")
Back to Journals » Clinical Epidemiology » Volume 12
Incidence of and Mortality Due to Human Prion Diseases in Taiwan: A Prospective 20-Year Nationwide Surveillance Study from 1998 to 2017
Authors Sun Y , Liu CC, Fan LY, Huang CT, Chen TF, Lu CJ, Guo WY , Chang YC, Chiu MJ
Received 27 July 2020
Accepted for publication 23 September 2020
Published 14 October 2020 Volume 2020:12 Pages 1073—1081
DOI https://doi.org/10.2147/CLEP.S274093
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Eyal Cohen
Yu Sun,1,2 Chih-Ching Liu,3,* Ling-Yun Fan,4,* Chung-Te Huang,5 Ta-Fu Chen,2 Chien-Jung Lu,1,2 Wan-Yuo Guo,6,7 Yang-Chyuan Chang,2,8 Ming-Jang Chiu2,9– 11
1Department of Neurology, En Chu Kong Hospital, New Taipei City, Taiwan; 2Department of Neurology, National Taiwan University Hospital, College of Medicine, National Taiwan University, Taipei, Taiwan; 3Department of Healthcare Administration, College of Medical and Health Science, Asia University, Taichung, Taiwan; 4Queensland Brain Institute, University of Queensland, St. Lucia, Brisbane, QLD, Australia; 5Center for Research, Diagnostics and Vaccine Development, Taiwan Centers for Disease Control, Taipei, Taiwan; 6Department of Radiology, Taipei Veterans General Hospital, Taipei, Taiwan; 7School of Medicine, National Yang-Ming University, Taipei, Taiwan; 8Department of Neurology, Min-Sheng General Hospital, Taoyuan, Taiwan; 9Graduate Institute of Brain and Mind Sciences, College of Medicine, National Taiwan University, Taipei, Taiwan; 10Graduate Institute of Psychology, College of Science, National Taiwan University, Taipei, Taiwan; 11Graduate Institute of Biomedical Electronics and Bioinformatics, National Taiwan University, Taipei, Taiwan
*These authors contributed equally to this work
Correspondence: Ming-Jang Chiu Tel +886-2-23123456 ext 65339
Fax +886-2-23418395
Email [email protected]
Introduction: Epidemiologic studies of Creutzfeldt-Jakob disease (CJD) have been undertaken worldwide since the new variant CJD outbreak in 1996 in the United Kingdom. A nationwide report system, the Creutzfeldt-Jakob Disease Surveillance Unit (CJDSU), directed by the Centers for Disease Control of Taiwan, was established in 1997 to identify human prion diseases.
Methods: From 1998 to 2017, 647 cases were referred to the committee for confirmation. The report to CJDSU included a structured questionnaire recording the clinical, demographic data, and potential iatrogenic exposure, and the results of the clinical and laboratory examination, including tests of blood and cerebrospinal fluid, electroencephalography, and brain magnetic resonance imaging.
Results: In total, 356 cases (women, n=178) were ascertained to be human prion diseases, and 97.4% (n=347) were sporadic CJD, including three definite, 314 probable, and 30 possible cases; one probable variant CJD and 8 cases of the genetic form human prion diseases. The age- and gender-specific average annual incidence were also significantly higher in the second decade (0.95/1,000,000) than in the first decade (0.63/1,000,000), with an incidence rate ratio of 1.51. The incidences increased with increasing age, reaching a peak at the age of 70– 79 years. The 10-year survival curve for sCJD patients showed that the 1-, 5-, and 10-year cumulative survival rate were 52%, 5%, and 1%, respectively. PRNP polymorphisms in 170 patients showed that 98.8% were M129M and 97.6% E219E.
Discussion: The significant increase in incidence after 2008 suggests the increase in the awareness of this rare disease among physicians. The longer disease duration in patients with sCJD in Taiwan than in other countries indicates that the comprehensive support of the health care system, as well as the end-of-life care culture in Taiwan, may prolong survival time in patients with such a progressive and fatal disease.
Keywords: human prion diseases, spongiform encephalopathy, incidence, mortality, disease duration
Introduction
Epidemiologic studies on the incidence of Creutzfeldt-Jakob disease (CJD) have been undertaken worldwide since the outbreak of transmissible spongiform encephalopathy when the new variant CJD (vCJD) was first described in 1996 in the United Kingdom.1 A nationwide hospital-based case report system, the Creutzfeldt-Jakob Disease Surveillance Unit (CJDSU), directed by the Centers for Disease Control of Taiwan, was established in 1997 to identify incident cases, including sporadic CJD (sCJD), genetic forms, and acquired forms, such as iatrogenic CJD (iCJD) and variant (vCJD).
The incidence of sCJD is commonly reported to be approximately 1 case per million per year worldwide.2 Our first long-term epidemiological study was published ten years ago, reporting the incidence of CJD in Taiwan from 1998 to 2007.3 The main findings are summarized as follows: (1) all the ascertained cases were sCJD; (2) the overall annual incidence rate remained in the range between 0.5 and 0.6 per million per year, which is lower than in other countries;4,5 (3) the incidence of onset after 80 years of age was still high, without a sharp decline, which was also different from other reports;6,7 and (4) the study of the prion protein gene (PRNP) polymorphism at codon 129 revealed that our patients were all homozygous for methionine.
