cancer. 2015;51(12):1611–1617. doi:10.1016/j.ejca.2015.05.009
3. Liu Q, Kong F, Zhou J, Dong M, Dong Q. Management of hemorrhage in gastrointestinal stromal tumors: a review. Cancer Manag Res. 2018;10:735–743. doi:10.2147/CMAR.S159689
4. Liu Q, Li Y, Dong M, Kong F, Dong Q. Gastrointestinal bleeding is an independent risk factor for poor prognosis in GIST patients. Biomed Res Int. 2017;2017:7152406.
5. Segales-Rojas P, Lino-Silva LS, Aguilar-Cruz E, Salcedo-Hernandez RA. Association of ki67 index with recurrence in gastrointestinal stromal tumors. J Gastrointest Cancer. 2018;49(4):543–547. doi:10.1007/s12029-018-0150-z
6. Li J, Gong JF, Li J, Gao J, Sun NP, Shen L. Efficacy of imatinib dose escalation in Chinese gastrointestinal stromal tumor patients. World J Gastroenterol. 2012;18(7):698–703. doi:10.3748/wjg.v18.i7.698
7. Li J, Shen L. The current status of and prospects in research regarding gastrointestinal stromal tumors in China. Cancer. 2020;126(Suppl 9):2048–2053. doi:10.1002/cncr.32684
8. Xia Y, Chen S, Luo M, et al. Correlations between imatinib plasma trough concentration and adverse reactions in Chinese patients with gastrointestinal stromal tumors. Cancer. 2020;126(Suppl 9):2054–2061. doi:10.1002/cncr.32751
9. Wang Y, Zhang P, Han Y, et al. Adherence to adjuvant imatinib therapy in patients with gastrointestinal stromal tumor in clinical practice: a cross-sectional study. Chemotherapy. 2020:1–8.
10. von Mehren M, Widmer N. Correlations between imatinib pharmacokinetics, pharmacodynamics, adherence, and clinical response in advanced metastatic gastrointestinal stromal tumor (GIST): an emerging role for drug blood level testing? Cancer Treat Rev. 2011;37(4):291–299. doi:10.1016/j.ctrv.2010.10.001
11. Zhang Q, Xu J, Qian Y, et al. Association of imatinib plasma concentration and single-nucleotide polymorphisms with adverse drug reactions in patients with gastrointestinal stromal tumors. Mol Cancer Ther. 2018;17(12):2780–2787. doi:10.1158/1535-7163.MCT-18-0498
12. Demlova R, Turjap M, Pes O, Kostolanska K, Jurica J. Therapeutic drug monitoring of sunitinib in gastrointestinal stromal tumors and metastatic renal cell carcinoma in adults-a review. Ther Drug Monit. 2020;42(1):20–32. doi:10.1097/FTD.0000000000000663
13. Verheijen RB, Yu H, Schellens JHM, Beijnen JH, Steeghs N, Huitema ADR. Practical recommendations for therapeutic drug monitoring of kinase inhibitors in oncology. Clin Pharmacol Ther. 2017;102(5):765–776. doi:10.1002/cpt.787
14. Zuidema S, Desar IME, van Erp NP, Kievit W. Optimizing the dose in patients treated with imatinib as first line treatment for gastrointestinal stromal tumours: a cost-effectiveness study. Br J Clin Pharmacol. 2019;85(9):1994–2001. doi:10.1111/bcp.13990
15. Peng B, Lloyd P, Schran H. Clinical pharmacokinetics of imatinib. Clin Pharmacokinet. 2005;44(9):879–894. doi:10.2165/00003088-200544090-00001
16. Peng B, Hayes M, Resta D, et al. Pharmacokinetics and pharmacodynamics of imatinib in a Phase I trial with chronic myeloid leukemia patients. J Clin Oncol. 2004;22(5):935–942. doi:10.1200/JCO.2004.03.050
17. Westerdijk K, Desar IME, Steeghs N, et al. Imatinib, sunitinib and pazopanib: from flat-fixed dosing towards a pharmacokinetically guided personalized dose. Br J Clin Pharmacol. 2020;86(2):258–273. doi:10.1111/bcp.14185
18. Demetri GD, Wang Y, Wehrle E, et al. Imatinib plasma levels are correlated with clinical benefit in patients with unresectable/metastatic gastrointestinal stromal tumors. J Clin Oncol. 2009;27(19):3141–3147. doi:10.1200/JCO.2008.20.4818
19. Haznedar JO, Patyna S, Bello CL, et al. Single- and multiple-dose disposition kinetics of sunitinib malate, a multitargeted receptor tyrosine kinase inhibitor: comparative plasma kinetics in non-clinical species. Cancer Chemother Pharmacol. 2009;64(4):691–706. doi:10.1007/s00280-008-0917-1
20. Eechoute K, Fransson MN, Reyners AK, et al. A long-term prospective population pharmacokinetic study on imatinib plasma concentrations in GIST patients. Clin Cancer Res. 2012;18(20):5780–5787. doi:10.1158/1078-0432.CCR-12-0490
