")
Back to Journals » Journal of Inflammation Research » Volume 14
In vivo Pharmacokinetic and Anticancer Studies of HH-N25, a Selective Inhibitor of Topoisomerase I, and Hormonal Signaling for Treating Breast Cancer
Authors Lawal B , Kuo YC, Sumitra MR, Wu ATH , Huang HS
Received 19 July 2021
Accepted for publication 11 September 2021
Published 22 September 2021 Volume 2021:14 Pages 4901—4913
DOI https://doi.org/10.2147/JIR.S329401
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Bashir Lawal,1,2,* Yu-Cheng Kuo,3,4,* Maryam Rachmawati Sumitra,1,2 Alexander TH Wu,5– 8 Hsu-Shan Huang1,2,8– 10
1PhD Program for Cancer Molecular Biology and Drug Discovery, College of Medical Science and Technology, Taipei Medical University and Academia Sinica, Taipei, 11031, Taiwan; 2Graduate Institute for Cancer Biology & Drug Discovery, College of Medical Science and Technology, Taipei Medical University, Taipei, 11031, Taiwan; 3Department of Pharmacology, School of Medicine, College of Medicine, Taipei Medical University, Taipei, 11031, Taiwan; 4School of Post-Baccalaureate Chinese Medicine, College of Chinese Medicine, China Medical University, Taichung, 40402, Taiwan; 5The PhD Program of Translational Medicine, College of Medical Science and Technology, Taipei Medical University, Taipei, 11031, Taiwan; 6Clinical Research Center, Taipei Medical University Hospital, Taipei Medical University, Taipei, 11031, Taiwan; 7TMU Research Center of Cancer Translational Medicine, Taipei Medical University, Taipei, 11031, Taiwan; 8Graduate Institute of Medical Sciences, National Defense Medical Center, Taipei, 11490, Taiwan; 9School of Pharmacy, National Defense Medical Center, Taipei, 11490, Taiwan; 10PhD Program in Drug Discovery and Development Industry, College of Pharmacy, Taipei Medical University, Taipei, 11031, Taiwan
*These authors contributed equally to this work
Correspondence: Alexander TH Wu; Hsu-Shan Huang Email [email protected]; [email protected]
Purpose: Breast cancer is the most frequently diagnosed cancer globally, and the leading cause of cancer-associated mortality among women. The efficacy of most clinical chemotherapies is often limited by poor pharmacokinetics and the development of drug resistance by tumors. In a continuing effort to explore small molecules as alternative therapies, we herein evaluated the therapeutic potential of HH-N25, a novel nitrogen-substituted anthra[1,2-c][1,2,5]thiadiazole-6,11-dione derivative.
Methods: We evaluated the in vivo pharmacokinetic properties and maximum tolerated dose (MTD) of HH-N25 in rats. We also characterized the compound for in vitro and in vivo anticancer activities and its inhibitory effects against DNA topoisomerases and hormonal signaling in breast cancer. Furthermore, we used molecular docking to analyse the ligand–receptor interactions between the compound and the targets.
Results: The maximum serum concentration (Cmax), half-life (t1/2 beta), mean residence time (MRT), oral clearance (CL/f), and apparent volume of distribution (VD/f) of HH-N25 were 1446.67 ± 312.05 ng/mL, 4.51 ± 0.27 h, 2.56 ± 0.16 h, 8.32 ± 1.45 mL/kg/h, and 1.26 ± 0.15 mL/kg, respectively, after single-dose iv administration at 3 mg/kg body weight. HH-N25 had potent anticancer activity against a panel of human breast cancer cell lines with 50% inhibitory concentrations (IC50) ranging 0.045± 0.01∼ 4.21± 0.05 μM. The drug also demonstrated marked in vivo anticancer activity at a tolerated dose and prolonged the survival duration of mice without unacceptable toxicities based on body weight changes in human tumor xenograft models. In addition, HH-N25 exhibited a dose-dependent inhibition of topoisomerase I and ligand-mediated activities of progesterone and androgen receptors.
Conclusion: HH-N25 represents a new molecular entity that selective suppressed TOP1 and hormonal signaling, and shows potent antitumor activities in human breast cancer cells in vitro and in vivo. HH-N25 thus represents a promising anticancer agent that warrants further preclinical and clinical exploration.
Keywords: pharmacokinetic, anticancer activities, HH-N25, topoisomerase inhibition, hormonal signaling
Introduction
Cancer, a major public health burden and the leading cause of global mortality, achieved a global prevalence of 19.3 million and a mortality rate of approximately 10.0 million deaths in 2020.1,2 With the trend of a 25%-per decade increase in prevalence rates, cancer was predicted to peak at up at 28.4 million cases by 2040.3,4 Such an expanding rate was predicted to occur due to increasing risk factors associated with globalization and a growing economy in addition to predisposing lifestyle and dietary factors.2,5 Female breast cancer alone constituted 11.7% (2.3 million) of new cases and was ranked as the most commonly diagnosed cancer in 2020. With an estimated mortality rate of 6.9%, breast cancer also topped the list of cancer-related deaths globally among women in 2020.1,4
Over the years, different therapeutic strategies, such as immunotherapy, radiotherapy, surgery, and chemotherapy, have been explored individually or in combination for treating breast cancer.6,7 However, the overall survival rate of patients with this cancer is disappointing.8 The efficacy of most available clinical drugs and developed anticancer small molecules is often limited by poor solubility or stability, pharmacokinetics (PKs), and tumor drug resistance.9–11 In addition, the development of a secondary malignancy is associated with the use of some chemotherapies.12 All these dilemmas associated with available chemotherapeutic agents have necessitated the search for alternative anticancer agents with good PK properties capable of eliciting antitumor effects in vitro and in vivo at tolerable doses.13
Determining drug concentrations and dispositions in animals is important for understanding drug transport and PK/pharmacodynamic relationships that can aid the process of drug development.14 The transition from preclinical to clinical drug development largely depends on preclinical toxicity data in animals from initial Phase I drug dosing.15 In modern drug discovery, high-throughput screening tools are commonly employed to identify less-toxic drug candidates, and standard toxicity criteria have limitations of identifying efficacious doses and regimens.14,16 A greater reliance on PK properties, such as drug clearance and the volume distribution in an animal, could provide a rationale for selecting efficacious drug dosing regimens.14,15
DNA topoisomerases (TOPs) are ubiquitous ribozymes, which play pivotal roles in maintaining topological homeostasis within cells during DNA replication, transcription, and chromosome segregation.17 TOP1 and TOP2 are the two recognized TOPs, with TOP2A being the main isoform of TOP2. TOP1 and TOP2A are oncogenic drivers that have been implicated in the development and progression of various cancers, and could serve as attractive therapeutic targets for anticancer agents.18–22 Several TOP inhibitors, including Adriamycin, doxorubicin, amsacrine, etoposide, and camptothecin, have been developed and are widely used in clinical settings.23–27 However, the loss of tumor sensitivity and undesirable adverse effects associated with the use of these drugs28–30 counter their antitumor potentials and necessitate a pursuit for novel TOP inhibitors.
Numerous preclinical studies have demonstrated the potentials of small molecules for treating various cancers,31–37 via directly or indirectly modulating hormonal and oncogenic signaling pathways. These low-molecular-weight compounds may be associated with a more-favorable toxicity profile than conventional cytotoxic chemotherapeutics.38 In a continuing effort to explore small molecules for development of drug candidates with potent anticancer activities at tolerated doses, we herein demonstrated that HH-N25, a novel nitrogen-substituted anthra[1,2-c][1,2,5]thiadiazole-6,11-dione derivative, has good PK properties suitable for in vivo therapeutic applications. We report that HH-N25 had potent antiproliferative activities against a panel of human breast cancer cell lines and marked in vivo anticancer activities in human tumor xenograft models. In addition, our study demonstrated that HH-N25 mediated the inhibition of TOPI activities and ligand-mediated activities of progesterone receptors (PRs) and androgen receptors (ARs) in a dose-dependent manner.
