")
Back to Journals » Drug Design, Development and Therapy » Volume 11
In vivo study of doxorubicin-loaded cell-penetrating peptide-modified pH-sensitive liposomes: biocompatibility, bio-distribution, and pharmacodynamics in BALB/c nude mice bearing human breast tumors
Authors Ding Y, Cui W, Sun D, Wang GL, Hei Y , Meng S, Chen JH, Xie Y, Wang ZQ
Received 23 August 2017
Accepted for publication 4 October 2017
Published 26 October 2017 Volume 2017:11 Pages 3105—3117
DOI https://doi.org/10.2147/DDDT.S149814
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Yuan Ding,1,* Wei Cui,2,* Dan Sun,1 Gui-Ling Wang,1 Yu Hei,1 Shuai Meng,1 Jian-Hua Chen,3 Ying Xie,1 Zhi-Qiang Wang4
1Beijing Key Laboratory of Molecular Pharmaceutics and New Drug Delivery Systems, Department of Pharmaceutics, School of Pharmaceutical Sciences, Peking University, 2School of Chemistry and Chemical Engineering, University of Chinese Academy of Sciences, Beijing, 3School of Medicine, Jianghan University, Wuhan, People’s Republic of China; 4Department of Chemistry and Biochemistry, Kent State University Geauga, Burton, OH, USA
*These authors contributed equally to this work
Abstract: In vivo evaluation of drug delivery vectors is essential for clinical translation. In BALB/c nude mice bearing human breast cancer tumors, we investigated the biocompatibility, pharmacokinetics, and pharmacodynamics of doxorubicin (DOX)-loaded novel cell-penetrating peptide (CPP)-modified pH-sensitive liposomes (CPPL) (referred to as CPPL(DOX)) with an optimal CPP density of 4%. In CPPL, a polyethylene glycol (PEG) derivative formed by conjugating PEG with stearate via acid-degradable hydrazone bond (PEG2000-Hz-stearate) was inserted into the surface of liposomes, and CPP was directly attached to liposome surfaces via coupling with stearate to simultaneously achieve long circulation time in blood and improve the selectivity and efficacy of CPP for tumor targeting. Compared to PEGylated liposomes, CPPL enhanced DOX accumulation in tumors up to 1.9-fold (p<0.01) and resulted in more cell apoptosis as a result of DNA disruption as well as a relatively lower tumor growth ratio (T/C%). Histological examination did not show any signs of necrosis or inflammation in normal tissues, but large cell dissolving areas were found in tumors following the treatment of animals with CPPL(DOX). Our findings provide important and detailed information regarding the distribution of CPPL(DOX) in vivo and reveal their abilities of tumor penetration and potential for the treatment of breast cancer.
Keywords: tumor targeting, TUNEL stain, hemolysis, therapy for breast cancer, pharmacokinetics
Abbreviations
AUC, area under the concentration–time curve; BBB, blood–brain barrier; BMCs, bone marrow cells; CCL, CPP-modified conventional liposomes; CL, conventional liposomes; CCL(DOX), doxorubicin-loaded CPP-modified conventional liposomes; CL(DOX), doxorubicin-loaded conventional liposomes; CPP, cell-penetrating peptide; CPPL, CPP-modified pH-sensitive PEGylated liposomes; CPPL(DOX), doxorubicin-loaded CPP-modified pH-sensitive PEGylated liposomes; DAU, daunomycin; DOX, doxorubicin; DSPE-PEG2000, poly(ethyleneglycol)-N-distearoyl-phosphatidylethanolamine; EDCI, 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride; EPR, enhanced permeability and retention; H&E, hematoxylin and eosin; HPLC, high-pressure liquid chromatography; NS, normal saline; PDI, polydispersity index; PEG, polyethylene glycol; PHS, mPEG2000-hydrazone-stearate (mPEG2000-Hz-stearate); PL, PEGylated liposomes; PL(DOX), doxorubicin-loaded PEGylated liposomes; RBCs, red blood cells; RV, relative tumor volume; SD, standard deviation; SPC, soybean phosphatidylcholine; SRB, sulfurhodamine B; TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling.
Introduction
The health burden of cancer is increasing worldwide, and breast cancer has become the second leading cancer-related cause of death in women.1 Common chemotherapeutics invariably disrupt normal tissues, which leads to irreversible toxic and adverse effects. Currently, nanomedicines such as liposomal formulation have become a successful drug delivery system in clinics for their capabilities of altering the behaviors of chemotherapeutics in vivo and reducing their toxicity on normal tissues.2
DOX hydrochloride liposome injection3 (Doxil®, Lipodox®, or Caelyx®) was approved in 1995 by the FDA as the first antitumor liposome preparation. With the presence of PEG on the surface of the liposomes, DOX hydrochloride liposome injection, named here as PL(DOX), has the ability to passively accumulate in tumor tissues through EPR effects4 and prolong the circulation time of DOX in blood. However, PEGylation reduces the extent of interactions of liposomes with target cells, resulting in improper cellular uptake as well as poor endosomal escape, a factor that ultimately leads to a significant loss of the pharmacological effect of a drug.5,6
At present, intracellular or organelle-specific targeting is becoming an emerging concept for improving nanomedicine actions.7,8 CPPs,9 small molecular transporters with the capabilities of cell penetration, internalization, and endosomal escape, due to the presence of large number of positively charged arginine residues in acidic environment, have the potential to be useful in drug delivery system.10,11 However, poor specificity for cells is one of the drawbacks of CPPs.12 By utilizing the function of hydration shell of PEG and acid sensitivity of hydrazone bond, we synthesized a kind of CPPL13 to improve the selectivity of these peptides for tumor targeting. In CPPL, CPP was directly attached to liposome surfaces via coupling with stearate to avoid the hindrance of PEG as a linker on the penetrating efficiency of CPP, and a PEG derivative was synthesized by conjugating PEG with stearate via acid-degradable hydrazone bond (PHS) and was incorporated into the lipid membrane. In vitro studies including HPLC, flow cytometry, ex vivo imaging, and confocal laser-scanning microscopy indicated that 8 mol% PHS was enough to cover 4 mol% CPPL at neutral pH and that CPPL was too stable to be captured by normal tissues which consequently led to a long circulation time in blood.13 When CPPL arrived at tumor cells, PEG on PHS was completely cleaved from liposome surface to expose CPP sufficiently under acidic environments in tumor for its penetration across tumor cells and helping entrapped cargoes such as DOX escape from lysosomes and gaining more opportunities to enter nucleus to exert pharmacological effects.13 CPPL not only balanced the interplay between cell binding and blood circulation of PL but also promised higher efficiency of CPP in internalizing the liposomes into targeted subcellular compartments.13
However, the presence of high serum concentrations and nonspecific binding to extracellular components can interfere with the in vivo performance of the delivery vector. In this study, we further explored the pharmacokinetics and pharmacodynamics of CPPL(DOX) in BALB/c nude mice bearing human breast tumors. PL were chosen as a comparative delivery vector. Preclinical in vitro and in vivo studies of CPPL for DOX loading, including formulation, mechanism of tumor cell uptake, pharmacokinetics, bio-distribution, pharmacodynamics, and biocompatibility, proved the potential of CPPL and provided the basis for entering clinical trials due to the non-immunogenicity of CPP and the stable quality of PHS which recommends their use in future CPPL manufacturing.
Materials and methods
Materials
SPC (99% purity) was purchased from Shanghai Taiwei Chemical Company (Shanghai, People’s Republic of China). Cholesterol was purchased from Alfa Aesar Co., Ltd. (Ward Hill, MA, USA). Stearate was obtained from Beijing Chemical Factory (Beijing, People’s Republic of China). EDCI was obtained from J&K Scientific, Ltd. (Beijing, People’s Republic of China). CPPs (GGRRRRRRRRR-amide) were provided by KTG Pharmaceuticals, Inc. (Wuhan, People’s Republic of China). DSPE-PEG2000 was purchased from Shanghai Advanced Vehicle Technology Pharmaceutical, Ltd. (Shanghai, People’s Republic of China). PHS was synthesized and characterized in our laboratory.13
Cell cultures
The MCF-7 human breast cancer cell line was purchased from the Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences (Beijing, People’s Republic of China). The cells were cultured in RPMI 1640 containing 2 mM L-glutamine and were supplemented with 10% (v/v) heat-inactivated fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. Cultures were maintained at 37°C in a humidified 5% CO2 incubator. The cells were subcultured approximately every 3 days throughout the experiments.
