")
Back to Journals » International Journal of Nanomedicine » Volume 11
In vivo near-infrared imaging for the tracking of systemically delivered mesenchymal stem cells: tropism for brain tumors and biodistribution
Authors Kim SM, Jeong CH, Woo JS, Ryu CH, Lee J, Jeun S
Received 24 September 2015
Accepted for publication 21 November 2015
Published 18 December 2015 Volume 2016:11 Pages 13—23
DOI https://doi.org/10.2147/IJN.S97073
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Seong Muk Kim,1 Chang Hyun Jeong,2 Ji Sun Woo,2 Chung Heon Ryu,1 Jeong-Hwa Lee,3 Sin-Soo Jeun1,2
1Postech-Catholic Biomedical Engineering Institute, College of Medicine, The Catholic University of Korea, Seoul, South Korea; 2Department of Neurosurgery, Seoul St Mary’s Hospital, The Catholic University of Korea, Seoul, South Korea; 3Department of Biochemistry, College of Medicine, The Catholic University of Korea, Seoul, Korea
Abstract: Mesenchymal stem cell (MSC)-based gene therapy is a promising tool for the treatment of various neurological diseases, including brain tumors. However, the tracking of in vivo stem cell migration, distribution, and survival need to be defined for their clinical application. The systemic routes of stem cell delivery must be determined because direct intracerebral injection as a cure for brain tumors is an invasive method. In this study, we show for the first time that near-infrared (NIR) imaging can reveal the distribution and tumor tropism of intravenously injected MSCs in an intracranial xenograft glioma model. MSCs were labeled with NIR fluorescent nanoparticles, and the effects of the NIR dye on cell proliferation and migratory capacity were evaluated in vitro. We investigated the tumor-targeting properties and tissue distribution of labeled MSCs introduced by intravenous injection and followed by in vivo imaging analysis, histological analysis, and real-time quantitative polymerase chain reaction. We observed no cytotoxicity or change in the overall growth rate and characteristics of labeled MSCs compared with control MSCs. NIR fluorescent imaging showed the organ distribution and targeted tumor tropism of systemically injected human MSCs. A significant number of MSCs accumulated specifically at the tumor site in the mouse brain. These results suggest that NIR-based cell tracking is a potentially useful imaging technique to visualize cell survival, migration, and distribution for the application of MSC-mediated therapies in the treatment of malignant gliomas.
Keywords: mesenchymal stem cells, near-infrared nanoparticles, glioma, systemic delivery, in vivo imaging
Introduction
Stem cell-mediated gene delivery is a promising strategy in anticancer therapy, including treatment of brain tumors. Among stem cells, mesenchymal stem cells (MSCs) have potential clinical use in cancer gene therapy because they have tumor-targeting properties, can be isolated easily, and can be engineered with viral vectors.1–3 Glioblastoma multiforme (GBM) is the most devastating of the brain tumors. Despite the use of conventional treatments such as surgical resection, radiation, and chemotherapy, the median survival of GBM patients is 14.6 months for radiotherapy plus temozolomide (3,4-dihydro-3-methyl-4-oxoimidazo-[5,1-d]-1,2,3,5-tetrazine-8-carboxamide) and 12.1 months for radiotherapy alone.4–6
To date, MSCs derived from a variety of tissues or organs that can migrate toward tumors have been used as vehicles for delivering therapeutic genes to treat brain tumors. Several therapeutic strategies for gene delivery by engineered MSCs have been developed using herpes simplex virus thymidine kinase, interferons, interleukins, apoptosis-inducing agents, or oncolytic viruses, and these engineered cells exhibit potent antitumor activity.7–12 However, several issues remain to be clarified before the clinical application of MSC-based gene therapy for the treatment of glioma, including questions about cell survival, migration, and distribution after transplantation. An appropriate in vivo imaging tool to evaluate the biology of transplanted cells in association with the therapeutic effects of gene therapy using MSCs is needed.13
In vivo live imaging plays an important role in biomedical research. Noninvasive imaging methods, such as magnetic resonance imaging (MRI) or positron emission tomography (PET), have contributed to advances in high-resolution in vivo imaging for stem cell tracking.14–16 MRI imaging provides high spatial resolution and anatomical information but has limited sensitivity. PET imaging has high sensitivity but low spatial resolution and does not provide anatomical data, and the radioisotopes have a short half-life. However, recently, a novel cell labeling agent (ie, Zirconium-89) has emerged as an attractive PET radionuclide for cell labeling application due to its high spatial resolution and 78.4-hour half-life that may allow monitoring of administered cells up to a 2- to 3-week period.17 Importantly, both MRI and PET provide low-resolution imaging at the cellular or subcellular level. Fluorescence imaging with nanoparticles is another noninvasive imaging method for in vivo tracking. Its advantages are the high sensitivity and resolution at the subcellular level with the use of microscopy, but it has a limited penetration depth through tissues. Near-infrared (NIR) fluorescence imaging has better penetration depth and provides more specific signals. NIR imaging offers new opportunities as a sensitive and noninvasive detection technique for diagnostics that allows deeper penetration into tissues with minimum background interference.18,19 The successful clinical application of MSC-based tumor therapies needs noninvasive imaging approaches for monitoring tumor progression and treatment outcomes in real time.
Intracranial injection in glioma therapy can bypass the blood–brain barrier (BBB) to directly deliver transplanted MSCs with the therapeutic genes to the tumor site. However, this method is invasive, damages surrounding normal brain tissue, and has limited capacity as a repeated treatment. Optimization of an effective stem cell delivery route is needed for clinical applications. Intravenous stem cell delivery for treatment is used increasingly in animal models and humans,20,21 although few stem cells reach the brain following injection because of trapping in the lungs or other organs. In the present study, we used NIR fluorescence imaging methods for the first time to monitor the movement of human bone marrow-derived MSCs toward tumors in a glioma xenograft mouse model. MSCs were labeled with fluorescent nanoparticles and administered through tail vein injection. We suggest that real-time in vivo imaging technologies using NIR nanoparticles could be applied to track the injected MSCs and to assess the effects of MSCs in the treatment of glioma.