Methods
Cell Lines and Cell Culture
Several breast cancer cell lines, including MCF7, MDB-MB-231/ATC, HS 578T, T-47D, and MDB-MB-468, were sourced from the US National Cancer Institute and cultured in Dulbecco’s modified Eagle medium (DMEM) containing 25 units/mL of penicillin, 25 units/mL of streptomycin, and 10% fetal bovine serum (FBS) at 37°C in a 5% CO2 and 95% humidity incubator. Culture media were replaced after 72 h, and cells were subcultured until 70%~80% confluence was achieved.
Drugs, Chemicals, and Enzyme Kits
HH-N25 was synthesized through an established protocol in our lab,39 while paclitaxel was procured from Selleckchem (Houston, TX, USA). TOPI and TOPII, supercoiled pRYG DNA, supercoiled pHOT1 DNA, camptothecin, and etoposide were purchased from TopoGEN. All other reagents and chemicals including DMEM (Gibco-Invitrogen, Grand Island, NY, USA), Matrigel (BD Biosciences, USA), Cremophor EL (Sigma, St. Louis, MO, USA), FBS (Hyclone, Logan, UT, USA), dimethylacetamide (DMA; Sigma), estradiol cyclopentyl propionate (Estol-depot injection, Astar, Taipei City, Taiwan), and phosphoric acid (Wako, Japan) were procured from reputable manufacturers.
Sulforhodamine B (SRB) Assay
The SRB assay protocols40 were used to evaluate the effect of HH-N25 on breast cancer cell viability. Cells were seeded in wells of a 96-well plate for 24 h. After additional incubation with HH-N25 (0, 0.1, 1.0, 10, and 100 μM), cells were washed and fixed with 10% trichloroacetic acid (TCA), while 0.4% SRB dye was added to stain the cells. Unbound dye was washed away with acetic acid (1%). The plates were air-dried, the contents were re-solubilized in 20 mmol/L Tris-base, and the absorbance was read at a wavelength of 570 nm.
DNA TOPI and TOPII Assays
The TOP assay kits (TopoGEN, USA) were used for the analysis of DNA TOPI and TOPII activities as described in previous studies.18,41,42 For TOPI, 0.5 μg of plasmid pHOT DNA was incubated with 4 units of recombinant human DNA TOPI (TopoGEN) in relaxation buffer (10 mM Tris-HCl at pH 7.9, 1 mM EDTA, 0.15 M NaCl, 0.1% bovine serum albumin (BSA), 0.1 mM spermidine, and 5% glycerol). For TOPII activity, 4 units of human TOPII were incubated with 0.5 μg of supercoiled pRYG DNA in cleavage buffer (50 mM KCl, 30 mM Tris-HCl at pH 7.8, 10 mM MgCl2, 15 mM mercaptoethanol, and 3 mM ATP), in the presence of varying concentrations of test compounds. Varying concentrations of HH-N25 were added to each of the reaction mixtures and incubated at 37°C for 60 min. The reaction was terminated by the addition of 1% sodium dodecyl sulfate (SDS) and 50 μg/mL proteinase K and then subjected to electrophoresis through a 0.8% agarose gel containing 0.5 mg/mL ethidium bromide in TBE buffer (90 mM Tris-borate and 2 mM EDTA). Gels were stained with ethidium bromide and photographed under UV light. Camptothecin (50 μM) and etoposide (VP-16) (50 μM) were, respectively, used as the positive controls for TOPI and TOPII.
In vivo PK Studies
Male Sprague-Dawley Great White rats (7 weeks) were obtained from Lesco Biotechnology. HH-N25 was administered to the rats at 3.0 mg/kg body weight (BW) (1.25 mL/kg BW) by an intravenous (iv) injection using a formulation of 0.5% (w/v) methylcellulose and 0.1% Tween-20. Blood (20 μL) was taken from the tail of each rat at 0.08 (5 min), 0.25, 0.5, 1.0, 2, 4, 6, 8, 10, and 24 h post-injection. Blood samples were centrifuged at 5000 rpm for 10 min, separated plasma samples were collected, and amounts of HH-N25 were quantified by internal standardization, protein precipitation, and high-performance liquid chromatography (HPLC)-tandem mass spectroscopy (MS/MS).43 Various PK parameters, such as the peak drug concentration (Cmax), time of peak concentration (Tmax), elimination half-life (t1/2), area under the concentration-time curve (AUC), and mean residence time (MRT), were computed by a non-compartmental analysis using WinNonlin software (Standard edition vers. 2, Pharsight, Mountain View, CA, USA).
Determination of the Maximum Tolerated Dose (MTD) of HH-N25
To estimate the appropriate dose level for the anticancer study, the MTD of HH-N25 was determined in BALB/c nude mice (three per group) by intraperitoneal (i.p.) administration of HH-N25 at various concentrations of 0, 5, 10, 20, 30, and 60 mg/kg BW in a 10-day toxicity study. MTD is defined as the highest concentrations that causes no more than a 10% BW decrement compared to an appropriate control group and produces no mortality or external signs of toxicity that would be predicted to shorten the natural lifespan of the animal.44,45 All animals were examined daily for signs of toxicity. At the end of 9 days, all animals were sacrificed, and their oral cavity, liver, colon, small intestines, kidneys, and stomach were examined for any abnormalities under a dissection microscope.
In vivo Anticancer Activity in a Human Breast Tumor Xenograft Model
Female nude (nu/nu) mice weighing 24.43±2.09 g and aged 6~7 weeks were procured from BioLasco Taiwan (Taipei city, Taiwan) under a Charles River Laboratories License. Animals were maintained under standard laboratory conditions (20~24°C temperature, 30%~70% humidity, and a 12-h light/dark cycle). They were allowed access to rat pellets and water ad libitum. All animal handling and experimentation were conducted in strict compliance with the Guide for the Care and Use of Laboratory Animals: Eighth Edition (National Academies Press, Washington, DC, 2011) in the AAALAC-accredited laboratory animal facility. In addition, the animal care and use protocol was reviewed and approved by the Institutional Care and Use Committee (IACUC) at Eurofins Panlabs Taiwan. Approximately 1.5×107 cells in a 0.2-mL mixture (medium: Matrigel = 1:1) of viable human breast tumor cells (MCF-7, ATCC HTB-22) were subcutaneously injected into the dorsal back region of a mouse, and estradiol cyclopentyl propionate at 100 µg/mouse was injected subcutaneously twice per week as a supplement during the study period.46 Following tumor development (tumor size ≥5 mm in diameter), mice were randomly divided into three groups (A~C) of six mice each. Group A mice were treated with 5 mg/kg BW HH-N25 for 45 days, and group B mice were treated with paclitaxel at 10 mg/kg, while group C served as the vehicle control. Tumor sizes, BWs, and overt signs of toxicity were observed and recorded every 4 days for 45 days.
Molecular Docking
PDB files of the crystal structure of human TOPI DNA complex (PDB:1EJ9), human PR ligand-binding domain (PDB:1A28), and human AR (PDB:1E3G) were obtained from the Protein Data Bank (https://www.rcsb.org/), while a mol2 file of HH-N25 was generated using the Avogadro molecular builder and visualization tool vers. 1.XX (http://avogadro.cc/),47 and were converted to PDB files using the PyMOL Molecular Graphics System, vers. 1.2r3pre (Schrödinger; https://pymol.org/edu/?q=educational/). All PDB files were converted to PDBQT files using AutoDock Vina (vers. 0.8, Scripps Research Institute, La Jolla, CA, USA).48 The ligands and receptors were prepared for docking, and docking was conducted using AutoDock Vina according to standard protocols described in previous studies.49–52 The docked complexes were visualized and analyzed using Discovery studio visualizer vers. 19.1.0.18287 (BIOVIA, San Diego, CA, USA).53
Data Analysis
Data analysis was conducted using GraphPad Prism vers. 6.04 for Windows (GraphPad Software, La Jolla, CA, USA). Results are presented as the mean ± standard deviation (SD), and results were assessed using Student’s t-test. A p value of <0.05 indicated a significant difference. Statistical differences between experimental groups were considered significant at p<0.05 (*), p<0.01 (**), and p<0.001 (***).