21. Judson I, Ma P, Peng B, et al. Imatinib pharmacokinetics in patients with gastrointestinal stromal tumour: a retrospective population pharmacokinetic study over time. EORTC soft tissue and bone sarcoma group. Cancer Chemother Pharmacol. 2005;55(4):379–386. doi:10.1007/s00280-004-0876-0
22. Gschwind HP, Pfaar U, Waldmeier F, et al. Metabolism and disposition of imatinib mesylate in healthy volunteers. Drug Metab Dispos. 2005;33(10):1503–1512. doi:10.1124/dmd.105.004283
23. Bello CL, Sherman L, Zhou J, et al. Effect of food on the pharmacokinetics of sunitinib malate (SU11248), a multi-targeted receptor tyrosine kinase inhibitor: results from a phase I study in healthy subjects. Anticancer Drugs. 2006;17(3):353–358. doi:10.1097/00001813-200603000-00015
24. Lankheet NA, Knapen LM, Schellens JH, Beijnen JH, Steeghs N, Huitema AD. Plasma concentrations of tyrosine kinase inhibitors imatinib, erlotinib, and sunitinib in routine clinical outpatient cancer care. Ther Drug Monit. 2014;36(3):326–334. doi:10.1097/FTD.0000000000000004
25. Buchdunger E, Cioffi CL, Law N, et al. Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. J Pharmacol Exp Ther. 2000;295(1):139–145.
26. Heinrich MC, Griffith DJ, Druker BJ, Wait CL, Ott KA, Zigler AJ. Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor. Blood. 2000;96(3):925–932. doi:10.1182/blood.V96.3.925
27. Keil C, Gotze L, Olbert P, Hofmann R, Nockher WA, Hegele A. [Metastasized renal cell carcinoma. Measurement of plasma levels of the tyrosine kinase inhibitors sunitinib, sorafenib and pazopanib]. Urol A. 2015;54(6):811–818. German. doi:10.1007/s00120-014-3711-1
28. Larson RA, Druker BJ, Guilhot F, et al. Imatinib pharmacokinetics and its correlation with response and safety in chronic-phase chronic myeloid leukemia: a subanalysis of the IRIS study. Blood. 2008;111(8):4022–4028. doi:10.1182/blood-2007-10-116475
29. Picard S, Titier K, Etienne G, et al. Trough imatinib plasma levels are associated with both cytogenetic and molecular responses to standard-dose imatinib in chronic myeloid leukemia. Blood. 2007;109(8):3496–3499. doi:10.1182/blood-2006-07-036012
30. Ijzerman NS, Groenland SL, Koenen AM, et al. Therapeutic drug monitoring of imatinib in patients with gastrointestinal stromal tumours - results from daily clinical practice. Eur J Cancer. 2020;136:140–148. doi:10.1016/j.ejca.2020.05.025
31. Bouchet S, Poulette S, Titier K, et al. Relationship between imatinib trough concentration and outcomes in the treatment of advanced gastrointestinal stromal tumours in a real-life setting. Eur J Cancer. 2016;57:31–38. doi:10.1016/j.ejca.2015.12.029
32. Gastrointestinal Stromal Tumor Meta-Analysis G. Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: a meta-analysis of 1640 patients. J Clin Oncol. 2010;28(7):1247–1253. doi:10.1200/JCO.2009.24.2099
33. Sun Y, Li J, Yang X, Zhang G, Fan X. The alternative 2/1 schedule of sunitinib is superior to the traditional 4/2 schedule in patients with metastatic renal cell carcinoma: a meta-analysis. Clin Genitourin Cancer. 2019;17(5):e847–e59. doi:10.1016/j.clgc.2019.05.003
34. George S, Blay JY, Casali PG, et al. Clinical evaluation of continuous daily dosing of sunitinib malate in patients with advanced gastrointestinal stromal tumour after imatinib failure. Eur J Cancer. 2009;45(11):1959–1968. doi:10.1016/j.ejca.2009.02.011
35. Di Gion P, Kanefendt F, Lindauer A, et al. Clinical pharmacokinetics of tyrosine kinase inhibitors: focus on pyrimidines, pyridines and pyrroles. Clin Pharmacokinet. 2011;50(9):551–603. doi:10.2165/11593320-000000000-00000
36. Goodman VL, Rock EP, Dagher R, et al. Approval summary: sunitinib for the treatment of imatinib refractory or intolerant gastrointestinal stromal tumors and advanced renal cell carcinoma. Clin Cancer Res. 2007;13(5):1367–1373. doi:10.1158/1078-0432.CCR-06-2328
37. Toyama Y, Ueyama J, Nomura H, et al. Contribution of plasma proteins, albumin and alpha 1-acid glycoprotein, to pharmacokinetics of a multi-targeted receptor tyrosine kinase inhibitor, sunitinib, in analbuminemic rats. Anticancer Res. 2014;34(5):2283–2289.