Materials and methods
Cell cultures
Human bone marrow-derived MSCs were obtained from the Catholic Institute of Cell Therapy (CIC; Seoul, Korea). Human bone marrow aspirates were obtained from the iliac crest of healthy donors aged 20 to 55 years after approval by the Institutional Review Board of Seoul St Mary’s Hospital. Bone marrow aspirates were obtained from healthy donors with written informed consent, and sent to the good manufacturing practice-compliant facility of the CIC for the isolation, expansion, and quality control of MSCs as described previously.22 The cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM)-low glucose (Thermo Fisher Scientific, Waltham, MA, USA) with 20% fetal bovine serum (Thermo Fisher Scientific) at a density of 5–8×103 cells/cm2. The human glioma cell line (U-87MG) was purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). Firefly luciferase (Luc)-expressing U-87MG cells (U87-Luc) were stably transduced using a lentivirus expressing Luc.23 Cells were grown in DMEM supplemented with antibiotics and 10% fetal bovine serum (Thermo Fisher Scientific). All cells were incubated at 37°C in a humidified atmosphere containing 5% CO2.
Cell labeling and characterization
Human MSCs were stained with fluorescent magnetic NEO-LIVE™-Magnoxide 675 nanoparticles (NIR675; Biterials, Seoul, Korea) at 0.4 mg/mL for the in vitro assay or in vivo injection. All procedures were performed according to the manufacturer’s instructions. MSCs were incubated in growth medium containing nanoparticles for 24 hours and washed with phosphate-buffered saline (PBS). To confirm the cellular internalization of NIR675 nanoparticles into MSCs, labeled MSCs cultured in the 4-well chamber slide were stained with anti-human nuclei antibody (1:200; EMD Millipore, Billerica, MA, USA) and then analyzed by confocal microscopy. Confocal images were obtained using a Zeiss laser scanning confocal microscope (LSM 510 Meta; Carl Zeiss Meditec AG, Jena, Germany) and Zeiss software. For the analysis of the detection limit and cell viability of this system, MSCs and NIR675-labeled MSCs (MSC-675) were plated in T75 flasks and maintained in culture. At different times after labeling, serial dilutions of cells were plated in 96-well plates. On day 3 after plating, fluorescent images were obtained using a Maestro Imaging System (CRI Inc., Woburn, MA, USA) for signal intensity and analysis. At that time, cell viability was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)-based cytotoxicity assay (Sigma-Aldrich Co., St Louis, MO, USA).
In vitro migration assay
The migratory ability of MSCs was determined using Transwell inserts with 8 μm pores (Corning Inc., Corning, NY, USA), as described previously.9 U-87MG cells (1×106) were incubated in serum-free medium (SFM) for 48 hours, and the resulting conditioned medium (CM) was used as the chemoattractant. MSCs or MSC-675 (2×104 cells) were plated in Transwell inserts, and SFM or CM was placed in the lower well. After 18 hours, non-migrating cells on the upper side of the filter were removed with a cotton swab. The filter was stained with the Three-Step Stain Set (Diff-Quik; Sysmex, Kobe, Japan), and the cells that migrated to the lower side of the filter were counted under a slide scanner (Pannoramic MIDI; 3DHISTECH Ltd., Budapest, Hungary) in five randomly chosen fields (×200).
Adenovirus (Ad) infection
The recombinant replication-deficient adenoviral vector encoding the gene for Ad-EGFP was constructed using the AdEasy vector system (QBioGene, Carlsbad, CA, USA). All Ad concentrations used for the transduction of cells are expressed as the multiplicity of infection in plaque-forming units per cell. For MSC transduction, a mixture of 50 multiplicity of infection of Ad and 50 μmol/L of Fe3+ was pre-incubated in SFM at room temperature for 30 minutes and then infected into MSCs for 30 minutes, as described previously.24
In vivo bioluminescence and fluorescence imaging
All animal experiments were performed in accordance with institutional guidelines and were approved by the Institutional Animal Care and Use Committee in The Catholic University of Korea. Orthotopic glioblastoma xenografts were established in male athymic nude mice (6–8 weeks of age; Charles River Laboratories, Wilmington, MA, USA). Mice received a stereotactically guided injection of human glioma cells (1×105 U87-Luc cells in 3 μL of PBS) in the right side of the brain, as described previously.9 In vivo bioluminescence and fluorescence images were obtained using the Maestro Imaging System for data acquisition and analysis. To analyze the detection limit of this system, serially diluted MSC-675 were injected into the skin, and the signal intensity was measured. Cell doses in the range of 1–20×104 MSC-675 were injected subcutaneously in 10 μL of PBS. A control animal was injected with PBS alone. To evaluate the duration of the signal intensity of the injected MSC-675 in the mouse brain, 2×105 cells were injected into the striatum and measured at different times until there was no detectable signal. To determine the distribution of systemically injected MSC-675, 1×106 MSC-675 in 100 μL of PBS were injected through the tail vein of normal or tumor-bearing mice. To evaluate the distribution of cell-free nanoparticles to verify MSC-specific tumor targeting, 0.4 mg of NIR675 alone in 100 μL PBS was injected. The first in vivo images of the whole body were captured 5 minutes after the injection (data not shown) for the detection of cells at the injection site and moving through the tail vein. Sequential in vivo fluorescence measurements were then taken at scheduled times and merged with the bioluminescence imaging for the detection of tumor sites. To assess tumor growth by direct visualization using the Maestro Imaging System, the substrate of Luc, D-luciferin (150 mg of luciferin/kg of body weight; PerkinElmer Inc., Waltham, MA, USA), was delivered by intraperitoneal injection 10 minutes before the bioluminescence imaging.
Quantitative real-time polymerase chain reaction (qPCR) analysis
To quantify the distribution of systemically injected MSCs, 1×106 MSCs transduced with EGFP (MSC-EGFP) in 100 μL of PBS were injected through the tail vein of normal or tumor-bearing mice. Mice (n=3/group) were sacrificed under deep anesthesia after MSC-EGFP injection, and the brains and other organs (lungs, liver, and spleen) were dissected at specific times. Total RNA was isolated using AccuZol (Bioneer, Daejeon, Korea), and first-strand cDNA was synthesized by reverse transcription to cDNA using a ReverTra Ace qPCR kit (Toyobo, Osaka, Japan) according to the manufacturer’s protocol. qPCR was performed to confirm the expression level of EGFP mRNA using SYBR® Premix Ex Taq™ (TaKaRa Bio, Shiga, Japan) on an Applied Biosystems 7300 machine (Thermo Fisher Scientific). The relative values for EGFP mRNA were calculated after normalization to the Ct value from β-actin in the same sample using the ΔΔCt method.25
Assessment of BBB integrity by extravasation of Evans blue (EB) dye
BBB permeability was evaluated by assessing EB (Sigma-Aldrich Co.) extravasation into brain tissue. EB dye (2%, 4 mL/kg) was injected through the tail vein of anesthetized mice 7 days after tumor inoculation. After 4 hours, animals were anesthetized and perfused with PBS to remove the intravascular dye. After decapitation, entire brain tissues were removed and dissected. The visualization of EB staining was then determined on photographs.
