")
Back to Journals » International Journal of Nanomedicine » Volume 15
In vivo Targeting of Liver Cancer with Tissue- and Nuclei-Specific Mesoporous Silica Nanoparticle-Based Nanocarriers in mice
Authors Ding Z, Wang D, Shi W , Yang X, Duan S, Mo F, Hou X, Liu A, Lu X
Received 21 July 2020
Accepted for publication 30 September 2020
Published 29 October 2020 Volume 2020:15 Pages 8383—8400
DOI https://doi.org/10.2147/IJN.S272495
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Ziqiang Ding,1,2,* Dujin Wang,1,2,* Wei Shi,2,3,* Xiaomei Yang,2,3 Siliang Duan,2 Fengzhen Mo,2 Xiaoqiong Hou,2,3 Aiqun Liu,2 Xiaoling Lu2,4
1National Center for International Research of Biological Targeting Diagnosis and Therapy, Guangxi Medical University, Nanning, Guangxi 530021, People’s Republic of China; 2International Nanobody Research Center of Guangxi, Guangxi Medical University, Nanning, Guangxi 530021, People’s Republic of China; 3School of Preclinical Medicine, Guangxi Medical University, Nanning, Guangxi 530021, People’s Republic of China; 4College of Stomatology, Guangxi Medical University, Nanning, Guangxi 530021, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xiaoling Lu
International Nanobody Research Center of Guangxi, Guangxi Medical University, Nanning, Guangxi 530021, People’s Republic of China
Tel/Fax +86 771-2387 518
Email [email protected]
Purpose: Cancer tissue-specific and nuclei-targeted drug delivery is ideal for the delivery of chemotherapy. However, it has only been achieved in in vitro studies mainly due to low efficiency in vivo. In this study, we aimed to establish an efficient dual-targeted system that targets liver cancer tissue as well as the nuclei of cancer cells in vivo.
Methods: We first synthesized TAT peptide (TATp)-mesoporous silica nanoparticle (MSN) complex (TATp-MSN) and generated liposomes that carried liver cancer-specific aptamer TLS11a (TLS11a-LB). We then generated the drug TLS11a-LB@TATp-MSN/doxorubicin (DOX) by mixing TLS11a-LB and DOX-loaded TATp-MSN. After physical and chemical characterization of the nanoparticles, DOX release from these formulations was evaluated at pH 5.0 and 7.4. Furthermore, we also evaluated nuclear localization and cytotoxicity of the drug in H22 cells in vitro and investigated the liver cancer targeting and antitumor activities of the nano-drug in vivo using a H22 tumor-bearing mice model.
Results: TLS11a-LB@TATp-MSN/DOX and its controls were confirmed as nano-drugs (< 100 nm) using transmission electron microscopy (TEM). The DOX release rate of TLS11a-LB@TATp-MSN/DOX was significantly faster at pH 5.0 than at pH 7.4. TLS11a-LB@TATp-MSN/DOX effectively targeted the nuclei of H22 cells and released DOX with a higher efficiency than that of the control groups. In addition, TLS11a-LB@TATp-MSN/DOX exhibited slight cytotoxicity, but not significantly more than controls. In vivo studies showed that TLS11a-LB@TATp-MSN accumulated in subcutaneous H22 tumors in the right axilla of BALB/c mice, reaching peak levels at 48 h after intravenous injection, respectively, and demonstrated that TLS11a-LB@TATp-MSN/DOX group enhanced tumor treatment efficacy while reducing systemic side effects.
Conclusion: TLS11a-LB@TATp-MSN/DOX can efficiently deliver DOX to the nuclei of liver cancer cells by dual targeting liver cancer tissue and the nuclei of the cancer cells in mice. Thus, it is a promising nano-drug for the treatment of liver cancer.
Keywords: targeted drug delivery, liver cancer treatment, MSN-based vehicles, doxorubicin, tissue- and nuclei-specific targeting
Introduction
Liver cancer, known as the sixth most commonly diagnosed cancer worldwide and the fourth leading cause of cancer death,1 is very difficult to treat because of its heterogeneity in terms of location, stage, and origin.2 As one of the most promising and convenient therapeutic approaches in the clinical setting, chemotherapy offers the best hopes for hepatoma patients. However, chemotherapy is still limited by many disadvantageous factors, such as poor target character, rapid drug metabolism and severe adverse effects.3 And sometimes, multidrug-resistant (MDR) cancer cells are easily recalcitrant to chemotherapy, which involves several mechanisms including P-glycoprotein-induced efflux and degradation of endosomes/lysosomes that prevent drugs from entering the nucleus.4–7 Therefore, targeted chemotherapy would be a more optimistic option to depress the unexpected side effects and enhance the therapeutic efficiency by delivering drugs to specific sub-cellular sites within cancer cells.
Doxorubicin (DOX) is a first-line drug for cancer chemotherapy that targets DNA, which is localized in the nuclei. The nuclear delivery efficiency of DOX is very low when it is used directly to treat liver cancer. Facilitated delivery can improve the efficacy of DOX for treating liver cancer. As a widely applied drug delivery system, liposome possesses several excellent features, including large drug loading, good biocompatibility, and biodegradability. For example, using modified liposomes such as galactosylated liposomes and lyso-thermosensitive liposomes as the drug carrier for DOX has been both reported to effectively improve the therapeutic efficiency.8–12 Application of superparamagnetic iron oxide (SPIO) nanoparticles,13,14 and hematoporphyrin (HP)-modified DOX-loaded nanoparticles (HP-NPs)15,16 to deliver DOX have also shown improved therapeutic efficacy for liver cancer in some preclinical experiments. However, there are still limitations of these approaches, such as their invasiveness, off-target effect, less biocompatibility, as well as relatively low efficiency. A more efficient and targeted precision-guided delivery system with DOX targeting liver cancer remains to be developed. Therefore, the development of nuclear-targeted drug delivery systems (DDSs), which is expected to kill cancer cells more directly and efficiently provides significantly enhanced anticancer efficiency of currently available drugs and is thus of great significance.
Among available nanomaterials for drug delivery, mesoporous silica nanoparticles (MSNs) with unique properties, including excellent biocompatibility, large specific surface area, and adjustable pore size (ranging from 2 to 50 nm), have been widely used in many biomedical fields especially for the delivery of anti-tumor drugs,17–22 gene therapy,23–27 and tumor imaging agents.28–31 Drug molecules can be confined in its mesopores through electrostatic adsorption while released responding to a given stimulus. However, the drug carried in the mesopores is dramatically lost in the complex body fluids and causes adverse reactions.32 Recent studies showed that the lipid bilayer (LB) can attach to the silicate surface in the mesopores and form liposomes. This enhanced biocompatibility of MSNs can extend their availability in circulation, reducing drug loss during delivery and avoiding adverse drug reactions. In addition, the lipid bilayer can be modified with specific targeting molecules.33–36
One key factor for the success of anti-cancer chemotherapy is the correct and sufficient arrival of chemical drug. Herein, enhancing the targeting specificity becomes a very essential issue to be solved when designs and constructs an efficient drug delivery system. In recent years, several approaches for targeted delivery of DNA-damaging chemotherapeutic drugs to the nuclei have been developed, including addition of nuclear localization signaling peptides such as TATp (GRKKRRQRRRPPQ) to the surface of nano-drugs. TATp can specifically bind with nuclear pore complex (NPC) and transport 30–40-kDa macromolecules to the nuclei, releasing nano-particle-carried drugs in the nuclei of cancer cells without triggering MDR mechanisms.37–42 TATp-mediated nuclear targeting has been achieved in vitro, but there has been few reports of an in vivo study testing this approach. This is probably because such drugs are unable to target the tumor in vivo such as nuclear localization signaling peptides without cell specificity. On the other hand, several single-stranded DNA or RNA aptamers, identified through SELEX (systematic enrichment of ligands by exponential enrichment), can recognize specific cell types.43–45 This provides an avenue for nano-drugs to target cancer cells. Aptamers have been widely used in targeted nano-drug delivery43,49 and examination of tumors.50–54 TLS11a is an aptamer identified using cell-SELEX. It can specifically recognize the BNL 1ME A.7R.1 (MEAR) mouse liver cancer cell line.55 TLS11a also has high affinity to the HepG2 and LH86 human liver cancer cells and the H22 mouse liver cancer cells.56,57
Inspired by the remarkable performance of cancer tissue-specific and nuclei-targeted drug delivery in vitro studies. In this study, we used the drug carriers MSNs and LBs and the targeting molecules TATp and TLS11a establish a facilitated in vivo drug delivery and targeting system for liver cancer treatment. As illustrated in Figure 1, the design of TLS11a-LB@TATp-MSN/DOX consists of liposomes carried the liver cancer-specific aptamer TLS11a (TLS11a-LB) and DOX-loaded the nuclear localization peptide (TATp)-MSNs (TATp-MSN/DOX). TLS11a-LB@TATp-MSN/DOX exhibited effectively targeted the nuclei of H22 cells, released DOX with a higher efficiency and potentiated strong antitumor effects with minimal adverse effects. Therefore, our results demonstrate that TLS11a-LB@TATp-MSN/DOX provide a promising nanoplatform for engineering tumor-targeting drug delivery systems.