Animals
BALB/c nude mice (female, 20±2 g) were provided by the Animal Center of Peking University Health Science Center. Animals were housed at 25°C and 55% humidity under natural light with free access to food and water. All animal-related experiments were performed in full compliance with institutional guidelines and approved by the Institutional Authority for Laboratory Animal Care of Peking University (No LA2016155).
Establishment of BALB/c nude mice bearing MCF-7 model
Breast tumors were grown in BALB/c nude mice by subcutaneous injections of 200 μL of MCF-7 suspension (1×107 cells/mL) in PBS into the right armpits.14 At the 7th day after tumor inoculation, the volume of tumor was measured with a vernier caliper and estimated using the formula: volume = length (cm) × width (cm2) ×0.5236.15 After 15 days of human breast tumor implanting, animals with tumor volume >100 mm3 were chosen for pharmacokinetic and pharmacodynamic studies.
Preparation of CPPL(DOX) and PL(DOX)
DOX was loaded into CPPL by a combination of the pH gradient16 and post-reaction methods13,17 as shown in Figure 1A. In brief, dried lipid films composed of SPC, cholesterol, and stearate at a molar ratio of 100:50:4 were hydrated with 300 mmol/L citrate buffer (pH 4.0) to prepare CL with a final lipid concentration of 1 mmol/L. After sonication for 10 min at 25°C, an aliquot of sodium carbonate solution (0.5 mmol/L) was added to the suspensions to create an external liposome medium of pH 7.8 and internal liposome medium of pH 4.0. To load DOX into liposomes, a mixture of the liposomal suspension and DOX at 100:8 molar ratio (lipid:drug) was incubated at 55°C–60°C for 30 min, cooled to room temperature in the dark, flushed with N2, and stored in the refrigerator overnight. Afterward, CPP was conjugated with CL via an activated stearate group. During the process, EDCI was first added into the CL(DOX) containing stearate, and the mixture was stirred for 1 min. S-NHS was then added at a stearate/EDCI/S-NHS molar ratio of 1:40:100. After stirring the mixture for 15 min, the pH was adjusted to 12. As the coupling efficiency of CPP with stearate was 75%,13 CPP were reacted with NHS-activated stearate group on CL at the molar ratio of 4:3 to form CCL for DOX loading. To obtain 4% CPPL for DOX loading, 8 mol% PHS of SPC was incubated with CCL at pH 7.0 for 12 h, and suspensions in ultrafilter tubes (MWCO, 30,000 Da) were centrifuged at 1,000× g for 30 min to remove unreacted EDCI, S-NHS, and CPP.
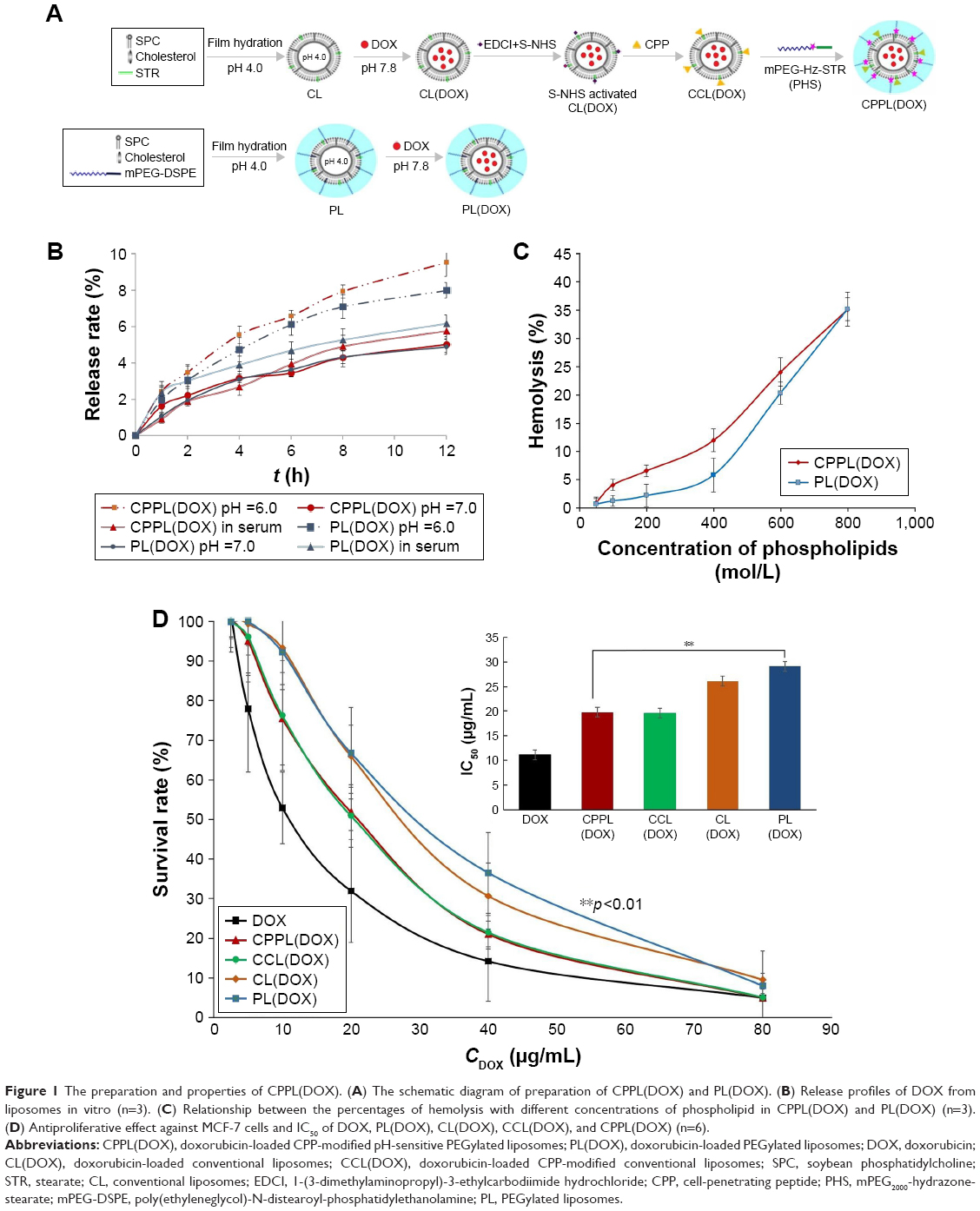 | Figure 1 The preparation and properties of CPPL(DOX). (A) The schematic diagram of preparation of CPPL(DOX) and PL(DOX). (B) Release profiles of DOX from liposomes in vitro (n=3). (C) Relationship between the percentages of hemolysis with different concentrations of phospholipid in CPPL(DOX) and PL(DOX) (n=3). (D) Antiproliferative effect against MCF-7 cells and IC50 of DOX, PL(DOX), CL(DOX), CCL(DOX), and CPPL(DOX) (n=6). |
DOX encapsulated into PL(DOX) served as the control, which was prepared using pH gradient method16 (Figure 1A). In brief, SPC/Chol/DSPE-PEG2000 (100:50:8 mol/mol/mol) was dissolved with medium containing chloroform and methanol at a volume ratio of 4:1 in a round-bottom flask. The solvent was evaporated at 37°C on a rotary evaporator until homogeneous lipid films formed on the glass wall. DOX was then loaded into PL as described.