Immunohistochemistry
Mice (n=3/group for the normal and tumor-bearing groups) were perfused with PBS followed by 4% paraformaldehyde under deep anesthesia 4 days after MSC-675 injection. The excised brains and other organs (lungs, liver, and spleen) were fixed, embedded, snap frozen in liquid nitrogen, and stored at −70°C until use. Tissues were cryo-sectioned at 14 μm, stained with anti-human mitochondria antibody (1:200; EMD Millipore), and exposed to the secondary antibody Alexa Fluor® 488 goat anti-mouse IgG (1:1,000; Molecular Probes, Eugene, OR, USA). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (Sigma-Aldrich Co.). Confocal images were obtained using a Zeiss laser scanning confocal microscope (LSM 510 Meta, Carl Zeiss Meditec) and Zeiss software.
Statistical analysis
All data are expressed as the mean ± standard error of the mean from at least three independent experiments. Significant differences between test conditions were identified using Student’s t-test. P<0.05 was considered significant.
Results
Characterization of MSC-675
We first used confocal microscopy to confirm the cellular internalization of NIR675 nanoparticles into MSCs. MSCs were incubated with NIR675 in culture medium for 24 hours and stained with anti-human nuclei antibody to detect the nuclei of human cells. NIR675 was internalized into the cells and resided mainly in the cytoplasm; there was no change in morphology in the MSC-675 (Figure 1A). We also determined the cell proliferation and survival capacity of MSC-675 compared with unlabeled (control) MSCs to check whether the internalized nanoparticles were stable in the cells and whether they induced cell cytotoxicity. We observed no cytotoxicity or change in the overall growth rate of MSC-675 compared with control MSCs at the serial dilutions used (Figure 1B).
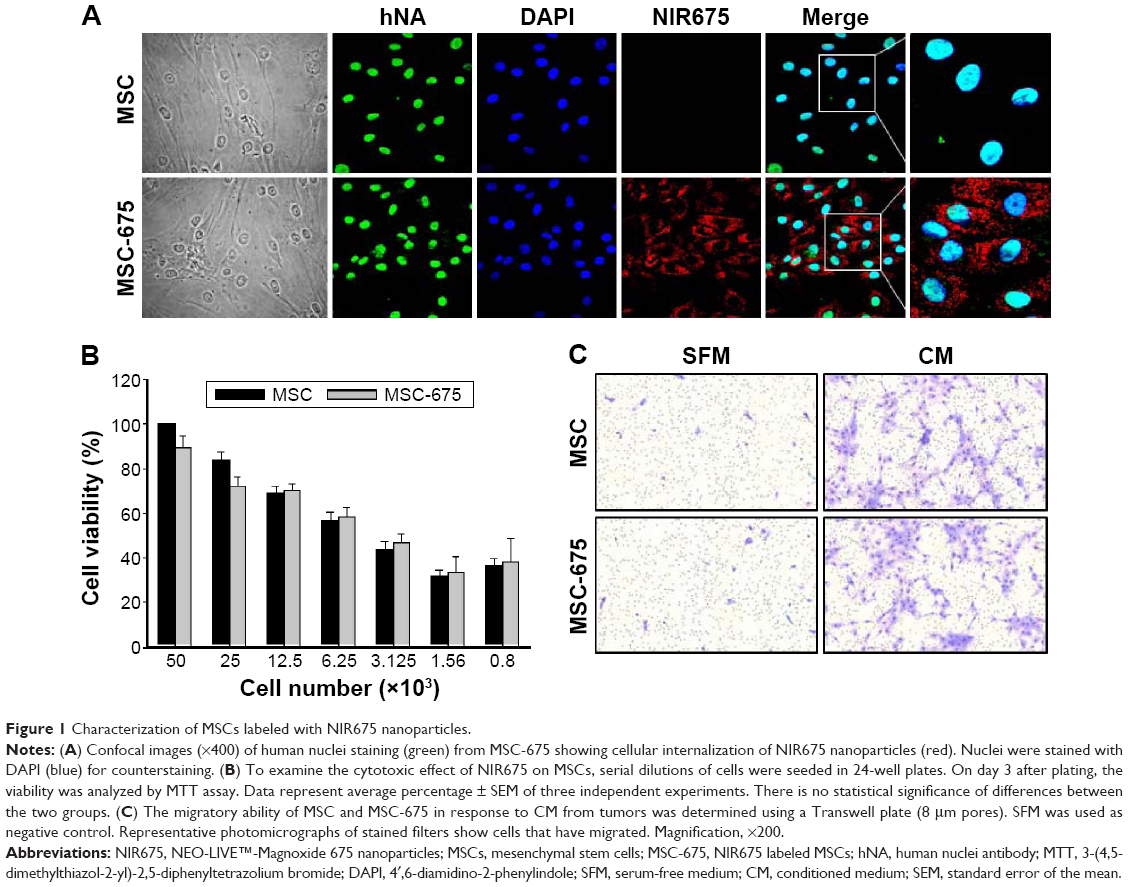 | Figure 1 Characterization of MSCs labeled with NIR675 nanoparticles. |
Among several characteristics of human MSCs, tumor tropism is the most important property in stem cell-based cancer therapy. Both cell populations maintained cell migratory activity toward CM from glioma cell culture (Figure 1C). The multi-differentiation potential (adipogenic, chondrogenic, and osteogenic differentiation) and cell surface antigens (CD90/CD73, 95% positive; CD34/CD45, 95% negative) did not change after the internalization of NIR675 into MSCs (data not shown). These results suggested that there were no changes in the characteristics of MSC-675 and that these cells could be used to study the MSC distribution after transplantation in an animal tumor model.
Detection limit and duration of MSC-675 in vitro and in vivo
Next, we assessed the fluorescent intensity of MSC-675 injected at various times in various cell numbers. Cells were labeled with NIR675 1 day before the experiment and the labeled MSCs were identified using the Maestro Imaging System before in vitro cell seeding and in vivo injection. It was important to determine whether and how the NIR675 signal diminished in MSCs over time as a result of cell division in vitro and to evaluate how the duration of the signal intensity was affected by death of transplanted cells in vivo. Serial dilutions of MSC-675 were seeded in 96-well plates at 3, 7, and 10 days after labeling and subculture, and the NIR signal intensity was measured. A quantifiable signal was detectable on day 3, and up to 5×103 MSC-675 were detected (Figure 2A and B). However, the intensity decreased and was not detectable in serial dilutions of MSC-675 on day 10. We also checked the survival of MSC-675 (2×105 cells) by measuring the fluorescent signal intensity at various times after intracranial injection. The fluorescent signals were observed up to 21 days after injection in live animals (Figure 2C and D). To determine the sensitivity of the in vivo detection of cells, we injected MSC-675 beneath the skin of mice in various cell numbers. The control animals displayed no detectable fluorescent activity, whereas up to 104 cells were detected in animals injected with MSC-675 (Figure 2E and F).