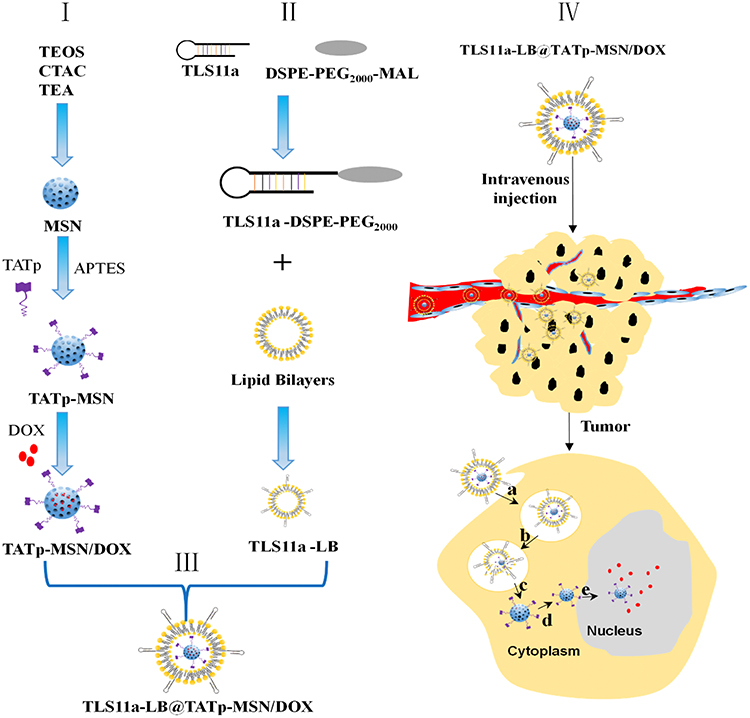 |
Figure 1 Schematic diagram for targeted liver cancer treatment using the nano-drug TLS11a-LB@TATp-MSN/DOX. The nuclear localization peptide (TATp)-mesoporous silica nanoparticle (MSN) complex (TATp-MSN) was synthesized and mixed with doxorubicin (DOX) (I). Liposomes that carried the liver cancer-specific aptamer TLS11a (TLS11a-LB) were produced (II). The nano-drug TLS11a-LB@TATp-MSN/DOX was generated by mixing TLS11a-LB and TATp-MSN/DOX (III). (IV) After intravenous injection, TLS11a-LB@TATp-MSN/DOX was designed to be distributed via blood circulation to liver cancer tissues through TLS11a targeting (a-b), to be localized to the nuclei of the cancer cells through TATp targeting, and to release DOX (c-e). |
Materials and Methods
Reagents
Tetraethyl orthosilicate (TEOS), triethanolamine (TEA), cetyltrimethylammonium chloride (CTAC, 25 wt%), 3-aminopropyltriethoxysilane (APTES), fluorescein isothiocyanate (FITC), N-hydroxysuccinimide (NHS), 1-ethyl-3-(3-dimethyl-aminopropyl) carbodiimide (NHS), α-tocopherol, cysteamine, and doxorubicin hydrochloride were purchased from Sigma-Aldrich (USA). 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), cholesterol, 1.2-distearoyl-sn-glycerol-3-phosphoethanolamine-N-[amino (polyethylene glycol)-2000] (ammonium salt, DSPE-PEG2000), dimethyldioctadecylammonium (bromide salt, DDAB), and 1.2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide (polyethylene glycol)-2000] (ammonium salt, DSPE-PEG2000-MAL) were purchased from Avanti Polar Lipids (Birmingham, AL, USA). 1,1ʹ-Dioctadecyl-3,3,3ʹ,3ʹ-tetramethylindocarbocyanineperch-lorate (DiI) was purchased from Beyotime Biotechnology (Jiangsu, China). TATp (YGRKKRRQRRR) and (FITC)-C6-TAT (TAT: YGRKKRRQRRR) were synthesized by Shanghai Sangon Biotech Co. Ltd. (Shanghai, China). SH- and FITC-modified TLS11a aptamer 3ʹ-SH-AAAAAAACAGCATCCCCATGTGAACAATCGCATTGTGATTGTTACGGTTTCCGCCTCATGGACGTGCTG-5ʹwere synthesized by Shanghai Sangon Biotech Co., Ltd. (Shanghai, China).
Preparation of the Nuclear Localization Peptide (TATp)-Mesoporous Silica Nanoparticle (MSN) Complex (TATp-MSN)
First, 10 g CTAC and 0.4g TEA were dissolved in 100 mL H2O and incubated at 95°C in a trimethyl silicone bath with condensation reflux and intensive magnetic stirring at 200 rpm for 1 h. The 7.5 mL TEOS was slowly added drop-wise to the above solution within 5 minutes, at a rate of one drop in two seconds and mixed by stirring for 1 h. After cooling down to room temperature, the MSN products were collected by centrifugation at 15,000 rpm for 15 min and washed with 100% ethanol three times. The MSN products were further washed several times with ethanolic and hydrochloric acid solution by condensation reflux for 6 h to remove CTAC. Finally, 100 mg MSN products were aliquoted and suspended in 20 mL H2O before mixing with 100 μL glacial acetic acid and 50 μL APTES by stirring at room temperature for 24 h. After washing with H2O and vacuum freezing-dried, MSN-NH2 nanoparticles were obtained.
TATp-MSNs were prepared from TATp and MSN-NH2 nanoparticles through cross-linking with EDC and NHS. Briefly, 3.12 mg TATp, 38.4 mg EDC, and 56 mg NHS were added to 20 mL PBS (pH 7.4) and mixed by intensive stirring at room temperature for 1 h. Then, 20 mg MSN-NH2 was added and mixed by intensively stirring at room temperature for 24 h. The TATp-MSN products were collected by centrifugation at 15,000 rpm for 15 min and washed with H2O. The TATp-MSN was stored at 4°C.
To label MSNs and TATp-MSN with FITC, a MSN or TATp-MSN solution was mixed with excess FITC by stirring in the dark at room temperature for 24 h and collected by centrifugation at 15,000 rpm for 15 min. After three washes with H2O, the FITC-labeled MSNs and FITC-labeled TATp-MSN were stored in the dark at 4°C.
Loading DOX into MSN or TATp-MSN
For DOX loading, 10 mg MSNs or TATp-MSN were suspended in 5 mL DOX PBS (pH 7.0) solution (0.5 mg/mL) and mixed by stirring for 24 h. The MSN/DOX or TATp-MSN/DOX products were collected by centrifugation at 12,000 rpm for 15 min and washed with PBS three times. The drug entrapment efficiency was determined using UV spectrophotometry. The DOX release rate was determined using fluorescence spectrophotometry. The drug loading content (DLC) was defined as the ratio of mass of the drug encapsulated within the nanoparticles to the total mass of drug-loaded nanoparticles. The drug loading efficiency (DLE) was the ratio of the mass of drug loaded into the nanoparticles to the mass of drug initially added. The DLC and DLE were calculated according to the following equations:
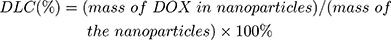
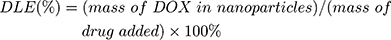
Generation of Liposomes Carrying Liver Cancer-Specific Aptamer TLS11a (TLS11a-LB)
First, 7.87 mg POPC, 3.2 mg cholesterol, 2.8 mg PEG2000-DSPE, 0.38 mg DDAB, 0.017 mg α-tocopherol, and 0.138 mg MAL-PEG2000-DSPE (molar ratio 51.8:40:5:3:0.2:0.3) were dissolved in chloroform and then vacuum-dried in a 5-mL round-bottomed flask using a rotary evaporator. The products were further vacuum-dried for 2 h for solvent removal and then suspended in 1 mL HEPES buffer (pH 6.5, 10 mmol/l) using an ultrasonic bath for 5 min. The suspension was loaded into a liposome generator and filtered with 200-nm and 100-nm polycarbonate membranes, 20 times each, and a 50-nm polycarbonate membrane 21 times. The product maleimide-modified liposomes (MAL-LB) were obtained and stored at 4°C.
To generate liposomes with a DiI-labeled phospholipid bilayer, DiI (5 mg/mL) (0.5% of the liposomes, molar ratio) and MAL-PEG2000-DSP were added at the same time during the production of MAL-LB in a round-bottomed flask. The DiI-labeled MAL-LB products were stored in the dark at 4°C.
TLS11a-LBs were generated by mixing MAL-LB and TLS11a and incubating the mixture in nitrogen gas for 24 h and purified to remove TLS11a by dialysis. The amount of TLS11a used in the mixture was equivalent to the 10% of MAL-PEG2000-DSPE in MAL-LB in a molar ratio.
Preparation of TLS11a-LB@TATp-MSN/DOX and Its Derivatives
TLS11a-LB@TATp-MSN/DOX was generated by mixing 10 mg TATp-MSN/DOX and 100 µL TLS11a-LB with pipette tips and storing the mixture at 4°C for 2 weeks. The products were collected by centrifugation at 15,000 rpm for 2 min and washed with PBS (pH 7.4) three times. Similarly, the derivatives such as TLS11a-LB@ MSN/DOX were generated by mixing MSN/DOX and TLS11a-LB.
Physical and Chemical Characterization of TLS11a-LB@TATp-MSN/DOX and Its Derivatives as Nanoparticles
TLS11a-LB@TATp-MSN/DOX and its derivatives as nanoparticles were characterized by transmission electron microscopy (TEM, TESCAN VEGA3 LMU, Tescan USA Inc.; Cranberry Twp., PA, USA) under H-7650, an electron beam accelerating voltage of 100 kV, along with a Fourier infrared spectrometer (VERTEX70, Bruker, Germany) and Zetasizer analyzer (Zetasizer Nano S, Malvern, UK). The sizes and potentials of the samples were determined by dynamic light scattering (DLS). The specific surface area and pore size distribution were determined using QUADRASORB S1 analyzer (Quantachrome, USA) at 77.3 K in liquid nitrogen. All samples were vacuumed at 200°C for 4 h before analysis. Specific surface area was calculated according to the Brunauer Emmett Teller (BET) method. The pore size was calculated according to Barrette Joyner Halenda (BJH) method.
Release Rates of DOX from TATp-MSN/DOX and TLS11a-LB@TATp-MSN/DOX
First, 500 μL TATp-MSN/DOX or TLS11a-LB@TATp-MSN/DOX containing 1 mg DOX was added to the bottom of a dialysis tube (MWCO 12,000). Then, 30 mL PBS (pH 5.0 and 7.4) was added and mixed by shaking in the dark at 150 rpm in a 37°C water bath. Next, 5 mL buffer was sampled from the dialysis tube and 5 mL fresh buffer was added to the dialysis tube at the designated time points. The level of DOX in the 5 mL sample buffer was determined using a fluorospectrophotometer (F7000, excitation = 485 nm, emission =560 nm).
Examination of the Nuclear DOX Delivery Efficiency and Intra-Cellular Localization of Nanoparticles in H22 Cells
Murine H22 hepatoma cell line was purchased from the China Center for Type Culture Collection (CCTCC, Wuhan, China), and maintained in DMEM media supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin and cultured at 37°C in 5% CO2.
To determine the nuclear DOX delivery efficiencies of the nanoparticles in H22 cells, H22 cells were seeded at 1×106 cells/well in 6-well plates and incubated overnight. DOX, LB@TATp-MSN/DOX, TLS11a-LB@MSN/DOX, and TLS11a-LB@TATp-MSN/DOX containing 5 μg/mL were added to the cell culture wells and incubated for 4 h. Alternatively, cells were incubated with nano-drugs at a concentration of 100 μg/mL for 24 h and 48 h and collected by centrifugation. After three washes with PBS, the cells were fixed using 4% paraformaldehyde for 30 min and washed with PBS three times. After the nuclei were stained with DAPI (1 mg/mL) for 5 min and washed with PBS three times, the cells were visualized and the images were recorded using an inverted fluorescence microscope (ECLIPSE 80i, Nikon, Japan).
To quantify the intensity of the fluorescence in the H22 cells after treatment with nano-drugs, H22 cells were seeded at 1×105 cells/well in 6-well plates and incubated overnight. After treatment with the nano-drugs carrying DOX (5 μg/mL) for 2 h, the cells were washed with PBS three times and suspended in 500 μL PBS. We then examined the intensity of the fluorescence using a FACS Caliburcytometer (Beckman Coulter, Brea, CA, USA).
Determination of Cytotoxicity of Nano-Drugs on H22 Cells Using CCK-8 Assay
H22 cells were seeded at 1×105 cells/well in 96-well plates and incubated for 24 h. DOX, TATp-MSN/DOX, LB-TATp-MSN/DOX, TLS11a-LB@TATp-MSN/DOX, and TLS11a-LB @MSN/DOX (1, 5, 10, 20 μg/mL DOX in each group) were added to the cell culture wells (6 replicated wells for each treatment) and incubated for 24 h or 48 h. After incubation, 10 μL CCK-8 (Cell Counting Kit-8) solution was added to each well and further incubated for 3 h. Optical absorption was determined at 450 nm using a multiscan spectrum microplate spectrophotometer (Thermo Scientific Multiskan GO). The toxic effect was indicated according to the cell viability, which was calculated by dividing the number of living cells in the treatment group by that of the control groups.
