")
Back to Journals » International Journal of Nanomedicine » Volume 12
In vivo evaluation of cetuximab-conjugated poly(γ-glutamic acid)-docetaxel nanomedicines in EGFR-overexpressing gastric cancer xenografts
Authors Sreeranganathan M, Uthaman S, Sarmento B , Mohan CG, Park IK , Jayakumar R
Received 8 June 2017
Accepted for publication 9 August 2017
Published 30 September 2017 Volume 2017:12 Pages 7165—7182
DOI https://doi.org/10.2147/IJN.S143529
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Maya Sreeranganathan,1 Saji Uthaman,2 Bruno Sarmento,3–5 Chethampadi Gopi Mohan,1 In-Kyu Park,1 Rangasamy Jayakumar2
1Centre for Nanosciences and Molecular Medicine, Amrita University, Kochi, India; 2Department of Biomedical Science, BK21 PLUS Center for Creative Biomedical Scientists, Chonnam National University Medical School, Gwangju, Republic of Korea; 3I3S – Instituto de Investigação e Inovação em Saúde, University of Porto, Porto, Portugal, 4INEB – Instituto de Engenharia Biomédica, University of Porto, Porto, Portugal, 5CESPU, Instituto de Investigação e Formação Avançada em Ciências e Tecnologias da Saúde, Instituto Universitário de Ciências da Saúde, Gandra, Portugal
Abstract: Epidermal growth factor receptor (EGFR), upregulated in gastric cancer patients, is an oncogene of interest in the development of targeted cancer nanomedicines. This study demonstrates in silico modeling of monoclonal antibody cetuximab (CET MAb)-conjugated docetaxel (DOCT)-loaded poly(γ-glutamic acid) (γ-PGA) nanoparticles (Nps) and evaluates the in vitro/in vivo effects on EGFR-overexpressing gastric cancer cells (MKN-28). Nontargeted DOCT-γ-PGA Nps (NT Nps: 110±40 nm) and targeted CET MAb-DOCT-γ-PGA Nps (T Nps: 200±20 nm) were prepared using ionic gelation followed by 1-Ethyl-3-(3-dimethyl aminopropyl)carbodiimide– N-Hydoxysuccinimide (EDC– NSH) chemistry. Increased uptake correlated with enhanced cytotoxicity induced by targeted Nps to EGFR +ve MKN-28 compared with nontargeted Nps as evident from MTT and flow cytometric assays. Nanoformulated DOCT showed a superior pharmacokinetic profile to that of free DOCT in Swiss albino mice, indicating the possibility of improved therapeutic effect in the disease model. Qualitative in vivo imaging showed early and enhanced tumor targeted accumulation of CET MAb-DOCT-γ-PGA Nps in EGFR +ve MKN-28–based gastric cancer xenograft, which exhibited efficient arrest of tumor growth compared with nontargeted Nps and free DOCT. Thus, actively targeted CET MAb-DOCT-γ-PGA Nps could be developed as a substitute to conventional nonspecific chemotherapy, and hence could become a feasible strategy for cancer therapy for EGFR-overexpressing gastric tumors.
Keywords: targeted nanoparticles, poly(γ-glutamic acid) nanoparticles, docetaxel, cetuximab, epidermal growth factor receptor, gastric cancer
Corrigendum for this paper has been published
Introduction
Gastric cancer or stomach carcinogenesis is an aggressively spreading disease with a poor prognosis and is reportedly the second most common cause of cancer death worldwide.1–3 A proper understanding is needed of gastric cancer biology and the shortfalls of conventional chemotherapeutic regimens that are directed toward the exploitation of molecular targets involved in its carcinogenesis. Epidermal growth factor receptor (EGFR) overexpression was observed in 27%–44% of primary gastric tumors, and is reported to be an indicator of poor prognostic outcome and, hence, an important therapeutic target.4 Cetuximab (CET; Erbitux®, Merck, Darmstadt, Germany) is a chimeric IgG monoclonal antibody (MAb) approved by the US Food and Drug Administration for colorectal carcinoma and head and neck cancers that has also shown clinical benefit toward advanced/metastatic gastric adenocarcinoma in combination with other chemotherapeutic agents as a first-line treatment.5–7
Researchers have been interested in exploring anticancer formulations in the form of nanomedicines, either as a combination of multiple drugs loaded into nanocarriers (combinatorial nanomedicines) or as drug-loaded nanocarriers that are actively targeted to specific overexpressed surface receptors such as EGFR (targeted nanomedicines) to improve the anticancer potential.8,9 Alternative cancer nanomedicines following molecularly targeted strategies have been developed to enhance the therapeutic efficiency of conventional chemotherapeutics. Our research group has previously reported the development of CET (MAb)-conjugated docetaxel (DOCT)-loaded poly(γ-glutamic acid) (γ-PGA) nanoparticles (Nps) and tested their in vitro targeting and therapeutic efficacy in EGFR +ve colorectal (HT-29) and A549 (non-small cell lung carcinoma) cell lines.10,11 These targeted Nps showed EGFR-specific cellular accumulation, thereby enhancing the availability of potent chemo drug DOCT to cause enhanced cancer cell death.
This study analyzed the potential of targeted Nps (CET MAb-DOCT-γ-PGA Nps) as an anticancer agent in vitro toward EGFR-overexpressing gastric carcinoma cells and in vivo in EGFR +ve MKN-28 cells-based gastric cancer xenograft in nude mouse models. Thus, CET MAb-conjugated γ-PGA Nps loaded with DOCT were found to be an effective targeted nanoformulation for EGFR-overexpressing gastric cancers.
Materials and methods
Materials
γ-PGA (molecular weight 400 kDa) was purchased from Vedan (Taichung, Taiwan), chitosan (molecular weight 100–150 kDa) from Koyo Chemical Co., Ltd. (Itami, Hyogo, Japan) and DOCT from AK Scientific, Inc. (Union City, CA, USA). Propidium iodide (PI) and RNAase for cell cycle analysis and fluorescein isothiocyanate (FITC) were purchased from Sigma-Aldrich (St Louis, MO, USA). CET (Erbitux) was obtained from Merck. Alexa fluor (AF)-647–conjugated anti-EGFR antibody (AF-647-anti-EGFR antibody) was purchased from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). The cell lines used for the study, namely, MKN-28 (human gastric carcinoma cells), were obtained from Dr Bruno Sarmento at Instituto de Engenharia Biomédica in Portugal (purchased from Japanese Collection of Research Bioresources Cell Bank Japan). RPMI medium for culturing MKN-28 cells was purchased from Sigma-Aldrich.
Preparation and characterization of CET MAb-DOCT-γ-PGA Nps
The preparation and characterization of the nontargeted DOCT-γ-PGA Nps and targeted CET MAb-DOCT-γ-PGA Nps were previously reported by our research group.10 Briefly, γ-PGA Nps were prepared through a simple polyionic complexation technique by cross-linking anionic γ-PGA with cationic chitosan during which DOCT was loaded, resulting in nontargeted DOCT-γ-PGA Nps. CET MAb conjugation onto DOCT-γ-PGA Nps was carried out via 1-Ethyl-3-(3-dimethyl aminopropyl)carbodiimide– N-Hydoxysuccinimide (EDC– NSH) chemistry, which resulted in targeted Nps. The developed Nps were characterized for their size distribution by dynamic light scattering (NanoZS Zeta sizer; Malvern Instruments, Malvern, UK) and scanning electron microscopy (JEOL JSM-6490LA), surface charge by zeta potential measurements (NanoZS Zeta sizer, Malvern Instruments) and the potential interaction between the components by Fourier Transform Infrared Spectroscopy (Perkin Elmer, Waltham, MA, USA). The amount of CET MAb conjugated to the CET MAb-DOCT-γ-PGA Nps was quantified using the bicinchoninic acid (BCA) assay following the protocol using the Pierce™ BCA Protein Assay Kit from Thermo Scientific (Waltham, MA, USA).10 High-performance liquid chromatography (HPLC) quantified the percentage of DOCT encapsulation and loading, along with the release kinetics of DOCT from the nanomatrix.
In silico modeling studies
Structure generation of γ-PGA Nps
Chemical structures of γ-PGA Nps formed via chitosan cross-linking were prepared using Chem BioUltra 11.0 software. γ-PGA Nps were prepared by the polyionic cross-linking reaction between cationic chitosan and anionic γ-PGA.10,12,13 DOCT drug interaction studies were analyzed by the formation of three-dimensional (3D) structures. Chitosan acts as an ionic cross-linking agent that folds and assembles the γ-PGA of different strands in spherical form, which mimics the experimental Nps. The 3D polymeric systems were optimized to the most stable structure using the MMF94 force field implemented in the software.
Molecular docking studies
Molecular docking studies of γ-PGA Nps and DOCT drug interaction studies were analyzed by AutoDock Vina software running on the Linux platform by calculating the grid maps, by adapting to the nature of the input systems, cluster and rank of different docked poses of the ligand in a straightforward manner. Binding free energy (BE) between hydrophobic drug DOCT and γ-PGA Nps was computed and visualized using PyMol software. Furthermore, the crystal structure of the CET MAb with PDB code 1YY814 was used for docking studies with the DOCT-γ-PGA Np complex systems to obtain the BE. The resulting CET MAb-conjugated DOCT-γ-PGA Np complex system was further analyzed by PyMol software.
AutoDock Tools assigned all of the atoms, and Gasteiger charges were added to prepare γ-PGA and chitosan polymer structure, which was further loaded in the AutoDock 4.0 program15 and saved in .pdbqt format. AutoDock Vina software performed blind docking by loading the system in the workspace and grid panel; the grid box option was opted, and then grid dimensions and spacing were set to completely occupy the entire γ-PGA Np surface.16 The γ-PGA Np surface was searched for possible drug binding to obtain its stable complex systems. Furthermore, using the same docking protocol, CET MAb was docked to the entire surface of the DOCT-γ-PGA Np complexes to obtain the antibody affinity toward the Np complex system.