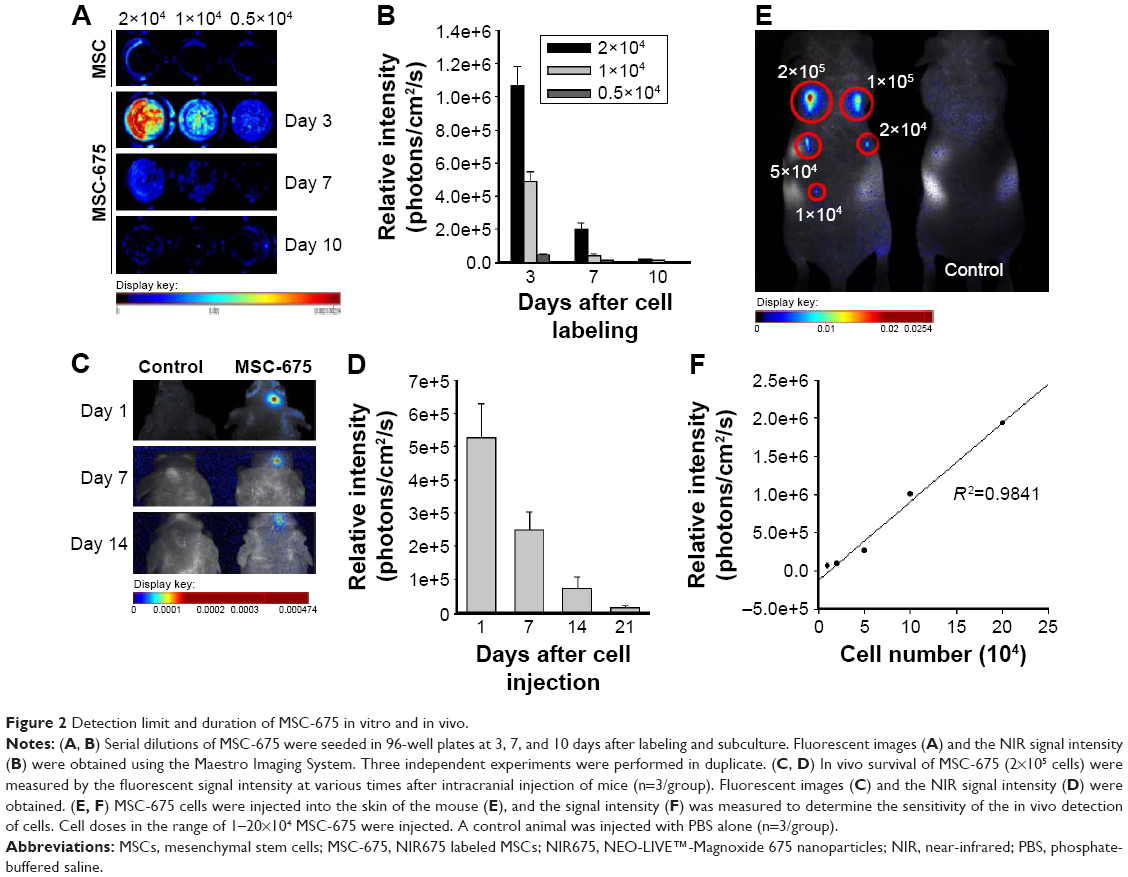 | Figure 2 Detection limit and duration of MSC-675 in vitro and in vivo. |
In vivo distribution of MSC-675 after systemic injection
It has been previously shown that MSCs have a tumor tropism to brain tumors in intracranial glioma xenografts.9,12,23 In this study, we confirmed that the migratory capacity of MSCs toward glioma cells after intravenous delivery could be visualized by in vivo NIR fluorescence imaging. We used noninvasive systemic delivery and in vivo imaging to examine the tumor-targeting property of MSCs. MSC-675 were injected into normal mice and tumor-bearing mice through the tail vein, and MSC localization was followed by fluorescent imaging at various times. MSCs delivered systemically via the tail vein initially resided predominantly in the lung, and the signal intensity decreased over time in both normal (Figure 3A) and tumor-bearing mice (Figure 3B). One day after tail vein injection, many cells moved out from the lung toward other organs (liver and spleen), and the signal remained stable in these regions for 14 days in both normal and tumor-bearing mice. Although normal mice showed a similar pattern of MSC migration, the period and signal intensity of lung residence of cells were greater in normal mice than in the tumor-bearing mice.