Globally, most countries reported increasing trends in incidence as CJD surveillance mechanisms have been optimized, the diagnostic testing capabilities have improved, and physicians have developed a greater awareness of this rare disease.8 Since the Taiwan CJDSU has been operating for more than 20 years, whether the incidence of and mortality due to CJD have temporal trends should be assessed. Besides, a very long disease duration was noted in some of our cases. Varied disease durations were reported among countries, with average survival times ranging from 5 to 17.4 months.9–11 Long-term survivors have been reported to have either sCJD or vCJD.9,12–14 The aims of this prospective study are (1) to report all forms of human prion diseases identified in Taiwan; (2) to calculate the temporal trends in incidence by gender and age; and (3) to assess the mortality rate and disease duration.
Method
Surveillance Methods
The operation of the Taiwan CJDSU was reported in our previous article.3 In brief, in 1997, all physicians throughout Taiwan were requested to notify the CJDSU of any clinically suspected CJD patients in their medical practice. Once a case was reported to CJDSU, the expert committee, composed of neurologists, neurosurgeons, neuroradiologists, and neuropathologists, would hold a case-identifying meeting to discuss the reported cases and determine the diagnosis of CJD. Cases ascertained by the CJDSU committee would then be reported to the Taiwan Centers for Disease Control (CDC). The CJDSU and the Taiwan CDC are responsible for following up on all the diagnosed and suspected CJD cases. The clinical diagnosis of definite, probable, or possible CJD was made according to the criteria recommended by the World Health Organization (WHO),15,16 and the updated clinical diagnostic criteria for sCJD.17 The reporting neurologist has to fill out a structured questionnaire recording the medical history of symptoms, clinical course, demographic data, and history of potential iatrogenic exposure such as dura matter implants, corneal grafts, or human cadaveric pituitary hormone, as well as the results of the physical examination and laboratory investigations including biochemistry tests of blood and cerebrospinal fluid (CSF), electroencephalography (EEG) and brain magnetic resonance imaging (MRI). Both EEG and MRI were reviewed by experienced EEG and neuroradiological specialists of the CJDSU committee on an individual basis. Recently, an increasing number of reports by neurologists in Taiwan included the results of tests for autoimmune encephalitis or paraneoplastic encephalopathy. The whole blood and CSF specimens of reported cases were sent to the CJDSU for further tests, including for the CSF 14-3-3 and total Tau proteins. The CSF 14-3-3 protein was measured by Western blot assay using polyclonal anti-14-3-3 pan (AB9748-I) as the primary antibody and goat anti-Rabbit IgG conjugated with HRP (AP307P) as the secondary antibody. The membranes were developed in Western Lightning ECL Pro (PerkinElmer) and visualized with VL FX7 (VILBER). The CSF total-tau protein concentration was assessed by enzyme-linked immunosorbent assay (KBH0042, Invitrogen) directed by the manufacturer with a cut-off value of 1200 pg/mL. The blood sample was used for the PRNP genetic analysis in some cases by the CJDSU.
Statistical Analysis
The crude annual incidence rates and mortality rates were calculated per million person-years with the number of incident cases divided by the total population at risk using the census data for each year in Taiwan. Incidence and mortality rates were provided for the entire population and every 10-year age group by gender and age at disease onset. The confidence intervals for the incidence and mortality rates were calculated according to the Poisson distribution. We adjusted for the gender and age distributions according to the census data for Taiwan in 2010. Poisson regressions were performed to analyze the changes in incidence and mortality rate within genders, age groups, residence areas, and calendar years of recording. Residence areas were classified into the north, middle, south, and east (including the surrounding islands) areas of Taiwan to ascertain CJD cases’ geographical distribution. We also performed trend tests to compare the temporal changes in incidence and mortality rates between the first and second decades. We further examined the survival duration from disease onset in the overall population and gender- and age-specific groups. The standardized mortality ratio (SMR) was calculated as the ratio of observed CJD deaths to expected deaths in the general population based on the age- and gender-specific mortality rates from 1998 to 2017.
Results
Diagnostic Classification and Genetic and Laboratory Studies
From 1998 to 2017, 647 cases were referred to the CJDSU for further confirmation. Among them, 356 cases (women, n=178) were ascertained to be human prion diseases, 97.4% (n=347) were sCJD, including three pathologically validated definite cases, 314 probable and 30 possible; one probable vCJD, and 8 cases of the genetic form human prion diseases. No direct cases of surgically acquired CJD had been documented as of the end of 2017 in Taiwan.