Characterization of CPPL(DOX) and PL(DOX)
CPPL(DOX) were diluted with distilled water to analyze the changes in size and zeta potential of liposomes caused by the 4 mol% CPP modification. After filtering each liposome with a 0.45 μm filter membrane, size distribution (nm), PDI, and zeta potential were determined using Nano ZS ZEN3600 Zetasizer (Malvern Instruments, Ltd., Malvern, UK) at 25°C. Assays were performed 15 times with an equilibrium time of 60 s. DOX encapsulation efficiency (ee%) in liposomes was determined by comparing the adsorption density at 485 nm of eluted liposome solutions after removing free drugs through a Sephadex G-50 column with the primary liposome suspensions in an equal volume.
Leakage stability of CPPL(DOX) in vitro
To simulate drug release profiles of CPPL(DOX) in blood and tumor environment in vitro, we chose three kinds of release media including PBS of pH 7.0, PBS of pH 6.0, and PBS of pH 7.0 containing 10% horse serum. In brief, 1.0 mL PL(DOX) and CPPL(DOX) were sealed into dialysis bags (MWCO, 50,000 Da), and incubated in 30 mL of release medium at 37°C in dark, with continuous stirring for 12 h. A 1 mL aliquot was removed at predetermined time points, and replaced with 1 mL of fresh medium. The released DOX concentrations (csample, μg/mL) were determined with a Cary Eclipse spectrofluorometer (Varian Corporation, Palo Alto, CA, USA) at excitation and emission wavelengths of 437 and 584 nm, respectively. The cumulative release proportion and the proportion of total DOX released from liposomes were calculated and plotted at given time points.
Evaluation of hemolytic activity
Hemolysis studies were performed to evaluate the safety of liposomes for in vivo applications. Rat RBCs were harvested by centrifugation at 1,000× g for 10 min following the collection of rat blood in EDTA-containing tubes. The plasma supernatant was discarded, and PBS was added to wash the RBC three times. The RBCs were resuspended in PBS to a concentration of 4×106 cells/mL. PL(DOX) and CPPL(DOX) were diluted with PBS at a phospholipid concentration of 50, 100, 200, 400, 600, and 800 μmol/L. An equal volume of liposomes and RBCs was incubated for 1 h in a water bath at 37°C followed by centrifugation at 1,000× g for 10 min. Absorbance of hemoglobin in the supernatant solution was measured at 570 nm using an ELISA Reader (Thermo Fisher Scientific, Waltham, MA, USA). RBC hemolysates in PBS solutions and in 1% Triton X-100 were used as negative and positive controls, respectively. The percent of hemolysis was calculated with the formula: percent hemolysis = (A[liposomes] − A[PBS])/A(Triton-100) ×100%. The sample with less than 10% hemolysis was considered nontoxic.18
In vitro cytotoxicity of CPPL(DOX)
In vitro cell viability was determined with MCF-7 cells using SRB stain method.19,20 Cells were seeded in a 96-well plate (1×104 cells/well) and incubated for 24 h at 37°C with 5% CO2. Cells were then washed with 100 μL/well PBS and incubated with dilutions of DOX, CPPL(DOX), CCL(DOX), CL(DOX), and PL(DOX) at pH 6.0. Equivalent DOX concentrations (2.5, 5.0, 10, 20, 40, 60, and 80 μg/mL) of each formulation were prepared by diluting with fresh serum-free RPMI 1640. Cell viability was measured via an SRB assay after an additional 24 h incubation at 37°C. Serum-free RPMI 1640 and 1% Triton X-100 culture solutions were used as negative and positive controls, respectively. Drug-administered cells were rinsed five times with deionized water, dried at 37°C after rinsing with ice-cold PBS, and fixed with 10% trichloroacetic acid at 4°C for 1 h. Then, 100 μL of 0.4% SRB in 1% acetum was added to each well at room temperature and incubated for 30 min to prepare for staining. Each well was then dried at the same temperature after rinsing five times again with 1% acetum. SRB in cells was dissolved with 200 μL of 10 mM Tris solution and shook at 37°C for 30 min. The optical density at 540 nm was recorded using Flexstation® 3 (Molecular Devices, Sunnyvale, CA, USA). Cytotoxicity was expressed as the percentage of the control. The IC50 was calculated using SPSS software (version 16.0; SPSS Inc., Chicago, IL, USA).
Pharmacokinetics and bio-distribution of CPPL(DOX) in nude mice bearing tumor
Thirty-six breast tumor-bearing BALB/c female nude mice were randomly divided into two groups. CPPL(DOX) and PL(DOX) were administered a dose of 5 mg DOX/kg via the tail vein.21 Subsequently, blood samples (0.5 mL) were collected into heparin-containing tubes at 30 min and 1, 2, 4, and 8 h from each animal. Plasma was extracted by centrifugation at 2,000× g for 10 min and stored at −20°C. Forty microliters of DAU (400 μg/mL) was used as an internal standard and added to 500 μL of plasma, and the drug was extracted using organic solvent (chloroform:methyl alcohol, 4:1, v/v) by centrifugation at 4,500× g for 15 min. The supernatant was dried with N2 gas and reconstituted with mobile phase before being measured using HPLC with the fluorescent detector at λex =437 nm and λem =584 nm (Agilent 1100 series combined with Agilent technologies 1260 infinity Detector; Kromasil C18-ODS 250×4.6 mm, 5 μm column; Dikma Co., Ltd, Lake Forest, CA, USA; mobile phase, 70% CH3OH:30% H2O; flow rate, 1.0 mL/min, injection volume, 20 μL). Pharmacokinetic parameters such as t1/2, AUC, volume of distribution, and clearance were calculated by fitting the concentrations of DOX to a noncompartmental model using DAS version 2.1.
In the bio-distribution study at time points of 30 min and 1, 2, 4, and 8 h, three tumor-bearing mice per group were sacrificed to obtain blood, major organs (the heart, liver, spleen, lung, and kidney), and tumors. Tissues were immediately washed twice with NS (0.9% NaCl), wiped with filter paper, weighed, and homogenized with citric acid buffer (pH 6.8). The internal standard substance DAU (100 μL, 400 μg/mL) was added to 2 mL of each homogenate, and samples were processed as described and detected using HPLC.
Therapy for breast tumor
Fifteen breast tumor-bearing BALB/c female nude mice were randomly divided into three groups (n=5 per group), and this was designated as day 0. From day 1, mice were treated with CPPL(DOX) and PL(DOX) via tail vein injection at a dose of 2.5 mg DOX/kg. NS served as control. Administration was done every other day for a total of four doses per mouse. Mice were weighted, and tumor sizes were monitored every day for 9 days. RV of each group was calculated with the formula: RV = Vt/V0, where Vt is the tumor volume at the monitoring time point during the experimental period and V0 is the original tumor volume prior to the first treatment. The relative tumor growth ratio was calculated with the formula:22 T/C% = (TRV/CRV) ×100%, where TRV is the RV after treating with drug and CRV is the RV after treating with NS as control. A T/C% of 40% indicated an effective treatment.22
H&E and TUNEL stain
On day 9, mice were sacrificed, and the tumor samples were used for H&E staining and TUNEL analysis.23,24
Tumor samples were fixed in 4% paraformaldehyde and embedded in paraffin. The sections were stained with Harris hematoxylin solution, washed in running tap water, and differentiated in 1% acid alcohol for 10 s. The stained tissue sections were then rinsed in tap water and stained with 1.36% lithium carbonate solution. The slides were then dehydrated in 95% alcohol and counterstained with eosin Y-phloxine B solution for 5–10 s. The stained tissue sections were cleared in xylene, mounted with Cytoseal 60 (Sigma), and observed under a light microscope.
For specific TUNEL staining, tissue samples of MCF-7 tumors were frozen in optimal cutting temperature embedding medium, cut into 4-μm-thick sections, and fixed in 4% (v/v) paraformaldehyde for 15 min at room temperature. The sectioned samples were washed three times with PBS and incubated with 1% (v/v) Triton X-100 (Sigma Co.) for 5 min on ice. The TUNEL reaction mixture was then added to the tissue sections, which was then incubated in the dark under humidified atmosphere at 37°C for 1 h. The samples were washed three times with PBS and treated with Hoechst 33258 (Molecular Probes Inc., Eugene, OR, USA) for 15 min prior to microscopic analysis using a confocal laser scanning microscope (TCS SP5; Leica, Heidelberg, Germany).