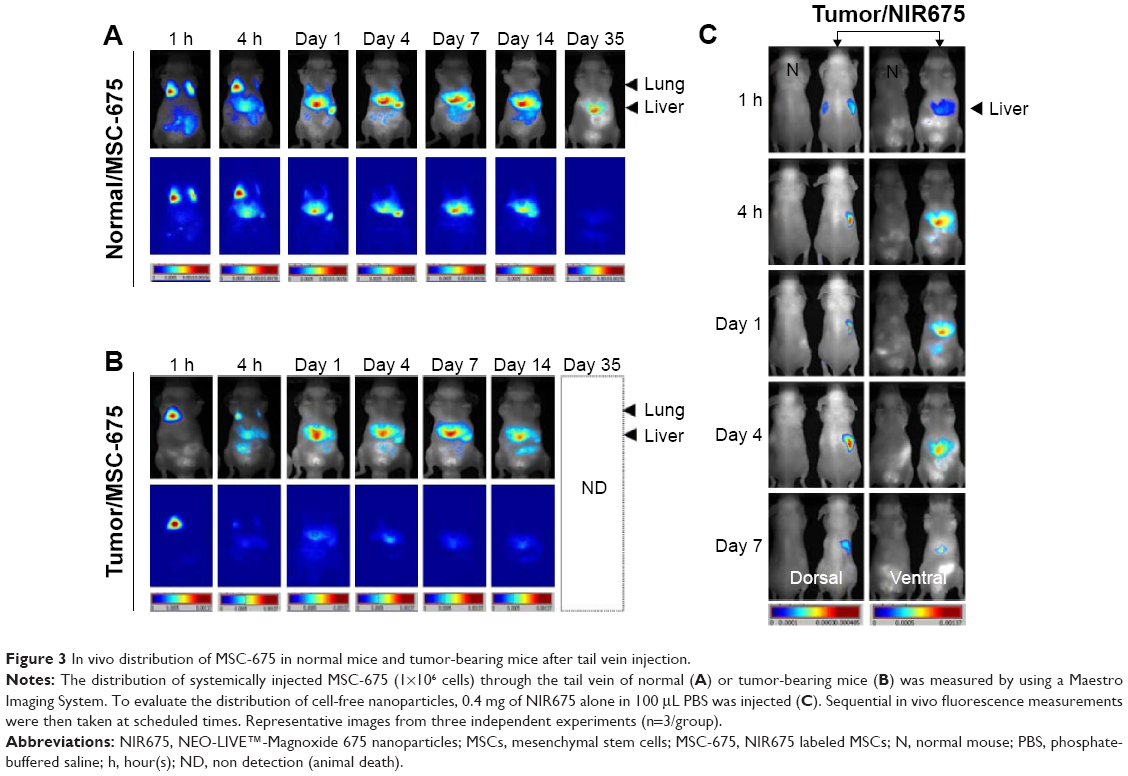 | Figure 3 In vivo distribution of MSC-675 in normal mice and tumor-bearing mice after tail vein injection. |
Because fluorescent dyes can transfer to neighboring cells, we also assessed whether dye transfer could have affected the in vivo distribution of MSC-675. We injected free NIR nanoparticles (NIR675 alone) into tumor-bearing animals in the same way as for MSC-675 injection (Figure 3C). NIR675 nanoparticles were not trapped within the lung region initially but moved to the liver immediately; this was a different pattern from that observed in mice injected with the MSC-675. NIR675 nanoparticles were cleared from the body by 7 days after tail vein injection. These results indicated that the NIR675 nanoparticles did not leak from labeled cells and transfer to other cells, and that it is possible to track the organ distribution of individual MSC-675.
Penetration of MSCs into the brain of tumor-bearing mice
We investigated whether MSCs could penetrate into and be tracked in the brain after systemic administration via intravenous delivery of MSC-675. MSC tracking was performed at various times after application of MSC-675 into the tail vein of U87-Luc glioma-bearing mice. We used bioluminescent tumor imaging of Luc-expressing human glioma cells over time to identify the conditions under which MSC tropism and selective engraftment in tumor sites could be monitored by NIR fluorescent imaging. Figure 4A shows that MSC-675 injected via the tail vein migrated to the right, tumor-bearing side of the brain. Although a number of MSCs remained in the lung and other organs up to 14 days after MSC administration (Figure 3B), small populations of injected cells migrated to the tumor in the right hemisphere. NIR fluorescence signal from the MSC-675 was detected at the location where tumor cells produced a bioluminescence signal on day 4 after intravenous injection of MSC-675 (Figure 4B). By contrast, no signal was observed in the normal mouse brain injected with MSC-675 and in the tumor-bearing mouse brain injected with NIR675 nanoparticles alone.
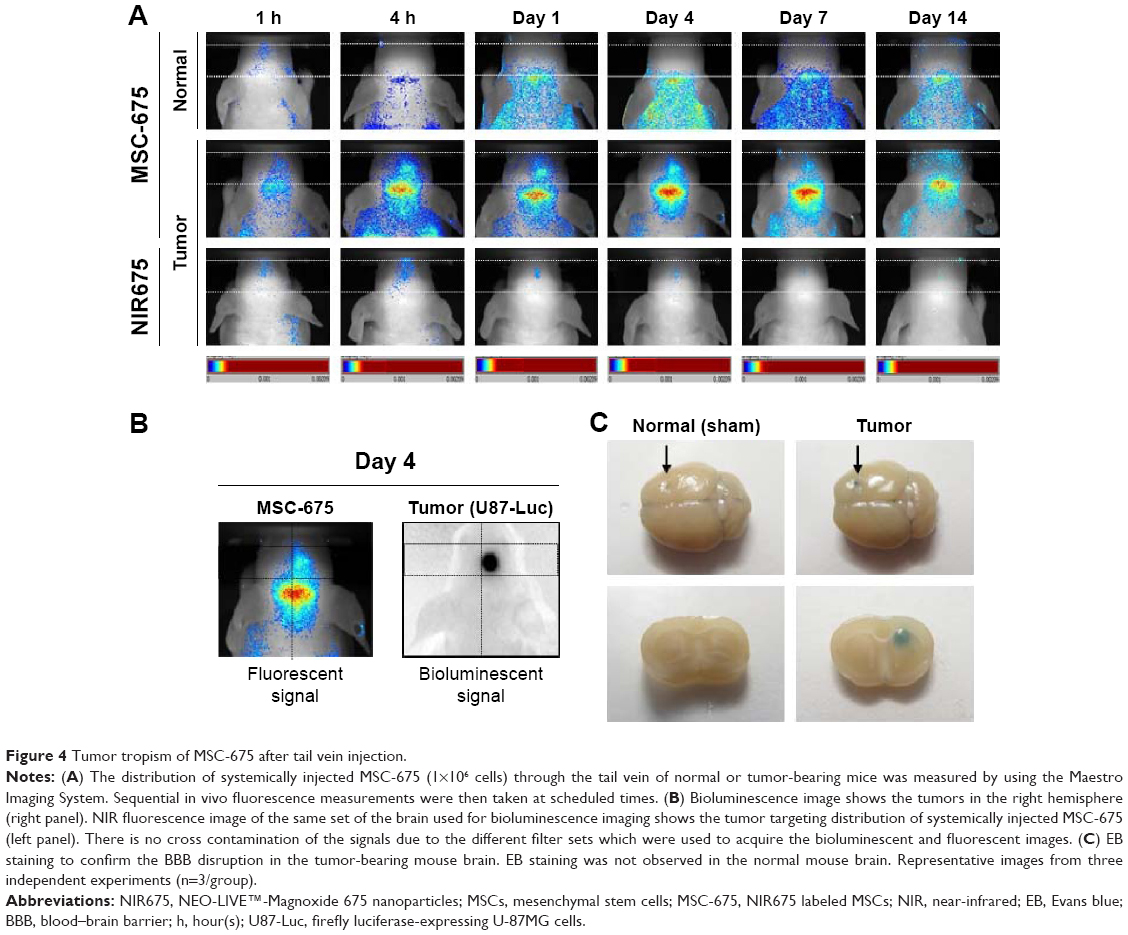 | Figure 4 Tumor tropism of MSC-675 after tail vein injection. |
Because the tumor-bearing brain showed leakage of the BBB, it was expected that intravenously injected MSCs could pass through the BBB and be detected in the brain. We used EB staining to confirm the BBB disruption in the tumor-bearing mouse brain (Figure 4C). No signal was observed in the normal mouse brain. Finally, the intensity of the fluorescent signal in organs of tumor-bearing mice after MSC-675 injection was quantified at each time point (Figure 5). The fluorescent signal of MSC-675 that had migrated into the tumor site in the brain (right hemisphere) was maintained for 7 days after injection. During this time, the intensity of the migrated MSCs in the lung decreased and MSCs accumulated in the liver or spleen.