The eight genetically confirmed cases included 4 cases of P102L-129M point mutation, which were endorsed by CJDSU; and 1 of a 72-base pair insertion-129M, and 3 additional cases of point mutations (2 cases of E196A-129M, 1 case of R148H-129M), they were included according to later genetic studies by authors. These four P102L inherited cases belonged to one family with a phenotype of Gerstmann–Sträussler–Scheinker syndrome (GSS). Their mean onset age was 51.5±17.6 years old (range: 29–67 years), with cerebellar ataxia as the onset symptom and a relatively chronic clinical course (mean survival time, 4.6±1.2 years, range, 2.9–5.5 years).18 Analyses of PRNP polymorphisms in 170 patients showed 168 cases of the methionine homozygosity at codon 129 (M129M) (98.8%), while there were 2 cases of methionine/valine (M129V) heterozygosity. Glutamic acid homozygosity at codon 219 (E219E) was found in 166 cases (97.6%), while there were 4 cases of glutamic acid/lysine (E219K) polymorphism heterozygosity. The only vCJD subject was a 34-year-old man who had lived in the UK between 1989 and 1997, with disease onset in 2008. He had early psychiatric and sensory symptoms followed by gait ataxia and cognitive impairment. He had typical pulvinar signs on the brain MRI. The patient developed akinetic mutism at 16 months and died 28 months after onset.19 A positive CSF 14-3-3 protein test was found in 66.7% (2/3) of definite cases, 67.0% (183/275) of probable cases, and 20% (5/25) of possible cases. The CSF 14-3-3 protein test’s sensitivity was 63%, and the specificity was 79.6%, which were calculated from the data after 2011 because the laboratory records of the excluded cases before then were incomplete. With a threshold of 1200 pg/mL, CSF total tau protein tests were positive in 66.7% (2/3) of definite cases, 78.0% (206/265) of probable cases, and 74% (17/23) of possible cases. The CSF total tau protein test’s sensitivity was 77%, and the specificity was 74.1%.
Incidence, Mortality, and Disease Duration
The age-adjusted annual incidences (95% CI) of CJD from 1998 to 2017 in men, women, and the total population were 0.77 (0.61–0.93), 0.82 (0.65–0.98), and 0.79 (0.68–0.91) per million persons, respectively, after adjustment with the Taiwan census data for 2010. There has been a positive trend in the annual number of suspected cases of CJD since 2008. In the first decade (1998 to 2007), the crude incidence remained within the range of 0.5 to 0.6 per million person-years. The crude incidence in the second decade (2008 to 2017) doubled to 1.03, compared to 0.52 in the previous decade. The highest incidence was 1.24 per million persons found between 2012 and 2013 (Table 1). The age- and gender-specific average annual incidence were also significantly higher in the second decade (0.95/1,000,000) than in the first decade (0.63/1,000,000), with an incidence rate ratio of 1.51 (95% CI 1.21–1.88, p<0.01). There was no significant difference in the incidences between men and women. The incidences increased with increasing age, reaching a peak at the age of 70–79 years in both men and women. We observed low incidence rates in those younger than 50 years. Although there was a mild decline, the incidence among patients aged 80 and above was still high (Figure 1). The incidences of sCJD in the middle and eastern regions (including the surrounding islands) were slightly higher than those in Taiwan’s southern region.