38. Goulooze SC, Galettis P, Boddy AV, Martin JH. Monte Carlo simulations of the clinical benefits from therapeutic drug monitoring of sunitinib in patients with gastrointestinal stromal tumours. Cancer Chemother Pharmacol. 2016;78(1):209–216. doi:10.1007/s00280-016-3071-1
39. Lankheet NAG, Desar IME, Mulder SF, et al. Optimizing the dose in cancer patients treated with imatinib, sunitinib and pazopanib. Br J Clin Pharmacol. 2017;83(10):2195–2204. doi:10.1111/bcp.13327
40. Noda S, Otsuji T, Baba M, et al. Assessment of sunitinib-induced toxicities and clinical outcomes based on therapeutic drug monitoring of sunitinib for patients with renal cell carcinoma. Clin Genitourin Cancer. 2015;13(4):350–358. doi:10.1016/j.clgc.2015.01.007
41. Faivre S, Delbaldo C, Vera K, et al. Safety, pharmacokinetic, and antitumor activity of SU11248, a novel oral multitarget tyrosine kinase inhibitor, in patients with cancer. J Clin Oncol. 2006;24(1):25–35. doi:10.1200/JCO.2005.02.2194
42. Zhang X, Zhou Y, Wu X, et al. Cytoreductive surgery for metastatic gastrointestinal stromal tumors followed by sunitinib compared to followed by imatinib-a multi-center cohort study. Eur J Surg Oncol. 2019;45(3):318–323. doi:10.1016/j.ejso.2018.08.001
43. Gao B, Yeap S, Clements A, Balakrishnar B, Wong M, Gurney H. Evidence for therapeutic drug monitoring of targeted anticancer therapies. J Clin Oncol. 2012;30(32):4017–4025. doi:10.1200/JCO.2012.43.5362
44. Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347(7):472–480. doi:10.1056/NEJMoa020461
45. Guilhot F, Hughes TP, Cortes J, et al. Plasma exposure of imatinib and its correlation with clinical response in the tyrosine kinase inhibitor optimization and selectivity trial. Haematologica. 2012;97(5):731–738. doi:10.3324/haematol.2011.045666
46. DeMatteo RP, Ballman KV, Antonescu CR, et al. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: a randomised, double-blind, placebo-controlled trial. Lancet. 2009;373(9669):1097–1104. doi:10.1016/S0140-6736(09)60500-6
47. Lipton JH, Chuah C, Guerci-Bresler A, et al. Ponatinib versus imatinib for newly diagnosed chronic myeloid leukaemia: an international, randomised, open-label, Phase 3 trial. Lancet Oncol. 2016;17(5):612–621. doi:10.1016/S1470-2045(16)00080-2
48. van Oosterom AT, Judson I, Verweij J, et al. Safety and efficacy of imatinib (STI571) in metastatic gastrointestinal stromal tumours: a phase I study. Lancet. 2001;358(9291):1421–1423. doi:10.1016/S0140-6736(01)06535-7
49. Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364(9440):1127–1134. doi:10.1016/S0140-6736(04)17098-0
50. Ben Ami E, Demetri GD. A safety evaluation of imatinib mesylate in the treatment of gastrointestinal stromal tumor. Expert Opin Drug Saf. 2016;15(4):571–578. doi:10.1517/14740338.2016.1152258