Examination of the Distribution of Nano-Drugs in Mice After Intravenous Injection
BALB/c mice (female, 6–8 weeks, and specific pathogen free (SPF)) were obtained from Guangxi Medical University Laboratory Animal Centre (Nanning, China). The protocols for using animals in this study were approved by the Animal Care & Welfare Committee of Guangxi Medical University (NO.201903023), and were carried out in compliance with the Laboratory animal-Guideline for ethical review of animal welfare issued by the National Standard GB/T35892-2018 of the People’s Republic of China.
The in vivo liver cancer mouse model was generated by seeding 2 × 106 H22 cells in the right axilla of BALB/c mice (6–8 weeks). When the diameter of the tumor reached 0.8–1.0 cm, the mice were divided into three groups (n = 4/group) and injected with DiR, DiR-labeled LB-TATp-MSN, and DiR-labeled TLS11a-LB@TATp-MSN, respectively, via the tail vein at a dose of 3 μg DiR in 100 μL PBS per mouse. The fluorescent images were taken at an excitation of 720 nm and an emission of 790 nm (exposure 20 s, 4×4 grading, 180 mm ×180 mm view field) using Bruker imaging system (FXPro, Carestream Health, Inc, USA). After the mice were fasted for 12 h, cleaned with anhydrous alcohol on the bodies, and subjected to anesthesia with 5% isoflurane inhalation, which was maintained with 3% isoflurane. After treatment with the drugs for 48 h, one mouse in each group was sacrificed, and its organs were collected. The fluorescence in the organs was examined using Bruker imaging system. The images were analyzed using Bruker MI SE software.
After the drug-carrying MSNs were labeled with green fluorescencing FITC, the nano-drugs TLS11a-LB@TATp-MSN/DOX, LB@TATp-MSN/DOX, TATp-MSN/DOX, and DOX were intravenously injected into the tumor-bearing mice via the tail vein. After 24-h treatment, tumor tissues were collected and used to prepare frozen sections. The distribution of all nano-drugs in the tumor tissues was visualized and recorded using a fluorescence microscope (ECLIPSE 80i, Nikon, Japan).
Evaluation of the Efficacy of Nano-Drugs for Treating BALB/c Mice with Liver Cancer in the Right Axilla
Model mice were divided into seven groups (n = 6/group) when the volumes of the tumor reached 100 mm3 and were then treated with PBS, TLS11a-LB@TATp-MSN, DOX, TATp-MSN/DOX, LB@TATp-MSN/DOX, TLS11a-LB@MSN/DOX, and TLS11a-LB@TATp-MSN/DOX, respectively, via the tail vein at a dose of 8 mg/kg DOX every other day and four times in total. The tumor volume (V) was calculated according to the short and long diameters (d, D) of the tumor tissue (V = d2 × D/2), which were measured on the first day after treatments were done and then measured every other day. At the same time, the body weights of mice were measured. The whole tumor tissues were collected from mice and weighed on the 12th treatment day.
To examine the survival rate after nano-drug treatment, model mice generated as described above divided the mice into seven groups (n = 8/group) when the volumes of the tumors reached 100 mm3. The seven groups of mice were treated with PBS, TLS11a-LB@TATp-MSN, DOX, TATp-MSN/DOX, LB@TATp-MSN/DOX, TLS11a-LB@MSN/DOX, and TLS11a-LB@TATp-MSN/DOX, respectively, via the tail vein at a dose of 8mg/kg DOX every other day and four times in total. The survival times of the mice in the different groups were recorded.
Statistical Analysis
Statistical analysis was performed using SPSS 16.0 (SPSS, Chicago, IL). The statistical significance of the differences was analyzed using analysis of variance (ANOVA) for multiple groups or Student’s t-test for two groups. P< 0.05 was considered significant.
Results
Physical and Chemical Characteristics of TLS11a-LB@TATp-MSN/DOX and Its Derivatives as Nanoparticles
To establish an in vivo drug delivery and targeting system for liver cancer treatment using TATp, MSN, TLS11a, and LB (Figure 1), we first synthesized or produced MSN, MSN-NH2, TATp-MSN, LB@TATp-MSN, and TLS11a-LB@TATp-MSN according to methods reported by Bein et al.58 These products were proven to be nanoparticles using TEM (Figure 2A and B, Figure S1) and DLS (Figure 2C and D). There was no significant change in the size of MSNs upon modification with TATp. The size was gradually increased from 50 nm to 100 nm after modification with LBs and loading of DOX (Figure S1 and Table S1). The specific surface areas of MSNs were around 631.47 m2/g, and the pore size of MSNs was about 2.6 nm in diameter according to the nitrogen adsorption desorption analysis (Figure 2E and F). The TATp-MSN was successfully generated and proved to be free of CTAC as indicated by FTIR analysis (Figure S2a and S2b). These results suggested that we generated the nano-drugs TLS11a-LB@TATp-MSN/DOX and its derivatives. Furthermore, after centrifugation, the characteristic absorption peak of FITC-labeled TATp measured by UV−vis absorbance spectrometry was disappeared (495 nm), and the color of the solution was changed in comparison (Figure S3), suggesting the successful conjugation of TATp onto the surface of the MSN. In addition, calculation based on spectrophotometric analysis of DOX concentration in the supernatant after centrifugation showed that the envelopment efficacy of TATp-MSN/DOX was 83.33% (Figure S4a and S4b).
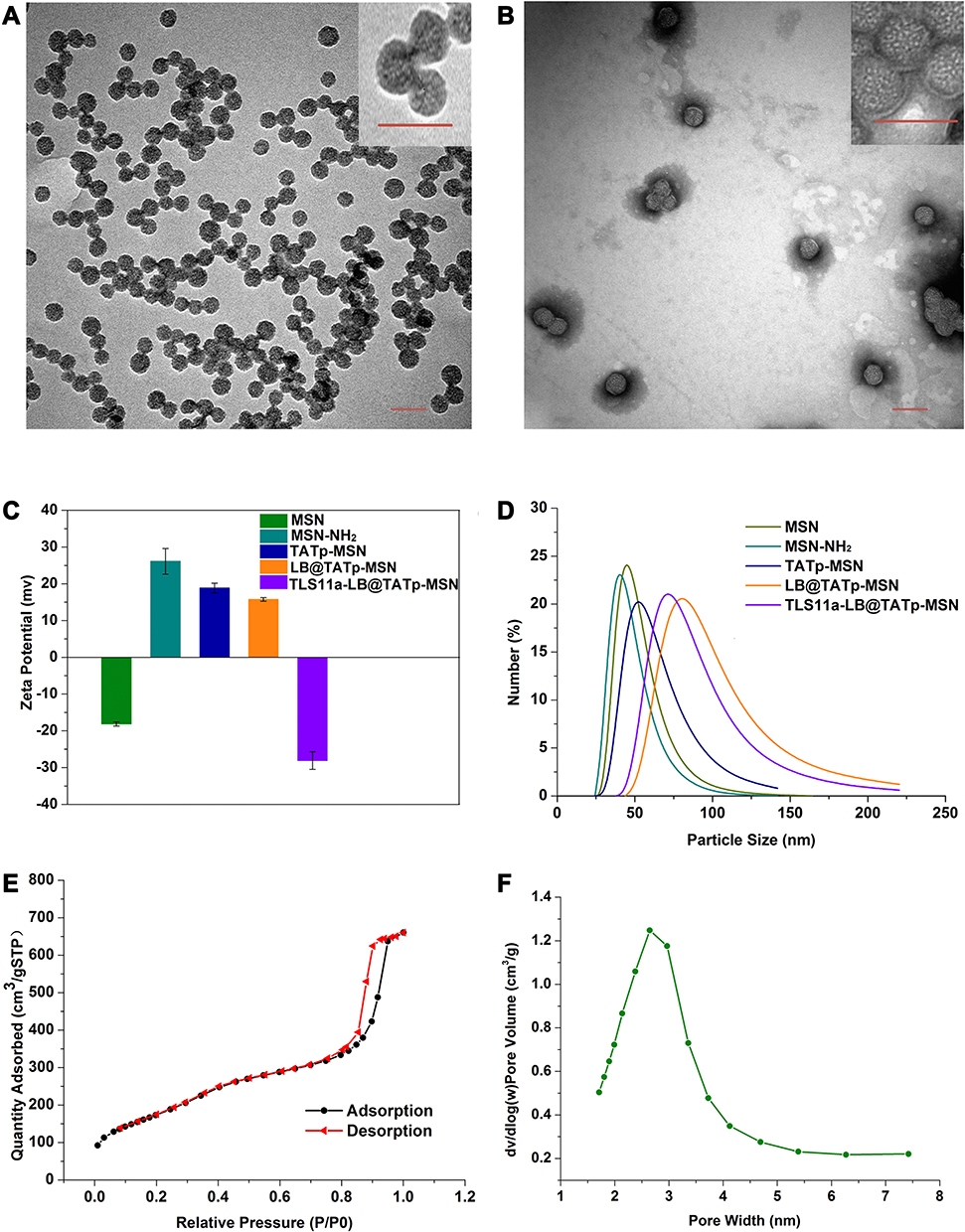 |
Figure 2 Physical and chemical characteristics of TLS11a-LB@TATp-MSN/DOX and its derivatives as nanoparticles. (A) Characterization of MSNs using transmission electron microscopy (TEM). Scale = 100 nm. (B) Characterization of TLS11a-LB@TATp-MSN using TEM. Scale = 100 nm. (C) Zeta potentials of the MSN, MSN-NH2, TATp-MSN, LB@TATp-MSN, and TLS11a-LB@TATp-MSN nano-particles. The size and potential of the samples were determined using dynamic light scattering (DLS). (D) The size profiles of the MSN, MSN-NH2, TATp-MSN, LB@TATp-MSN, and TLS11a-LB@TATp-MSN particles. (E) Specific surface area of the MSN nanoparticles. Specific surface area was determined using nitrogen adsorption desorption analysis. (F) Pore size distribution of the MSN nanoparticles. |
Release Rates of DOX from TLS11a-LB@TATp-MSN/DOX and TATp-MSN/DOX in PBS at pH 5.0 and 7.4
Tumors grow in a slightly acidic environment.59–61 To examine the DOX release rates from TLS11a-LB@TATp-MSN/DOX and TATp-MSN/DOX in tumors, we determined the DOX release rates from these two drugs in PBS at pH 5.0 and 7.4 (standard curve for DOX release see Figure 3A). The results showed that both TATp-MSN/DOX and TLS11a-LB@TATp-MSN/DOX quickly released DOX in PBS and released >60% DOX within 72 h at pH 5.0, whereas they released less DOX into PBS at pH 7.4 (Figure 3B), suggesting that both TLS11a-LB@TATp-MSN/DOX and TATp-MSN/DOX released less DOX in the normal tissues and more DOX in tumors. The results also showed that TLS11a-LB@TATp-MSN/DOX released about the same amount of DOX as TATp-MSN/DOX did at pH 5.0. TLS11a-LB@TATp-MSN/DOX released <30% DOX within 72 h, significantly less than TATp-MSN/DOX did at pH 7.4 (Figure 3B), suggesting that TLS11a-LB@TATp-MSN/DOX released more DOX in tumors than TATp-MSN/DOX did because it released less DOX in normal tissues.