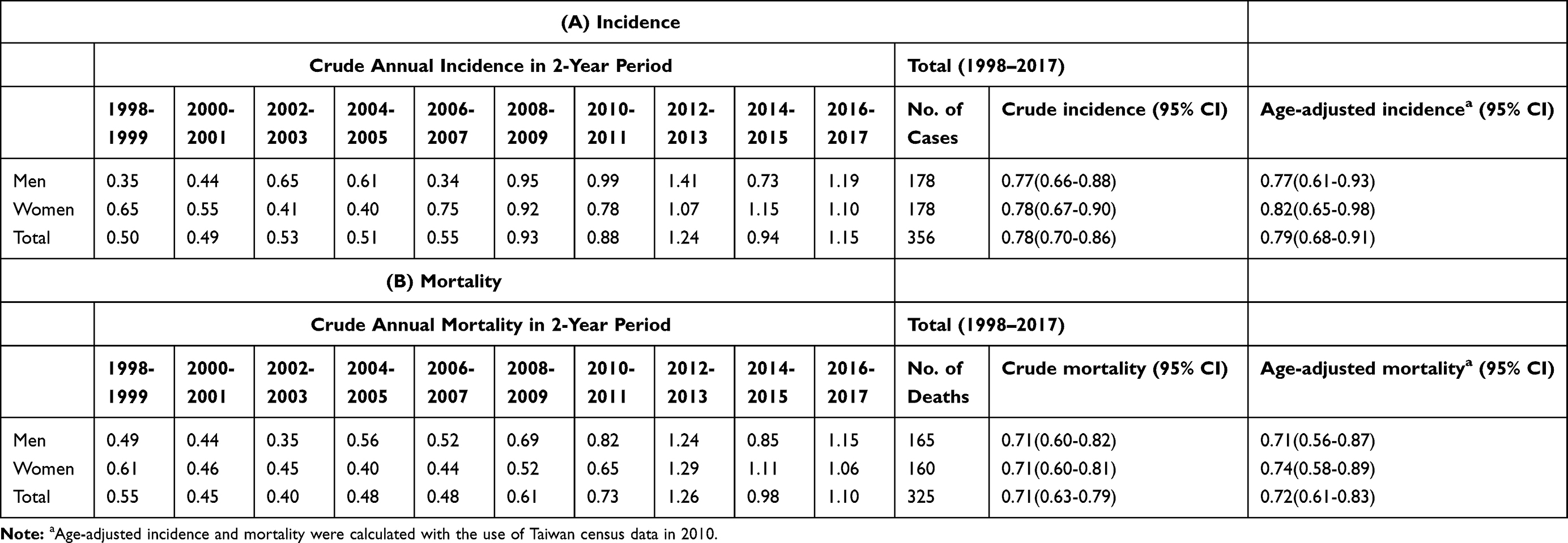 |
Table 1 Biennial Incidence (A) and Mortality Rate (B) per 1,000,000 Persons of CJD in Taiwan from 1998 to 2017 |
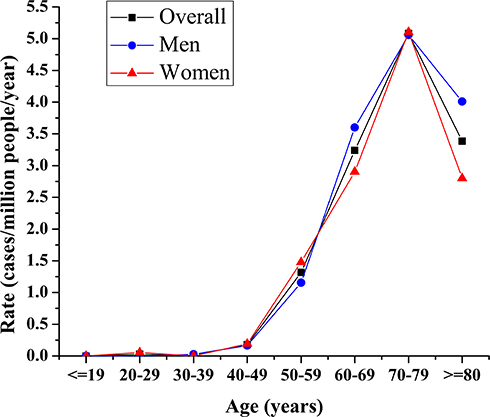 |
Figure 1 Annual Incidence Rate of all CJD Patients by Gender and Age. The incidences increased with increasing age, reaching a peak at the age of 70–79 years in both men and women; the incidence among patients aged 80 and above was still high; low incidence rate observed below 50 years. |
The temporal trend in the mortality rate during the 20 years was similar to the change in incidence. The mortality rates increased after 2008, with the highest rate of 1.26 per million noted between 2012 and 2013. The age-adjusted mortality rates (95% CI) due to CJD from 1998 to 2017 in men, women, and the total population were 0.71 (0.56–0.87), 0.74 (0.58–0.89), and 0.72 (0.61–0.83) per million persons, respectively, after adjustment with the census data from Taiwan in 2010 (Table 1).
The higher incidence of CJD in the second decade resulted in a higher mortality rate than that in the first decade. The average age- and gender-adjusted annual mortality rates in the periods from 1998 to 2007 and 2008 to 2017 were 0.58 and 0.86 per million persons. The adjusted mortality rate ratio of the second decade to the first decade was 1.52 (p=0.01). There was no significant difference in mortality rates between men and women or among geographical areas in Taiwan. Similar to incidence, the highest mortality rate was found in the 70- to 79-year-old age group. The Kaplan–Meier survival curve with a 10-year follow-up after disease onset (onset date before December 31, 2007) is plotted in Figure 2. The cumulative survival rates at the end of the 1st, 5th, and 10th years after onset were 52%, 5%, and 1%, respectively. The median survival time was 13.5 months. The median survival times for patients younger than 50 years and those aged 50–59 years were 37.6 and 22.9 months. The mean (±SD) disease durations were 19.1 (±20.2) months in the total population, 14.3 (±15.6) months in men and 22.3 (±22.8) months in women. Women and patients with younger ages at onset had longer survival times (both p<0.01) (Figure 2).
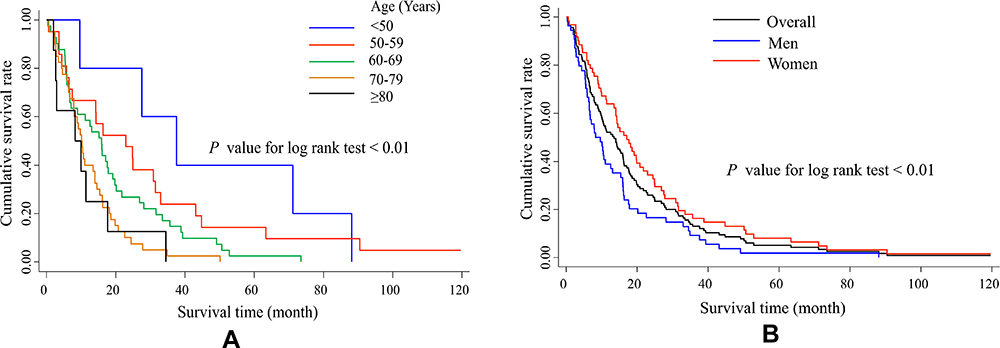 |
Figure 2 Kaplan-Meier Survival Curves for Patients with Sporadic CJD by Age (A) and Gender (B). Kaplan-Meier curves display time from disease onset to death or to the end of 120 months. The 10-year cumulative survival rates for men and women were 0% and 2%, respectively. Longer survival time was noted in patients with younger age at onset (A), and in women than men (B). |
Discussion
Our previous epidemiological research of CJD from 1998 to 2007 has been reported 10 years ago.3 In current analysis, we rechecked the information of all the ascertained cases of the first decade in the registry and found that 9 cases before 2005 should be excluded because they could not meet the diagnostic criteria with some missing data. Moreover, 2 cases that were identified in 2008 actually had their disease onset in 2007 by tracing their history again.