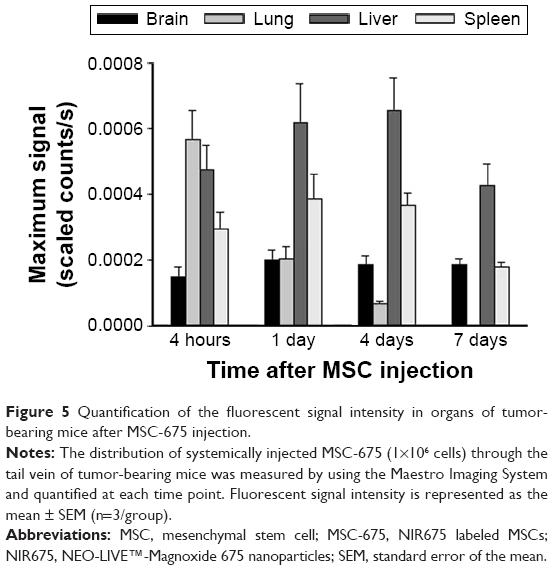 | Figure 5 Quantification of the fluorescent signal intensity in organs of tumor-bearing mice after MSC-675 injection. |
Histological and qPCR analysis of MSC-675 distribution
Based on the data from the in vivo imaging analysis, we used qPCR and histological analysis to quantify the in vivo distribution and the fate of intravenously injected MSCs. We injected EGFP-tagged MSCs into normal or tumor-bearing mice through the tail vein and used an EGFP-specific PCR probe to identify the injected human MSCs in the mouse lung, liver, spleen, and brain. To examine the organ distribution of the transplanted cells, organs were extracted at each time point after injection. MSCs moved out from the lung (Figure 6A) and accumulated mainly in the brain and liver on day 1 (Figure 6B), and almost all transplanted cells disappeared by day 7 in all organs. This pattern differed from the pattern obtained by in vivo imaging of the signal intensity, which showed that the signal in the liver was maintained up to 14 days after injection. The fluorescent signal in the liver at this time was probably the signal from nanoparticles from dead cells accumulated in the liver before being washed from the body. These data suggested that, for a more accurate evaluation of the distribution in animals after MSC injection, the fluorescent signal intensity should be compared with the result from analysis of living cells in the body, such as qPCR. The MSC distribution was also determined by histological analysis of tissues from the sacrificed animals to find MSC-675 in mouse organs after tail vein injection (Figure 6C). MSCs were found in the brain tissues of tumor-bearing mice, but not in normal mice. The distribution pattern of the migrated MSCs in the lung and liver was similar in normal and tumor-bearing mice on day 4 after intravenous injection of MSC-675.
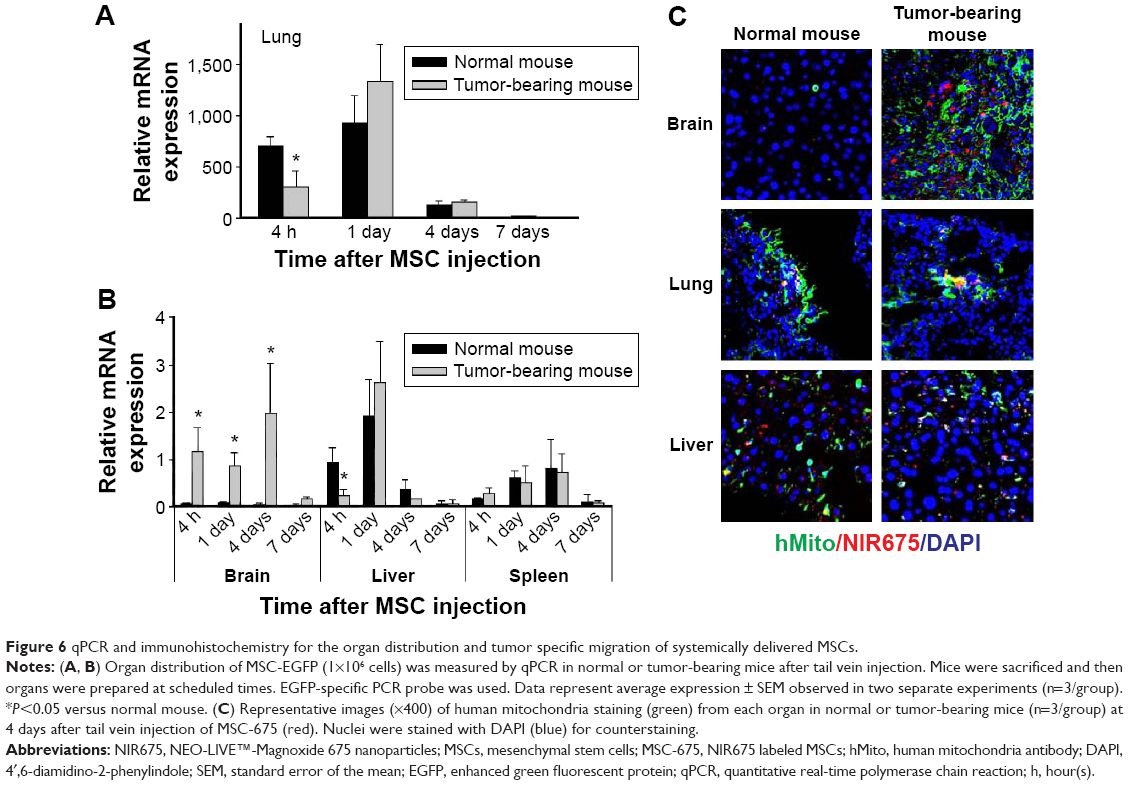 | Figure 6 qPCR and immunohistochemistry for the organ distribution and tumor specific migration of systemically delivered MSCs. |
Discussion
GBM is one of the most common and aggressive primary brain tumors, with a median survival of only 9–15 months despite the use of extensive surgical resection followed by radiation therapy and chemotherapy.4,5 The migratory capacity of glioma cells and their ability to infiltrate into normal brain parenchyma distant from the main tumor mass makes this glioma resistant to chemotherapeutics and eventually results in tumor recurrence. Therefore, for several years, one novel therapeutic strategy targeted the infiltrated tumor cells using MSCs as cell-based gene vehicles for the delivery of therapeutic genes because of the intrinsic homing property of MSCs, which enables them to migrate directly to sites of injury, inflammation, and tumors.26–28 These cells can be used to track the invasive tumor cells and secrete the therapeutic protein at the tumor site.