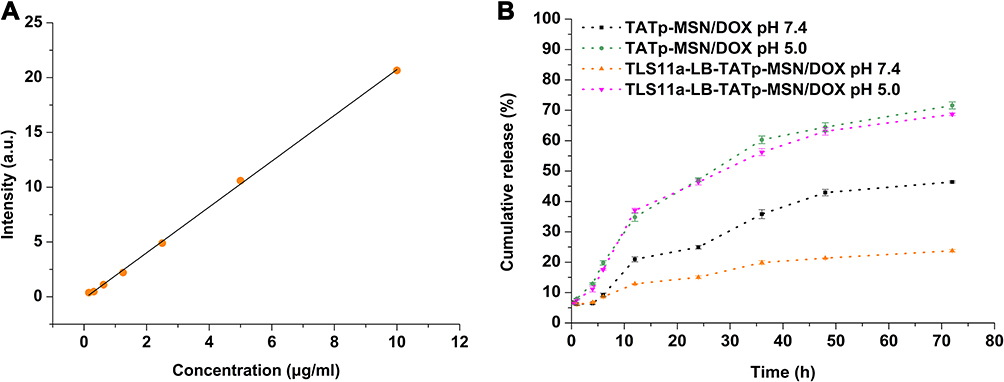 |
Figure 3 In vitro DOX release rates. (A) DOX standard curve. (B) DOX release rates of TLS11a-LB@TATp-MSN/DOX and TATp-MSN/DOX in PBS at pH 5.0 and 7.4. |
Nuclear Localization of TLS11a-LB@MSN and TLS11a-LB@TATp-MSN in H22 Cells
To determine whether TLS11a-LB@MSN and TLS11a-LB@TATp-MSN were localized in the nuclei of H22 cells, we labeled LB with DiI dye and MSN with FITC dye, treated H22 cells with these nanoparticles, and examined the fluorescence in H22 cells. The results showed that there was green (FITC) fluorescence in the plasma but not in the nuclei of H22 cells after treatment with TLS11a-LB@MSN and TLS11a-LB@TATp-MSN for 4 h (Figure 4A and Figure S5). After treatment for 24 h, there was green (FITC) and red fluorescence in the nuclei of H22 cells treated with TLS11a-LB@TATp-MSN and TLS11a-LB@MSN (Figure 4B). More obvious green (FITC) and red (DiI) fluorescence was seen in the nuclei of H22 cells treated with TLS11a-LB@TATp-MSN compared to TLS11a-LB@MSN (Figure 4B and Figure S7). Green fluorescence was not observed in the nuclei of the other groups (Figure S6). After treatment for 48 h, a large number of TATp-MSN accumulated in the nuclei in the TLS11a-LB@TATp-MSN group as seen the green fluorescence, but much less in the TLS11a-LB@MSN group (Figure 4C and Figure S7). These results suggested that TATp played a key role in localizing TLS11a-LB@TATp-MSN into the nuclei of H22 cells.
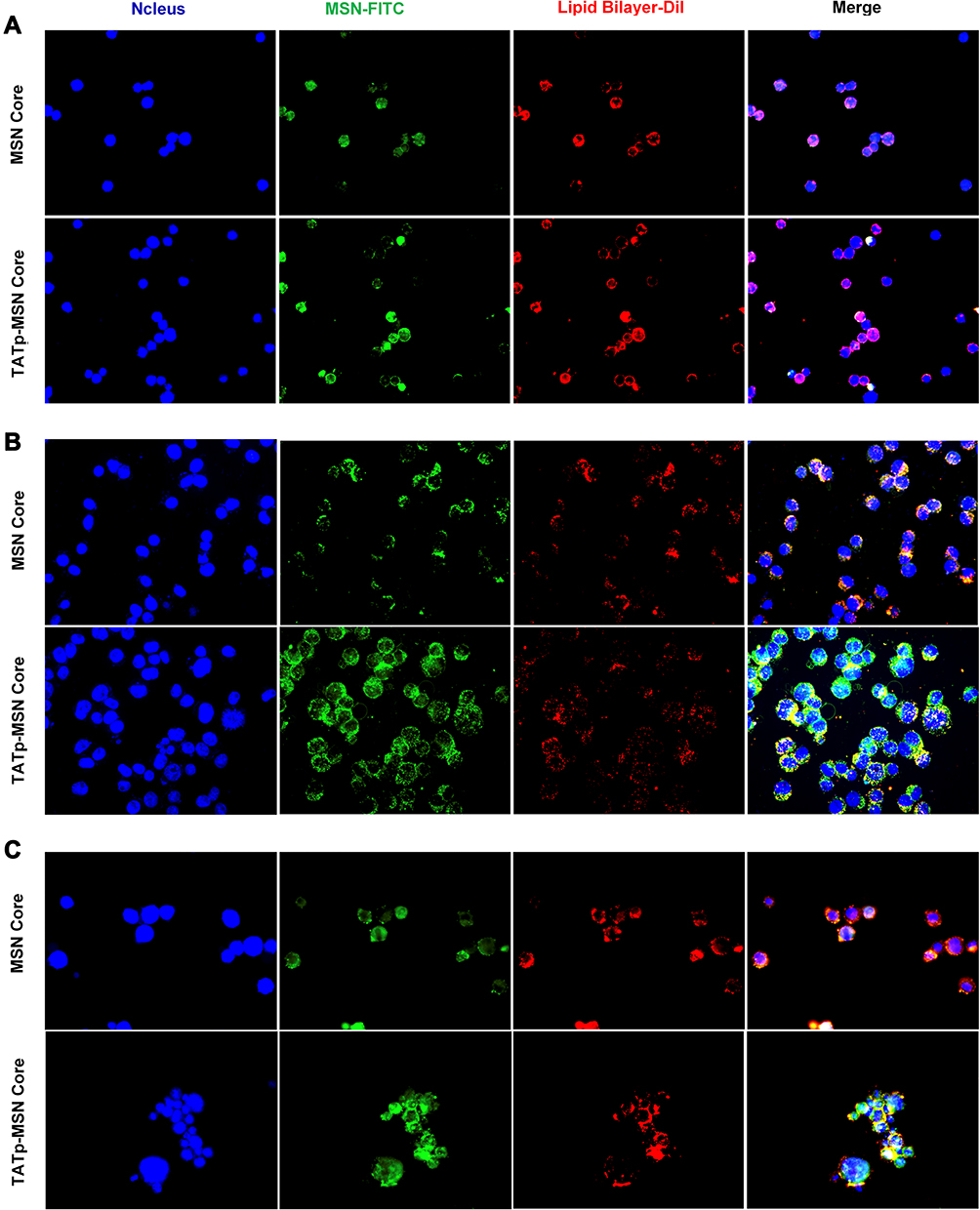 |
Figure 4 Nuclear localization efficiency of TLS11a-LB@TATp-MSN (TATp-MSN core) and TLS11a-LB@MSN (MSN core) in H22 cells. H22 cells were exposed to TLS11a-LB@MSN and TLS11a-LB@TATp-MSN for 4 h (A), 24 h (B), and 48 h (C). The cells were visualized under a fluorescent microscope. Blue: DAPI-stained nuclei. Green: FITC-labeled MSN. Red: DiI-labeled LB. |
Nuclear DOX Delivery Efficiencies of LB@TATp-MSN, TLS11a-LB@MSN, and TLS11a-LB@TATp-MSN in H22 Cells
To determine the nuclear DOX delivery efficiencies of LB@TATp-MSN, TLS11a-LB@MSN, and TLS11a-LB@TATp-MSN in liver cancer, we treated H22 cells with these nano-drugs and examined the fluorescence in the cells using fluorescence microscopy. The results showed that the nuclear concentration of DOX was almost the highest in the DOX treatment group as indicated by the intensity of red fluorescence. There was no significant difference in the nuclear DOX concentration between the TLS11a-LB@TATp-MSN/DOX group and the TLS11a-LB@MSN/DOX group under microscopy (Figure 5A). The nuclear DOX content was significantly higher in the TLS11a-LB@TATp-MSN/DOX group than in the TLS11a-LB@MSN/DOX group (p < 0.05), and they were both higher than that of the LB@TATp-MSN/DOX group using flow cytometry (Figure 5B and C). These results suggested that TLS11a played a key role in improving the nuclear DOX delivery efficiencies of TLS11a-LB@TATp-MSN/DOX and TLS11a-LB@MSN/DOX.
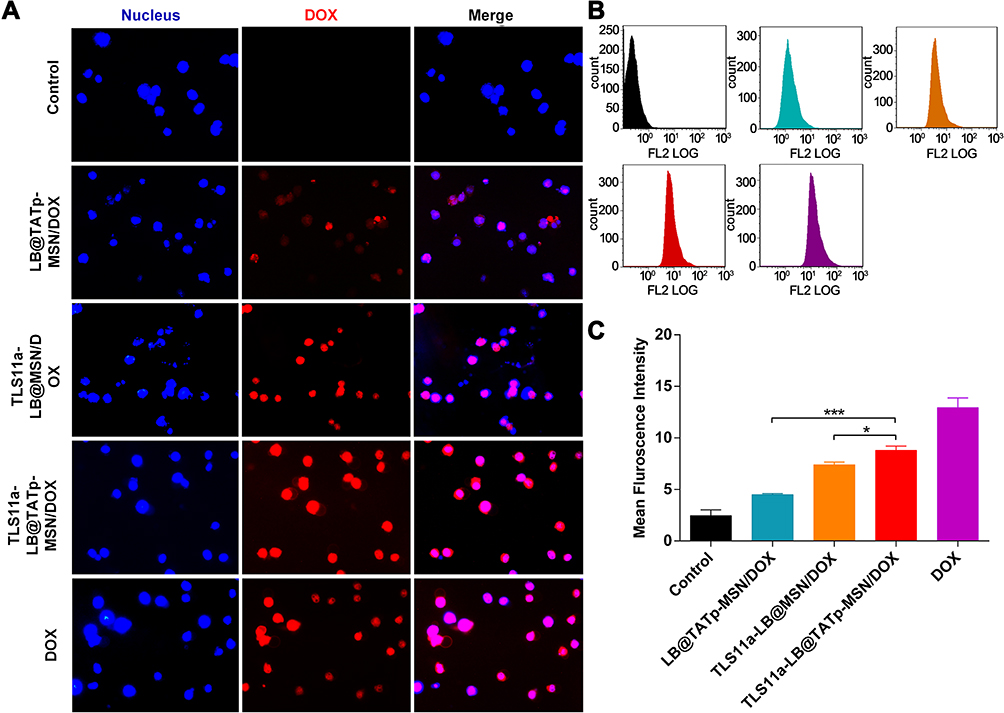 |
Figure 5 Nuclear DOX delivery efficiencies of LB@TATp-MSN, TLS11a-LB@MSN, and TLS11a-LB@TATp-MSN in H22 cells. PBS/DOX, LB@TATp-MSN/DOX, TLS11a-LB@MSN/DOX, TLS11a-LB@TATp-MSN/DOX, and DOX were incubated with H22 cells for 2 h. (A) The cells were visualized under a fluorescent microscope for DOX in H22 cells. (B) The intensities of the intra-cellular fluorescence signals were determined using flow cytometry. (C) The mean intensities of the intra-cellular fluorescence were calculated. *P < 0.05, ***P < 0.001. |
Toxic Effects of TATp-MSN/DOX, LB@TATp-MSN/DOX, TLS11a-LB@TATp-MSN/DOX, and TLS11a-LB@ MSN/DOX on H22 Cells
To examine the toxic effects of TLS11a-LB@TATp-MSN/DOX and its derivatives on H22 cells, we treated H22 cells with TLS11a-LB@TATp-MSN/DOX and its derivatives and determined the cell viability using the CCK-8 assay. The results showed that there were marginal changes in the cell viability of H22 cells after treatment with TATp-MSN and TLS11a-LB@TATp-MSN for 24 h (Figure 6A). Treatment of H22 cells with DOX, TATp-MSN/DOX, LB@TATp-MSN/DOX, TLS11a-LB@TATp-MSN/DOX, or TLS11a-LB@MSN/DOX for 24 h and 48 h resulted in significant decreases in the cell viability in a dose-dependent manner. The cell viability was lower at 48 h than at 24 h (Figure 6B and C). The cell viability was higher in the TLS11a-LB@TATp-MSN/DOX group than in the TATp-MSN/DOX group, but nearly the same as that in the DOX group (Figure 6B and C). These results suggested that TLS11a-LB@TATp-MSN/DOX and its derivatives had anti-tumor effects through DOX and the nanoparticle carriers did not have anti-tumor effects.