The overall incidence rate of CJD from 1998 to 2017 in Taiwan was 0.79 per million persons. The incidence significantly increased after 2008 to approximately 1 case per million persons per year, comparable to the worldwide incidence, typically reported to be approximately 1–2 per million person-years based on surveillance from 2005 onward.2,20 The increase in incidence for the second decade could be explained by the enhanced awareness of clinical physicians, the improved diagnostic tests, and the increase in the aging population in Taiwan, as the incidence of CJD cases, peaked in the 70- to 79-year-old age group similar distribution to the USA study.21 The incidence of CJD onset after 80 years of age was still high, without a sharp decline, which was different from other reports.6,22 Since the start of the CJDSU in 1997, several national or international symposiums focused on human transmissible spongiform encephalopathy have been held by the Taiwan Neurological Society to educate neurologists and general physicians about the updated diagnostic criteria and management of CJD patients. The Taiwan CDC has regularly revised the workbook.23 In 2008, an imported case of vCJD was reported to CJDSU.19 This first acquired case of CJD in Taiwan was a critical public health concern and might have helped raise awareness of the disease among neurologists, leading to an increase in the number of reported cases afterward. In 2009, an updated set of criteria, including MRI findings, were proposed and soon adopted by the CJDSU.17 The pattern of high signal intensity had high sensitivity and specificity for the differential diagnosis, assisting in distinguishing sCJD from other neurological diseases and helping clinicians detect suspected cases early; this, in part, resulted in the increase in the number of referrals in subsequent years.
The polymorphism at codon 129 (M129V) of PRNP is a recognized genetic marker for susceptibility to CJD in Caucasians.24 In Europe, 51% of the general population has methionine/valine (MV) heterozygosity, while 37% has methionine homozygosity (MM).25,26 In East Asia, the MM genotype was found in 94% of Koreans27 and 92% of Japanese individuals.26 Ethnic Han Chinese, who account for over 95% of the Taiwanese population, has a remarkably high frequency (98%) of methionine homozygosity in the general population.28 Methionine homozygosity is high in both healthy individuals and diseased patients, with 98.8% of CJD patients in our study having the MM genotype and less than 2% carrying the MV genotype. Prion susceptibility and protective alleles can exhibit marked geographic differences, with the effects being different in the Asian and Caucasian populations.26
Because of the short disease duration in most cases, the increase in mortality with time parallels the temporal trend in incidence, as the mortality rate was significantly higher in the second decade than in the first decade. There was no difference in the incidence and mortality rates between men and women in our study. Some countries reported a higher incidence in women than men because of the larger number of women among older populations.21 The disease duration varied based on CJD types, with a relatively chronic course in some genetic forms. The mean survival time was 56.4 (range, 35.1–67.4) months in four GSS patients (P102L), 15.4 (9.0, 21.7) months in two E196A patients, and 11.9 months in an R148H patient, while a substantially shorter disease duration of 2.4 months was noted in a patient with a 72-base pair insertion. Because of the small number of gCJD patients in our study, it is challenging to compare Taiwan with other countries regarding these patients’ survival time.
Among the patients with sCJD, women and those with younger ages at onset had longer disease durations. This finding is comparable with the results from a collaborative multi-national CJD surveillance program (EUROCJD) conducted by the European Union and allied countries. The precise mechanisms underlying the effects of age and gender effects on survival time are not known. Age-related variations in care or resistance to terminal infections have been proposed as possible explanations.29 Whether gender-specific factors influence the disease duration needs to be studied. In this national cohort, we found some long-term survivors of sCJD, with 3-, 5- and 10-year survival rates of 13%, 5%, and 1%, respectively. The median survival time was 13.5 months (mean: 19.1 months), and 48% died within one year of onset. Our data were similar to those in the report from Japan, in which the mean survival time of sCJD patients was 15.7 (range: 1–126) months, and 46.0% of all patients with prion disease died within one year.29 However, reports from other counties revealed a much shorter survival time. The study by the EUROCJD involving 2,451 sCJD patients showed that the median survival time was five months (range: 1–81), and 85.8% died within one year of onset.11 The median duration in a study involving 150 definite or probable sCJD cases in Argentina was 4.6 (range: 1–70) months,30 while a Swedish study involving 123 patients with prion disease found that 74.6% of patients died within one year.31 A study from China also showed a short survival time, with a median duration of 7.1 months (range: 1.0–23.3), and 78.5% of patients died within one year of onset.10
The survival times of patients with sCJD in Taiwan and Japan were relatively longer than those reported in most of the other countries in the world. Japanese researchers explained the prolonged survival as the effects of their robust public medical insurance system and a culture allowing patients with end-stage neurological disease to receive intensive life-sustaining treatments such as tube feeding and intravenous high-calorie infusion.9,32 Taiwan adopted a single-payer National Health Insurance (NHI) system in 1995 that also provides comprehensive healthcare support.33 Patients with human prion disease do not need to pay any co-payment for outpatient or inpatient care. In addition, Taiwanese also share this end-of-life care culture.