In vitro targeting efficiency
In vitro evaluation was performed in the human gastric adenocarcinoma cellline MKN-28 which was cultured using RPMI media. Expression of EGFR by MKN-28 cells was analyzed using AF-647-anti-EGFR antibody (Santa Cruz Biotechnology Inc.) as reported earlier.11 Cells bound to the AF-647-anti-EGFR antibody were analyzed by flow cytometer FACS Aria II (Beckton Dickinson, San Jose, CA, USA) with an excitation of 633 nm and confirmed using confocal microscopy.17,18 The binding efficiency of FITC-labeled Nps to the EGFR +ve MKN-28 cancer cells was quantified using flow cytometry, which was performed to evaluate the in vitro targeting efficiency of CET MAb-DOCT-γ-PGA Nps. The procedure performed was mentioned in our previous article.11 MKN-28 cells were treated with 0.1 mg/mL of CET MAb-FITC-γ-PGA Nps and FITC-γ-PGA Nps for 1 h with and without CET MAb pretreatment for confirming the targeting efficiency of CET MAb-γ-PGA Nps. Following the treatment, fluorescence intensity emitted from the cells bound with Nps was measured using flow cytometer. The flow cytometer measured the fluorescence intensity after excitation with a 488 nm argon laser using a FACS Aria II. Nontargeted Nps (FITC-γ-PGA Nps) were used as a control.11 The experiment was conducted in triplicate (n=3) for checking the consistency.
In vitro cellular internalization of targeted CET MAb-γ-PGA Nps by EGFR binding was qualitatively analyzed by confocal microscopic examination. 4% paraformaldehyde (PFA)-fixed MKN-28 cells incubated with CET MAb-FITC-γ-PGA Nps (0.1 mg/mL) for 6 h were mounted on glass slides using DPX and viewed under a confocal microscope (Leica SP 5 II).10,11
In vitro cell cycle analysis
Cell cycle analysis was performed by measuring the DNA content in each phase using PI stain as reported previously.10,11 The percentage of MKN-28 cells in each cell cycle phase (G0/G1, S, G2/M, apoptotic and dead cells) was analyzed by a flow cytometer19 following 24 h treatment with DOCT, DOCT-γ-PGA Nps and CET MAb-DOCT-γ-PGA Nps (0.25 mg/mL). The experiment was conducted in triplicate (n=3) for checking the consistency.
In vitro cytotoxicity evaluation
The cytotoxic potential of free DOCT, DOCT-γ-PGA Nps and CET MAb-DOCT-γ-PGA Nps against EGFR +ve MKN-28 cancer cells was evaluated by MTT assay following a reported protocol.10,11,20 The assay protocol involved incubation of the sample-treated cells with MTT reagent for 4 h, solubilization of the formazan crystals and measurement of the absorbance at 570 nm using a Beckmann Coulter ELISA plate reader (BioTek Power Wave XS). Cell viability was calculated relative to the absorbance obtained from the negative control or cells alone:
|
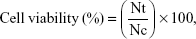
where Nt is the absorbance of the sample-treated cells and Nc is the absorbance of the untreated cells.
The efficiency of these targeted Nps to induce EGFR-specific cancer cell death was further confirmed via flow cytometry analysis in terms of mitochondrial membrane potential depletion and apoptosis induction. The assay procedure was reported previously.10,11 Flow cytometric assay based on 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1; BD Biosciences, San Diego, CA, USA) was performed to evaluate mitochondrial membrane potential (Δψm)21 and apoptosis11,22 from Annexin V/PI assay (Molecular probes, Eugene, OR, USA) according to the instruction manual. The experiment was conducted in triplicate (n=3) for checking the consistency.
In vivo studies
In vivo experiments were carried out following the approved protocols for the pharmacokinetic and organ distribution analysis of DOCT. Male Swiss Albino mice aged 4–6 weeks and weighing 20 g were obtained from the small animal housing facility and all animals were maintained according to the Guide for the Care and Use of Laboratory Animals provided by the Committee for the Purpose of Control and Supervisor of Experiments on Animals. The procedures and protocols used for conducting in vivo evaluations were approved by the Institutional Animal Ethical Committee (Reference No IAEC/2013/3/3) conducted at the Amrita Institute of Medical Sciences and Research Center under Amrita University, Kochi, India.
The efficiency of the developed targeted nanoformulation was evaluated by performing pharmacodynamic analysis in a gastric cancer xenograft model in athymic (nu/nu-ncr) male Balb/c mice aged 5–6 weeks and weighing 20–25 g. Animal experiments were conducted in compliance with relevant institutional laws and ethical guidelines of Chonnam National University Medical School and Chonnam National University Hwasun Hospital in South Korea, and the procedures were approved by the Institutional Animal Ethical Committee (CNU IACUC-H-2015–47). Mice were housed in a laminar flow caging system (Thoren Caging Systems, Inc., Hazleton, PA, USA), and the food, bedding and water given to the animals were autoclaved. Three test groups and one control group were subjected to intravenous injection as follows free DOCT (group 1, n=4); nontargeted, DOCT-γ-PGA Nps (group 2, n=4); targeted, CET MAb-DOCT-γ-PGA Nps (group 3, n=4) and finally sterile saline (control, n=4). The dose of administration with respect to the pre-existing reported preclinical studies was fixed at 10 mg/kg of DOCT.23–25
In vivo pharmacokinetics and organ distribution studies
Pharmacokinetics of the prepared Nps in Swiss albino mice was performed following ethically approved protocols in four animals in each group (n=4) and four animals in the control group. Samples at a dose of 10 mg/kg of DOCT (reconstituted as prescribed26 using ethanol/polysorbate 80/saline) and Nps (with an equivalent dose of DOCT) in sterile 0.9% saline were injected intravenously through the lateral tail vein. At specific time points, blood was drawn through the retro-orbital sinuses to Acid Citrate Dextrose vials. The blood samples were centrifuged at 4,000 rpm for 5 min to collect the plasma. The plasma samples were processed for protein precipitation using 150 mM ammonium acetate in methanol, followed by incubation at −20°C overnight. For organ distribution studies, the organs (brain, heart, lungs, liver, kidney and spleen) were excised after the animals were euthanized, washed in saline, dried in blotting paper and then weighed. The organs were homogenized in 0.9% saline (1 mL/g of organ), which was followed by protein precipitation. The resulting samples were centrifuged (8,000 rpm, 15 min) and the supernatants were collected, which were further analyzed using HPLC with the calibrated protocols. The procedures were also performed for the control group treated with saline to remove background signals from the HPLC chromatogram. The concentration of DOCT from the plasma samples at each time point was calculated using the calibration graph based on the area under the curve (AUC) values. Pharmacokinetic parameters such as concentration maximum (Cmax), mean resident time (MRT), AUC, time of maximum concentration (Tmax), elimination rate constant (Kel) and half-life (t1/2) were determined using the Microsoft add-in tool, PK solver.
Development of gastric cancer xenografts
The in vivo targeting and therapeutic efficacy of the CET MAb-targeted Nps were evaluated in EGFR +ve MKN-28 cell-based gastric cancer xenografts. MKN-28 cells were maintained in RPMI medium as previously described.27–29 Development of MKN-28 tumor xenografts followed Matrigel® (Corning Lifesciences, Corning, NY, USA)-based tumor formation as per the reported protocol. The MKN-28 gastric cancer xenografts were developed by subcutaneous injection of 2×106 cells/100 μL of sterile PBS with 50 μL of Matrigel on the right flank of the male nude mice. Tumors were allowed to reach sufficient volume, and the tumor volumes were calculated as:
|

Qualitative in vivo targeting using in vivo optical imaging
To analyze the distribution pattern of the prepared Nps, a hydrophobic near infrared (NIR) dye IR780 was loaded following a similar protocol to that described previously for loading DOCT and FITC. IR780-loaded targeted (CET MAb-IR780-γ-PGA Nps) and nontargeted (IR780-γ-PGA Nps) Nps at a dose equal to that of the dose used for verifying the pharmacokinetics and therapeutic efficiency (Nps containing 10 mg/kg of DOCT) dispersed in saline were administered intravenously through the lateral tail vein of MKN-28 tumor-bearing nude mice. A single dose was given, and imaging using a fluorescence-labeled organism bioimaging instrument (fluorescence-labeled organism bioimaging instrument [FOBI]; NEO science, Gyeonggi, Korea) was performed at different time points starting from 15 min postinjection.
In vivo DOCT quantification
The targeting potential of CET MAb-DOCT-γ-PGA Nps for gastric cancer was analyzed in the MKN-28 xenograft gastric cancer model in nude mice by quantifying the amount of DOCT from the tumors and other organs posttreatment using HPLC. Three groups with four animals each were tested at two time points (shorter duration: day 1 and longer duration: day 4): group 1: free DOCT (n=4); group 2: nontargeted DOCT-γ-PGA Nps (n=4) and group 3: targeted CET MAb-DOCT-γ-PGA Nps (n=4). Samples at a dose of 10 mg/kg of DOCT were administered intravenously via the tail vein to mice bearing tumors with an average tumor volume of 100±12 mm3 and body weights of around 22–25 g. Four mice were injected with sterile 0.9% saline as untreated controls. At 1 and 4 days postinjection, mice from each group were euthanized; ~1 mL of blood was removed by cardiac puncture into a vacutainer and centrifuged (4,500 rpm for 5 min) to collect the plasma. The organs were isolated from the body, cleaned in saline, dried and homogenized in saline. Then, 150 mM ammonium acetate in methanol was added at a ratio of 1:4 to plasma and 1:2.5 to the organ homogenates, vortexed well for a minute and then stored overnight at −20°C for extracting the drug and precipitating out the proteins. After overnight incubation, the samples were thawed, vortexed again and centrifuged (8,000 rpm for 15 min). Supernatants with the extracted DOCT were given to Chonnam Medical hospital for HPLC.
In vivo anticancer efficacy in gastric cancer xenografts
The anticancer potential of CET MAb-DOCT-γ-PGA Nps for gastric cancer was analyzed in the MKN-28 xenograft gastric cancer model in nude mice by measuring the tumor volume and tumor weight posttreatment.23,30,31 The four groups tested were: group 1: control saline (n=4); group 2: free DOCT (n=4); group 3: nontargeted DOCT-γ-PGA Nps (n=4) and group 4: targeted CET MAb-DOCT-γ-PGA Nps (n=4). Three doses (10 mg/kg of DOCT and Nps with equivalent DOCT) were administered intravenously for 3 weeks (1 dose/week) to mice bearing tumors of an average volume of 150 mm3. Tumor volume was measured with Vernier calipers every 2 days for a period of 20 days. The animals were euthanized after 20 days of treatment, and the tumors were excised, weighed and photographed.
Statistical analysis
All results were obtained from a minimum of triplicate samples. Data are expressed as mean ± standard deviation. Student’s two-tailed t-tests were used for comparing the in vitro effect of targeted Nps and nontargeted Nps on EGFR +ve cancer cells. One-way analysis of variance was utilized to compare differences among free DOCT and nontargeted and targeted Nps in tumor accumulation, pharmacokinetics and therapeutic efficacy in the in vivo models. Values representing p≤0.05 were considered to be statistically significant.