Results
PKs of HH-N25 in Male Sprague-Dawley Rats
Representative PK parameters of HH-N25 following single iv dosing of HH-N25 to male Sprague-Dawley rats are presented in Table 1 and Figure 1. HH-N25 was detected to have reached a maximum concentration (Cmax) in plasma of 1446.67 ± 312.05 ng/mL after 0.14 ± 0.06 h (tmax) with a mean residence time (MRT) of 2.56 ± 0.16 h. The stability of the compound in plasma is reflected by its half-life (t1/2) that was estimated to be 4.51 ± 0.27 h. To gain insights into its absorption following oral dosing, iv bioavailability of HH-N25 was determined, and we found that the mean oral clearance (CL/f) and apparent volume of distribution (Vd/f) averaged 8.32 ± 1.45 mL/h/kg and 1.26 ± 0.15 mL/kg, respectively.
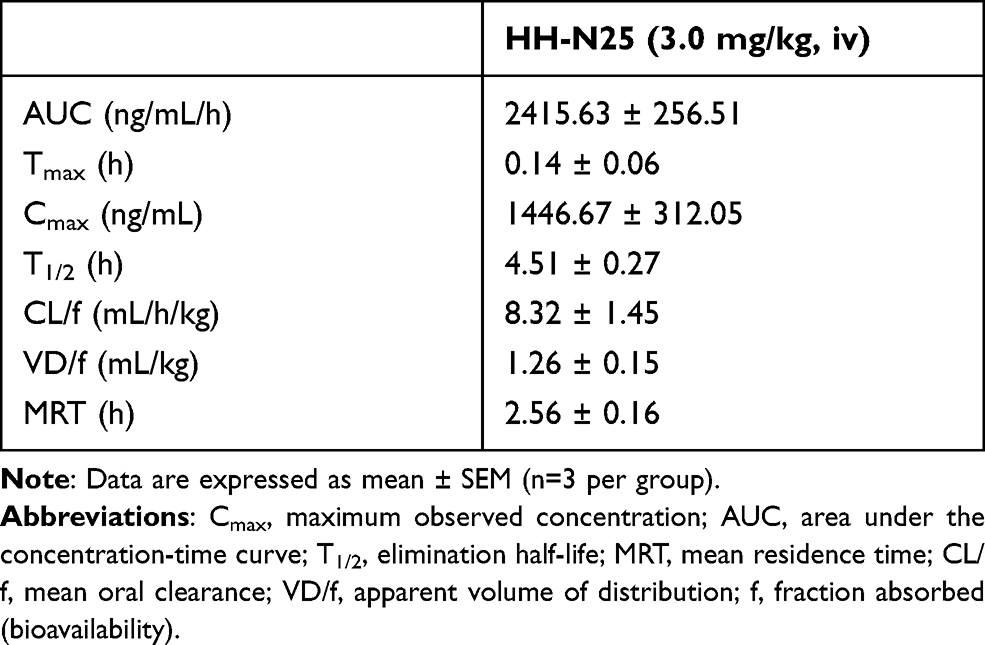 |
Table 1 Pharmacokinetic Parameters of HH-N25 After an iv Injection (3 mg/kg Body Weight) in Male Sprague Dawley Rats |
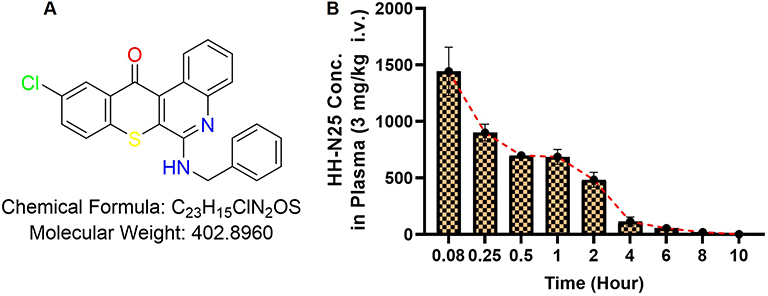 |
Figure 1 Pharmacokinetic properties of HH-N25. (A) Chemical structure of HH-N25. (B) Time-dependent concentration of HH-N25 in plasma of male Sprague-Dawley rats after injection (3.0 mg/kg body weight, iv) to rats. Data are expressed as the mean ± SEM (n=3 per group). |
In vivo Analysis of the MTD of HH-N25 in Mice
We examined the MTD of various doses of HH-N25, after 7 days’ administration to BALB/c nude mice. The MTD was estimated based on the threshold at which all animals survived with no more than a 10% BW loss. Interestingly, all mice treated with 5, 10, 20, 30, and 60 mg/kg BW tolerated these doses. To further define the MTD, the overall toxicity as revealed by BWs was monitored, and we found that none of the mice exhibited weight loss throughout the study period. During the observation period, no mortality and no deterioration in health were observed in mice treated with HH-N25 at all concentrations. However, severe to moderate writhing, and decreases in spontaneous activity, abdominal tone, and deep respiration were observed 30 min after each treatment at 60, 30, and 20 mg/kg BW (Table 2). These activities, however, disappeared after 1 h of observation. Mice treated with 5 mg/kg BW of HH-N25 were completely devoid of overt toxicity, and this was selected as the dose for in vivo evaluation of HH-N25’s anticancer activities.
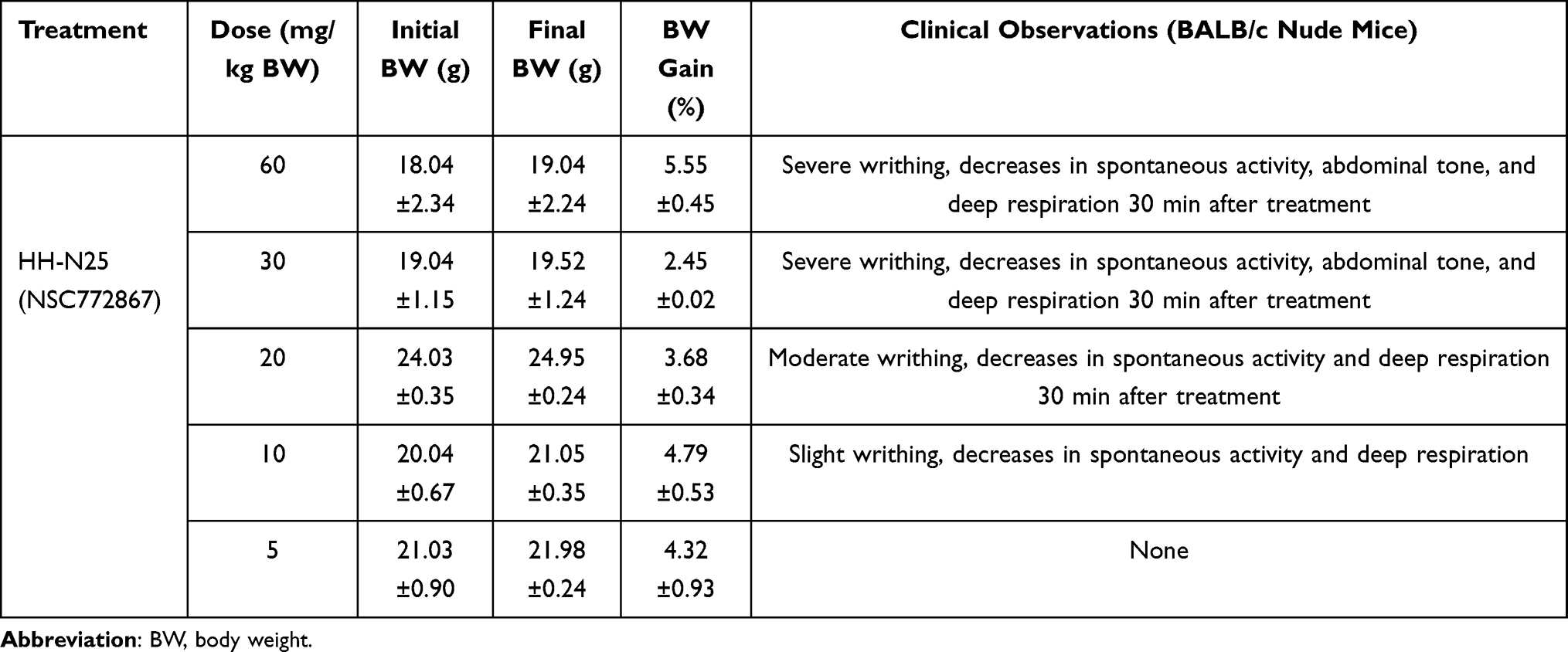 |
Table 2 Maximum Tolerated Dose (MTD) and Acute Toxicity Profile of HH-N25 |
In vitro Anticancer Activities of HH-N25
HH-N25 was analyzed at the National Cancer Institute (NCI) for anticancer activities against a panel of NCI60 human cancer cell lines representing renal, prostate, breast, melanoma, leukemia, non-small cell lung, prostate, brain, and ovarian cancers. Interestingly, we found that HH-N25 demonstrated antiproliferative activities against all of the NCI60 human cancer cell lines (Figure 2A) but exhibited distinct cytotoxic activities against breast cancer cell lines. In addition, it demonstrated dose-dependent activities (Figure 2B) against breast cancer lines (MCF7, MDA-MB-231/ATCC, HS 578T, BT-549, T-47D, and MDA-MB-468) with 50% inhibitory concentrations (IC50) ranging 0.045±0.01~4.21±0.05 µM (Figure 2C). Our mechanistic correlation analysis using the DTP-COMPARE algorithm revealed that HH-N25 shared an anticancer mechanism with cisplatin, paclitaxel, and a number of DNA TOP inhibitors including topotecan, VM-26 (teniposide), m-AMSA (amsacrine), doxorubicin (Adriamycin), and VP-16 (etoposide). Collectively, the present findings indicated that HH-N25 exhibited antiproliferative activities against various human cancer cell lines and exhibited a distinct cytotoxic preference for breast cancer cell lines, with inhibition of DNA-TOP and hormonal signaling the most likely mechanisms of action (Figure 2D).