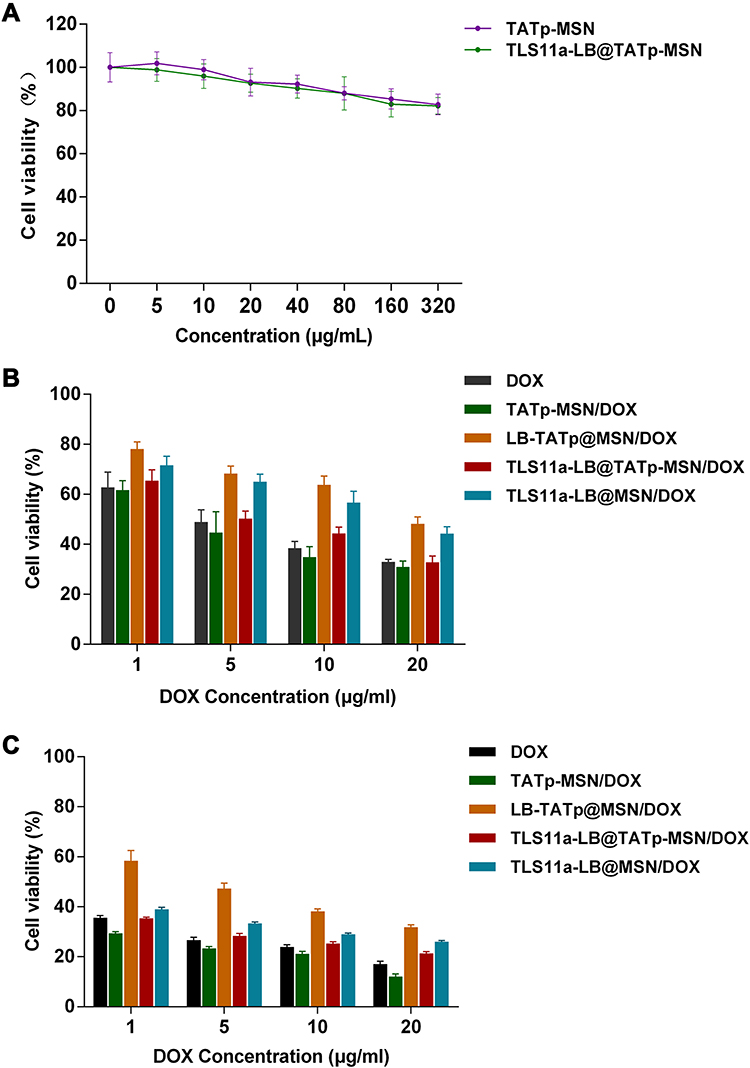 |
Figure 6 Toxic effects of TLS11a-LB@TATp-MSN/DOX and its derivatives on H22 cells. (A) Toxic effects of TATp-MSN and TLS11a-LB@TATp-MSN on H22 cells. H22 cells were incubated with TATp-MSN and TLS11a-LB@TATp-MSN for 24 h. The cell viability was determined using CCK-8 assay. (B and C) The toxic effects of DOX, TATp-MSN/DOX, LB@TATp-MSN/DOX, TLS11a-LB@TATp-MSN/DOX, and TLS11a-LB@ MSN/DOX on H22 cells. H22 cells were incubated with DOX, TATp-MSN/DOX, LB@TATp-MSN/DOX, TLS11a-LB@TATp-MSN/DOX, and TLS11a-LB@ MSN/DOX for 24 h (B) and 48 h (C). The cell viability was determined using CCK-8 assay. |
TLS11a-LB@TATp-MSN and LB@TATp-MSN Were Specifically Distributed in H22 Tumors in the Right Axilla of BALB/c Mice After Intravenous Injection
To determine whether TLS11a-LB@TATp-MSN/DOX and LB@TATp-MSN/DOX would be distributed to liver cancer tissues in vivo, we treated groups of BALB/c mice carrying H22 tumors in the right axilla with DiR, DiR-labeled TLS11a-LB@TATp-MSN, or DiR-labeled LB@TATp-MSN via tail vein injection and examined the intensity of fluorescence in the H22 tumors and organ systems of mice. In the DiR treatment group, the results showed that fluorescence was present in the H22 tumors and liver, and the intensity of the fluorescence in the H22 tumors was very low but increased over time. In the DiR-labeled LB@TATp-MSN group, the fluorescence was only present in the H22 tumors, and the intensity of fluorescence in the tumor tissues increased during the first 24 h, reaching a peak at 24 h and then gradually decreasing to a stable level during 24–120 h. In the DiR-labeled TLS11a-LB@TATp-MSN group, the fluorescence was only present in the tumor tissues, and the intensity of fluorescence in the tumor tissues increased during the first 48 h, reaching a peak at 48 h and then gradually decreasing from 48 to 120 h (Figure 7A). Notably, the intensity of the fluorescence in the H22 tumors of the DiR-labeled TLS11a-LB@TATp-MSN group was significantly higher than that in those of the DiR-labeled LB@TATp-MSN group, although the intensity in both groups was much higher than that of the DiR group (Figure 7C). The results also showed that fluorescence was most significantly enriched in the H22 tumors in both the DiR-labeled LB@TATp-MSN group and the DiR-labeled TLS11a-LB@TATp-MSN group, while fluorescent signal accumulated in liver in the DiR group 48 h after treatment among H22 tumors and all organs examined (Figure 7B and D).
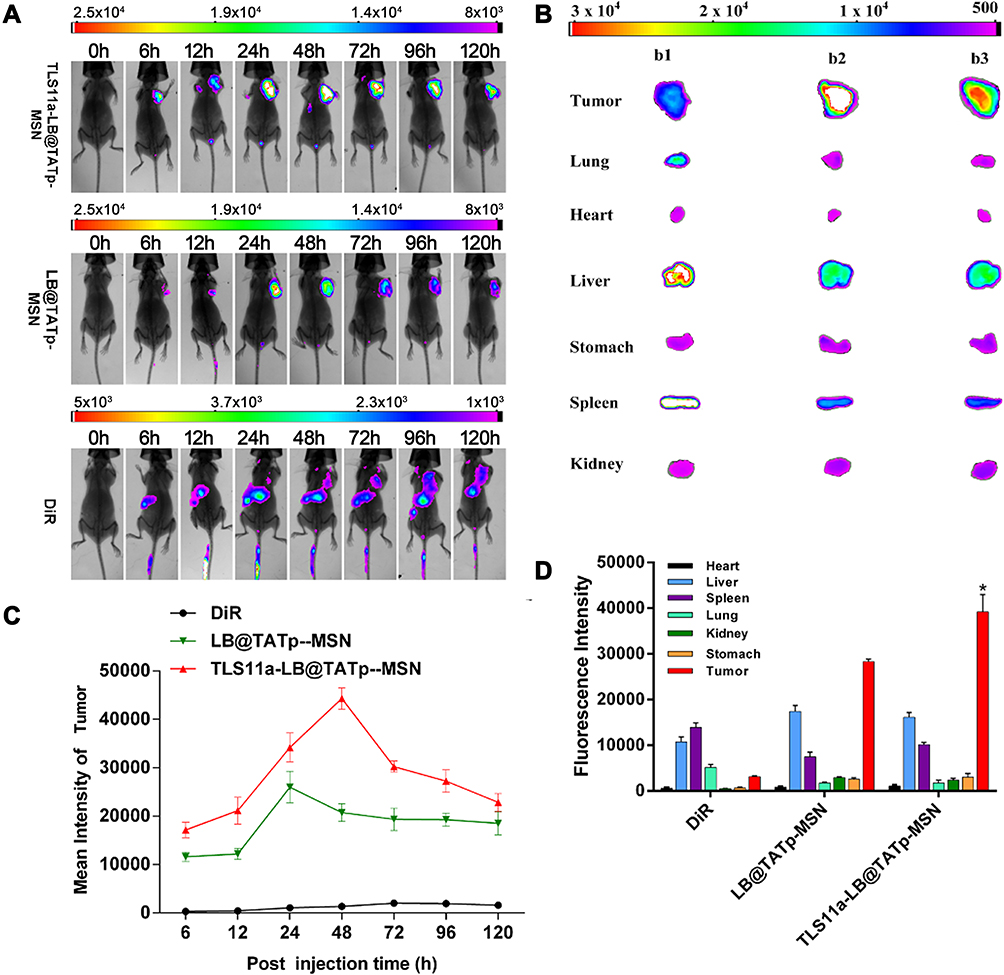 |
Figure 7 Specific distribution of intravenously injected TLS11a-LB@TATp-MSN and LB@TATp-MSN to H22 tumors in the right axilla of BALB/c mice. DiR-labeled TLS11a-LB@TATp-MSN, LB@TATp-MSN, and DiR were intravenously injected via the tail vein into BALB/c mice carrying H22 tumors in the right axilla. (A) Fluorescence in the mice was examined using an in vivo imaging system for small animals. (B) The fluorescence in the heart, liver, spleen, lung, kidney, stomach, and tumor tissues of the DiR-labeled (b1) TLS11a-LB@TATp-MSN, (b2) LB@TATp-MSN, and (b3) DiR mice. (C) The fluorescence intensities of tumor tissues. Bruker MI SE software was used for quantification. (D) The fluorescence intensities of tissues of the TLS11a-LB@TATp-MSN, LB@TATp-MSN, and DiR mice after 48-h treatment. *P < 0.05 compared with the LB@TATp-MSN group. |
We further examined the localization of nano-drugs labeled with FITC through visualizing the fluorescence in tumor tissue sections and found that in the TLS11a-LB-TATp-MSN/DOX group, the nanomaterials with green fluorescence and DOX with red fluorescence were the most widely distributed in tumor tissues, indicating that TLS11a-LB-TATp-MSN/DOX had a superior tumor-specific targeting effect (Figure 8). The TLS11a-LB-TATp-MSN/DOX could accumulate in individual tumor cells (Figure S8). These results suggested that TLS11a-LB@TATp-MSN and LB@TATp-MSN were specifically distributed within H22 tumors in the right axilla of BALB/c mice after intravenous injection. TLS11a played a critical role in enrichment of TLS11a-LB@TATp-MSN in H22 tumors in the right axilla of BALB/c mice.