Results
Preparation and characterization of CET MAb-DOCT-γ-PGA Nps
Our research group previously reported the development of CET MAb-targeted Nps, which were found to be effective in targeted therapy toward EGFR +ve colon and lung cancer cell lines in vitro.10 The Np synthesis followed the technique of ionic gelation for nontargeted Nps and further EDC–NHS chemistry for targeted Nps. DOCT-γ-PGA Nps were synthesized by ionic gelation of anionic γ-PGA using the polycationic chitosan as a cross-linker, during which DOCT was loaded within the nanomatrix. CET MAb was conjugated through EDC–NHS chemistry between the free carboxylic groups of the Nps and the amine group from the antibody. Targeted CET MAb-DOCT-γ-PGA Nps (200±20 nm) and nontargeted DOCT-γ-PGA Nps (110±40 nm) were synthesized and were found to have −28±8 and −17±5 mV zeta potential values, respectively. Around 42% of CET MAb conjugation efficiency was observed from the BCA assay, and the developed nanoformulation exhibited controlled release of DOCT.10,11
In silico modeling studies
In silico studies were performed using γ-PGA and chitosan structures generated using ChemDraw software. The polymeric system containing 18 γ-PGA units cross-linked with six chitosan units was generated and is presented in Figure 1A. For molecular docking studies, this molecular system was tested as an optimal Np system with the aim of reducing the computational time and cost, and the putative binding sites were also identified for DOCT within the γ-PGA Nps. The BE between γ-PGA Nps and DOCT was −6.2 kcal/mol.
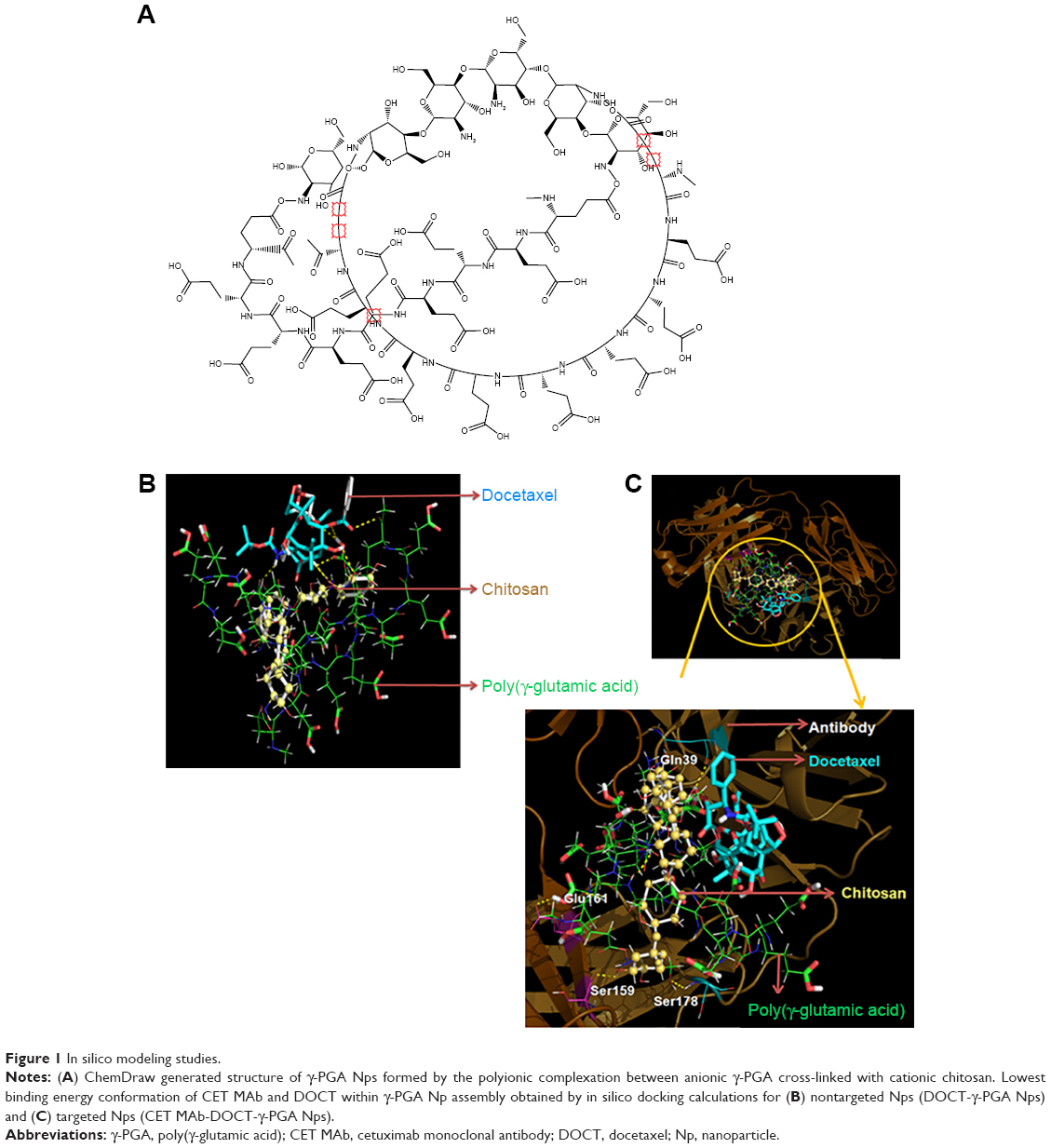 | Figure 1 In silico modeling studies. |
Furthermore, DOCT-γ-PGA Np complexes were docked with the CET MAb antibody using the same protocol. The 3D crystal structure of the antigen binding (Fab) fragment of CET MAb was solved at 2.8 Å resolution with PDB code 1YY8.14 This 3D structure was used for docking studies to understand the antibody-Np atomic level interactions. Figure 1B represents the physical interactions between DOCT and γ-PGA Nps, as analyzed from the atomic level binding using PyMol software. Different hydrogen bonding interactions between DOCT and those of γ-PGA and linker chitosan (γ-PGA Nps) are depicted as yellow dotted lines. Furthermore, CET MAb-targeted atomic level binding toward DOCT-γ-PGA Nps is presented in Figure 1B. We observed that targeted CET MAb binds directly to the γ-PGA Np system, and the possible hydrogen bonding formation is shown as yellow dotted lines (Figure 1C) and is tabulated in Table 1. As the residue Ser178 has an amide group, it also mediated hydrogen bonding with the PGA ligand in the DOCT-γ-PGA Np system. BE between DOCT-γ-PGA Nps and CET MAb was −10.6 kcal/mol.
 | Table 1 Molecular interactions between CET MAb and the Np system |
In vitro targeting efficiency
Figure 2A represents the histograms obtained from flow cytometry and the images from confocal microscopic analysis of MKN-28 cells treated with anti-EGFR antibody (AF-647). Histograms suggest that 90%±2% of cancer cells exhibited anti-EGFR antibody binding, which was qualitatively confirmed from red fluorescence observed on the cells using confocal microscopy. The targeting ability of CET MAb-γ-PGA Nps against cancer cells was evaluated from the binding of FITC-labeled targeted Nps (CET MAb-FITC-γ-PGA Nps) by MKN-28 cells in comparison to that of nontargeted (FITC-γ-PGA Nps) Nps by flow cytometry. It was observed that EGFR +ve MKN-28 cells (89%±2%) showed increased antibody binding compared with that of FITC-γ-PGA Nps (48%±4%), as revealed by the representative flow cytometry histograms (Figure 2B) based on the fluorescence intensity. CET MAb pretreatment may have utilized the available EGFR, as a result of which, the percentage of MKN-28 cells bound to the FITC-CET MAb-γ-PGA Nps was significantly reduced (22%±2%). This was not observed in the nontargeted Nps (42%±5%) group. The study indicated EGFR-specific binding of the Nps.32
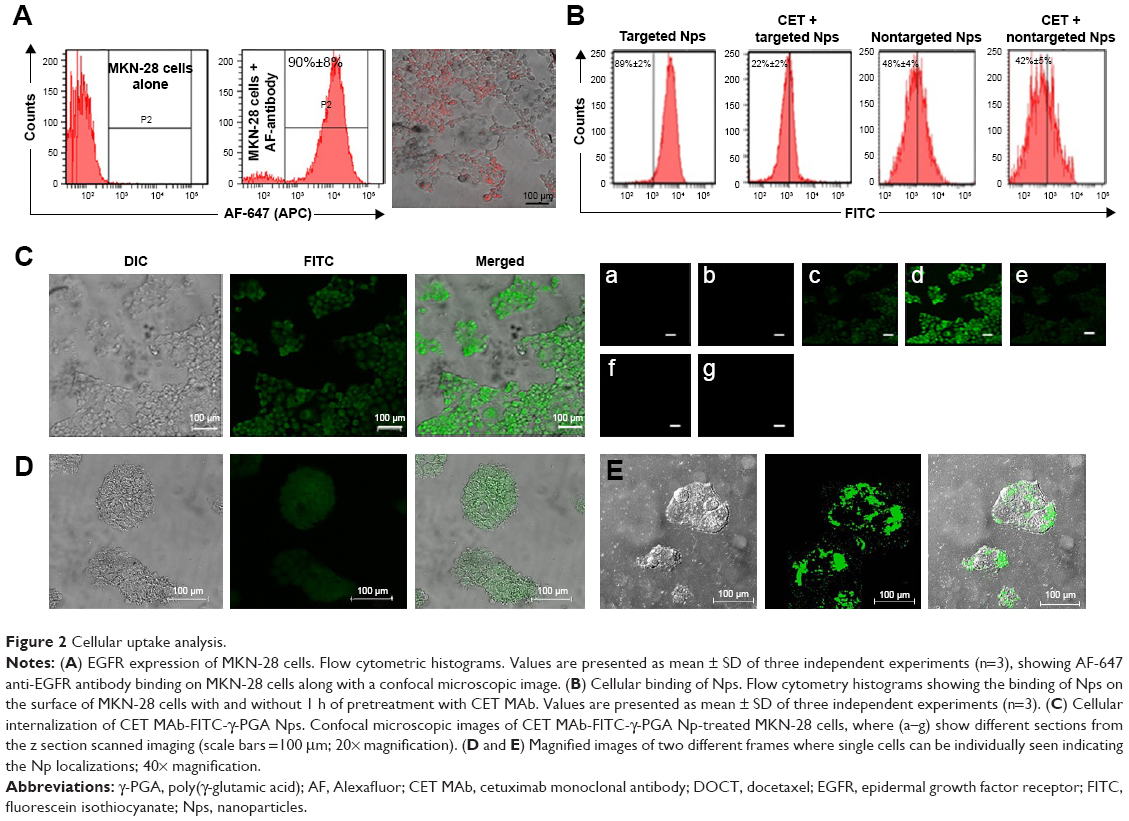 | Figure 2 Cellular uptake analysis. |
In vitro cellular internalization
The targeting ability of CET MAb-γ-PGA Nps was studied by the above experiment, where it was found that CET MAb-conjugated targeted Nps bind to a higher number of EGFR +ve cancer cells. Confocal microscopy was used for the qualitative analysis in order to determine whether these targeted Nps were only bound on the surface or whether they were being taken up into the intracellular compartments (Figure 2C). The confocal microscopic images showed enhanced fluorescence intensity in the intracellular sections of MKN-28, indicating the targeting and internalization ability of CET MAb-γ-PGA Nps. The Z-scan images (Figure 2Ca–g) tracked the high fluorescence signal of FITC-CET MAb-γ-PGA Nps in the internal cell sections, when the optical slices were scanned from top to bottom. Figure 2D, E shows magnified area (60×) of single cells for better understanding of Np localization within the cancer cells. This, in turn, indicated the intracellular localization of FITC-CET MAb-γ-PGA Nps via EGFR-specific receptor-mediated uptake by EGFR +ve cancer cells.33,34
In vitro cell cycle analysis
Flow cytometry analysis was used to test the targeted and nontargeted Np effects on the cell cycle progression following 24 h treatment of CET MAb-DOCT-γ-PGA Nps and DOCT-γ-PGA Nps and to compare them with those of free DOCT. Figure 3A represents the flow cytometry analyses of different phases of the cell cycle. It was observed that DOCT treatment arrested the cells in the mitotic stage, resulting in a peak in the P5 (G2/M) population. DOCT is a microtubule destabilizer; as a result, the cells will be arrested in the G2/M phase and progress to apoptosis or necrosis.35,36 All three formulations, free DOCT and nontargeted and targeted Nps, followed the same pattern by accumulating the cells in the G2/M phase, indicating the presence of the active form of DOCT even after nanoencapsulation. The G2/M phase population was higher in the case of CET MAb-DOCT-γ-PGA Nps (39.7%±1.6%) compared with that of DOCT-γ-PGA Nps (12.8%±1.1%). The G2/M phase cell population was higher for EGFR +ve MKN-28 cells treated with CET MAb-DOCT-γ-PGA Nps compared with DOCT-γ-PGA Np treatment, indicating that targeted Nps induced G2/M phase arrest followed by cancer cell death.