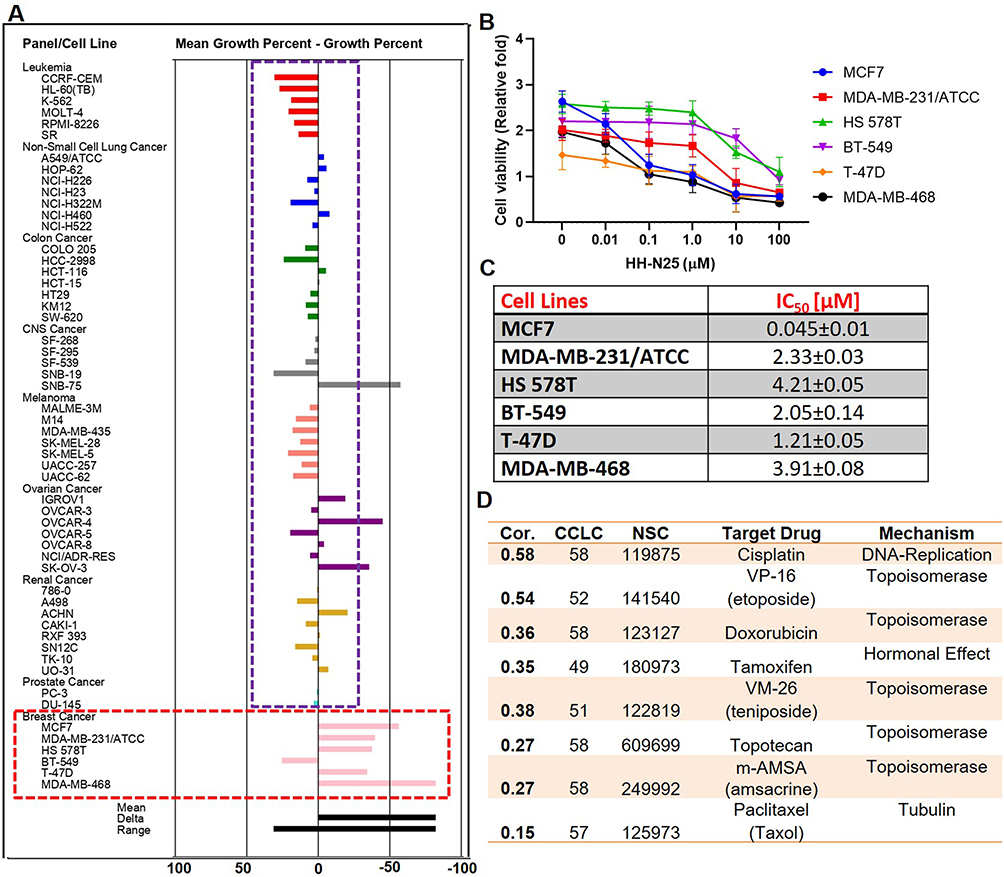 |
Figure 2 In vitro anti-breast cancer activity of HH-N25. (A) Antiproliferative activities of HH-N25 against NCI60 human cancer cell lines. The percentage growth inhibition of each cell line relative to the mean is represented by values under 100, whereas those values below 0 indicate cell death. (B) Line graph showing the effect of HH-N25 on the viability of breast cancer cell lines. (C) 50% inhibitory concentration (IC50) values of HH-N25 against a panel of breast cancer cell lines. (D) NCI standard anticancer agent shared similar anticancer fingerprints and mechanistic correlations with HH-N25. Abbreviations: CCLC, common cell line count; Cor, Pearson’s correlation coefficient. |
TOP Inhibition Activities of HH-N25
Having established the association between the anticancer fingerprint of HH-N25 and a number of TOP inhibitors, we evaluated the inhibitory effect of HH-N25 on DNA TOPI and TOPII. Interestingly, our analysis revealed that HH-N25 significantly inhibited the TOPI enzyme (Figure 3A) but had no inhibitory activities against TOPII (Figure 3B). The TOPI inhibitory activities of HH-N25 occurred in a dose-dependent manner and were comparable to activities demonstrated by camptothecin, a clinical TOP1 inhibitor (Figure 3C). Furthermore, our molecular docking analysis revealed that HH-25 docked well to the binding cavity of TOPI (Figure 3D) by several H-bonds, pi-interactions, alkyl interactions, hydrophobic contacts, and Van der Waal forces, and with a strong binding affinity of −8.3 kcal/mol (Table 1). The binding affinity and interaction between HH-N25 and TOP1 were very similar to the interactions and affinity that camptothecin has for TOP1 (Figure 3D, Table 3). Taken together, these findings demonstrated that HH-N25 is a potent and selective inhibitor of TOPI.