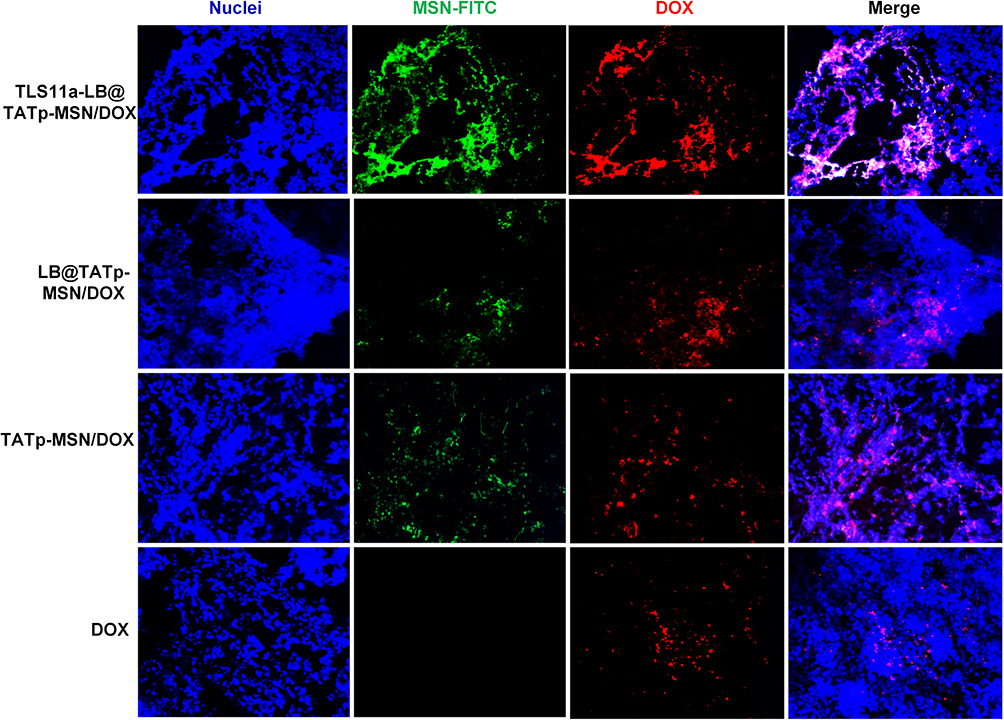 |
Figure 8 Localization of TLS11a-LB and TATp-MSN-derived nano-drugs in tumor tissues in the right axilla of BALB/c mice. MSNs were labeled with FITC. The nano-drugs drugs were intravenously injected via the tail vein into BALB/c mice carrying H22 tumors in the right axilla. After 24 h, the tumor tissues were collected, and frozen sections were prepared and visualized under a fluorescent microscope. Red, DOX; green, MSN; blue, DAPI. |
Anti-Tumor Effect of TLS11a-LB@TATp-MSN/DOX and Its Derivatives in vivo
To determine the anti-tumor effect of the TLS11a-LB@TATp-MSN/DOX and its derivatives in vivo, we treated BALB/c mice carrying H22 tumors in the right axilla with TLS11a-LB@TATp-MSN/DOX or its derivatives via tail vein injection and examined the tumor volume and weight, body weight, survival time of mice, and fresh whole tumor tissues. The results showed that the tumor volumes increased with time extended after drug treatment. The increases in the tumor volume were the lowest in the TLS11a-LB@TATp-MSN/DOX group, followed by the TLS11a-LB@ MSN/DOX, LB@TATp-MSN/DOX, DOX, TATp-MSN/DOX, TLS11a-LB@TATp-MSN, and PBS groups (Figure 9A). The changes in tumor weight after treatment followed a trend similar to that for tumor volume (Figure 9B). The median survival times for mice carrying H22 tumors were 37, 40, 65, 49.5, 66.5, 85, and 94.5 days for the PBS, TLS11a-LB@TATp-MSN, DOX, TATp-MSN/DOX, LB@TATp-MSN/DOX, TLS11a-LB@MSN/DOX, and TLS11a-LB@TATp-MSN/DOX groups, respectively (Figure 9C). The body weight was increased in the TLS11a-LB@TATp-MSN and PBS groups, and it was decreased in the LB@TATp-MSN/DOX, DOX, and TATp-MSN/DOX groups. There were no significant changes in the TLS11a-LB@TATp-MSN/DOX and TLS11a-LB@ MSN/DOX groups (Figure 9D). The sizes of fresh whole tumor tissues taken from the mice after treatment followed a trend similar to those for the volumes and weight of the H22 tumors (Figure 9E). These results suggested that TLS11a-LB@TATp-MSN/DOX and its derivatives had an in vivo anti-tumor effect, and TLS11a-LB@TATp-MSN/DOX had the strongest effect.
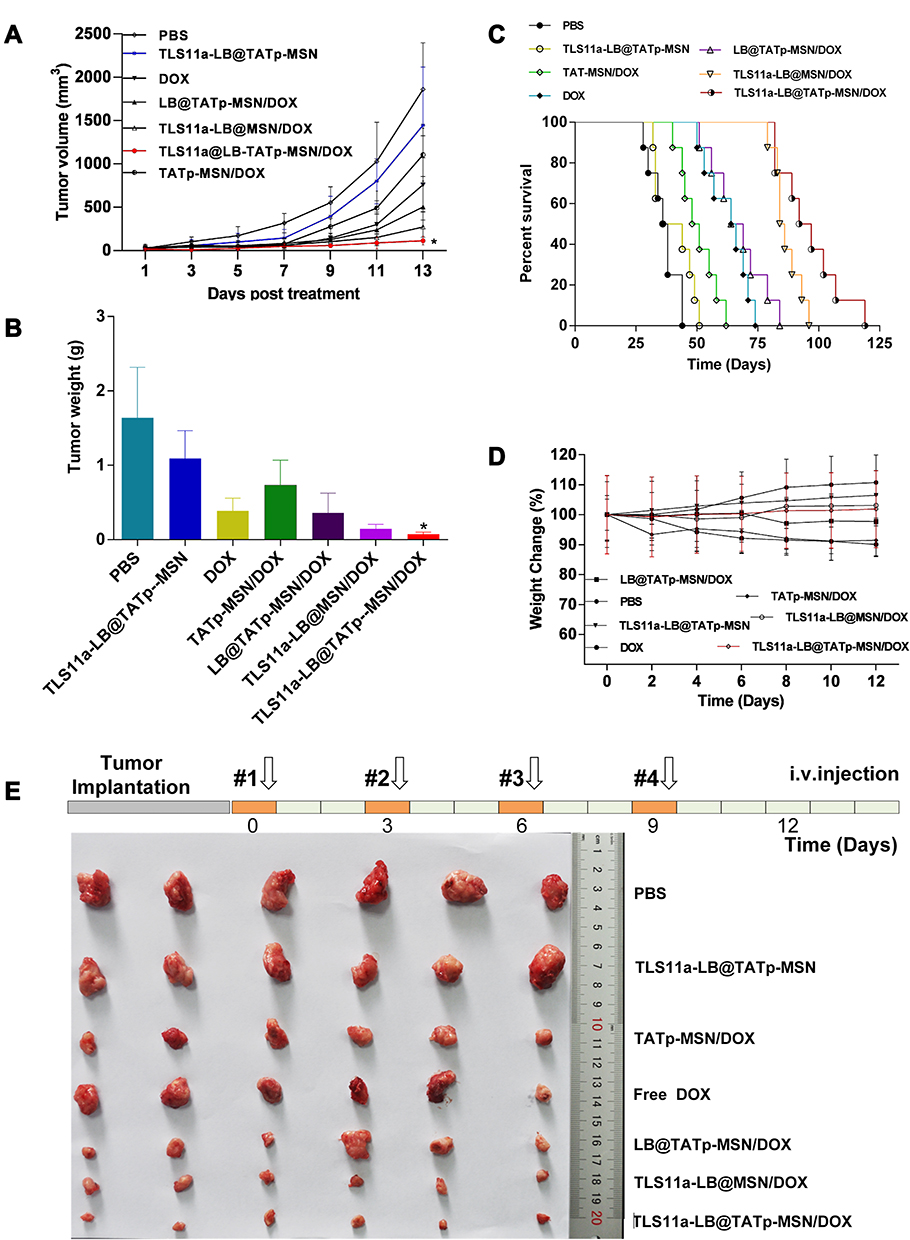 |
Figure 9 The in vivo anti-tumor effect of TLS11a-LB@TATp-MSN/DOX and its derivatives. BALB/c mice carrying H22 tumors in the right axilla were treated with the TLS11a-LB@TATp-MSN/DOX or its derivatives via tail vein injection. The tumor volume and weight, body weight, survival rate of mice, and fresh whole tumor tissues were examined. (A) Changes in the tumor volumes after treatment with the TLS11a-LB@TATp-MSN/DOX and its derivatives for 12 days. *P < 0.05. (B) Changes in the tumor weight after treatment with the TLS11a-LB@TATp-MSN/DOX and its derivatives for 12 days. *P < 0.05. (C) The survival rates of mice after treatment with the TLS11a-LB@TATp-MSN/DOX and its derivatives. (D) The changes in the body weight during treatment with the TLS11a-LB@TATp-MSN/DOX and its derivatives. (E) The whole H22 tumor tissues taken from the mice after treatment with the TLS11a-LB@TATp-MSN/DOX and its derivatives. |
Discussion
In this study, we have developed a novel drug delivery platform that targets liver cancer tissue and the nuclei of the cancer cells in vivo. The drug delivery system that was constructed by conjugating TAT peptide onto the surface of TATp-MSN and with the liver cancer-specific aptamer TLS11a-modified LBs. We first generated the drug TLS11a-LB@TATp-MSN/DOX by mixing TLS11a-LB and TATp-MSN carrying DOX, which confirmed the formation of nano-drug (<100 nm) using scanning electron microscopy, and performed physical and chemical characterization.