Biocompatibility of CPPL(DOX)
The in vivo biocompatibility of liposomes was evaluated by histological examination of tissue sections from normal mice. CPPL(DOX), PL(DOX), and DOX were intravenously administered to normal BALB/c mice at a dose of 2.5 mg DOX/kg every other day with a total of four doses per mouse. On the 9th day of administration, the mice were sacrificed, and the hearts, livers, lungs, and kidneys were separated for H&E staining. Since the livers and spleen were exposed to the highest concentration of liposomes, these tissues were carefully examined for any signs of toxicity. The liver sections were observed for ballooning of hepatocytes, enlargement of nuclei or Kupffer cells, and granulomas. Similarly, the splenic tissue was evaluated for necrosis and inflammation. The hearts and lungs were also examined for signs of necrosis and extravasation of RBCs and thickening of alveoli, respectively. Kidneys were observed for inflammation and necrosis.
The myelosuppressive effect was monitored using BMCs.25 After the mice were euthanized on day 9, a thigh bone was dissected from each mouse, and the cavum ossis was washed with 1 mL of PBS. The rinse solution was collected, and the BMCs were obtained using centrifugation. The BMCs were counted using a white blood cell counting chamber.
Statistical analysis
Statistical data were processed using Microsoft Excel 2013 software, and presented as mean ± SD of at least three independent experiments. The differences/correlations between two groups were analyzed using Student’s t-test, in which data were considered statistically significant if p-value was less than 0.05 (*) and very significant if p-value was less than 0.01 (**).
Results and discussion
Characterization of CPPL(DOX) and PL(DOX)
CPPL(DOX) and PL(DOX) suspension demonstrated red opalescence. As shown in Table 1, their average sizes were approximately 120 nm with PDI <0.2. Compared with PL(DOX), the zeta potential of CPPL(DOX) significantly changed in a positive direction due to the arginine-rich residues of CPP (p<0.01), which may be beneficial for improving tumor cell penetration and endosomal escape. The ee% of CPPL(DOX) prepared using the post-reaction method was about 90%, which was consistent with the ee% of PL(DOX). The post-reaction method is a preferable preparation technique for loading DOX into CPPL because it avoided the interaction between DOX and CPP in the external medium of liposomes which impeded DOX from entering the internal water phase of liposomes.26
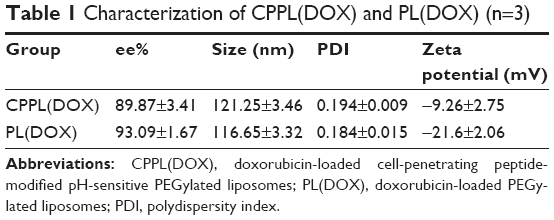 | Table 1 Characterization of CPPL(DOX) and PL(DOX) (n=3) |
Leakage behavior of CPPL(DOX) in vitro
The profiles of DOX release from liposomes in vitro are shown in Figure 1B. In the release medium of pH 7.0 PBS containing horse serum, there were no significant differences between CPPL(DOX) and PL(DOX) (p>0.05), indicating that CPPL(DOX) were as stable in blood as PL(DOX). However, when the pH of the release PBS medium decreased to 6.0, the release rate of DOX from CPPL was significantly higher than that of PL. This demonstrated that CPP modification of liposomes promoted DOX leakage caused by DOX and CPP interactions, which included cation–pi interaction and the formation of hydrogen bonds when guanidinium group of CPP was parallel to the planar aromatic chromophore of DOX.26 The lower pH caused more protonation of CPP amino groups, which then led to more DOX release from CPPL. The leakage behavior of CPPL(DOX) showed the advantages of DOX escape from endosomes in an acid microenvironment and stability in neutral blood environment.13
Evaluation of hemolytic activity
CPP normally exhibits toxicity because of its cell penetrating and nonselective properties. Based on a single hemolysis experiment, although there was a little significant difference in hemolysis between CPPL(DOX) and PL(DOX) when phospholipid concentrations were <400 μmol/L (Figure 1C), CPPL(DOX) were considered nontoxic due to their low hemolysis (<10%). The dose of DOX hydrochloride liposome injection used for clinical intravenous drip administration is 1.2 mg/kg, the ratio of DOX and phospholipid in formulation is 1:10 (w:w),27 and the blood volume in adults is about 86 mL/kg.28 Thus, the phospholipid concentration in the blood is about 186 μmol/L when DOX hydrochloride liposome injection is administrated via intravenous injection, which is far less than the threshold concentration of 400 μmol/L. That is, the hemolysis of CPPL(DOX) is about 5% when they are exposed to tumors via intravenous injection at the dosage of DOX hydrochloride liposome injection available in the market. But in fact, CPPL(DOX) are preferably administrated by intravenous drip, and the hemolytic activity of CPPL(DOX) is too low to be mentioned. These findings suggest that CPPL(DOX) are safe for clinical application.
In vitro cytotoxicity of CPPL(DOX)
The IC50 calculated from the viability of MCF-7 cells after 24 h incubation with DOX and DOX-loaded liposomes at pH 6.0 (Figure 1D) showed that the cytotoxicity of DOX encapsulated into liposomes was lower than that of free DOX in direct contact with cells. The lower IC50 values of CPPL(DOX) revealed that CPPL at pH 6.0 significantly promoted the efficacy of DOX, which suggests they are a potential delivery system for anticancer drugs.
These findings were in great agreement with the structure design of CPPL, in which CPP directly attached to liposomal surfaces via coupling with stearate and a PEG derivative PHS formed by conjugating PEG with stearate via an acid-degradable hydrazone bond covered the liposomal surface. We have confirmed that PHS is highly sensitive to the acidic environment.13 It stabilizes on the surface of liposomes to shield CPP and prevents its penetration into normal cells at pH 7.0. The same IC50 value as CCL(DOX) (Figure 1D) demonstrated that PHS on CPPL leaves liposomes completely to expose CPP on their surface which is beneficial for promoting penetration and endosomal escape at pH 6.0.13 Entrapped cargo such as DOX gain more opportunities to enter the nucleus to inhibit DNA synthesis and initiate DNA cleavage by type II topoisomerases.29
Pharmacokinetics and bio-distribution of CPPL(DOX) in nude mice bearing tumor
We determined in vivo DOX concentration by HPLC using the internal standard. The resulting chromatograms were essentially free from endogenous artifacts. Representative chromatograms are shown in Figure 2, which includes a blank plasma sample (Figure 2A), a plasma sample (Figure 2B), and a tumor sample (Figure 2C) obtained 30 min after the intravenous injection of 5 mg DOX/kg of CPPL(DOX). The typical retention time for DOX and the internal standard (DAU) was 5.4 min and 9.2 min, respectively, and the peaks were satisfactory and suitable for quantitative analysis.