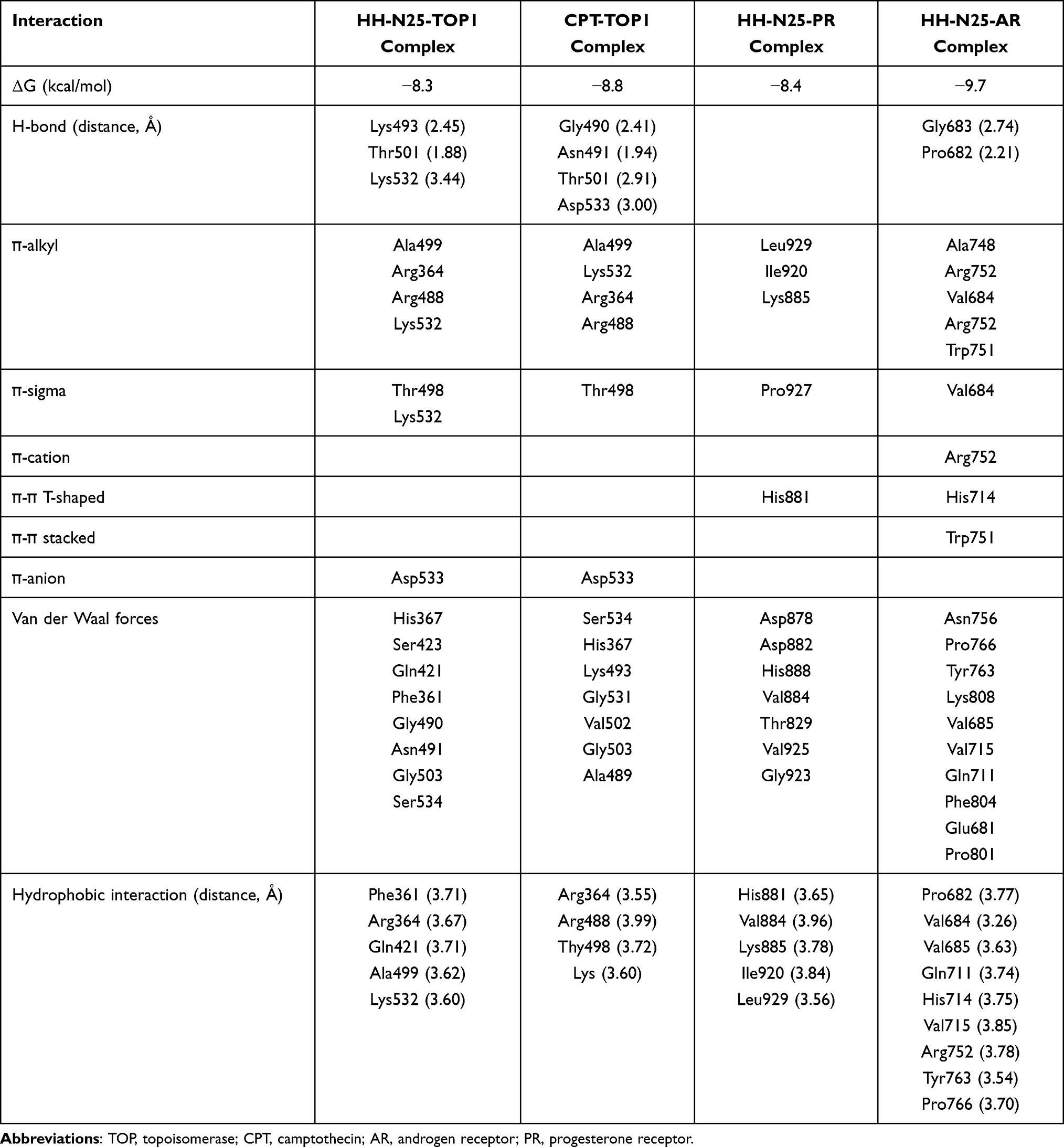 |
Table 3 Docking Profiles of HH-N25 with Progesterone, Aldosterone, and Topoisomerase 1 Activities |
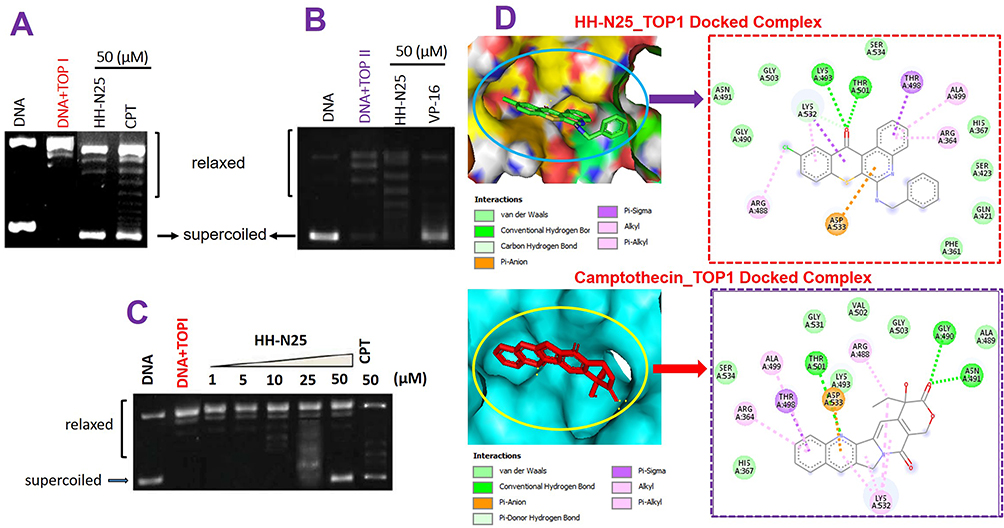 |
Figure 3 Topoisomerase inhibition activities of HH-N25. Effect of a single dose of HH-N25 on (A) DNA topoisomerase I (Top I) and (B) topoisomerase II (Top II). (C) Dose-dependent effect of HH-N25 on DNA topoisomerase I. (D) Solid surface representation of the binding-site flap of topoisomerase 1 accommodating the ligands (HH-N25 and camptothecin) and 2D representations of ligand–receptor complexes, showing the interacting amino acid residues and the types of interactions between the ligands (HH-N25 and camptothecin). |
Effects of HH-N25 on Hormonal Signaling in Breast Cancer
We investigated the effects of HH-N25 on hormonal signaling including the PR, AR, and mineralocorticoid receptor (MR) signaling pathways using a luciferase reporter gene assay in T47D cells. Our results revealed that HH-N25 produced dose-dependent inhibition of ligand-mediated activities of PRs (Figure 4A) and ARs (Figure 4B). However, a reversed effect of the drug on the receptors was observed in the absence of the ligands (Figure 4E). In contrast, HH-N25 demonstrated no significant effect on the activity of the MR in the presence or absence of aldosterone (Figure 4C). Furthermore, the molecular docking analysis revealed that HH-25 bonded well to the binding cavity of PRs and ARs (Figure 4D and E). However, a stronger binding affinity and higher number of H-bonds, pi-interactions, hydrophobic contacts, and Van der Waal forces created on the HH-N25 backbone with a higher number of amino acid residues were observed between the HH-N25-AR complex than those observed for the HH-N25-PR complex (Table 3).