51. Francis J, Dubashi B, Sundaram R, Pradhan SC, Chandrasekaran A. A study to explore the correlation of ABCB1, ABCG2, OCT1 genetic polymorphisms and trough level concentration with imatinib mesylate-induced thrombocytopenia in chronic myeloid leukemia patients. Cancer Chemother Pharmacol. 2015;76(6):1185–1189. doi:10.1007/s00280-015-2905-6
52. Zhuang W, Xie JD, Zhou S, et al. Can therapeutic drug monitoring increase the safety of Imatinib in GIST patients? Cancer Med. 2018;7(2):317–324. doi:10.1002/cam4.1286
53. Qian Y, Sun LN, Liu YJ, et al. Genetic polymorphisms and adverse events on unbound imatinib and its active metabolite concentration in patients with gastrointestinal stromal tumors. Front Pharmacol. 2019;10:854. doi:10.3389/fphar.2019.00854
54. Ibrahim EM, Kazkaz GA, Abouelkhair KM, Bayer AM, Elmasri OA. Sunitinib adverse events in metastatic renal cell carcinoma: a meta-analysis. Int J Clin Oncol. 2013;18(6):1060–1069. doi:10.1007/s10147-012-0497-2
55. Kollmannsberger C, Soulieres D, Wong R, Scalera A, Gaspo R, Bjarnason G. Sunitinib therapy for metastatic renal cell carcinoma: recommendations for management of side effects. Can Urol Assoc J. 2007;1(2 Suppl):S41–S54.
56. Izzedine H, Massard C, Spano JP, Goldwasser F, Khayat D, Soria JC. VEGF signalling inhibition-induced proteinuria: mechanisms, significance and management. Eur J Cancer. 2010;46(2):439–448. doi:10.1016/j.ejca.2009.11.001
57. Li J, Wang M, Zhang B, et al. Chinese consensus on management of tyrosine kinase inhibitor-associated side effects in gastrointestinal stromal tumors. World J Gastroenterol. 2018;24(46):5189–5202.
58. Houk BE, Bello CL, Poland B, Rosen LS, Demetri GD, Motzer RJ. Relationship between exposure to sunitinib and efficacy and tolerability endpoints in patients with cancer: results of a pharmacokinetic/pharmacodynamic meta-analysis. Cancer Chemother Pharmacol. 2010;66(2):357–371. doi:10.1007/s00280-009-1170-y
59. Takasaki S, Kawasaki Y, Kikuchi M, et al. Relationships between sunitinib plasma concentration and clinical outcomes in Japanese patients with metastatic renal cell carcinoma. Int J Clin Oncol. 2018;23(5):936–943. doi:10.1007/s10147-018-1302-7
60. Thomas-Schoemann A, Blanchet B, Bardin C, et al. Drug interactions with solid tumour-targeted therapies. Crit Rev Oncol Hematol. 2014;89(1):179–196.
61. Sica DA. Angiogenesis inhibitors and hypertension: an emerging issue. J Clin Oncol. 2006;24(9):1329–1331. doi:10.1200/JCO.2005.04.5740
62. Liu J, Chen Z, Chen H, et al. Genetic polymorphisms contribute to the individual variations of imatinib mesylate plasma levels and adverse reactions in Chinese GIST patients. Int J Mol Sci. 2017;18(3).