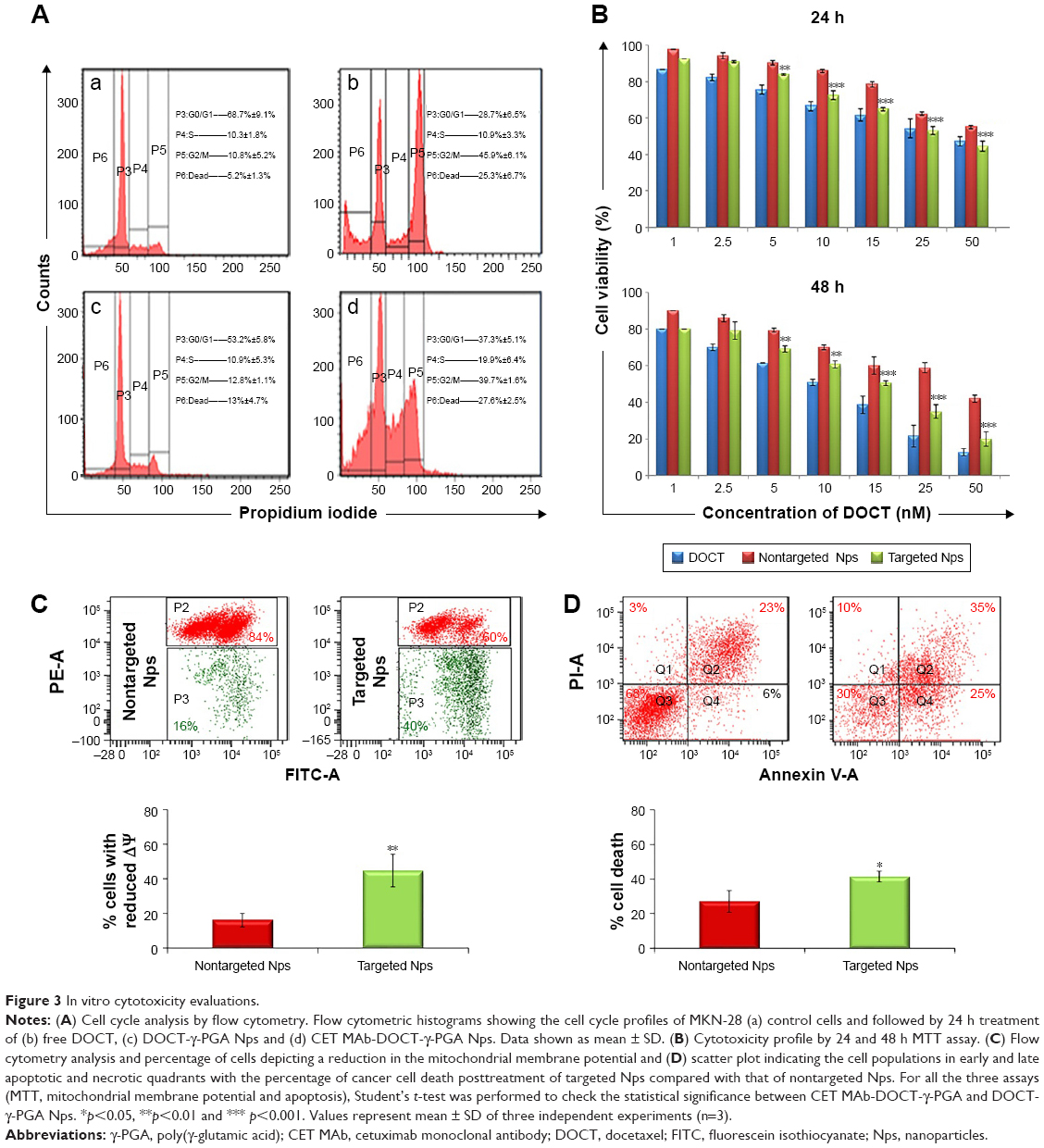 | Figure 3 In vitro cytotoxicity evaluations. |
In vitro cytotoxicity evaluations
The cytotoxicity effects of free DOCT, DOCT-γ-PGA Nps and CET MAb-γ-DOCT-γ-PGA Nps were quantified by MTT assay on MKN-28 cells. Cell viability profiles of MKN-28 for 24 and 48 h (Figure 3B), as determined by MTT assay, exhibited time- and dose-dependent cytotoxicity in all three drug formulations (free DOCT, DOCT-γ-PGA Nps and CET MAb-DOCT-γ-PGA Nps). Targeted CET MAb-γ-DOCT-γ-PGA Nps demonstrated significant toxicity potential in achieving lower cell viability compared with that of nontargeted DOCT-γ-PGA Nps in EGFR +ve MKN-28 cells. The IC50 value of CET MAb-DOCT-γ-PGA Nps (33.39±6.85 nM) was reduced for DOCT-γ-PGA Nps (56.9±3.25 nM) upon 24 h treatment and was three times lower upon 48 h treatment (from 36.81±0.194 to 13.11±2.88 nM). It was reported that DOCT can disturb the mitochondrial pathway, and apoptosis is featured by deregulated mitochondrial membrane depolarization.37 Flow cytometric analysis of apoptosis and mitochondrial membrane potential was conducted using JC-1 and Annexin V/PI assays, respectively. The flow cytometry scatter plot and corresponding graphical representation (Figure 3C) indicated that targeted CET MAb-DOCT-γ-PGA Nps disrupted ΔΨm in significantly higher percentages compared with those of nontargeted DOCT-γ-PGA Nps. The apoptosis or necrosis stage of cells treated with CET MAb-DOCT-γ-PGA Nps and DOCT-γ-PGA Nps has been represented in Figure 3D, along with the percentage of dead cells upon Np treatment. The apoptotic profiles also indicated higher cell death in the case of targeted Nps, compared with those of nontargeted samples. Thus, the results referred to EGFR-specific uptake of targeted Nps by MKN-28 cells, resulting in mitochondrial membrane potential disruption leading to apoptosis in a higher number of cells compared with that by nontargeted Nps, thereby enhancing cancer cell death.