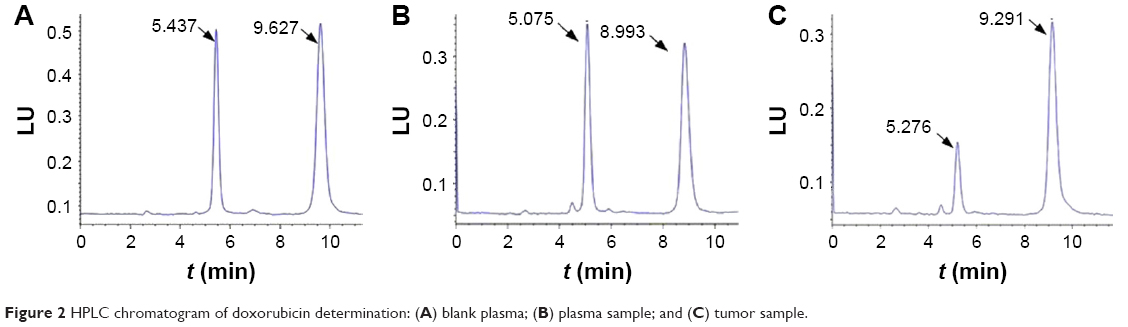 | Figure 2 HPLC chromatogram of doxorubicin determination: (A) blank plasma; (B) plasma sample; and (C) tumor sample. |
Recovery of DOX from mouse plasma and tissue samples showed acceptable ratios of DOX to DAU (Table 2). The DOX concentration was ADOX/ADAU =1.3231c (μg/mL) +0.0365; R2=0.9999. Linearity covered a range of 3.125 ng/mL–4.00 μg/mL, and the lower limits of quantification were 0.625 ng/mL. The intra- and inter-day precisions of 0.0625, 0.5, and 2.0 μg/mL DOX in blank and sample tissues were assessed by calculating the relative SD and were <5% indicating that the method was reliable and reproducible.
 | Table 2 Recoveries of DOX from plasma and tissue samples (n=3) |
After administration of formulation via the tail vein to the nude mice bearing breast tumors, the profile of concentration versus time of the plasma with two formulations was analyzed using DAS software. The results showed that the pharmacokinetics of the two formulations complied with the two-compartment model. The pharmacokinetic parameters are summarized in Table 3. Compared with PL(DOX), the half-life of CPPL(DOX) did not change significantly, demonstrating that CPPL(DOX) remained in the circulation for a longer period and provided an effective distribution of drugs to tumors. In the bloodstream, CPPL(DOX) were stable at a pH >7.0 because of the stable hydrazone bond in PHS, and CPP was masked completely by the PEG. CPPL constructed in the study overcame the drawbacks of CPP-SSL30 we reported before, which was cleared from the blood quickly due to the exposure of CPP on the surface of liposome.
 | Table 3 The pharmacokinetic parameters of PL(DOX) and CPPL(DOX) (n=6) |
In addition, as shown in Figure 3, DOX plasma concentrations were lower in nude mice bearing tumors that were administered CPPL(DOX) than those administered PL(DOX); this led to higher clearance of CPPL(DOX), lower AUC, and lower mean resident time, which may have been the result of higher distribution in other tissues.
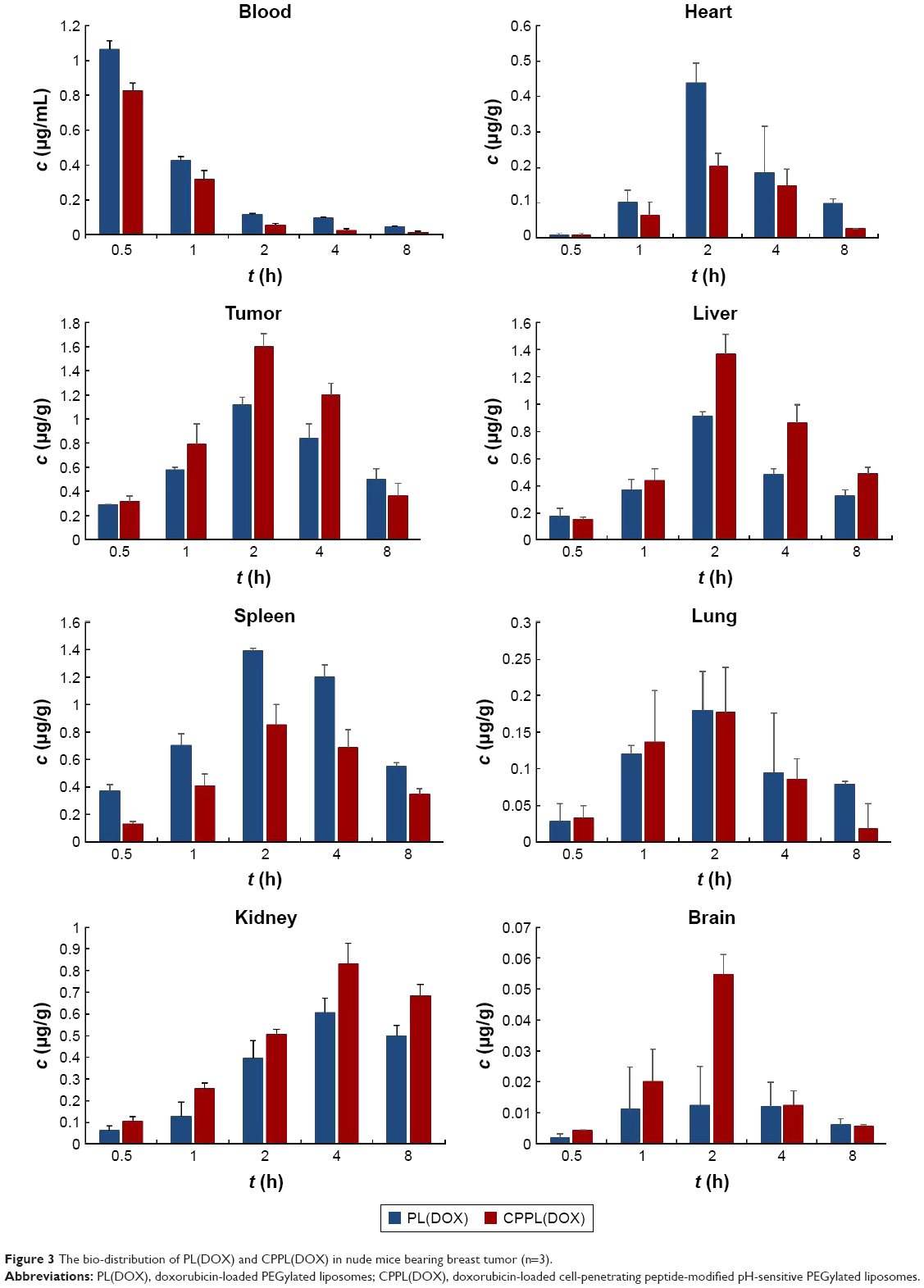 | Figure 3 The bio-distribution of PL(DOX) and CPPL(DOX) in nude mice bearing breast tumor (n=3). |
The mean concentration–time profile of each tissue for the two formulations is also presented in Figure 3. The pharmacokinetic parameters are summarized in Table 4. Compared with PL(DOX), the AUC of CPPL(DOX) in tumor and liver increased significantly (p<0.05) but decreased in the spleen; Cmax of CPPL(DOX) increased in tumor and brain but decreased in the heart and spleen. The ratio of AUC in the tumor to blood of CPPL(DOX) was calculated to be 1.84-fold more than that of PL(DOX), indicating CPPL(DOX) are useful for tumor targeting. In addition, the lower distribution in the heart and spleen indicated that CPPL(DOX) had less cardiac toxicity and accelerated blood clearance31 at the second dosage. PL(DOX) and CPPL(DOX) showed minimal distribution in the lung and brain due to their size and the existence of the BBB. The mechanism that caused the difference in in vivo distribution between PL(DOX) and CPPL(DOX) should be examined in future studies.
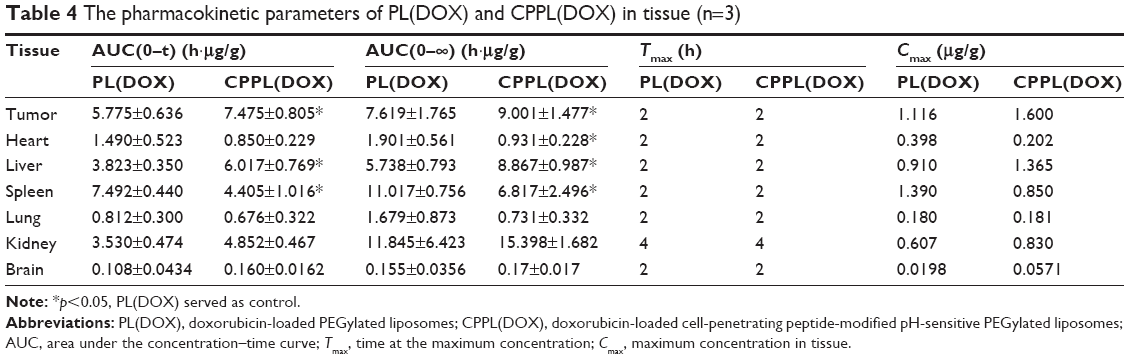 | Table 4 The pharmacokinetic parameters of PL(DOX) and CPPL(DOX) in tissue (n=3) |
Therapy for breast tumors
During the administration period, although there were no animal deaths in any group, the survival status of the CPPL(DOX) group was significantly better than that of the PL(DOX) and NS groups. As shown in the body weight change curve (Figure 4A), the weight of CPPL(DOX) group of nude mice did not decline, but showed an increasing trend, whereas the weight of the nude mice in the PL(DOX) and NS groups decreased with the growth of tumor.