There are several limitations to the current study. First, the percentage of definite cases of human spongiform encephalopathies was relatively small. Because of the traditional ethical values among the general population in Taiwanese society, the very low autopsy rate has long been acknowledged as an unfortunate and unavoidable reality. In recent years, using the real-time quaking-induced conversion assay to detect the pathological prion protein in the CSF or other tissues has been developed and has high diagnostic accuracy.34 However, this technique was not used by the CJDSU in Taiwan during our study period. Without obtaining tissue specimens or testing for the pathological prion protein, we are not only unable to make a definite diagnosis but also are unable to assess the various molecular subtypes of sCJD, which is useful for phenotypic classification. Second, underreporting and underestimation of CJD are inevitable. Because of the older age at onset, patients with CJD may be misdiagnosed with other diseases with the rapid progression of neurological symptoms such as stroke,35 encephalitides,36 degenerative dementia,37 and epilepsy.38 A retrospective archival survey published in 1995 showed that only approximately 60% of prion disease patients with pathologically spongiform encephalopathy were diagnosed clinically while alive. In recent decades, diagnostic accuracy has been improved by the use of CSF biomarkers and brain MRI.17 Not infrequently, the committee recommended that the primary care physician follow-up during the clinical course and perform MRI/EEG in case of uncertainty. They were obliged to report back at the next relevant meeting.
Furthermore, due to our NHI system’s comprehensive coverage, patients with spongiform encephalopathy received complete financial support covering all necessary MRI or EEG follow-up. There is nearly a consensus in clinical practice among neurologists in Taiwan that for those patients with rapid cognitive decline, in addition to CSF studies, MRI and EEG examination, tests to exclude autoimmune encephalitis or paraneoplastic encephalopathy should usually be performed. Third, the finding of 2% (8/356) of gCJD among all CJD forms was far lower than the corresponding figures elsewhere in the world, which range from 10–14%.25,39 Although the incidence of gCJD varies considerably among countries, we speculate that the lower proportion of gCJD in our study is likely because the PRNP gene sequencing test was not routinely performed before 2009, only 170 of the 356 CJD cases tested. Approximately 60% of genetic CJD cases were reported in patients with no family history, suggesting that they could have been misclassified in the absence of the PRNP genetic analysis.39
In conclusion, this study reports the 20-year epidemiologic features of CJD in Taiwan. The second decade’s incidence rate was comparable with the corresponding figures in most other countries in the world. The significant increase in incidence after 2008 suggests the increase in awareness of this rare disease among clinical physicians and the increase in Taiwan’s aging population. The longer disease duration in patients with sCJD in Taiwan than in Western countries indicates that the healthcare system and end-of-life care culture in Taiwan may prolong survival time in patients with such a rapidly progressive and fatal disease.
Availability of data and material
The data and raw material could be available after an application to and approval from the Taiwan CDC.
Ethics Approval
The Research Ethics Committee approved the study of the National Taiwan University Hospital (201801019RINB). The written informed consent form was waived because the study was done retrospectively on the National Surveillance Data Bank of Taiwan Centers for Disease Control.
Consent to Participate
All participants received PRNP gene sequencing had provided written informed consent by their proxy on reporting to CDC.
Acknowledgment
We give our sincere thanks to colleagues of Taiwan CDC for their full administrative support and also to our lab colleague Hsu HF for her great endurance in coordination and data managing.
Author Contributions
YS: manuscript drafting, data analysis, and statistical analysis
CCL: statistical analysis
LYF: clinical and laboratory data preprocessing and encoding
CTH: PRNP gene sequence, prion protein Western Blot, tau ELISA measurement, and analysis
TFC: assisting project coordination
CJL: assisting manuscript drafting
WYG: neuroimages ascertain and interpretation
YCC: EEG and clinical data ascertain and interpretation
MJC: manuscript finalization, coordination and integration of all sorts of data acquisition, analysis and interpretation
All the authors have read and approved the manuscript in its final form.
CC Liu and LY Fan contribute equally to this paper.
Funding
This report was supported by a grant (MOHW108-CDC-C- 114-123502) from the Ministry of Health and Welfare Taiwan.
Disclosure
Prof. Dr. Ming-Jang Chiu reports grants from Ministry of Health and Welfare, during the conduct of the study. All authors declare no other potential conflicts of interest.