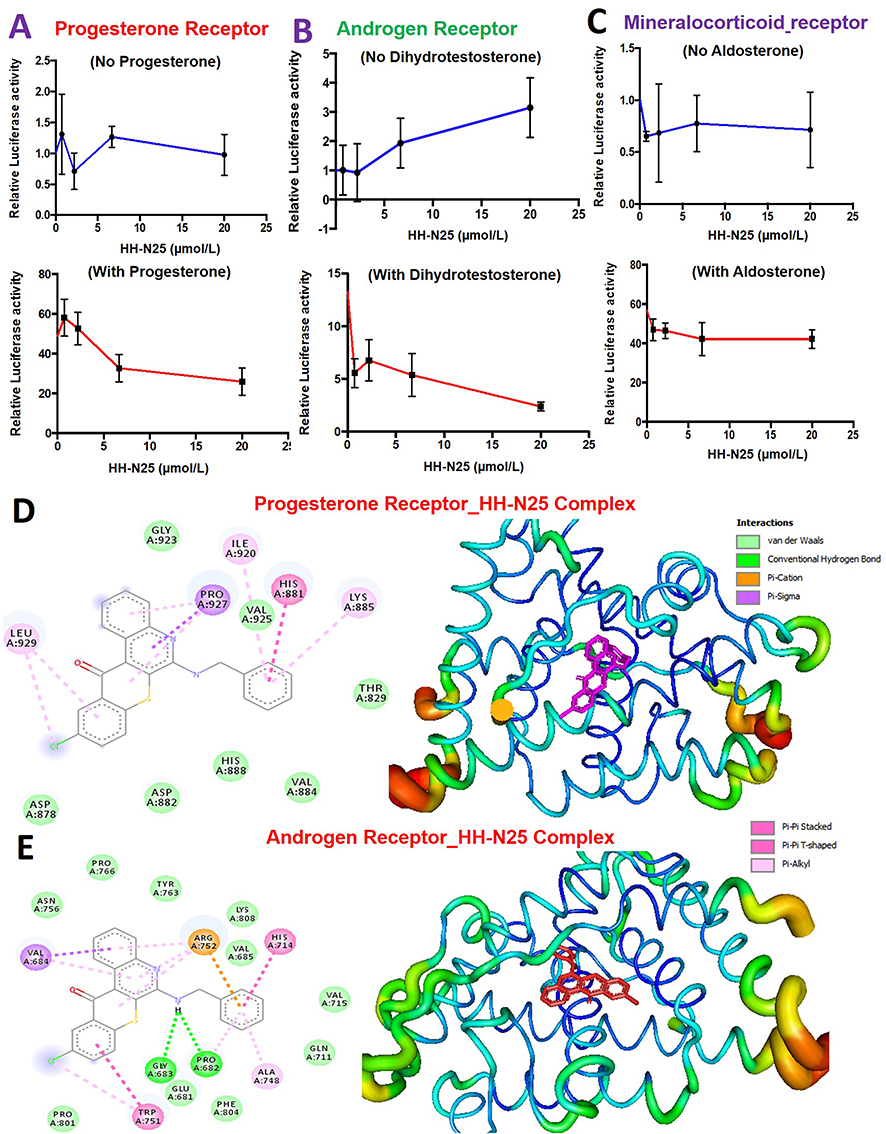 |
Figure 4 Effects of HH-N25 on hormonal signaling in breast cancer. Dose-dependent plot of the effects of HH-N25 on (A) the progesterone receptor (PR), (B) androgen receptor (AR), and (C) mineralocorticoid receptor (MR) signaling pathways in the absence (upper panel) and presence of the respective receptors. (D) Docking profiles of HH-N25 with the (D) progesterone receptor and (E) androgen receptor. |
In vivo Anti-Breast Cancer Activity of HH-N25 in Balb/c Mice
The in vivo anticancer activities of HH-N25 are presented in Figure 4. HH-N25 (5 mg/kg BW) produced a progressive and significant (p<0.001) inhibition of tumor growth (Figure 5A and B) compared to the untreated counterpart, which showed a progressive increase in tumor growth (Figure 5A). The in vivo antitumor effects demonstrated by HH-N25 were comparable and did not significantly differ (p>0.05) from those elicited by paclitaxel (10 mg/kg BW). In addition, treatment of tumor-bearing mice with HH-N25 prolonged the survival period (Figure 5C) of animals and caused significant (p<0.05) improvement in BW (Figure 5D) of animals compared to the untreated counterpart.
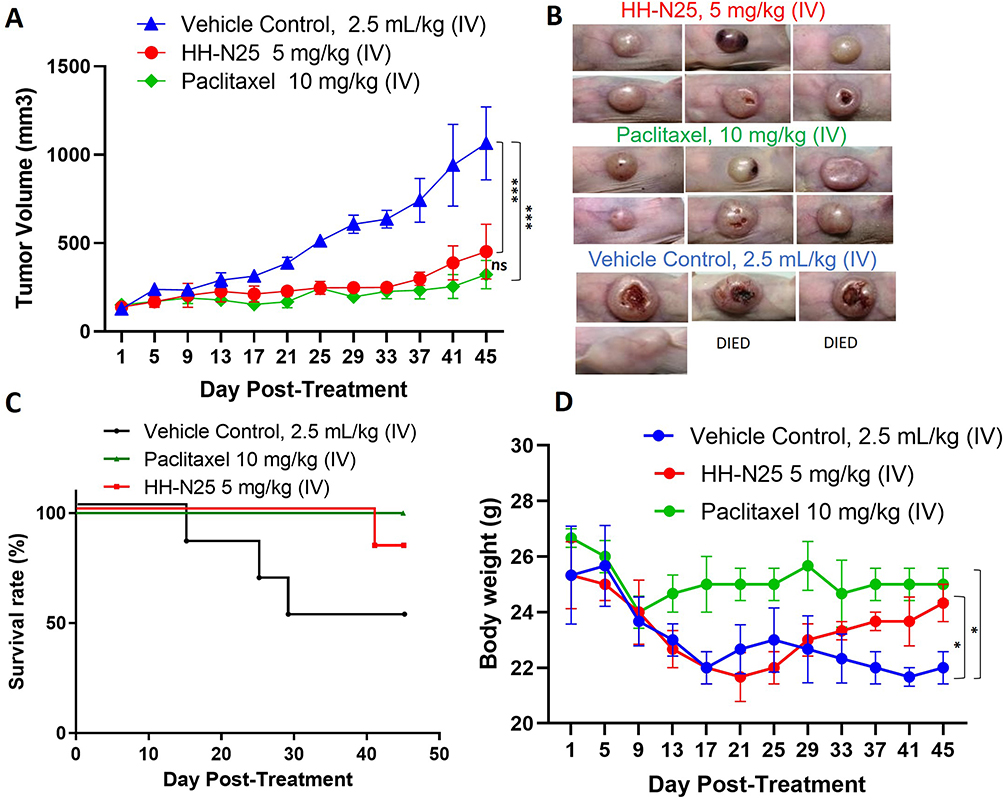 |
Figure 5 In vivo anti-breast cancer activity of HH-N25 in Balb/c mice. (A) Average tumor volume versus time curve shows that treatments with HH-N25 and paclitaxel significantly (p<0.001) inhibited tumor growth and the tumor burden. (B) Graphical representation of the tumor burden and ameliorative effects on HH-N25 on the tumor size. (C) Survival curve of mice treated with HH-N25. (D) Graph of body weight changes of mice; HH-N25 produced improvements in body weight gain in animals over the treatment course, suggesting no apparent systemic toxicity. *p<0.05, ***p<0.001. Abbreviation: ns, not significant. |
Discussion
Preclinical evaluation of drug PKs can aid the process of drug development by providing a rationale for selecting efficacious drug doses and treatment schedules.14,15 The PKs, metabolism, and distribution of HH-NS25 were examined in rats. Upon iv infusion at 3 mg/kg BW in rats, HHN25 produced an area under the plasma drug concentration-time curve (AUC) value of 2415.63 ± 256.51 ng/mL/h, which is a measure of the actual body exposure to HH-N25. This AUC was dependent on the rate of drug elimination; consequently, HHN25 exhibited a short plasma half-life (t1/2 = 4.51 ± 0.27 h) with rapid elimination. Because the highest concentration of HH-N25 was detected at the earliest time point sampled (5 min after dosing) with a Cmax of 1446.67 ± 312.05 ng/mL at exactly 0.14 ± 0.06 h, it is clear that the metabolism of this drug occurs very rapidly. This suggests rapid drug metabolism by phase I and Phase II enzymes primarily localized in the liver. Furthermore, the plasma concentration versus time plots of HH-N25 revealed that concentrations of the drug consistently decreased from 1443.33±314.24 ng/mL at 5 min to 2.84±0.19 ng/mL at 1440 min (10 h), suggesting that exposure to HH-N25 induced no apparent drug accumulation, and that it was rapidly cleared from the circulation.54 The lack of accumulation of HH-N25 is consistent with the shorter t1/2 (4.51 ± 0.27 h) noted following the administration of HH-N25. This is also supported by the mean residence time (2.56 ± 0.16 h), representing the average time the drug molecules stay in the body.
As most drugs are orally administered, oral clearance (CL/f) is an important PK parameter that plays a pivotal role in the safety and tolerability of a drug. The t1/2 values obtained in this study are within the range reported for some clinical drugs. However, the calculated VD value (1.26 ± 0.15 mL/kg) suggested a low tissue distribution of HH-N25.
Our analysis of the anticancer effect revealed that HH-N25 was potent against all six breast cancer cell lines tested, inhibiting the in vitro growth with IC50 values ranging 0.045±0.01~4.21±0.05 µM. HH-N25 not only demonstrated potent in vitro anticancer activities but also exhibited in vivo antitumor effects by reducing tumor progression relative to the vehicle and improved survival of animals at the MTD. The in vivo antitumor activities of HH-N25 were further strengthened by the absence of apparent systemic toxicity during the experimental period, suggesting the safety and tolerability of HH-N25. The remarkable in vivo anticancer activities observed in this study at the MTD are an indication that the t1/2 (4.51 ± 0.27 h) and MRT of HH-N25 are sufficient to elicit inhibition of tumor growth in mice.