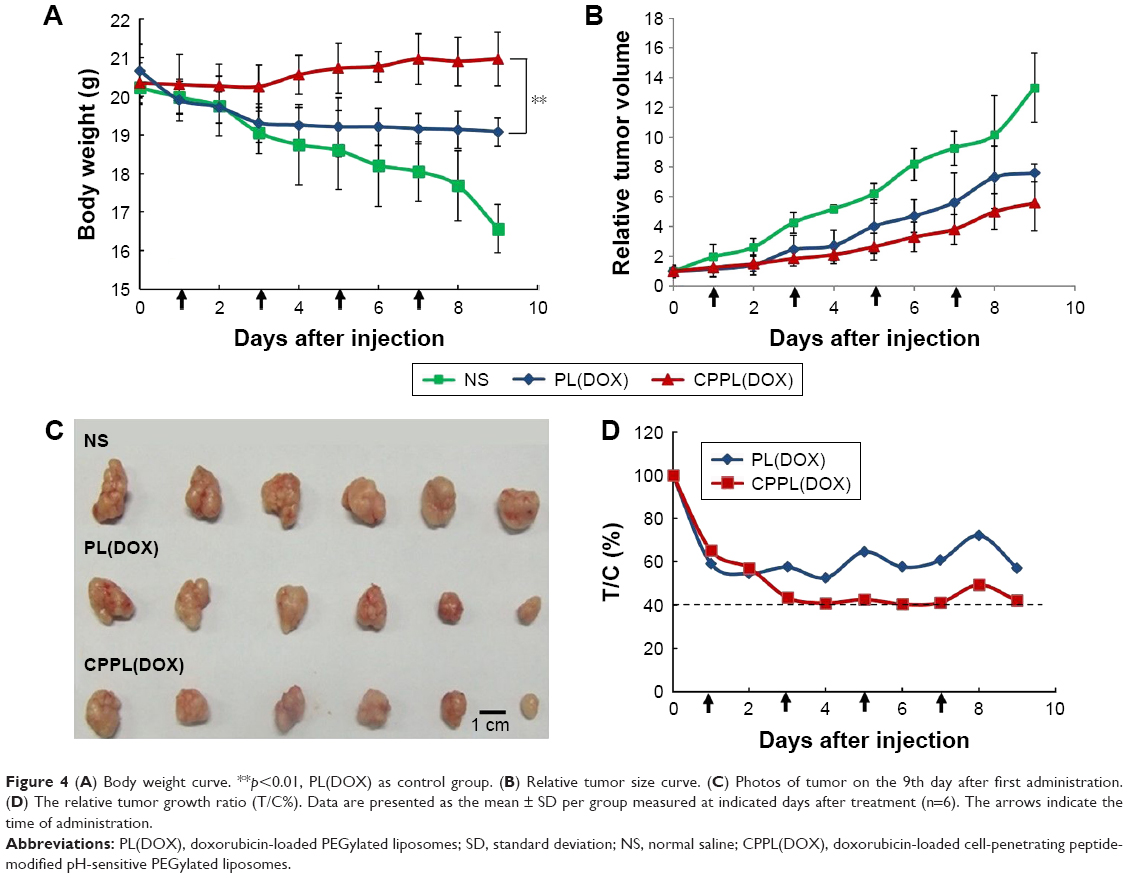 | Figure 4 (A) Body weight curve. **p<0.01, PL(DOX) as control group. (B) Relative tumor size curve. (C) Photos of tumor on the 9th day after first administration. (D) The relative tumor growth ratio (T/C%). Data are presented as the mean ± SD per group measured at indicated days after treatment (n=6). The arrows indicate the time of administration. |
The relative tumor size of each group and the photos of tumors separated from nude mice on the treatment day 9 are shown in Figure 4B and C, respectively. Although the tumor size increased in each group when compared to that measured pretreatment, the tumors in the mice in the CPPL(DOX) and PL(DOX) groups grew more slowly than that in the NS group.
From the antitumor efficacy evaluation index (Figure 4D), the tumor inhibition by CPPL(DOX) was found to be significantly greater than that by PL(DOX), indicating that CPPL(DOX) is a better choice for breast cancer therapy. The results showed that although PL(DOX) accumulated in tumor tissues due to the EPR effect32,33 and inhibited tumor growth rate, they failed to achieve the antitumor efficacy index of 40%. However, CPPL(DOX) accumulated in some tumor sites depending not only on the EPR effect but also on the PEG cleavage from liposomal surface and internal CPP exposure that promotes penetration and endocytosis.13 According to these criteria, CPPL(DOX) have achieved the therapeutic goal for treating breast tumors. Successful CPPL synthesis may provide more opportunities for the development of therapies for other tumors, including brain gliomas, by combining the positive characteristics of CPPL and receptors expressed on the BBB.
H&E and TUNEL staining
Tumors were separated from the euthanized animals at 48 h after the last administration and stained with H&E. As shown in Figure 5, the tissue structure of the tumor in the NS treatment group was complete and clear with bright-stained nuclei, indicating that cells were in the vigorous growth period. After treatment with PL(DOX), the cells had shrunk, which caused a large gap between tissues. Particularly, large areas of exfoliative cells and tissue necrosis appeared in the CPPL(DOX) group, indicating tumor cells were in severe apoptosis. To analyze DNA strand breaks, the TUNEL assay was extensively employed in the assessment of tumor cell apoptosis. As seen in Figure 5, NS did not significantly induce cell apoptosis as demonstrated by the absence of detectable TUNEL-positive tumor cells (green). In contrast, exposure to CPPL(DOX) induced significant apoptosis in cells compared to PL(DOX). The observed trend for apoptosis was consistent with the results of in vivo antitumor efficacy.