In vivo pharmacokinetics and organ distribution studies
The pharmacokinetics and biodistribution profile of free DOCT, DOCT-γ-PGA Nps and CET MAb-DOCT-γ-PGA Nps are represented in Figure 4. The maximum plasma concentration (Cmax) of free DOCT and the nanoformulations (nontargeted and targeted Nps) with their corresponding peak time (Tmax) were analyzed from the plasma concentration–time profiles of the drug. The plasma concentrations of DOCT over the time course of 1 week are plotted in Figure 4A, which exhibited remarkable differences between free DOCT and nanoformulated DOCT. The maximum concentration of DOCT was obtained from the initial time point of 5–10 min postadministration of free DOCT (Cmax: 10.026±0.973 μg/mL) with a steep decline within 1–2 h and it was completely eliminated in 8–10 h. On the other hand, nanoformulated DOCT (both DOCT-γ-PGA Nps and CET MAb-DOCT-γ-PGA Nps) followed a different pattern; maximum DOCT concentration was observed at 24 h (nontargeted Nps, Cmax: 6.75±1.3 μg/mL and targeted Nps, Cmax: 7.09±0.509 μg/mL) and existed until day 4, but in very low concentrations. This pattern is pointing toward the slow release of DOCT from the Nps at a physiologic pH of 7.4. We could not observe any significant difference between nontargeted and targeted Np behavior. The noticeable difference between free DOCT and the nanoformulations occurred within the time period during which the DOCT dose was sustained with in the therapeutic range.23,38,39
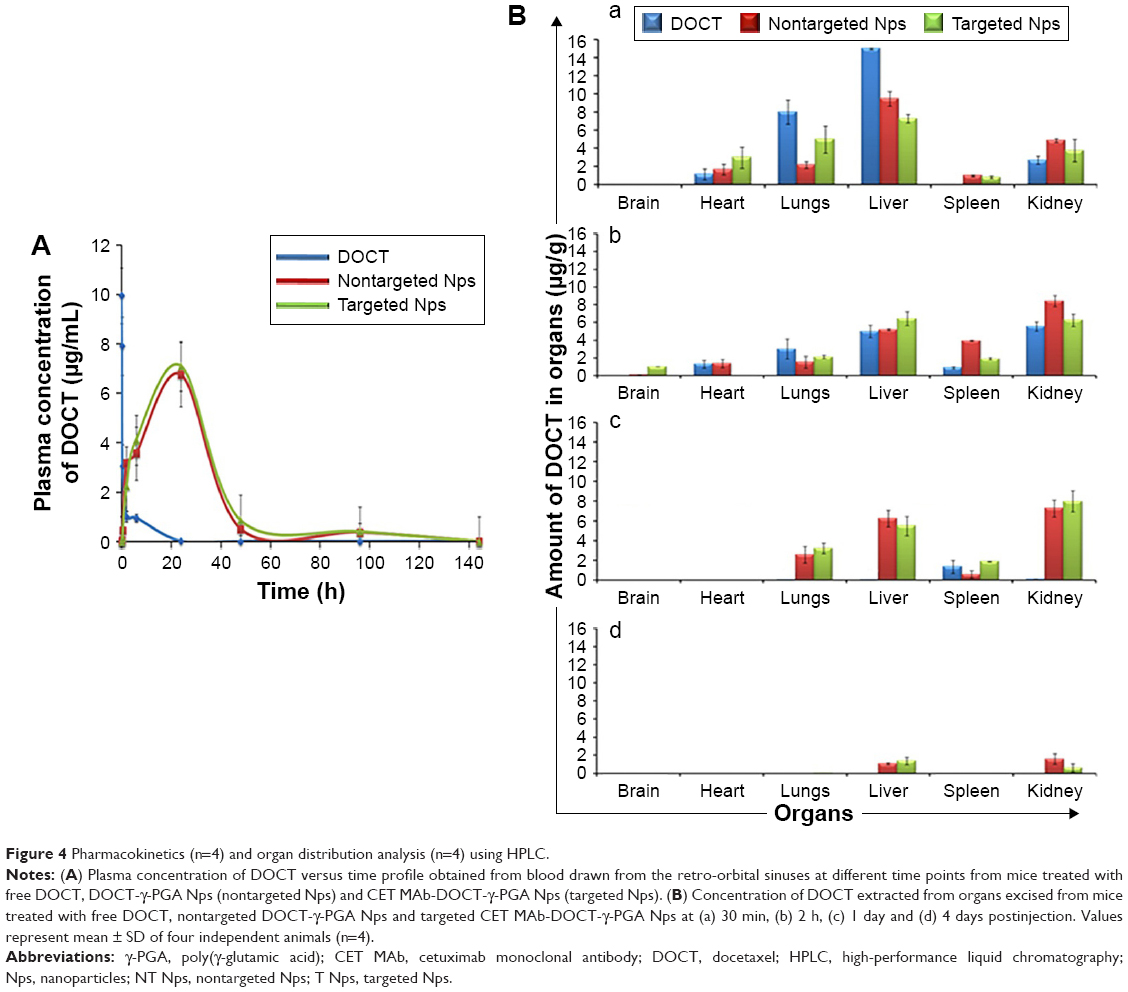 | Figure 4 Pharmacokinetics (n=4) and organ distribution analysis (n=4) using HPLC. |
The biodistributions (Figure 4B) of free DOCT, nontargeted DOCT-γ-PGA Nps and targeted CET MAb-DOCT-γ-PGA Nps were analyzed at different time points (30 min, 2 h, 24 h and 4 days) postadministration by extracting DOCT and quantified using HPLC from various organs (brain, heart, lungs, liver, spleen and kidney). All three drug formulations followed the same pattern of distribution. However, the accumulation and elimination times were different between free and nanoformulated DOCT. Free DOCT showed an earlier liver accumulation (30 min) compared with that of Nps (Figure 4Ba). Nps accumulated in highly perfused organs such as the heart and lungs. By 2 h (Figure 4Bb), all three drug formulations followed a similar accumulation pattern, and by 24 h (Figure 4Bc), free DOCT became undetectable. At the end of day 4 (Figure 4Bd), nanograms of DOCT were quantified from the livers and kidneys of mice treated with Nps.
Development of gastric cancer xenografts
Gastric cancer xenografts were developed following reported protocols with modifications by subcutaneous implantation of EGFR +ve MKN-28 gastric cancer cell lines.28,29,40 Different trials have been performed by varying the cell density to obtain optimized tumor growth, which could be used for further studies. Finally, Matrigel-based tumor induction followed, which resulted in sustained growth of the tumor, which reached an average size of 180±22 mm3 in 3 weeks. The protocol was confirmed to verify the repeatable tumor formation (Figure 5). Figure 5A represents the time frame indication of the tumor growth with the point of initiation and the degree of tumor growth within 3 weeks. Figure 5B and C represents the tumor burden in all the four mice which have been consistently used for the study.
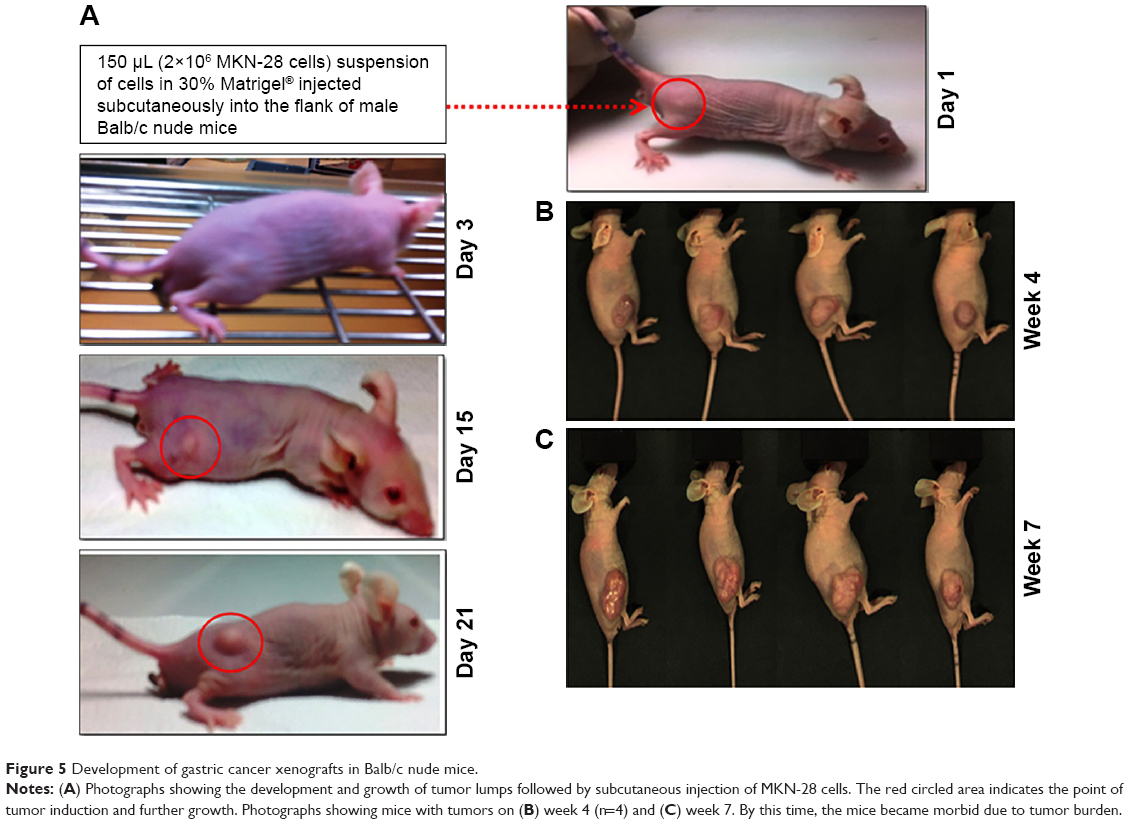 | Figure 5 Development of gastric cancer xenografts in Balb/c nude mice. |
Qualitative in vivo targeting using in vivo optical imaging