Anticancer agents targeting TOPI and TOPII have been developed and employed in clinical chemotherapy; however, although proven to be effective in arresting tumor growth, significant side effects associated with their use have spurred a global search for novel efficacious and safe TOP inhibitors.55 By comparing the anticancer fingerprint of HH-N25 with those of NCI standard drugs, we found a significant correlation with inhibitors of TOPs. Interestingly, our in vitro assay successfully validated our COMPARE analysis. Selectivity for inhibition of TOPI but not TOPII was observed for HH-N25. HH-N25 exhibited the ability to regulate TOPI-mediated relaxation of supercoiled pHOT DNA in a concentration-dependent manner at doses of 1, 5, 10, 25, and 50 µM. HH-N25 not only exhibited more-potent inhibitory activity than camptothecin, but almost completely blocked TOP-mediated DNA relaxation at 50 mM. These findings led us to conjecture that HH-N25 represents a new class of anticancer agents which target TOPI as their cellular mode of action.
Our results showed that HH-N25 is a potent dual inhibitor of DNA TOPI and hormonal signaling in vitro. Our findings represent the first attempt to show tumor regression and selective suppression of TOP1, and hormonal signaling in human breast cancer cell lines by treatment with a single small-molecular compound. These double inhibition features of HH-N25 highlight one of the clear benefits of HH-N25 over other anticancer drugs that exhibit their effect via inhibition of TOP only.
Conclusions
In conclusion, HH-N25 is a new first-in-class molecular entity that suppresses TOP1 and shows potent antitumor activities in human breast cancer cells in vitro and in vivo. Our findings represent the first attempt to document tumor regression and selective suppression of TOP1, and hormonal signaling in human breast cancer cell lines by treatment with a single small-molecular compound. Further extensive investigations of HH-N25 in various cancer types to develop this novel therapeutic approach seem worthwhile.
Data Sharing Statement
The datasets generated and analyzed in this study will be available upon reasonable request.
Acknowledgments
We acknowledged the NCI Developmental Therapeutics Program (DTP) of the National Cancer Institute, National Institutes of Health (NIH‐NCI) for the 60‐cancer‐cell line screening of HH-N25.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
Hsu-Shan Huang was funded by the Ministry of Science and Technology (MOST 110-2314-B-038-120).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249.
2. Ferlay J, Colombet M, Soerjomataram I, et al. Cancer statistics for the year 2020: an overview. Int J Cancer. 2021;149(4):778–789.
3. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
4. Cao W, Chen H-D, Yu Y-W, Li N, Chen W-Q. Changing profiles of cancer burden worldwide and in China: a secondary analysis of the global cancer statistics 2020. Chin Med J. 2021;134(7):783. doi:10.1097/CM9.0000000000001474
5. Chapek MA, Martindale RG. Nutrition in cancer therapy: overview for the cancer patient. J Parenteral Enteral Nutr. 2021;10:21
6. Pasha N, Turner NC. Understanding and overcoming tumor heterogeneity in metastatic breast cancer treatment. Nature Cancer. 2021;4:1–13.
7. Waks AG, Winer EP. Breast cancer treatment: a review. JAMA. 2019;321(3):288–300. doi:10.1001/jama.2018.19323
8. Fredholm H, Eaker S, Frisell J, Holmberg L, Fredriksson I, Lindman H. Breast cancer in young women: poor survival despite intensive treatment. PLoS One. 2009;4(11):e7695. doi:10.1371/journal.pone.0007695
9. Saneja A, Khare V, Alam N, Dubey RD, Gupta PN. Advances in P-glycoprotein-based approaches for delivering anticancer drugs: pharmacokinetic perspective and clinical relevance. Expert Opin Drug Deliv. 2014;11(1):121–138. doi:10.1517/17425247.2014.865014
10. Felici A, Verweij J, Sparreboom A. Dosing strategies for anticancer drugs: the good, the bad and body-surface area. Eur J Cancer. 2002;38(13):1677–1684. doi:10.1016/S0959-8049(02)00151-X
11. Keizer RJ, Huitema AD, Schellens JH, Beijnen JH. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49(8):493–507. doi:10.2165/11531280-000000000-00000
12. Akindele AJ, Wani ZA, Sharma S, et al. In vitro and in vivo anticancer activity of root extracts of Sansevieria liberica Gerome and labroy (Agavaceae). Evidence-Based Compl Alternative Med. 2015;2015:560404. doi:10.1155/2015/560404
13. Haider MS, Ahmad T, Groll J, Scherf-Clavel O, Kroiss M, Luxenhofer R. The challenging pharmacokinetics of mitotane: an old drug in need of new packaging. Eur J Drug Metab Pharmacokinet. 2021;1–19.
14. Gallo JM, Vicini P, Orlansky A, et al. Pharmacokinetic model-predicted anticancer drug concentrations in human tumors. Clin Cancer Res. 2004;10(23):8048–8058. doi:10.1158/1078-0432.CCR-04-0822
15. White RE, Manitpisitkul P. Pharmacokinetic theory of cassette dosing in drug discovery screening. Drug Metab Dispos. 2001;29(7):957–966.
16. Graham M, Kaye S. New approaches in preclinical and clinical pharmacokinetics. Cancer Surv. 1993;17:27–49.
17. Ogino M, Fujii T, Nakazawa Y, et al. Implications of Topoisomerase (TOP1 and TOP2α) expression in patients with breast cancer. In Vivo. 2020;34(6):3483–3487. doi:10.21873/invivo.12188
18. Bielawski K, Winnicka K, Bielawska A. Inhibition of DNA topoisomerases I and II, and growth inhibition of breast cancer MCF-7 cells by ouabain, digoxin and proscillaridin A. Biol Pharm Bull. 2006;29(7):1493–1497. doi:10.1248/bpb.29.1493
19. Oksuzoglu E, Tekiner-Gulbas B, Alper S, et al. Some benzoxazoles and benzimidazoles as DNA topoisomerase I and II inhibitors. J Enzyme Inhib Med Chem. 2008;23(1):37–42. doi:10.1080/14756360701342516
20. Ahn B-Z, Baik K-U, Kweon G-R, Lim K, Hwang B-D. Acylshikonin analogs: synthesis and inhibition of DNA topoisomerase-I. J Med Chem. 1995;38(6):1044–1047. doi:10.1021/jm00006a025
21. Walker JV, Nitiss JL. DNA topoisomerase II as a target for cancer chemotherapy. Cancer Invest. 2002;20(4):570–589. doi:10.1081/CNV-120002156
22. Solary E, Bertrand R, Pommier Y. Apoptosis induced by DNA topoisomerase I and II inhibitors in human leukemic HL-60 cells. Leuk Lymphoma. 1994;15(1–2):21–32. doi:10.3109/10428199409051674
23. Onishi Y, Azuma Y, Sato Y, Mizuno Y, Tadakuma T, Kizaki H. Topoisomerase inhibitors induce apoptosis in thymocytes. Biochim Biophys Acta. 1993;1175(2):147–154. doi:10.1016/0167-4889(93)90017-J
24. Utsugi T, Mattern MR, Mirabelli CK, Hanna N. Potentiation of topoisomerase inhibitor-induced DNA strand breakage and cytotoxicity by tumor necrosis factor: enhancement of topoisomerase activity as a mechanism of potentiation. Cancer Res. 1990;50(9):2636–2640.