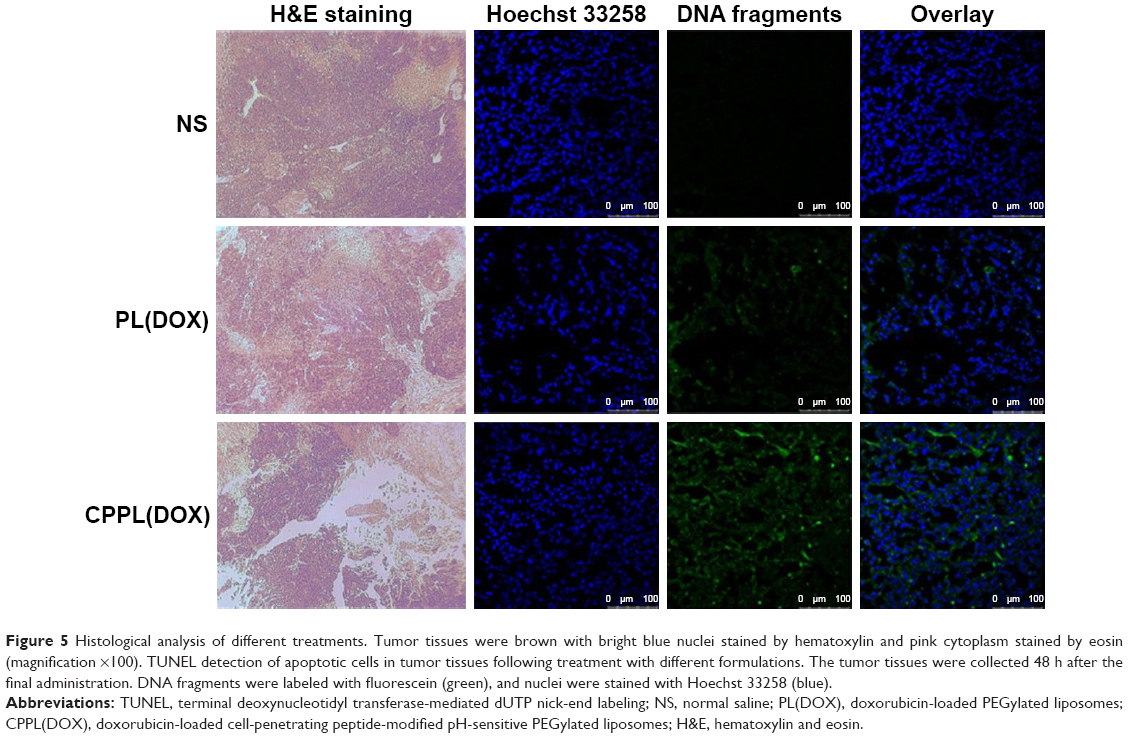 | Figure 5 Histological analysis of different treatments. Tumor tissues were brown with bright blue nuclei stained by hematoxylin and pink cytoplasm stained by eosin (magnification ×100). TUNEL detection of apoptotic cells in tumor tissues following treatment with different formulations. The tumor tissues were collected 48 h after the final administration. DNA fragments were labeled with fluorescein (green), and nuclei were stained with Hoechst 33258 (blue). |
Overall, these results strongly support our hypothesis that the combination of pH responsiveness and CPP penetration by multifunctional liposomes should enhance MCF-7 cell recognition and uptake and reduce nonspecific uptake. pH-responsive acid cleavage appears to enhance specific cellular uptake through CPP by selectively unmasking PEG in tumor tissues. Subsequently, DOX could escape from endosomes into the nucleus to disrupt DNA synthesis.
Biocompatibility of CPPL(DOX)
DOX is widely used in clinical therapy for acute and chronic leukemia, malignant lymphomas, breast, ovarian, and non-small-cell lung cancers, and sarcomas.34–37 The most adverse effects of DOX for clinical application are bone marrow suppression and cardiotoxicity.38 After liposomal encapsulation, the pharmacokinetics of DOX changed and cardiotoxicity was reduced. Although the encapsulation efficiency of DOX in liposomes was >95%, the therapeutic efficacy did not dramatically increase, thereby indicating the need for further improvement of its formulation. In this study, we constructed novel CPPL and evaluated CPPL(DOX) biocompatibility by histological examination and bone marrow suppression after four rounds of administration to healthy mice. PL(DOX) and DOX served as negative and positive controls, respectively. H&E staining (Figure 6A) revealed that tissue sections from different organs did not show any necrosis, inflammation, or enlargement of nuclei in either CPPL(DOX) or PL(DOX) groups, but showed serious signs of necrosis or extravasation of RBCs in the DOX group. Since bio-distribution studies demonstrated higher accumulation of liposomes in the liver and spleen, these organs were carefully examined for any histological changes. In the CPPL(DOX) group, no ballooning of hepatocytes or signs of inflammation was observed in the liver sections, and no signs of necrosis were evident in the tissue sections obtained from the spleen, indicating normal organs and tissues.
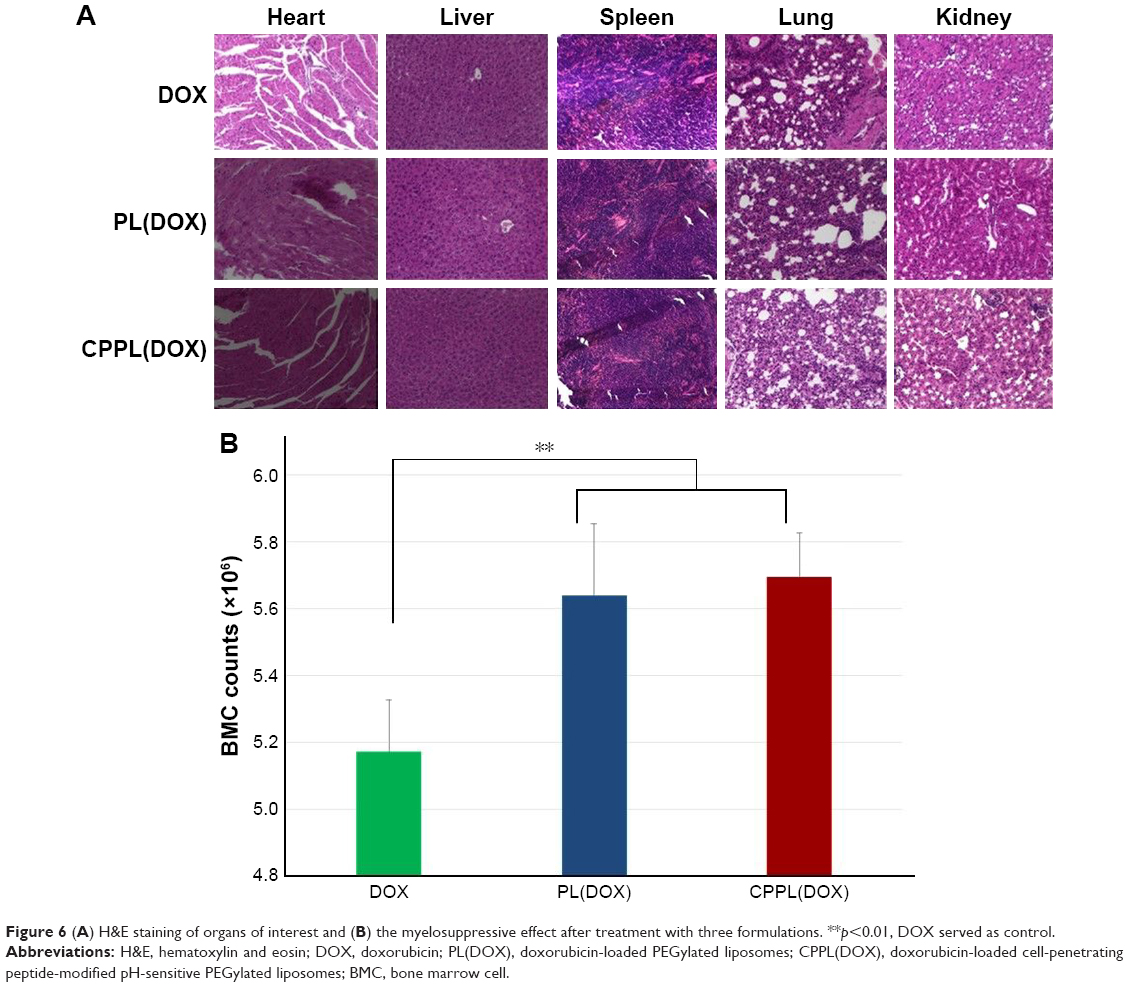 | Figure 6 (A) H&E staining of organs of interest and (B) the myelosuppressive effect after treatment with three formulations. **p<0.01, DOX served as control. |
The myelosuppressive effect (Figure 6B) showed that the number of BMCs in the CPPL(DOX) administration group was similar to that in the PL(DOX) group and significantly increased when compared with DOX (p<0.01), indicating CPPL(DOX) was safe even in the liposomes modified with CPP, a non-cell-selective peptide with positive charges.
Conclusion
DOX-encapsulated CPPL, obtained via post-reaction method, exhibited a high encapsulation efficiency of about 90%, leakage stability in serum, and minimal hemolysis, and thus found to be safe for tumor treatment. In in vivo evaluations, CPPL(DOX) demonstrated longer blood circulation, high tumor accumulation, and good biocompatibility in addition to more cell apoptosis induced by DNA disruption and lower relative tumor growth ratio. These findings indicated that the goals of delivering DOX by CPPL to tumor cell nucleus and effective treatment of breast cancer were accomplished.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (grant no 81202469).