The targeting effect of CET MAb-conjugated targeted Nps was initially confirmed by in vivo optical imaging studies, which used NIR dye (IR780) as a tracker for analyzing the distribution of CET MAb-IR780-γ-PGA Nps and IR780-γ-PGA Nps in the gastric cancer xenograft model using the FOBI imaging system. Figure 6 contains the images obtained using FOBI at various time points after sample injection. It was observed that the distribution pattern remained similar for both CET MAb-IR780-γ-PGA Nps and IR780-γ-PGA Nps; the difference was in the intensity of tumor accumulation and the time point of maximum accumulation. From these images, it could be said that the nontargeted Nps (Figure 6A) accumulated nonspecifically in other organs along with tumors, whereas the targeted Nps (Figure 6B) reduced the nonspecific accumulation after 24 h. The accumulation rate of the Nps into the tumors was also enhanced upon targeting. The mean fluorescence intensity based on a region of interest in tumors measured using the FOBI machine was plotted against time (Figure 6C). Even though both targeted and nontargeted Nps present a high level of IR780 fluorescence signal, CET MAb-IR780-γ-PGA Nps induced sustainable exposure throughout the study. Initial time points showed a complete distribution of both samples, and by 24 h, specific and differentiated accumulation in tumors was observed in the case of nontargeted Nps, whereas for targeted Nps, early enhanced accumulation was observed from 6 h onward. The biodistribution study thus pointed toward the circulation and tumor retention of targeted Nps.41,42
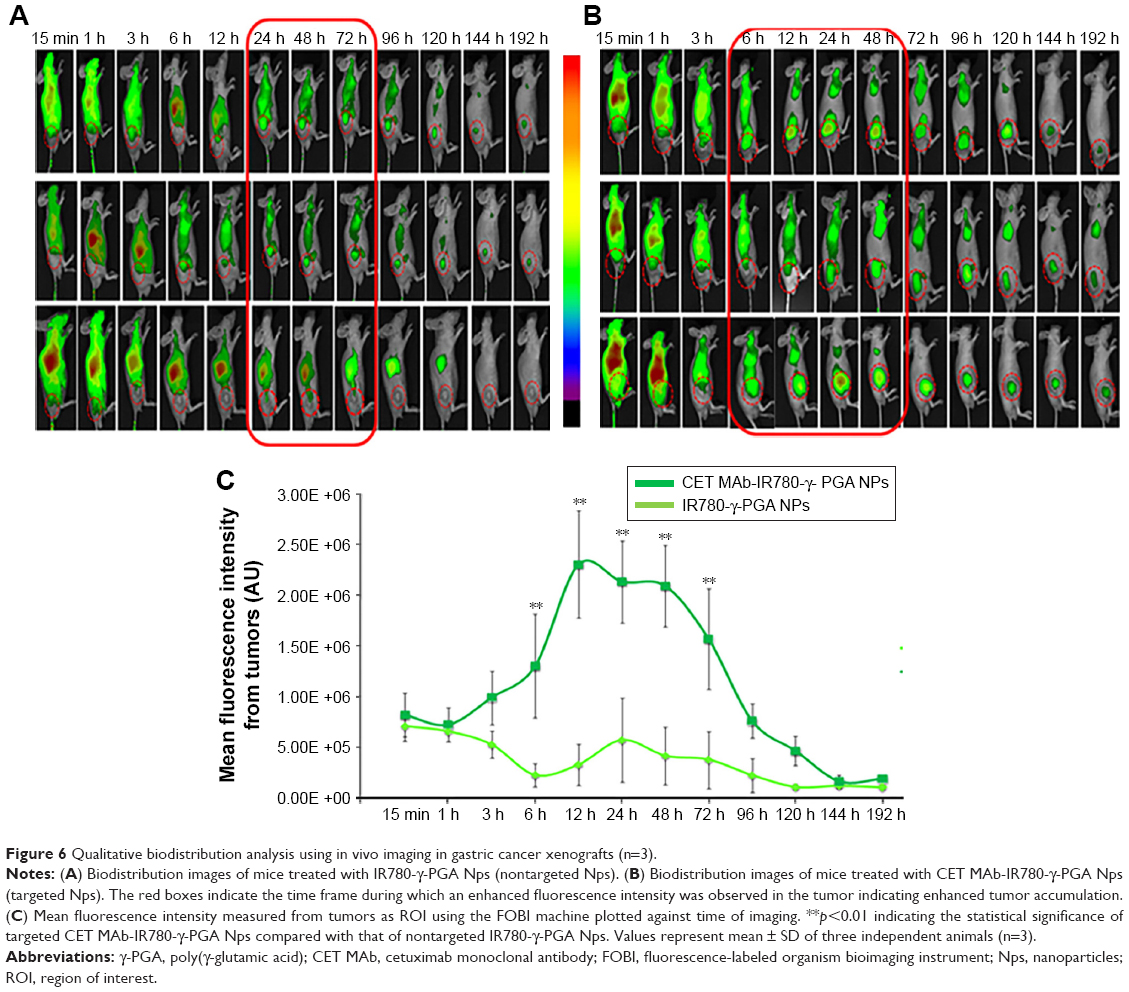 | Figure 6 Qualitative biodistribution analysis using in vivo imaging in gastric cancer xenografts (n=3). |
In vivo DOCT quantification
The targeting potential, which in turn refers to the tumor accumulation capability of CET MAb-DOCT-γ-PGA Nps for gastric cancer, was analyzed in the MKN-28 xenograft gastric cancer model in nude mice by quantifying the amount of DOCT from the tumors and other organs posttreatment using HPLC. The amount of DOCT accumulated per milligram of each organ was calculated from the concentration value obtained using HPLC analysis. From Figure 7A, it is clear that the plasma concentration of DOCT was enhanced significantly after the nanoformulation was injected. Twenty-four hours postinjection, a 20-fold higher concentration of DOCT was found in the plasma of Np-treated mice, compared with that of free DOCT. There was no significant difference between plasma DOCT concentrations of mice treated with nontargeted and targeted Nps. A time-dependent reduction was observed in the plasma DOCT concentration, where DOCT was completely undetected. However, nanoformulated DOCT was also observed in low concentrations in plasma on day 4, even though by day 4, the concentration was considerably reduced, indicating that the Nps were cleared from the body. Figure 7B presents the amount of DOCT extracted from the tumors, where an average concentration of 496±91 ng/mg was observed after 24 h in the case of mice treated with targeted Nps, which is statistically significantly different from that of free DOCT (10.4±0.41 ng/mg) and nontargeted Nps (84±0.52 ng/mg). The DOCT quantified from tumors after 4 days was found to be around 47±9.6 ng/mg for nontargeted Nps and 103±16 ng/mg for targeted Nps. However, the free drug was completely cleared, indicating the retention of nanoformulated DOCT within the tumors. This result indicated that a combination of enhanced permeation and retention (EPR) effect enhancing the accumulation of nanoformulated drug and CET MAb-conjugated targeted Nps improved EGFR-mediated tumor accumulation.42–44
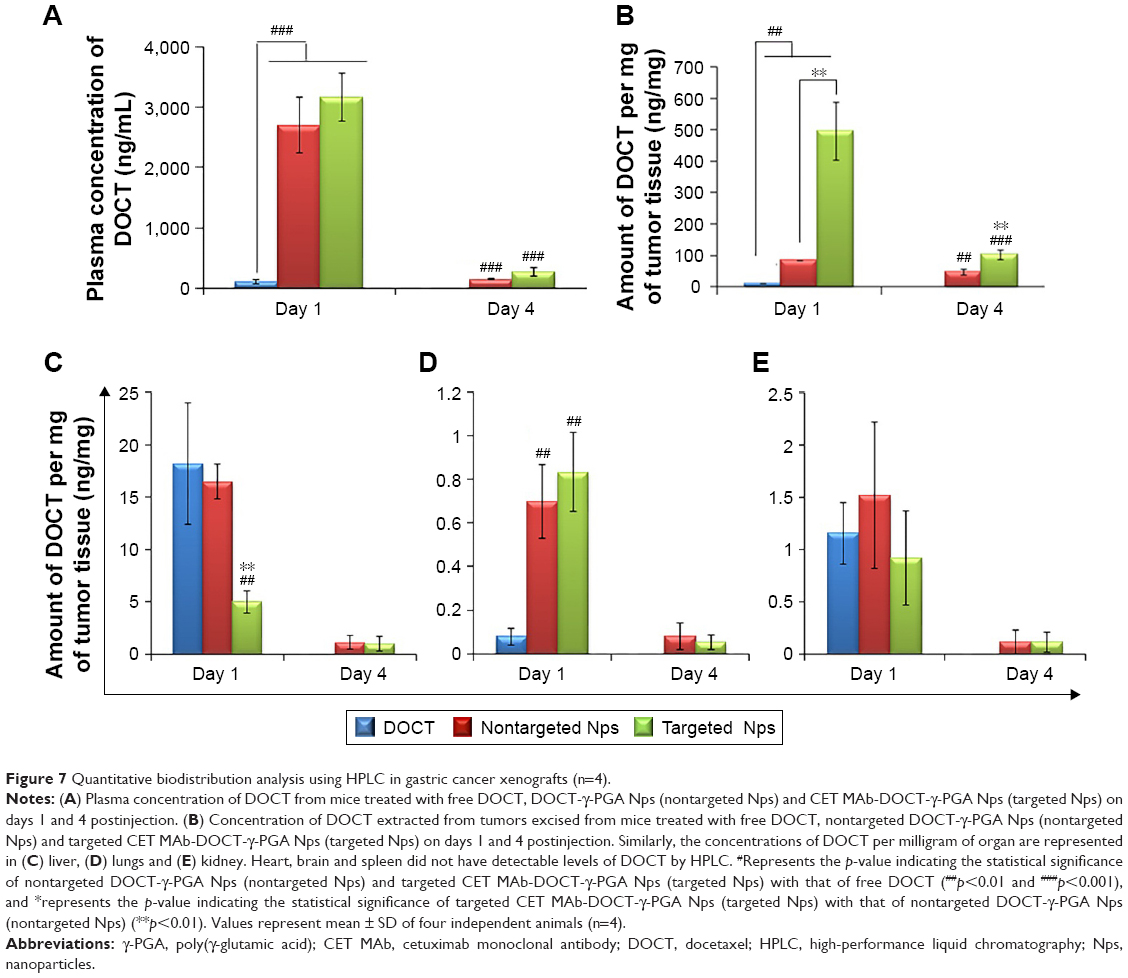 | Figure 7 Quantitative biodistribution analysis using HPLC in gastric cancer xenografts (n=4). |
Additionally, DOCT was quantified in the liver (Figure 7C), lungs (Figure 7D) and kidneys (Figure 7E) to understand the distribution profile. Considering other organs, DOCT was significantly accumulated in the liver, lungs and kidneys of mice treated with free DOCT and nontargeted Nps, suggesting the nonspecific affinity or accumulation of intravenously administered drugs or Nps.41 On the other hand, CET MAb-conjugated targeted Nps reduced the nonspecific accumulation to some extent. This could be due to the vast distribution of targeted Nps in areas where EGFR was expressed, rather than concentrating on specific organs. Thus, quantitative biodistribution analysis of free DOCT, nontargeted DOCT-γ-PGA Nps and targeted CET MAb-DOCT-γ-PGA Nps proved that the availability of the drug to the target tumor tissue was significantly enhanced using CET MAb conjugation, resulting in EGFR-targeted DOCT delivery.