We and others have previously shown that transplanted MSCs engineered to contain therapeutic genes display tropism for the tumors and that these MSCs exert significant antitumor effects in a human glioma xenograft mouse model.7–12 However, in previous animal experiments, the migratory activities of labeled MSCs toward tumors could be monitored by immunohistochemistry staining and fluorescent evaluation of tissue sections, or PCR only after the animal had been sacrificed. These methods require numerous animals to be sacrificed at multiple time points and are limited to certain parts of the tissue that can be prepared and analyzed. To overcome these problems, we used noninvasive in vivo live imaging to sequentially monitor the migration and distribution of labeled MSCs in the same animal at a range of time points. Noninvasive imaging methods, such as MRI and PET, have different advantages and limitations in terms of sensitivity, spatial resolution, and penetration depth through tissues. For this reason, fluorescence imaging with nanoparticles can be used as an imaging tool complementary to MRI and PET.29,30 It can provide images of high sensitivity and resolution at the subcellular level with the use of microscopy. However, it has a limited penetration depth through tissues. Recently, NIR fluorescence imaging has better penetration depth up to several centimeters and provides more specific signals.31,32 Photons in the NIR region in the range 650–900 nm provide low light scattering and generate less auto-fluorescence from tissues than visible light, but NIR imaging has the limitation of photo-bleaching. To overcome the limitations of each single modality, multimodal imaging methods have been reported.16,33 This is the first study to use NIR imaging technology to track the cell fate, migration, and distribution of systemically administered MSCs in a glioma xenograft mouse model.
In this study, we found that MSCs injected systemically were distributed to the brain and other organs in glioma-bearing mice. The MSCs were first detected in the lungs within 30 minutes and remained there for 3–4 days. The signal intensity decreased gradually in the lung and increased as the cells migrated into the liver and spleen 4 hours after administration, where it remained detectable for up to 14 days. Normal mice exhibited a similar pattern of MSC migration, but the period and signal intensity of lung residence were greater in the normal mice than in the tumor-bearing mice, as shown by the greater number of MSCs that migrated to the tumor site in the tumor-bearing mice. In both animals, NIR fluorescent signal intensity decreased compared with the initial signal, a finding that agreed with that from other tissues. A quantifiable NIR signal is detectable from the live cells in the animal tissues; thus, the decrease of fluorescent signal represents the decrease in the number of live MSCs. Several studies have reported the death of transplanted stem cells in vivo with time starting immediately after the injection of cells.20,34 We also found evidence of the cell death of NIR-labeled MSCs transplanted into the mouse brain, as shown by the decrease in signal intensity (Figure 2C). It is important to find a way of improving the conditions to increase the survival rate of transplanted cells.
MSCs delivered systemically localize preferentially to sites of injury, ischemic lesions, and tumors in the brain despite their predominant entrapment in the lung vasculature.35–37 In the present study, we found that MSCs are trapped transiently in the lungs but migrate over time to the brain, which means that MSCs transplanted systemically via the tail vein can cross the BBB and accumulate selectively in brain tumors. These results suggest that MSCs possess leukocyte-like, active homing mechanisms that enable them to interact with and migrate across the BBB under conditions of injury or inflammation.38 However, the integrity of the cerebral vasculature is probably compromised following injury or inflammation, which can lead to passive MSC accumulation in the brain via entrapment.38 It is not clear whether MSCs can actively “home” or are passively “captured” at sites of inflamed and disrupted vessels. Therefore, the exact mechanisms to explain how MSCs actively cross the BBB remain to be determined.39
To understand the fate of transplanted MSCs in vivo, noninvasive imaging techniques are needed to monitor the cells in both the brain and other tissues. For our study, MSC labeling was achieved by direct incubation in culture medium without transfection agents or electroporation techniques. In addition, in vitro and in vivo migration studies showed that the labeled MSCs retained their migratory ability and that the cell viability, tumor tropism, and other characteristics were not affected by NIR nanoparticle labeling. These NIR-labeled MSCs can be used for noninvasive monitoring to visualize cell survival, distribution, and behavior, and may have potential use in preclinical protocols.
Conclusion
In this study, we showed that NIR fluorescent imaging can reveal the distribution and tropism of systemically injected human MSCs toward tumor sites in glioma-bearing mice. The systemic delivery of stem cell-based therapeutics is a feasible and highly effective treatment modality and allows for noninvasive and repeated application to target malignant gliomas. NIR-based cell tracking has potential as an imaging technique to monitor MSC-mediated therapies and to improve the treatment of malignant gliomas.
Acknowledgments
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2013R1A1A2A10059399) and by the Ministry of Science, ICT and Future Planning (2014R1A2A2A01004525). The Catholic MASTER Cells supplied by Catholic Institute of Cell Therapy (CIC, Seoul, Korea) were derived from human bone marrow donated by healthy donors after informed consent.
Disclosure
The authors report no conflicts of interest in this work.