25. Chen GL, Yang L, Rowe TC, Halligan BD, Tewey KM, Liu LF. Nonintercalative antitumor drugs interfere with the breakage-reunion reaction of mammalian DNA topoisomerase II. J Biol Chem. 1984;259(21):13560–13566. doi:10.1016/S0021-9258(18)90729-5
26. Hsiang YH, Hertzberg R, Hecht S, Liu LF. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J Biol Chem. 1985;260(27):14873–14878. doi:10.1016/S0021-9258(17)38654-4
27. Nelson EM, Tewey KM, Liu LF. Mechanism of antitumor drug action: poisoning of mammalian DNA topoisomerase II on DNA by 4ʹ-(9-acridinylamino)-methanesulfon-m-anisidide. Proc Nat Acad Sci. 1984;81(5):1361–1365. doi:10.1073/pnas.81.5.1361
28. Di X, Gennings C, Bear HD, et al. Influence of the phosphodiesterase-5 inhibitor, sildenafil, on sensitivity to chemotherapy in breast tumor cells. Breast Cancer Res Treat. 2010;124(2):349–360. doi:10.1007/s10549-010-0765-7
29. Masuda N, Matsui K, Negoro S, et al. Combination of irinotecan and etoposide for treatment of refractory or relapsed small-cell lung cancer. J Clin Oncol. 1998;16(10):3329–3334. doi:10.1200/JCO.1998.16.10.3329
30. Joel SP, Shah R, Clark PI, Slevin ML. Predicting etoposide toxicity: relationship to organ function and protein binding. J Clin Oncol. 1996;14(1):257–267. doi:10.1200/JCO.1996.14.1.257
31. He S, Dong G, Wang Z, et al. Discovery of novel multiacting topoisomerase I/II and histone deacetylase inhibitors. ACS Med Chem Lett. 2015;6(3):239–243. doi:10.1021/ml500327q
32. Huang HS, Chen TC, Chen RH, et al. Synthesis, cytotoxicity and human telomerase inhibition activities of a series of 1,2-heteroannelated anthraquinones and anthra[1,2-d]imidazole-6,11-dione homologues. Bioorg Med Chem. 2009;17(21):7418–7428. doi:10.1016/j.bmc.2009.09.033
33. Huang H-S, Chiou J-F, Fong Y, et al. Activation of human telomerase reverse transcriptase expression by some new symmetrical bis-substituted derivatives of the anthraquinone. J Med Chem. 2003;46(15):3300–3307. doi:10.1021/jm020492l
34. Huang H-S, Chiu H-F, Lee A-L, Guo C-L, Yuan C-L. Synthesis and structure–activity correlations of the cytotoxic bifunctional 1,4-diamidoanthraquinone derivatives. Bioorg Med Chem. 2004;12(23):6163–6170. doi:10.1016/j.bmc.2004.09.001
35. Shen C-J, Lin P-L, Lin H-C, Cheng Y-W, Huang H-S, Lee H. RV-59 suppresses cytoplasmic Nrf2-mediated 5-fluorouracil resistance and tumor growth in colorectal cancer. Am J Cancer Res. 2019;9(12):2789–2796.
36. Huang HS, Yu DS, Chen TC. Thiochromeno [2, 3-c] quinolin-12-one derivatives, preparation method and application thereof. Google Patents; 2015.
37. Lawal B, Wang Y-C, Wu ATH, Huang H-S. Pro-oncogenic c-Met/EGFR, biomarker signatures of the tumor microenvironment are clinical and therapy response prognosticators in colorectal cancer, and therapeutic targets of 3-phenyl-2H-benzo[e][1,3]-oxazine-2,4(3H)-dione derivatives. Front Pharmacol. 2021;12:2300. doi:10.3389/fphar.2021.691234
38. Thurston DE, Pysz I. Chemistry and Pharmacology of Anticancer Drugs. CRC press; 2021.
39. Chen T-C, Wu C-L, Lee -C-C, Chen C-L, Yu D-S, Huang H-S. Structure-based hybridization, synthesis and biological evaluation of novel tetracyclic heterocyclic azathioxanthone analogues as potential antitumor agents. Eur J Med Chem. 2015;103:615–627. doi:10.1016/j.ejmech.2014.09.050
40. Orellana EA, Kasinski AL. Sulforhodamine B (SRB) assay in cell culture to investigate cell proliferation. Bio-Protocol. 2016;6(21):e1984. doi:10.21769/BioProtoc.1984
41. Chen T-C, Yu D-S, Chen S-J, et al. Design, synthesis and biological evaluation of tetracyclic azafluorenone derivatives with topoisomerase I inhibitory properties as potential anticancer agents. Arabian J Chem. 2019;12(8):4348–4364. doi:10.1016/j.arabjc.2016.06.014
42. Das P, Jain CK, Roychoudhury S, Majumder HK, Das S. Design synthesis and in vitro anticancer activity of a Cu(II) complex of carminic acid: a novel small molecule inhibitor of human DNA topoisomerase I and topoisomerase II. Chem Select. 2016;1(21):6623–6631. doi:10.1002/slct.201601152
43. Doudka N, Giocanti M, Basso M, et al. Development and validation of a simple and rapid ultrahigh-performance liquid chromatography tandem spectrometry method for the quantification of hydroxychloroquine in plasma and blood samples in the emergency context of SARS-CoV-2 pandemic. Ther Drug Monit. 2021;43(4):570. doi:10.1097/FTD.0000000000000836
44. Dhar S, Kolishetti N, Lippard SJ, Farokhzad OC. Targeted delivery of a cisplatin prodrug for safer and more effective prostate cancer therapy in vivo. Proc Nat Acad Sci. 2011;108(5):1850–1855. doi:10.1073/pnas.1011379108
45. Rao CV, Reddy BS, Steele VE, et al. Nitric oxide–releasing aspirin and indomethacin are potent inhibitors against colon cancer in azoxymethane-treated rats: effects on molecular targets. Mol Cancer Ther. 2006;5(6):1530–1538. doi:10.1158/1535-7163.MCT-06-0061
46. Traphagen NA, Hosford SR, Jiang A, et al. High estrogen receptor alpha activation confers resistance to estrogen deprivation and is required for therapeutic response to estrogen in breast cancer. Oncogene. 2021;40(19):3408–3421. doi:10.1038/s41388-021-01782-w
47. Marcus D, Hanwell DEC, Lonie DC, Vandermeersch T, Zurek E, Hutchison GR. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J Cheminform. 2012;4:17.
48. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461.
49. Lawal B, Liu Y-L, Mokgautsi N, et al. Pharmacoinformatics and preclinical studies of NSC765690 and NSC765599, potential STAT3/CDK2/4/6 inhibitors with antitumor activities against NCI60 human tumor cell lines. Biomedicines. 2021;9(1):92. doi:10.3390/biomedicines9010092
50. Lawal B, Lee C-Y, Mokgautsi N, et al. mTOR/EGFR/iNOS/MAP2K1/FGFR/TGFB1 are druggable candidates for N-(2,4-difluorophenyl)-2′,4′-difluoro-4-hydroxybiphenyl-3-carboxamide (NSC765598), with consequent anticancer implications. Front Oncol. 2021;11:932. doi:10.3389/fonc.2021.656738
51. Wu S-Y, Lin K-C, Lawal B, Wu TH, Wu C-Z. MXD3 as an onco-immunological biomarker encompassing the tumor microenvironment, disease staging, prognoses, and therapeutic responses in multiple cancer types. Comput Struct Biotechnol J. 2021;19:4970–4983. doi:10.1016/j.csbj.2021.08.047
52. Chen J-H, Wu ATH, Lawal B, et al. Identification of cancer hub gene signatures associated with immune-suppressive tumor microenvironment and ovatodiolide as a potential cancer immunotherapeutic agent. Cancers. 2021;13(15):3847. doi:10.3390/cancers13153847
53. Visualizer DS. BIOVIA, Dassault systèmes, BIOVIA workbook, release 2020; BIOVIA pipeline pilot, release 2020. San Diego: Dassault Systèmes; 2020.
54. Clausen DM, Guo J, Parise RA, et al. In vitro cytotoxicity and in vivo efficacy, pharmacokinetics, and metabolism of 10074-G5, a novel small-molecule inhibitor of c-Myc/Max dimerization. J Pharmacol Exp Ther. 2010;335(3):715–727. doi:10.1124/jpet.110.170555
55. You F, Gao C. Topoisomerase inhibitors and targeted delivery in cancer therapy. Curr Top Med Chem. 2019;19(9):713–729. doi:10.2174/1568026619666190401112948
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.