Disclosure
The authors have no conflicts of interest to disclose in this work.
References
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30. | ||
Lucas AT, White TF, Deal AM, et al. Profiling the relationship between tumor-associated macrophages and pharmacokinetics of liposomal agents in preclinical murine models. Nanomedicine. 2017;13(2):471–482. | ||
Barenholz Y. Doxil® – the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160(2):117–134. | ||
Forssen EA, Coulter DM, Proffitt RT. Selective in vivo localization of daunorubicin small unilamellar vesicles in solid tumors. Cancer Res. 1992;52(12):3255–3261. | ||
Koren E, Apte A, Jani A, Torchilin VP. Multifunctional PEGylated 2C5-immunoliposomes containing pH-sensitive bonds and TAT peptide for enhanced tumor cell internalization and cytotoxicity. J Control Release. 2012;160(2):264–273. | ||
Kale AA, Torchilin VP. “Smart” drug carriers: PEGylated TATp-modified pH-sensitive liposomes. J Liposome Res. 2007;17(3–4):197–203. | ||
Biswas S, Dodwadkar NS, Piroyan A, Torchilin VP. Surface conjugation of triphenylphosphonium to target poly(amidoamine) dendrimers to mitochondria. Biomaterials. 2012;33(18):4773–4782. | ||
Zhou J, Zhao WY, Ma X, et al. The anticancer efficacy of paclitaxel liposomes modified with mitochondrial targeting conjugate in resistant lung cancer. Biomaterials. 2013;34(14):3626–3638. | ||
Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55(6):1189–1193. | ||
Mudhakir D, Akita H, Harashima H. Topology of octaarginines (R8) or IRQ ligand on liposomes affects the contribution of macropinocytosis- and caveolae-mediated cellular uptake. J React Funct Polym. 2011;71:340–343. | ||
El-Sayed A, Futaki S, Harashima H. Delivery of macromolecules using arginine-rich cell-penetrating peptides: ways to overcome endosomal entrapment. AAPS J. 2009;11(1):13–22. | ||
Mai JC, Shen H, Watkins SC, Cheng T, Robbins PD. Efficiency of protein transduction is cell type-dependent and is enhanced by dextran sulfate. J Biol Chem. 2002;277(33):30208–30218. | ||
Ding Y, Sun D, Wang GL, et al. An efficient PEGylated liposomal nanocarrier containing cell penetrating peptide and pH-sensitive hydrazone bond for enhancing tumor-targeted drug delivery. Int J Nanomedicine. 2015;10:6199–6124. | ||
Zeng F, Ju RJ, Liu L, et al. Application of functional vincristine plus dasatinib liposomes to deletion of vasculogenic mimicry channels in triple-negative breast cancer. Oncotarget. 2015;6(34):36625–36642. | ||
Zhao BX, Zhao Y, Huang Y, et al. The efficiency of tumor-specific pH-responsive peptide-modified polymeric micelles containing paclitaxel. Biomaterials. 2012;33(8):2508–2520. | ||
Yu KF, Zhang WQ, Luo LM, et al. The antitumor activity of a doxorubicin loaded, iRGD-modified sterically-stabilized liposome on B16-F10 melanoma cells: in vitro and in vivo evaluation. Int J Nanomedicine. 2013;8:2473–2485. | ||
Ying X, Wen H, Lu WL, et al. Dual-targeting daunorubicin liposomes improve the therapeutic efficacy of brain glioma in animals. J Control Release. 2010;141(2):183–192. | ||
Sharma G, Modgil A, Layek B, et al. Cell penetrating peptide tethered bi-ligand liposomes for delivery to brain in vivo: biodistribution and transfection. J Control Release. 2013;167(1):1–10. | ||
Skehan P, Storeng R, Scudiero D, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst. 1990;82(13):1107–1112. | ||
León-González AJ, López-Lázaro M, Espartero JL, Martín-Cordero C. Cytotoxic activity of dihydrochalcones isolated from Corema album leaves against HT-29 colon cancer cells. Nat Prod Commun. 2013;8(9):1255–1256. | ||
Ren S, Li C, Dai Y, et al. Comparison of pharmacokinetics, tissue distribution and pharmacodynamics of liposomal and free doxorubicin in tumour-bearing mice following intratumoral injection. J Pharm Pharmacol. 2014;66(9):1231–1239. | ||
Teicher BA, Andrews PA. Anticancer Drug Development Guide: Preclinical Screening, Clinical Trials, and Approval. 2nd ed. Totowa, NJ: Humana Press; 2004. | ||
Wang B, Ma Y, Kong X, et al. NAD(+) administration decreases doxorubicin-induced liver damage of mice by enhancing antioxidation capacity and decreasing DNA damage. Chem Biol Interact. 2014;212:65–71. | ||
Shi C, Guo X, Qu Q, Tang Z, Wang Y, Zhou S. Actively targeted delivery of anticancer drug to tumor cells by redox-responsive star-shaped micelles. Biomaterials. 2014;35(30):8711–8722. | ||
Tong SW, Xiang B, Dong DW, Qi XR. Enhanced antitumor efficacy and decreased toxicity by self-associated docetaxel in phospholipid-based micelles. Int J Pharm. 2012;434(1–2):413–419. | ||
Liu C, Luo Q, Tu YF, Wang GL, Liu YC, Xie Y. Drug-carrier interaction analysis in the cell penetrating peptide-modified liposomes for doxorubicin loading. J Microencapsul. 2015;32(8):745–754. | ||
Lee RJ, Low PS. Folate-mediated tumor cell targeting of liposome-entrapped doxorubicin in vitro. Biochim Biophys Acta. 1995;1233(2):134–144. | ||
Hallenbeck JM, Dutka AJ, Tanishima T, et al. Polymorphonuclear leukocyte accumulation in brain regions with low blood flow during the early postischemic period. Stroke. 1986;17(2):246–253. | ||
Momparler RL, Karon M, Siegel SE, Avila F. Effect of adriamycin on DNA, RNA, and protein synthesis in cell-free systems and intact cells. Cancer Res. 1976;36(8):2891–2895. | ||
Tu YF, Liu C, Ju R, Xie Y. In vivo study on pharmacokinetics and pharmacodynamics of P167 modified liposomal doxorubicin in rats. Chin J Clin Pharm. 2014;23(1):9–14. | ||
Ishida T, Kiwada H. Accelerated blood clearance (ABC) phenomenon upon repeated injection of PEGylated liposomes. Int J Pharm. 2008;354(1–2):56–62. | ||
Kontogiannopoulos KN, Tsermentseli SK, Assimopoulou AN, Papageorgiou VP. Sterically stabilized liposomes as a potent carrier for shikonin. J Liposome Res. 2014;24(3):230–240. | ||
Foged C, Nielsen HM, Frokjaer S. Phospholipase A2 sensitive liposomes for delivery of small interfering RNA (siRNA). J Liposome Res. 2007;17(3–4):191–196. | ||
Allen TM, Sapra P, Moase E. Use of the post-insertion method for the formation of ligand-coupled liposomes. Cell Mol Biol Lett. 2002;7(3):889–894. | ||
Hira SK, Mishra AK, Ray B, Manna PP. Targeted delivery of doxorubicin-loaded poly (epsilon-caprolactone)-b-poly (N-vinylpyrrolidone) micelles enhances antitumor effect in lymphoma. PLoS One. 2014;9(4):e94309. | ||
Iranshahi M, Barthomeuf C, Bayet-Robert M, et al. Drimane-type sesquiterpene coumarins from Ferula gummosa fruits enhance doxorubicin uptake in doxorubicin-resistant human breast cancer cell line. J Tradit Complement Med. 2014;4(2):118–125. | ||
Staropoli N, Ciliberto D, Botta C, et al. Pegylated liposomal doxorubicin in the management of ovarian cancer: a systematic review and metaanalysis of randomized trials. Cancer Biol Ther. 2014;15(6):707–720. | ||
Zhang S, Liu X, Bawa-Khalfe T, et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. 2012;18(11):1639–1642. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.