We next evaluated the efficiency of TLS11a-LB@TATp-MSN/DOX drug delivery and the efficacy of its anti-tumor effect. The DOX release rate from TLS11a-LB@TATp-MSN/DOX was higher at pH 5.0 than at pH 7.4, which represented the acidic tumor environment and normal tissue environment, respectively. Moreover, TLS11a-LB@TATp-MSN/DOX was effectively localized to the nuclei of H22 cells and released DOX with a higher efficiency than TLS11a-LB@MSN/DOX or LB@TATp-MSN/DOX. TLS11a-LB@TATp-MSN/DOX but not TLS11a-LB@TATp-MSN exhibited cytotoxicity against H22 cells. TLS11a-LB@TATp-MSN accumulated in the subcutaneous H22 tumors in the right axilla of BALB/c mice following tail vein injection. After treatment with TLS11a-LB@TATp-MSN/DOX, the volume and weight of growing tumors and the fresh whole H22 tumor tissues of the TLS11a-LB@TATp-MSN/DOX group were significantly less than those of the control groups. The median survival time of the TLS11a-LB@TATp-MSN/DOX group was significantly longer than those of the control groups. These data indicate that TLS11a-LB@TATp-MSN/DOX can efficiently release DOX into the nuclei of liver cancer cells in vivo by targeting liver cancer tissue and the nuclei of the cancer cells, and therefore, it is a promising nano-drug for the treatment of liver cancer.
In the current study, we combined MSN and liposomes to deliver DOX to overcome the drawbacks of their individual use. MSNs and liposomes have been widely used in drug delivery.17–22,33 However, there are several disadvantages using only one of these as a drug carrier, including premature loss of drugs from MSNs and the instability of liposomes in extreme pH and circulation.32,33 We observed that the cell viability after treatment of H22 cells with TLS11a-LB@TATp-MSN/DOX was higher than that after treatment with TATp-MSN/DOX, which is probably due to less favorable delivery of the larger TLS11a-LB@TATp-MSN/DOX nanoparticles than that of the relatively small TATp-MSN/DOX nanoparticles in vitro. In addition, TATp as a nucleotide could also easily penetrate the cellular membrane other than targeting the nuclear, and rapidly transport nanoparticles into the cytoplasm. Moreover, the presence of TLS11a helped more TLS11a-LB@TATp-MSN carrying the drug entering the tumor cell membrane through active transport, then led the drug transporting to the nucleus to play its role, in comparison with LB@TATp-MSN (lacking TLS11a) that largely relied on phagocytosis to enter the cells. Apparently, TLS11a played very critical role in tumor cell targeting, entering and killing for TLS11a-LB@TATp-MSN/DOX. On the other hand, based on the H22 tumor xenografic mice as a model to assess the in vivo antitumor potency of TLS11a-LB@TATp-MSN/DOX, we observed that TLS11a-LB@TATp-MSN/DOX showed higher tumor regression than that of TATp-MSN/DOX according to the changes in tumor volume, tumor size, tumor weight, body weight, median survival time, and fresh whole tumor tissues (Figure 9). This is consistent with recent studies showing that LBs can attach to the silicate surface in the mesopores and form liposomes, enhancing the biocompatibility of MSNs and extending their availability in circulation.34–36 Therefore, the combination of MSNs and LBs offers more efficient delivery of DOX for the treatment of liver cancer.
To facilitate delivery of DOX and improve the delivery efficiency, we modified MSNs with TATp and modified LBs with TLS11a. TATp is a nuclear localization signaling peptide that has been used in nuclei-targeted drug delivery within cancer cells without triggering MDR mechanisms.37–42 TLS11a is an aptamer that can specifically recognize the mouse liver cancer cell lines BNL 1ME A.7R.1 (MEAR) and H22 as well as human liver cancer cells HepG2 and LH86.55–57 We found that the in vivo anti-tumor efficacy of TLS11a-LB@TATp-MSN/DOX is higher than that of LB@TATp-MSN/DOX and TLS11a-LB@MSN/DOX (Figure 9), suggesting that TLS11a and TATp are critical in targeted therapy for liver cancer using TLS11a-LB@TATp-MSN/DOX. This is consistent with our observation of fluorescent microscopy formerly described that both TLS11a and TATp promoted the subcellular localization of TLS11a-LB@TATp-MSN to the nuclei of H22 cells. As shown in Figure 4A, LB and MSN of both TLS11a-LB@TATp-MSN and TLS11a-LB@MSN remained in a union status at 4 h post incubation with H22 cells, evidenced by the co-localization of red (DiI-LB) and green (FITC-MSN) fluorescence signals merging into pink color. As time went on, we observed that green fluorescence and red fluorescence were gradually separated in TLS11a-LB@TATp-MSN group (Figure 4B and C, lower) but not in the TATp-free TLS11a-LB@MSN control group (Figure 4B and C, upper) at 24 h and 48 h post incubation with H22, indicating the release of TATp-MSN (green) from TLS11a-LB (red) in the cells. Moreover, we also found bright green fluorescence in the nucleus of H22 after TLS11a-LB@TATp-MSN was incubating with the cells for 24 h and 48 h, which was hardly observed in other control groups (Figure S6 and S7). TLS11a and/or TATp played a key role in the increased cytotoxicity of TLS11a-LB@TATp-MSN/DOX in H22 cells, compared with LB@TATp-MSN/DOX and TLS11a-LB@ MSN/DOX (Figure 6). TLS11a played a critical role in enrichment of TLS11a-LB@TATp-MSN in H22 tumors in the right axilla of BALB/c mice (Figures 7 and 8). These data support that TLS11a and TATp are critical in targeted therapy for liver cancer using TLS11a-LB@TATp-MSN/DOX.
DOX has been used to treat liver cancer for nearly 40 years.62 Currently, DOX is still a first-line drug for cancer chemotherapy via the targeting of DNA, which is localized in the nuclei. The traditional ways, such as liposomes,8 lipid nanoparticles,12 and superparamagnetic iron oxide nanoparticles,13 can improve the therapeutic efficacy for liver cancer. However, there are several problems in these approaches, such as their invasiveness, off-target effects, biocompatibility, and low efficiency. In the current study, we have established a TLS11a/TATp double-targeted and LB-MSN–mediated DOX delivery through intravenous injection, providing a novel approach to treating liver cancer.
In summary, we have generated a new anti-cancer agent TLS11a-LB@TATp-MSN/DOX by mixing TLS11a-LB and TATp-MSN that carried DOX. The size of TLS11a-LB@TATp-MSN/DOX has been confirmed to be a nano-drug (<100 nm). This nano-drug can be effectively localized to the nuclei of H22 cells and released with DOX with high efficiency. TLS11a-LB@TATp-MSN/DOX but not TLS11a-LB@TATp-MSN exhibits cytotoxicity against H22 cells. TLS11a-LB@TATp-MSN/DOX can be enriched in liver cancer tissues and efficiently release DOX into the nuclei of liver cancer cells in vivo by targeting liver cancer tissue and the nuclei of the cancer cells. Our results in the present study demonstrate the great potential of TLS11a-LB@TATp-MSN/DOX as a promising strategy of cancer tissue-specific and nuclei-targeted nano-drug delivery system for cancer treatment in the future.
Funding
This work was supported, in part, by grants from Programs for Changjiang Scholars and Innovative Research Team in University (No. IRT1119), National Natural Scientific Foundation of China (No. 81372452), the Project for International Nanobody Research Center of Guangxi (No. GuiKe-AD17195001) and Guangxi Bagui Honor Scholars.
Disclosure
The authors declare no potential conflicts of interest.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Liu CY, Chen KF, Chen PJ. Treatment of Liver Cancer. Csh Perspect Med. 2015;5:9.
3. Shi B, Huang K, Ding J, et al. Intracellularly swollen polypeptide nanogel assists hepatoma chemotherapy. Theranostics. 2017;7(3):703–716.
4. Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5(4):275–284.
5. Bar-Zeev M, Livney YD, Assaraf YG. Targeted nanomedicine for cancer therapeutics: towards precision medicine overcoming drug resistance. Drug Resist Updat. 2017;31:15–30. doi:10.1016/j.drup.2017.05.002
6. Sui MH, Liu WW, Shen YQ. Nuclear drug delivery for cancer chemotherapy. J Control Release. 2011;155(2):227–236. doi:10.1016/j.jconrel.2011.07.041
7. Larsen AK, Escargueil AE, Skladanowski A. Resistance mechanisms associated with altered intracellular distribution of anticancer agents. Pharmacol Ther. 2000;85(3):217–229. doi:10.1016/S0163-7258(99)00073-X
8. Zhao C, Feng Q, Dou Z, et al. Local targeted therapy of liver metastasis from colon cancer by galactosylated liposome encapsulated with doxorubicin. PLoS One. 2013;8(9):e73860. doi:10.1371/journal.pone.0073860
9. Poon RT, Borys N. Lyso-thermosensitive liposomal doxorubicin: an adjuvant to increase the cure rate of radiofrequency ablation in liver cancer. Future Oncol. 2011;7(8):937–945. doi:10.2217/fon.11.73
10. Wood BJ, Poon RT, Locklin JK, et al. Phase I study of heat-deployed liposomal doxorubicin during radiofrequency ablation for hepatic malignancies. J Vasc Interv Radiol. 2012;23(2):248–255 e247. doi:10.1016/j.jvir.2011.10.018
11. Tak WY, Lin SM, Wang YJ, et al. Phase III HEAT study adding lyso-thermosensitive liposomal doxorubicin to radiofrequency ablation in patients with unresectable hepatocellular carcinoma lesions. Clin Cancer Res. 2018;24(1):73–83. doi:10.1158/1078-0432.CCR-16-2433
12. Zhao X, Chen Q, Liu W, et al. Codelivery of doxorubicin and curcumin with lipid nanoparticles results in improved efficacy of chemotherapy in liver cancer. Int J Nanomedicine. 2015;10:257–270.
13. Maeng JH, Lee DH, Jung KH, et al. Multifunctional doxorubicin loaded superparamagnetic iron oxide nanoparticles for chemotherapy and magnetic resonance imaging in liver cancer. Biomaterials. 2010;31(18):4995–5006. doi:10.1016/j.biomaterials.2010.02.068
14. Liu Y, Zhao JT, Jiang JL, Chen FF, Fang XD. Doxorubicin delivered using nanoparticles camouflaged with mesenchymal stem cell membranes to treat colon cancer. Int J Nanomed. 2020;15:2873–2884. doi:10.2147/IJN.S242787
15. Chang JE, Shim WS, Yang SG, et al. Liver cancer targeting of doxorubicin with reduced distribution to the heart using hematoporphyrin-modified albumin nanoparticles in rats. Pharm Res-Dordr. 2012;29(3):795–805. doi:10.1007/s11095-011-0603-6
16. Chang JE, Yoon IS, Sun PL, Yi E, Jheon S, Shim CK. Anticancer efficacy of photodynamic therapy with hematoporphyrin-modified, doxorubicin-loaded nanoparticles in liver cancer. J Photoch Photobio B. 2014;140:49–56. doi:10.1016/j.jphotobiol.2014.07.005
17. Meng H, Mai WX, Zhang HY, et al. Codelivery of an optimal drug/siRNA combination using mesoporous silica nanoparticles to overcome drug resistance in breast cancer in vitro and in vivo. Acs Nano. 2013;7(2):994–1005. doi:10.1021/nn3044066
18. Meng H, Liong M, Xia T, et al. Engineered design of mesoporous silica nanoparticles to deliver doxorubicin and P-glycoprotein siRNA to overcome drug resistance in a cancer cell line. Acs Nano. 2010;4(8):4539–4550. doi:10.1021/nn100690m
19. Qu QY, Ma X, Zhao YL. Anticancer effect of alpha-tocopheryl succinate delivered by mitochondria-targeted mesoporous silica nanoparticles. Acs Appl Mater Inter. 2016;8(50):34261–34269.