References
1. Caramelli M, Ru G, Acutis P, Forloni G. Prion diseases: current understanding of epidemiology and pathogenesis, and therapeutic advances. CNS Drugs. 2006;20(1):15–28. doi:10.2165/00023210-200620010-00002
2. Uttley L, Carroll C, Wong R, Hilton DA, Stevenson M. Creutzfeldt-Jakob disease: a systematic review of global incidence, prevalence, infectivity, and incubation. Lancet Infect Dis. 2020;20(1):e2–e10. doi:10.1016/S1473-3099(19)30615-2
3. Lu CJ, Sun Y, Chen SS. Incidence of Creutzfeldt-Jakob disease in Taiwan: a prospective 10-year surveillance. Eur J Epidemiol. 2010;25(5):341–347. doi:10.1007/s10654-010-9446-4
4. Doi Y, Yokoyama T, Sakai M, Nakamura Y. Creutzfeldt-Jakob disease mortality in Japan, 1979–2004: analysis of national death certificate data. J Epidemiol. 2007;17(4):133–139. doi:10.2188/jea.17.133
5. Ruegger J, Stoeck K, Amsler L, et al. A case-control study of sporadic Creutzfeldt-Jakob disease in Switzerland: analysis of potential risk factors with regard to an increased CJD incidence in the years 2001–2004. BMC Public Health. 2009;9:18. doi:10.1186/1471-2458-9-18
6. Cousens SN, Zeidler M, Esmonde TF, et al. Sporadic Creutzfeldt-Jakob disease in the United Kingdom: analysis of epidemiological surveillance data for 1970–96. BMJ. 1997;315(7105):389–395. doi:10.1136/bmj.315.7105.389
7. Brown P, Cathala F, Raubertas RF, Gajdusek DC, Castaigne P. The epidemiology of Creutzfeldt-Jakob disease: conclusion of a 15-year investigation in France and review of the world literature. Neurology. 1987;37(6):895–904. doi:10.1212/WNL.37.6.895
8. Li CH, Fan SP, Chen TF, Chiu MJ, Yen RF, Lin CH. Frontal variant of Alzheimer’s disease with asymmetric presentation mimicking frontotemporal dementia: case report and literature review. Brain Behav. 2020;10(3). doi:10.1002/brb3.1548
9. Nagoshi K, Sadakane A, Nakamura Y, Yamada M, Mizusawa H. Duration of prion disease is longer in Japan than in other countries. J Epidemiol. 2011;21(4):255–262. doi:10.2188/jea.JE20100085
10. Chen TF, Tang MC, Chou CH, Chiu MJ, Huang RFS. Dose-dependent folic acid and memantine treatments promote synergistic or additive protection against A beta((25–35)) peptide-induced apoptosis in SH-SY5Y cells mediated by mitochondria stress-associated death signals. Food Chem Toxicol. 2013;62:
11. Collins SJ, Sanchez-Juan P, Masters CL, et al. Determinants of diagnostic investigation sensitivities across the clinical spectrum of sporadic Creutzfeldt-Jakob disease. Brain: J Neurol. 2006;129(Pt 9):2278–2287. doi:10.1093/brain/awl159
12. D’Arcy CE, Bitnun A, Coulthart MB, et al. Sporadic Creutzfeldt-Jakob disease in a young girl with unusually long survival. J Neuropathol Exp Neurol. 2019;78(4):373–378. doi:10.1093/jnen/nlz013
13. Hayashi Y, Iwasaki Y, Waza M, et al. Clinicopathological findings of a long-term survivor of V180I genetic Creutzfeldt-Jakob disease. Prion. 2020;14(1):109–117. doi:10.1080/19336896.2020.1739603
14. Wang J, Xiao K, Zhou W, Shi Q, Dong XP. Analysis of 12 Chinese patients with proline-to-leucine mutation at codon 102-associated Gerstmann-Straussler-Scheinker disease. J Clin Neurol. 2019;15(2):184–190. doi:10.3988/jcn.2019.15.2.184
15. WHO. Global surveillance, diagnosis and therapy of human transmissible spongiform encephalopathies: report of a WHO consultation, Geneva, Switzerland, 9–11 February 1998. 1998. Available from: https://appswhoint/iris/handle/10665/65516.
16. World Health Organization (WHO). WHO manual for surveillance of human transmissible spongiform encephalopathies including variant Creuzfeldt-Jakob disease. Geneva: World Health Organization; 2003.
17. Zerr I, Kallenberg K, Summers DM, et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain: J Neurol. 2009;132(10):2659–2668. doi:10.1093/brain/awp191
18. Chi NF, Lee YC, Lu YC, Wu HM, Soong BW. Transmissible spongiform encephalopathies with P102L mutation of PRNP manifesting different phenotypes: clinical, neuroimaging, and electrophysiological studies in Chinese kindred in Taiwan. J Neurol. 2010;257(2):191–197. doi:10.1007/s00415-009-5290-4
19. Yang CW, Fuh JL, Wang SJ, Lirng JF, Yang CC, Cheng SJ. Probable variant Creutzfeldt-Jakob disease in Asia: a case report from Taiwan and review of two prior cases. Psychiatry Clin Neurosci. 2010;64(6):652–658. doi:10.1111/j.1440-1819.2010.02151.x
20. Creutzfeldt-Jakob Disease International Surveillance Network CJD surveillance data 1993–2018. 2018. Available from: https://www.eurocjd.ed.ac.uk/surveillance data 1.html - scjd-deaths.