63. Wu X, Li J, Zhou Y, et al. Relative factors analysis of imatinib trough concentration in Chinese patients with gastrointestinal stromal tumor. Chemotherapy. 2018;63(6):301–307. doi:10.1159/000493195
64. Narjoz C, Cessot A, Thomas-Schoemann A, et al. Role of the lean body mass and of pharmacogenetic variants on the pharmacokinetics and pharmacodynamics of sunitinib in cancer patients. Invest New Drugs. 2014;33(1):257–268. doi:10.1007/s10637-014-0178-2
65. Chae JW, Teo YL, Ho HK, et al. BSA and ABCB1 polymorphism affect the pharmacokinetics of sunitinib and its active metabolite in Asian mRCC patients receiving an attenuated sunitinib dosing regimen. Cancer Chemother Pharmacol. 2016;78(3):623–632. doi:10.1007/s00280-016-3104-9
66. Houk BE, Bello CL, Kang D, Amantea M. A population pharmacokinetic meta-analysis of sunitinib malate (SU11248) and its primary metabolite (SU12662) in healthy volunteers and oncology patients. Clin Cancer Res. 2009;15(7):2497–2506. doi:10.1158/1078-0432.CCR-08-1893
67. Khosravan R, Motzer RJ, Fumagalli E, Rini BI. Population pharmacokinetic/pharmacodynamic modeling of sunitinib by dosing schedule in patients with advanced renal cell carcinoma or gastrointestinal stromal tumor. Clin Pharmacokinet. 2016;55(10):1251–1269. doi:10.1007/s40262-016-0404-5
68. Crombag MBS, van Doremalen JGC, Janssen JM, et al. Therapeutic drug monitoring of small molecule kinase inhibitors in oncology in a real-world cohort study: does age matter? Br J Clin Pharmacol. 2018;84(12):2770–2778. doi:10.1111/bcp.13725
69. Ramanathan RK, Egorin MJ, Takimoto CHM, et al. Phase I and pharmacokinetic study of imatinib mesylate in patients with advanced malignancies and varying degrees of liver dysfunction: a study by the national cancer institute organ dysfunction working group. J Clin Oncol. 2008;26(4):563–569. doi:10.1200/JCO.2007.11.0304
70. Bello CL, Garrett M, Sherman L, Smeraglia J, Ryan B, Toh M. Pharmacokinetics of sunitinib malate in subjects with hepatic impairment. Cancer Chemother Pharmacol. 2010;66(4):699–707. doi:10.1007/s00280-009-1213-4
71. Gibbons J, Egorin MJ, Ramanathan RK, et al. Phase I and pharmacokinetic study of imatinib mesylate in patients with advanced malignancies and varying degrees of renal dysfunction: a study by the National Cancer Institute Organ Dysfunction Working Group. J Clin Oncol. 2008;26(4):570–576.
72. Franke RM, Sparreboom A. Inhibition of imatinib transport by uremic toxins during renal failure. J Clin Oncol. 2008;26(25):
73. Terada T, Noda S, Inui K. Management of dose variability and side effects for individualized cancer pharmacotherapy with tyrosine kinase inhibitors. Pharmacol Ther. 2015;152:125–134. doi:10.1016/j.pharmthera.2015.05.009
74. Kobuchi S, Ito Y, Okada K, Imoto K, Takada K. Pharmacokinetics of 5-fluorouracil and increased hepatic dihydropyrimidine dehydrogenase activity levels in 1,2-dimethylhydrazine-induced colorectal cancer model rats. Eur J Drug Metab Pharmacokinet. 2013;38(3):171–181.
75. Yoo C, Ryu MH, Kang BW, et al. Cross-sectional study of imatinib plasma trough levels in patients with advanced gastrointestinal stromal tumors: impact of gastrointestinal resection on exposure to imatinib. J Clin Oncol. 2010;28(9):1554–1559. doi:10.1200/JCO.2009.26.5785
76. Zhuang W, Qiu HB, Chen XM, et al. Simultaneous quantification of imatinib and its main metabolite N-demethyl-imatinib in human plasma by liquid chromatography-tandem mass spectrometry and its application to therapeutic drug monitoring in patients with gastrointestinal stromal tumor. Biomed Chromatogr. 2017;31(12). doi:10.1002/bmc.4022.
77. Cardoso E, Csajka C, Schneider MP, Widmer N. Effect of adherence on pharmacokinetic/pharmacodynamic relationships of oral targeted anticancer drugs. Clin Pharmacokinet. 2018;57(1):1–6. doi:10.1007/s40262-017-0571-z
78. Marin D, Bazeos A, Mahon FX, et al. Adherence is the critical factor for achieving molecular responses in patients with chronic myeloid leukemia who achieve complete cytogenetic responses on imatinib. J Clin Oncol. 2010;28(14):2381–2388. doi:10.1200/JCO.2009.26.3087
79. Hurria A, Lichtman SM. Pharmacokinetics of chemotherapy in the older patient. Cancer Control. 2007;14(1):32–43. doi:10.1177/107327480701400105
80. Grunewald S, Klug LR, Muhlenberg T, et al. Resistance to avapritinib in PDGFRA-driven GIST is caused by secondary mutations in the PDGFRA kinase domain. Cancer Discov. 2020.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.