20. Sarkar A, Ghosh S, Chowdhury S, Pandey B, Sil PC. Targeted delivery of quercetin loaded mesoporous silica nanoparticles to the breast cancer cells. Bba-Gen Subjects. 2016;1860(10):2065–2075. doi:10.1016/j.bbagen.2016.07.001
21. Zhao Q, Liu J, Zhu W, et al. Dual-stimuli responsive hyaluronic acid-conjugated mesoporous silica for targeted delivery to CD44-overexpressing cancer cells. Acta Biomater. 2015;23:147–156. doi:10.1016/j.actbio.2015.05.010
22. Liu J, Li QL, Zhang JX, et al. Safe and effective reversal of cancer multidrug resistance using sericin-coated mesoporous silica nanoparticles for lysosome-targeting delivery in mice. Small. 2017;13:9.
23. Park IY, Kim IY, Yoo MK, Choi YJ, Cho MH, Cho CS. Mannosylated polyethylenimine coupled mesoporous silica nanoparticles for receptor-mediated gene delivery. Int J Pharm. 2008;359(12):280–287. doi:10.1016/j.ijpharm.2008.04.010
24. Ashley CE, Carnes EC, Epler KE, et al. Delivery of small interfering RNA by peptide-targeted mesoporous silica nanoparticle-supported lipid bilayers. Acs Nano. 2012;6(3):2174–2188. doi:10.1021/nn204102q
25. Xiong L, Bi JX, Tang YH, Qiao SZ. Magnetic core-shell silica nanoparticles with large radial mesopores for siRNA delivery. Small. 2016;12(34):4735–4742. doi:10.1002/smll.201600531
26. Shen J, Liu H, Mu C, et al. Multi-step encapsulation of chemotherapy and gene silencing agents in functionalized mesoporous silica nanoparticles. Nanoscale. 2017;9(16):5329–5341. doi:10.1039/C7NR00377C
27. Shen J, Kim HC, Su H, et al. Cyclodextrin and polyethylenimine functionalized mesoporous silica nanoparticles for delivery of siRNA cancer therapeutics. Theranostics. 2014;4(5):487–497. doi:10.7150/thno.8263
28. Huang XL, Zhang F, Lee S, et al. Long-term multimodal imaging of tumor draining sentinel lymph nodes using mesoporous silica-based nanoprobes. Biomaterials. 2012;33(17):4370–4378. doi:10.1016/j.biomaterials.2012.02.060
29. Su JH, Sun HP, Meng QS, Zhang PC, Yin Q, Li YP. Enhanced blood suspensibility and laser-activated tumor-specific drug release of theranostic mesoporous silica nanoparticles by functionalizing with erythrocyte membranes. Theranostics. 2017;7(3):523–537. doi:10.7150/thno.17259
30. Chen Y, Ai KL, Liu JH, Sun GY, Yin Q, Lu LH. Multifunctional envelope-type mesoporous silica nanoparticles for pH-responsive drug delivery and magnetic resonance imaging. Biomaterials. 2015;60:111–120. doi:10.1016/j.biomaterials.2015.05.003
31. Samykutty A, Grizzle WE, Fouts BL, et al. Optoacoustic imaging identifies ovarian cancer using a microenvironment targeted theranostic wormhole mesoporous silica nanoparticle. Biomaterials. 2018;182:114–126. doi:10.1016/j.biomaterials.2018.08.001
32. Liu JW, Jiang XM, Ashley C, Brinker CJ. Electrostatically mediated liposome fusion and lipid exchange with a nanoparticle-supported bilayer for control of surface charge, drug containment, and delivery. J Am Chem Soc. 2009;131(22):7567. doi:10.1021/ja902039y
33. Roggers RA, Lin VSY, Trewyn BG. Chemically reducible lipid bilayer coated mesoporous silica nanoparticles demonstrating controlled release and hela and normal mouse liver cell biocompatibility and cellular internalization. Mol Pharmaceut. 2012;9(9):2770–2777. doi:10.1021/mp200613y
34. Dengler EC, Liu JW, Kerwin A, et al. Mesoporous silica-supported lipid bilayers (protocells) for DNA cargo delivery to the spinal cord. J Control Release. 2013;168(2):209–224. doi:10.1016/j.jconrel.2013.03.009
35. Ashley CE, Carnes EC, Phillips GK, et al. The targeted delivery of multicomponent cargos to cancer cells by nanoporous particle-supported lipid bilayers. Nat Mater. 2011;10(5):389–397.
36. Li PC, Li D, Zhang LX, Li GP, Wang EK. Cationic lipid bilayer coated gold nanoparticles-mediated transfection of mammalian cells. Biomaterials. 2008;29(26):3617–3624. doi:10.1016/j.biomaterials.2008.05.020
37. Mei L, Fu L, Shi KR, et al. Increased tumor targeted delivery using a multistage liposome system functionalized with RGD, TAT and cleavable PEG. Int J Pharmaceut. 2014;468(12):26–38. doi:10.1016/j.ijpharm.2014.04.008
38. Pan LM, He QJ, Liu JN, et al. Nuclear-targeted drug delivery of tat peptide-conjugated monodisperse mesoporous silica nanoparticles. J Am Chem Soc. 2012;134(13):5722–5725. doi:10.1021/ja211035w
39. Pan LM, Liu JA, He QJ, Wang LJ, Shi JL. Overcoming multidrug resistance of cancer cells by direct intranuclear drug delivery using TAT-conjugated mesoporous silica nanoparticles. Biomaterials. 2013;34(11):2719–2730. doi:10.1016/j.biomaterials.2012.12.040
40. Liu JN, Bu WB, Pan LM, et al. Simultaneous nuclear imaging and intranuclear drug delivery by nuclear-targeted multifunctional upconversion nanoprobes. Biomaterials. 2012;33(29):7282–7290. doi:10.1016/j.biomaterials.2012.06.035
41. Pan ZZ, Wang HY, Zhang M, et al. Nuclear-targeting TAT-PEG-Asp (8)-doxorubicin polymeric nanoassembly to overcome drug-resistant colon cancer. Acta Pharmacol Sin. 2016;37(8):1110–1120. doi:10.1038/aps.2016.48
42. Liu JH, Zhao YX, Guo QQ, et al. TAT-modified nanosilver for combating multidrug-resistant cancer. Biomaterials. 2012;33(26):6155–6161. doi:10.1016/j.biomaterials.2012.05.035
43. Tan WH, Wang H, Chen Y, et al. Molecular aptamers for drug delivery. Trends Biotechnol. 2011;29(12):634–640. doi:10.1016/j.tibtech.2011.06.009
44. Zhou JH, Rossi J. Aptamers as targeted therapeutics: current potential and challenges. Nat Rev Drug Discov. 2017;16(3):181–202. doi:10.1038/nrd.2016.199
45. Zhuo ZJ, Yu YY, Wang ML, et al. Recent advances in selex technology and aptamer applications in biomedicine. Int J Mol Sci. 2017;18:10. doi:10.3390/ijms18102142
46. Liu J, Wei T, Zhao J, et al. Multifunctional aptamer-based nanoparticles for targeted drug delivery to circumvent cancer resistance. Biomaterials. 2016;91:44–56.
47. Liao J, Liu B, Liu J, Zhang JN, Chen K, Liu HX. Cell-specific aptamers and their conjugation with nanomaterials for targeted drug delivery. Expert Opin Drug Del. 2015;12(3):493–506. doi:10.1517/17425247.2015.966681
48. Gray BP, Kelly L, Ahrens DP, et al. Tunable cytotoxic aptamer-drug conjugates for the treatment of prostate cancer. P Natl Acad Sci USA. 2018;115(18):4761–4766. doi:10.1073/pnas.1717705115
49. Li LL, Xie MY, Wang J, et al. A vitamin-responsive mesoporous nanocarrier with DNA aptamer-mediated cell targeting. Chem Commun. 2013;49(52):5823–5825. doi:10.1039/c3cc41072b
50. Kang HZ, O’Donoghue MB, Liu HP, Tan WH. A liposome-based nanostructure for aptamer directed delivery. Chem Commun. 2010;46(2):249–251. doi:10.1039/B916911C
51. Lee JH, Yigit MV, Mazumdar D, Lu Y. Molecular diagnostic and drug delivery agents based on aptamer-nanomaterial conjugates. Adv Drug Deliver Rev. 2010;62(6):592–605. doi:10.1016/j.addr.2010.03.003
52. Shen QL, Xu L, Zhao LB, et al. Specific capture and release of circulating tumor cells using aptamer-modified nanosubstrates. Adv Mater. 2013;25(16):2368–2373. doi:10.1002/adma.201300082
53. Jung YK, Kim TW, Park HG, Soh HT. Specific colorimetric detection of proteins using bidentate aptamer-conjugated polydiacetylene (PDA) liposomes. Adv Funct Mater. 2010;20(18):3092–3097. doi:10.1002/adfm.201001008
54. Sun HG, Tan WH, Zu YL. Aptamers: versatile molecular recognition probes for cancer detection. Analyst. 2016;141(2):403–415. doi:10.1039/C5AN01995H
55. Shangguan DH, Meng L, Cao ZHC, et al. Identification of liver cancer-specific aptamers using whole live cells. Anal Chem. 2008;80(3):721–728. doi:10.1021/ac701962v
56. Kashefi-Kheyrabadi L, Mehrgardi MA, Wiechec E, Turner APF, Tiwari A. Ultrasensitive detection of human liver hepatocellular carcinoma cells using a label-free aptasensor. Anal Chem. 2014;86(10):4956–4960. doi:10.1021/ac500375p
57. Meng L, Yang L, Zhao XX, et al. Targeted delivery of chemotherapy agents using a liver cancer-specific aptamer. PLoS One. 2012;7:4.
58. Kobler J, Moller K, Bein T. Colloidal suspensions of functionalized mesoporous silica nanoparticles. Acs Nano. 2008;2(4):791–799. doi:10.1021/nn700008s
59. Gatenby RA, Gillies RJ. Hypoxia and metabolism - Opinion - A microenvironmental model of carcinogenesis. Nat Rev Cancer. 2008;8(1):56–61. doi:10.1038/nrc2255
60. Hashim AI, Zhang XM, Wojtkowiak JW, Martinez GV, Gillies RJ. Imaging pH and metastasis. NMR Biomed. 2011;24(6):582–591. doi:10.1002/nbm.1644
61. Vaupel P, Kallinowski F, Okunieff P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res. 1989;49(23):6449–6465.
62. Vilaseca J, Guardia J, Bacardi R, Monne J. Doxorubicin for liver cancer. Lancet. 1978;1(8078):1367. doi:10.1016/S0140-6736(78)92448-0
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.