21. Holman RC, Belay ED, Christensen KY, et al. Human prion diseases in the United States. PLoS One. 2010;5(1):e8521. doi:10.1371/journal.pone.0008521
22. Gao C, Shi Q, Tian C, et al. The epidemiological, clinical, and laboratory features of sporadic Creutzfeldt-Jakob disease patients in China: surveillance data from 2006 to 2010. PLoS One. 2011;6(8):e24231. doi:10.1371/journal.pone.0024231
23. Ta AT, Huang SE, Chiu MJ, et al. Age-related vulnerabilities along the hippocampal longitudinal axis. Hum Brain Mapp. 2012;33(10):2415–2427. doi:10.1002/hbm.21364
24. Mitrová E, Mayer V, Jovankovicová V, Slivarichová D, Wsólová L. Creutzfeldt-Jakob disease risk and PRNP codon 129 polymorphism: necessity to revalue current data. Eur J Neurol. 2005;12(12):998–1001. doi:10.1111/j.1468-1331.2005.01110.x
25. Ladogana A, Puopolo M, Croes EA, et al. Mortality from Creutzfeldt-Jakob disease and related disorders in Europe, Australia, and Canada. Neurology. 2005;64(9):1586–1591. doi:10.1212/01.WNL.0000160117.56690.B2
26. Kobayashi A, Teruya K, Matsuura Y, et al. The influence of PRNP polymorphisms on human prion disease susceptibility: an update. Acta Neuropathol. 2015;130(2):159–170. doi:10.1007/s00401-015-1447-7
27. Jeong BH, Nam JH, Lee YJ, et al. Polymorphisms of the prion protein gene (PRNP) in a Korean population. J Hum Genet. 2004;49(6):319–324. doi:10.1007/s10038-004-0150-7
28. Yu SL, Jin L, Sy MS, et al. Polymorphisms of the PRNP gene in Chinese populations and the identification of a novel insertion mutation. Eur J Hum Genet. 2004;12(10):867–870. doi:10.1038/sj.ejhg.5201245
29. Pocchiari M, Puopolo M, Croes EA, et al. Predictors of survival in sporadic Creutzfeldt-Jakob disease and other human transmissible spongiform encephalopathies. Brain: J Neurol. 2004;127(Pt 10):2348–2359. doi:10.1093/brain/awh249
30. Begué C, Martinetto H, Schultz M, et al. Creutzfeldt-Jakob disease surveillance in Argentina, 1997–2008. Neuroepidemiology. 2011;37(3–4):193–202. doi:10.1159/000331907
31. Lundberg PO. Creutzfeldt-Jakob disease in Sweden. J Neurol Neurosurg Psychiatry. 1998;65(6):836–841. doi:10.1136/jnnp.65.6.836
32. Iwasaki Y, Akagi A, Mimuro M, Kitamoto T, Yoshida M. Factors influencing the survival period in Japanese patients with sporadic Creutzfeldt-Jakob disease. J Neurol Sci. 2015;357(1–2):63–68. doi:10.1016/j.jns.2015.06.065
33. Chen YH, Ho CH, Huang CC, et al. Comparison of healthcare utilization and life-sustaining interventions between elderly patients with dementia and those with cancer near the end of life: a nationwide, population-based study in Taiwan. Geriatr Gerontol Int. 2017;17(12):2545–2551. doi:10.1111/ggi.13084
34. Fiorini M, Iselle G, Perra D, et al. High diagnostic accuracy of RT-QuIC assay in a prospective study of patients with suspected sCJD. Int J Mol Sci. 2020;21(3):880. doi:10.3390/ijms21030880
35. Sharma DK, Boggild M, van Heuven AW, White RP. Creutzfeldt-Jakob disease presenting as stroke: a case report and systematic literature review. The Neurologist. 2017;22(2):48–53. doi:10.1097/NRL.0000000000000107
36. Zuhorn F, Hubenthal A, Rogalewski A, et al. Creutzfeldt-Jakob disease mimicking autoimmune encephalitis with CASPR2 antibodies. BMC Neurol. 2014;14:227.
37. Miyazawa N. Creutzfeldt-Jakob disease mimicking alzheimer disease and dementia with lewy bodies-findings of FDG PET with 3-dimensional stereotactic surface projection. Clin Nucl Med. 2017;42(5):e247–e248. doi:10.1097/RLU.0000000000001602
38. Hsiao PS, Lee YM, Chu FS, et al. Probable sporadic Creutzfeldt-Jakob disease mimicking focal epilepsy. Epilepsy Behav Case Rep. 2019;11:77–80. doi:10.1016/j.ebcr.2019.01.006
39. Ladogana A, Puopolo M, Poleggi A, et al. High incidence of genetic human transmissible spongiform encephalopathies in Italy. Neurology. 2005;64(9):1592–1597. doi:10.1212/01.WNL.0000160118.26865.11
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.