In vivo anticancer efficacy in gastric cancer xenografts
After demonstrating the ability of nanoformulated DOCT to accumulate in tumors, the potential of the same nanoformulations to reduce tumor growth in vivo has been analyzed by evaluating the tumor growth profile of MKN-28 xenograft-bearing nude mice with respect to various treatments. Based on the distribution profile observed in these xenografts from imaging and HPLC studies, it was observed that the Nps circulate for 3–4 days in the animals and then are completely cleared. Thus, multiple doses (three doses) of samples were administered as one dose/week for 20 days of analysis. Figure 8A is a photograph of one mouse representing each of the four (n=4) test groups on the day of treatment initiation with tumors of the hind limb. The tumor volume versus time profile (Figure 8B) showed that the control tumors (mice treated with saline) grew significantly compared with the sample-treated tumors. All mice were euthanized on day 20; the tumors were excised, and their dimensions and weight were measured. Figure 8C presents the photograph of the excised tumors, the corresponding weight (mg) of these tumors is presented in Figure 8D. From Figure 8 B and D it is observed that the nanoformulated DOCT significantly reduced the tumor volume and weight compared to that of free DOCT Targeted Nps were found to significantly prevent tumor growth, compared with free DOCT and nontargeted Nps.45–47
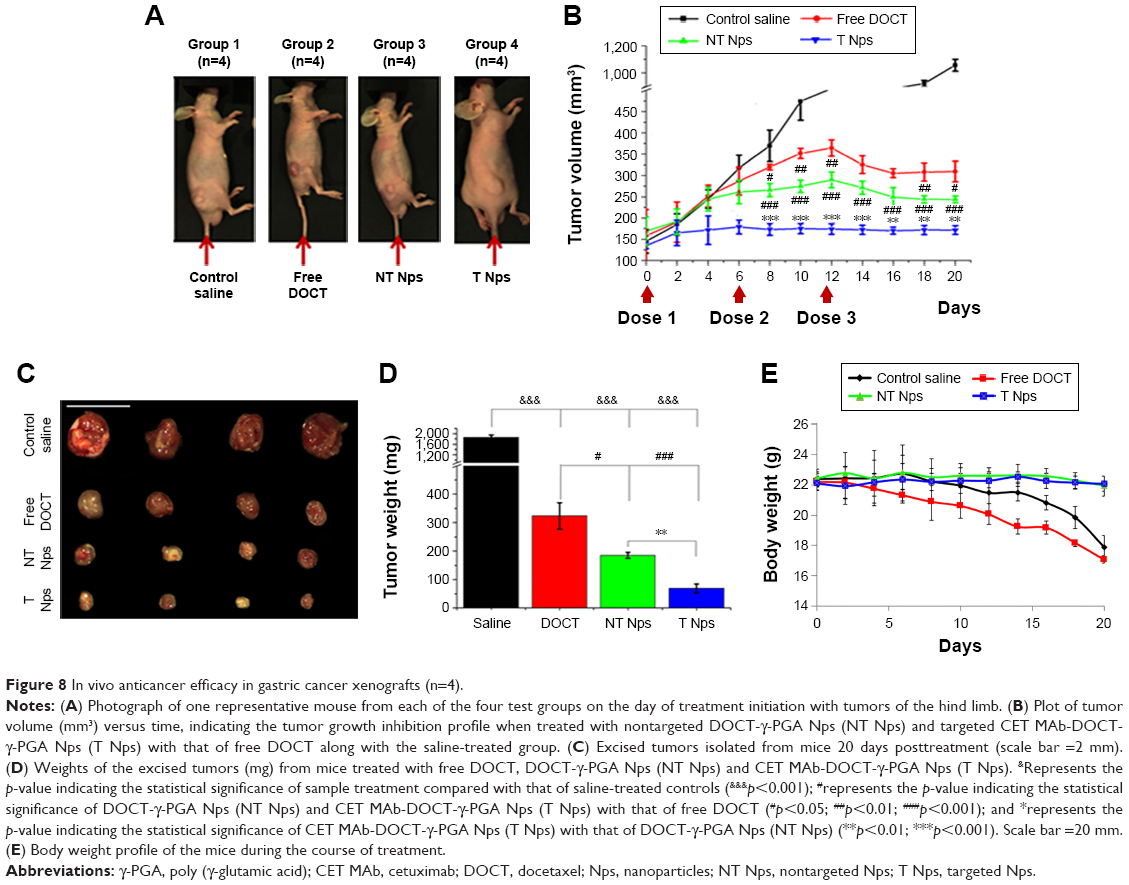 | Figure 8 In vivo anticancer efficacy in gastric cancer xenografts (n=4). |
Preliminary evaluation was done based on the animal behavior and body weight to know if the administered formulations cause any health issues. Figure 8E shows the body weight profile of mice in each group (n=4) during the course of treatment for 20 days. The body weight of mice in the saline-treated controls and free drug-treated groups reduced gradually and the mice became morbid. The Np-treated groups (both nontargeted and targeted) were observed to be healthy enough, maintaining their normal behavior and body weight. Even though the amount of DOCT accumulated in the organs was not differentiated as free form or Np bound form, the formulation did not affect the health of the mice compared to the free drug. This could be due to the fact that nanoencapsulation reduces direct exposure of the tissues to the high toxic concentration of the drug. This observation pointed toward the safety and therapeutic profiles of the nanoformulated DOCT compared to that of free drug formulations.
Discussion
A prerequisite for the development of cancer nanomedicine is its specificity toward the target tumor tissue. Researchers have exploited the ability of passively targeted Nps (EPR effect) and actively targeting (by specific ligands) Nps for enhancing the availability of anticancer drugs specifically at the tumor site, and thereby improving the therapeutic efficacy. This study aimed to evaluate the in vivo fate of previously reported EGFR-targeted Nps by analyzing the pharmacokinetics, biodistribution profile, tumor targeting efficiency and in vivo anticancer effect in EGFR +ve MKN-28-based gastric cancer xenograft models. In silico studies provided considerable insight into the strength, chemical nature and interacting sites of CET MAb at the atomic level and DOCT toward γ-PGA Nps.
Flow cytometric analysis tested the receptor-binding capability of the targeted Nps by using EGFR +ve MKN-28 cells with nontargeted Nps as control and also by competitively blocking the receptors with free antibody. The data clearly prove the specificity of targeted Nps toward EGFR after CET MAb conjugation; furthermore, the binding affinity of CET MAb was not diminished upon conjugation to Nps. This was well correlated with other studies that have reported the development of targeted Nps.32,48–50 Thus, the tumor uptake of these Nps would take place specifically via an EGFR-mediated uptake mechanism. Cell cycle analysis indicated that the EGFR-specific uptake of the targeted CET MAb-DOCT-γ-PGA Nps was more efficient than that of the nontargeted DOCT-γ-PGA Nps by MKN-28 cells, thereby arresting the cells in the G2/M phase, disturbing the process of cell division, and further progressing the cancer cells into the death phase.35,36,51 The in vitro assays demonstrated EGFR-specific uptake of targeted Nps, making DOCT available to the cells and inducing apoptotic cancer cell death. Altogether, the results from MTT assays, cell cycle analysis, mitochondrial membrane potential analysis and apoptosis analysis provide strong support to the idea that CET MAb conjugation could improve the therapeutic potential of Nps for EGFR +ve cancers.52–55
The pharmacokinetic parameters derived for free DOCT, nontargeted DOCT-γ-PGA Nps and targeted CET MAb-DOCT-γ-PGA Nps subsequent to intravenous administration are summarized in Table 2. The rate of drug absorption is indicated by Tmax (peak time); Cmax is the peak plasma concentration systematically indicating sufficient drug absorption to provide an effective therapeutic response. The measure of the extent of absorption or the drug amount in systemic circulation was indicated by AUC0–t. Here, the increased AUC0–t values for both nontargeted (218.62±48.6 μg/mL/h) and targeted (246.25±17.94 μg/mL/h) Nps indicated enhanced systemic availability of the drugs, which was not achieved by the free drug solutions (free DOCT: 19.31±1.32 μg/mL/h). This along with the significantly higher mean resident time values for the Nps collectively suggested an increased and prolonged pharmacokinetic effect when administered intravenously. The enhanced t1/2 and Kel values support the fact that the Nps resulted in longer circulation in vivo.9,56
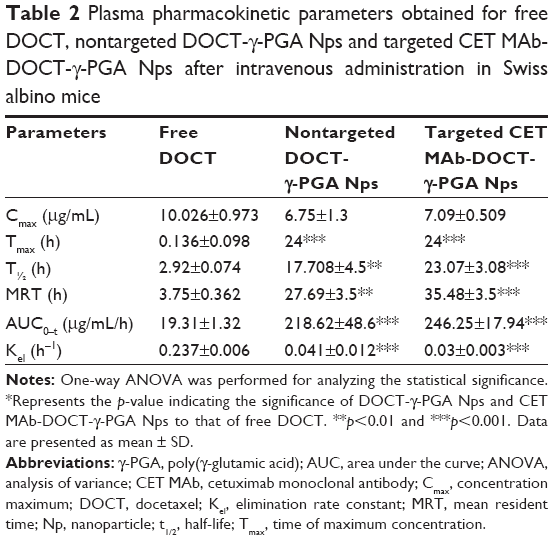 | Table 2 Plasma pharmacokinetic parameters obtained for free DOCT, nontargeted DOCT-γ-PGA Nps and targeted CET MAb-DOCT-γ-PGA Nps after intravenous administration in Swiss albino mice |
The organ distribution data showed early liver accumulation for free DOCT and it was reduced over time, which suggested hepatic metabolism of DOCT. The nanoformulated DOCT was observed to accumulate in all of the tested organs, but at a reduced level compared with that of free DOCT. The presence of DOCT was detected even after 24 h in the organs, indicating the presence of Nps in circulation, which could slowly release the drugs. DOCT accumulation was observed in reticulo endothelial system (RES) organs (liver and spleen), kidneys and in highly perfused organs such as the lungs and heart.25,57 No significant difference in the pattern of distribution between targeted and nontargeted Nps was observed. These nanoformulations could be validated in a suitable cancer model to obtain a clear understanding of their fate.
Based on the FOBI imaging analysis of MKN-28 xenograft-bearing nude mice intravenously treated with IR780-labeled Nps, it was qualitatively proven that the Nps accumulated in the tumor, and targeting with an EGFR-specific antibody resulted in enhanced tumor accumulation and retention. Thus, the qualitative biodistribution images further suggested the retention of Nps in the circulation and preferential tumor accumulation upon CET MAb targeting. This is well correlated with previously reported studies.58,59
In the HPLC quantification of DOCT from the MKN-28 cancer-bearing mice, a time-dependent reduction in plasma concentration of DOCT was observed, indicating eventual clearance of the Nps or the released DOCT from the circulation following the hepatic or renal route as indicated by liver and kidney accumulation, respectively. Studies have reported that even PEG modification could not enhance the circulation time, and that the Nps were cleared by the RES system following liver and spleen accumulation. Thus, in the case of the current Np system, this reduction in the plasma concentration of DOCT was supported by the clearance mechanisms as reported in the literature.41,60,61 However, nanoformulations have significantly improved the circulation, tumor accumulation and retention of hydrophobic DOCT, compared with those of the free drug formulation. HPLC quantified a significantly higher amount of DOCT from tumors than those of other organs on day 1, whereas the concentration of DOCT was reduced considerably with respect to time and was still prominent in the tumors on day 4, with undetectable concentration in the other organs. Another notable factor is that, considering the initial injected dose of 10 mg/kg, a maximum of only 10% of DOCT was recovered. This could be attributed to many factors as follows: 1) In our study, only the major organs (heart, lungs, liver, kidney, spleen, tumor and whole blood) were isolated for extracting the drug. The possibility of Np distribution to other organs (brain, intestine, pancreas and stomach), other tissues (muscle, skin) and excretory products (urine, feces) was not explored; 2) The study was performed in such a way that the DOCT was quantified using the extraction procedures for DOCT in free form. Thus, there is no demarcation between the released and Np-bound DOCT, and it should be considered that only the released DOCT from the Nps existing in a free form has been quantified.