References
Hamada H, Kobune M, Nakamura K, et al. Mesenchymal stem cells (MSC) as therapeutic cytoreagents for gene therapy. Cancer Sci. 2005;96(3):149–156. | ||
Bovenberg MS, Degeling MH, Tannous BA. Advances in stem cell therapy against gliomas. Trends Mol Med. 2013;19(5):281–291. | ||
Shah K. Mesenchymal stem cells engineered for cancer therapy. Adv Drug Deliv Rev. 2012;64(8):739–748. | ||
Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. | ||
Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–466. | ||
Lefranc F, Brotchi J, Kiss R. Possible future issues in the treatment of glioblastomas: special emphasis on cell migration and the resistance of migrating glioblastoma cells to apoptosis. J Clin Oncol. 2005;23(10):2411–2422. | ||
Nakamizo A, Marini F, Amano T, et al. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 2005;65(8):3307–3318. | ||
Miletic H, Fischer Y, Litwak S, et al. Bystander killing of malignant glioma by bone marrow-derived tumor-infiltrating progenitor cells expressing a suicide gene. Mol Ther. 2007;15(7):1373–1381. | ||
Kim SM, Lim JY, Park SI, et al. Gene therapy using TRAIL-secreting human umbilical cord blood-derived mesenchymal stem cells against intracranial glioma. Cancer Res. 2008;68(23):9614–9623. | ||
Sasportas LS, Kasmieh R, Wakimoto H, et al. Assessment of therapeutic efficacy and fate of engineered human mesenchymal stem cells for cancer therapy. Proc Natl Acad Sci U S A. 2009;106(12):4822–4827. | ||
Uchibori R, Okada T, Ito T, et al. Retroviral vector-producing mesenchymal stem cells for targeted suicide cancer gene therapy. J Gene Med. 2009;11(5):373–381. | ||
Kim SM, Oh JH, Park SA, et al. Irradiation enhances the tumor tropism and therapeutic potential of tumor necrosis factor-related apoptosis-inducing ligand-secreting human umbilical cord blood-derived mesenchymal stem cells in glioma therapy. Stem Cells. 2010;28(12):2217–2228. | ||
Li SC, Tachiki LM, Luo J, Dethlefs BA, Chen Z, Loudon WG. A biological global positioning system: considerations for tracking stem cell behaviors in the whole body. Stem Cell Rev. 2010;6(2):317–333. | ||
Villa C, Erratico S, Razini P, et al. Stem cell tracking by nanotechnologies. Int J Mol Sci. 2010;11(3):1070–1081. | ||
Ferreira L, Karp JM, Nobre L, Langer R. New opportunities: the use of nanotechnologies to manipulate and track stem cells. Cell Stem Cell. 2008;3(2):136–146. | ||
Kim JS, Kim YH, Kim JH, et al. Development and in vivo imaging of a PET/MRI nanoprobe with enhanced NIR fluorescence by dye encapsulation. Nanomedicine (Lond). 2012;7(2):219–229. | ||
Bansal A, Pandey MK, Demirhan YE, et al. Novel (89)Zr cell labeling approach for PET-based cell trafficking studies. EJNMMI Res. 2015;5:19. | ||
Ha S, Ahn S, Kim S, et al. In vivo imaging of human adipose-derived stem cells in Alzheimer’s disease animal model. J Biomed Opt. 2014;19(5):051206. | ||
Bossolasco P, Cova L, Levandis G, et al. Noninvasive near-infrared live imaging of human adult mesenchymal stem cells transplanted in a rodent model of Parkinson’s disease. Int J Nanomedicine. 2012;7:435–447. | ||
Fischer UM, Harting MT, Jimenez F, et al. Pulmonary passage is a major obstacle for intravenous stem cell delivery: the pulmonary first-pass effect. Stem Cells Dev. 2009;18(5):683–692. | ||
Krause U, Harter C, Seckinger A, et al. Intravenous delivery of autologous mesenchymal stem cells limits infarct size and improves left ventricular function in the infarcted porcine heart. Stem Cells Dev. 2007;16(1):31–37. | ||
Kim SM, Woo JS, Jeong CH, Ryu CH, Jang JD, Jeun SS. Potential application of temozolomide in mesenchymal stem cell-based TRAIL gene therapy against malignant glioma. Stem Cells Transl Med. 2014;3(2):172–182. | ||
Kim SM, Kim DS, Jeong CH, et al. CXC chemokine receptor 1 enhances the ability of human umbilical cord blood-derived mesenchymal stem cells to migrate toward gliomas. Biochem Biophys Res Commun. 2011;407(4):741–746. | ||
Kim SW, Kim SJ, Park SH, et al. Complete regression of metastatic renal cell carcinoma by multiple injections of engineered mesenchymal stem cells expressing dodecameric TRAIL and HSV-TK. Clin Cancer Res. 2013;19(2):415–427. | ||
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. | ||
Loebinger MR, Janes SM. Stem cells as vectors for antitumour therapy. Thorax. 2010;65(4):362–369. | ||
Spaeth E, Klopp A, Dembinski J, Andreeff M, Marini F. Inflammation and tumor microenvironments: defining the migratory itinerary of mesenchymal stem cells. Gene Ther. 2008;15(10):730–738. | ||
Doucette T, Rao G, Yang Y, et al. Mesenchymal stem cells display tumor-specific tropism in an RCAS/Ntv-a glioma model. Neoplasia. 2011;13(8):716–725. | ||
Olson ES, Jiang T, Aguilera TA, et al. Activatable cell penetrating peptides linked to nanoparticles as dual probes for in vivo fluorescence and MR imaging of proteases. Proc Natl Acad Sci U S A. 2010;107(9):4311–4316. | ||
Jeon YH, Kim YH, Choi K, et al. In vivo imaging of sentinel nodes using fluorescent silica nanoparticles in living mice. Mol Imaging Biol. 2010;12(2):155–162. | ||
Luker GD, Luker KE. Optical imaging: current applications and future directions. J Nucl Med. 2008;49(1):1–4. | ||
Aswathy RG, Yoshida Y, Maekawa T, Kumar DS. Near-infrared quantum dots for deep tissue imaging. Anal Bioanal Chem. 2010;397(4):1417–1435. | ||
Tennstaedt A, Aswendt M, Adamczak J, Hoehn M. Noninvasive multimodal imaging of stem cell transplants in the brain using bioluminescence imaging and magnetic resonance imaging. Methods Mol Biol. 2013;1052:153–166. | ||
Kidd S, Spaeth E, Dembinski JL, et al. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem Cells. 2009;27(10):2614–2623. | ||
Zhang L, Li Y, Romanko M, et al. Different routes of administration of human umbilical tissue-derived cells improve functional recovery in the rat after focal cerebral ischemia. Brain Res. 2012;1489:104–112. | ||
Gutova M, Frank JA, D’Apuzzo M, et al. Magnetic resonance imaging tracking of ferumoxytol-labeled human neural stem cells: studies leading to clinical use. Stem Cells Transl Med. 2013;2(10):766–775. | ||
Osanai T, Kuroda S, Sugiyama T, et al. Therapeutic effects of intra-arterial delivery of bone marrow stromal cells in traumatic brain injury of rats – in vivo cell tracking study by near-infrared fluorescence imaging. Neurosurgery. 2012;70(2):435–444. | ||
Lee RH, Pulin AA, Seo MJ, et al. Intravenous hMSCs improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti-inflammatory protein TSG-6. Cell Stem Cell. 2009;5(1):54–63. | ||
Liu L, Eckert MA, Riazifar H, Kang DK, Agalliu D, Zhao W. From blood to the brain: can systemically transplanted mesenchymal stem cells cross the blood-brain barrier? Stem Cells Int. 2013;2013:435093. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.