On separately analyzing the growth profile of the tumors of the mice treated with the drug formulations, it was observed that there was no considerable change in the tumor size when treated with targeted Nps. The first dose of free DOCT and nontargeted Nps did not influence the tumor growth, whereas after the second dose of targeted Nps, the growth seemed to arrest, which was followed by a slight reduction after the third dose. However, overall, rather than a reduction potential, these targeted Nps showed a cytostatic ability that prevented tumor growth and the tumors became static. CET MAb is reported to elicit its therapeutic activity by competing with normal ligand EGF and binding to the extracellular domain of EGFR, thereby blocking transduction of the EGFR signaling cascade, resulting in internalization and downregulation of EGFR, cell cycle arrest, apoptosis, inhibition of angiogenesis and metastasis, which improve the sensitivity of cancer cells to radiochemotherapy.45 Much of the preclinical data reported the tumor cytostatic property of CET MAb when administered alone, rather than proving it to be an effective cytotoxic agent.46,47 Despite its therapeutic activity, for this study, the targeting potential of CET MAb was exploited, so as to maximize the intratumor DOCT concentration. The tumor volume profile with respect to time indicated an additive effect of the cytostatic potential of CET MAb and the cytotoxic potential of DOCT. CET is approved in the clinic to be used as a monotherapy or in combination with other chemotherapeutic drugs and radiotherapy. One of the studies reported that the growth inhibition rate of tumor xenografts treated with CET MAb in combination with DOCT was higher than that of those treated with CET MAb and DOCT alone in an non-small cell lung cancer xenograft model,62 which shows the effectiveness of combining mAbs with cytotoxic agents. Such a combination, when incorporated into a nanoformulation, would result in an improved therapeutic outcome, which was the main hypothesis of this study. Thus, the current experimental results supported the hypothesis that CET MAb conjugation improved the accumulation of DOCT-γ-PGA Nps in tumors through EGFR-mediated targeting in addition to the EPR effect, which further elicited a combined therapeutic effect of cytostatic CET MAb and cytotoxic DOCT.
Conclusion
In summary, CET-conjugated DOCT-loaded γ-PGA Nps were shown to induce EGFR-specific cellular internalization and cancer cell death in vitro and further tumor growth inhibition in vivo in the MKN-28 gastric cancer xenograft model. The in vitro results correlated well with existing literature reports, confirming significant cancer cell death induced by active targeting. The in vivo pharmacokinetic analysis in Swiss albino mice, tumor accumulation and tumor reduction studies in the gastric cancer xenograft model showed improved systemic circulation-enhanced tumor accumulation by passive targeting due to the EPR effect of the tumor environment and EGFR-mediated cellular internalization, ultimately enhancing the drug availability at the tumor site, resulting in tumor growth arrest, compared with nontargeted and free drug formulations. On the whole, the combination of targeting agent, CET, and the therapeutic agent, DOCT, with the γ-PGA nanomatrix proved to be an efficient cancer nanomedicine for EGFR-overexpressing gastric cancers.
Acknowledgments
The authors acknowledge the Department of Science & Technology (DST), India and Portuguese Science Foundation (FCT), Portugal for providing financial support under the Indo-Portugal Joint Program (Ref No INT/PORTUGAL/P-05/2009). IKP acknowledges financial support from Basic Science Research Program (No 2016R1A2B4011184) and the Bio & Medical Technology Development Program (No NRF-2017M3A9F5030940), and the Pioneer Research Center Program through the National Research Foundation of Korea, Ministry of Science, ICT & Future Planning, contract grant number (2014M3C1A3053035) and a grant (CRI16071-3) of the CNUH-GIST. The in vivo aspect of this study has been well supported by the animal handling facilities of Chonnam National University Medical School, South Korea. This work was also financed by Fundo Europeu de Desenvolvimento Regional (FEDER) funds through the COMPETE 2020 – Operational Programme for Competitiveness and Internationalisation (POCI), Portugal 2020, and by Portuguese funds through Fundação para a Ciência e a Tecnologia (FCT)/Ministério da Ciência, Tecnologia e Inovação in the framework of the projects “Institute for Research and Innovation in Health Sciences” (POCI-01–0145-FEDER-007274) and UID/BIM/04293/2013. This work was also supported by NORTE-01-0145-FEDER-000012, supported by Norte Portugal Regional Operational Programme (NORTE 2020), under the PORTUGAL 2020 Partnership Agreement, through the European Regional Development Fund (ERDF) CGM also acknowledges the Amrita Centre for Nanosciences and Molecular Medicine, Amrita Institute of Medical Sciences and Research Centre, Kochi for computational infrastructural support. MS is thankful to the Council of Scientific and Industrial Research (CSIR), India for providing a Senior Research Fellowship (SRF Award No: 9/963 (0012) 2K11-EMR-I).
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Power DG, Kelsen DP, Shah MA. Advanced gastric cancer–Slow but steady progress. Cancer Treat Rev. 2010;36(5):384–392. | ||
Wagner AD, Unverzagt S, Grothe W, et al. Chemotherapy for advanced gastric cancer. Cochrane Database Syst Rev. 2010;3(3):CD004064. | ||
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. | ||
Fuse N, Kuboki Y, Kuwata T, et al. Prognostic impact of HER2, EGFR, and c-MET status on overall survival of advanced gastric cancer patients. Gastric Cancer. 2016;19(1):183–191. | ||
Sakai K, Mori S, Kawamoto T, et al. Expression of epidermal growth factor receptors on normal human gastric epithelia and gastric carcinomas. J Natl Cancer Inst. 1986;77(5):1047–1052. | ||
Takehana T, Kunitomo K, Suzuki S, et al. Expression of epidermal growth factor receptor in gastric carcinomas. Clin Gastroenterol Hepatol. 2003;1(6):438–445. | ||
Pinto C, Di Fabio F, Siena S, et al. Phase II study of cetuximab in combination with FOLFIRI in patients with untreated advanced gastric or gastroesophageal junction adenocarcinoma (FOLCETUX study). Ann Oncol. 2007;18(3):510–517. | ||
Zhang Y, Cao Y, Luo S, Mukerabigwi JF, Liu M. Nanoparticles as drug delivery systems of combination therapy for cancer. In: Nanobiomaterials in Cancer Therapy. Seventh Ed. Elsevier; 2016:253–280. | ||
Narayanan S, Pavithran M, Viswanath A, et al. Sequentially releasing dual-drug-loaded PLGA-casein core/shell nanomedicine: design, synthesis, biocompatibility and pharmacokinetics. Acta Biomater. 2014;10(5):2112–2124. | ||
Maya S, Sarmento B, Lakshmanan VK, Menon D, Jayakumar R. Actively targeted cetuximab conjugated γ-poly(glutamic acid)-docetaxel nanomedicines for epidermal growth factor receptor over expressing colon cancer cells. J Biomed Nanotechnol. 2014;10(8):1416–1428. | ||
Maya S, Sarmento B, Lakshmanan VK, Menon D, Seabra V, Jayakumar R. Chitosan cross-linked docetaxel loaded EGF receptor targeted nanoparticles for lung cancer cells. Int J Biol Macromol. 2014;69:532–541. | ||
Lin YH, Mi FL, Chen CT, et al. Preparation and characterization of nanoparticles shelled with chitosan for oral insulin delivery. Biomacromolecules. 2007;8(1):146–152. | ||
Tang D, Yu S, Ho Y, Mi F, Kuo P, Sung H. Heparinized chitosan/poly(γ-glutamic acid) nanoparticles for multi-functional delivery of fibroblast growth factor and heparin. Biomaterials. 2010;31(35):9320–9332. | ||
Li S, Schmitz KR, Jeffrey PD, Wiltzius JJW, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. 2005;7(4):301–311. | ||
Steffen C, Thomas K, Huniar U, Hellweg A, Rubner O, Schroer A. TmoleX–a graphical user interface for TURBOMOLE. J Comput Chem. 2010;31(16):2967–2970. | ||
Trott O, Olson AJ. Software news and update AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. | ||
Kikuchi O, Ohashi S, Horibe T, et al. Novel EGFR-targeted strategy with hybrid peptide against oesophageal squamous cell carcinoma. Sci Rep. Epub March 9, 2016. | ||
Kralj S, Rojnik M, Romih R, Jagodič M, Kos J, Makovec D. Effect of surface charge on the cellular uptake of fluorescent magnetic nanoparticles. J Nanoparticle Res. 2012;14(10):1151. | ||
Schwartz GK. Targeting the Cell Cycle: A New Approach to Cancer Therapy. J Clin Oncol. 2005;23(36):9408–9421. | ||
Stockert JC, Blázquez-Castro A, Cañete M, Horobin RW, Villanueva Á. MTT assay for cell viability: Intracellular localization of the formazan product is in lipid droplets. Acta Histochem. 2012;114(8):785–796. | ||
Ly JD, Grubb DR, Lawen A. The mitochondrial membrane potential (Δψm) in apoptosis; an update. APOPTOSIS. 2003;8(2):115–128. | ||
Maya S, Sarmento B, Lakshmanan VK, Menon D, Jayakumar R. Actively targeted cetuximab conjugated γ-poly(glutamic acid)-docetaxel nanomedicines for epidermal growth factor receptor over expressing colon cancer cells. J Biomed Nanotechnol. 2014;10(8):1416–1428. | ||
Hrkach J, Von Hoff D, Mukkaram Ali M, et al. Preclinical development and clinical translation of a PSMA-targeted docetaxel nanoparticle with a differentiated pharmacological profile. Sci Transl Med. 2012;4(128):128ra39. | ||
Werner ME, Cummings ND, Sethi M, et al. Preclinical evaluation of Genexol-PM, a nanoparticle formulation of paclitaxel, as a novel radiosensitizer for the treatment of non small cell lung cancer. Int J Radiat Oncol. 2013;86(3):463–468. | ||
Ernsting MJ, Murakami M, Undzys E, Aman A, Press B, Li SD. A docetaxel-carboxymethylcellulose nanoparticle outperforms the approved taxane nanoformulation, Abraxane, in mouse tumor models with significant control of metastases. J Control Release. 2012;162(3):575–581. | ||
Engels FK, Sparreboom A, Mathot RAA, Verweij J. Potential for improvement of docetaxel-based chemotherapy: a pharmacological review. Br J Cancer. 2005;93(2):173–177. | ||
Benton G, Arnaoutova I, George J, Kleinman HK, Koblinski J. Matrigel: from discovery and ECM mimicry to assays and models for cancer research. Adv Drug Deliv Rev. 2014;79:3–18. | ||
Mullen P. The use of matrigel to facilitate the establishment of human cancer cell lines as xenografts. Methods Mol Med. 2004;88:287–292. | ||
Okochi-Takada E, Hattori N, Tsukamoto T, et al. ANGPTL4 is a secreted tumor suppressor that inhibits angiogenesis. Oncogene. 2014;33(17):2273–2278. | ||
Teicher BA. Tumor models for efficacy determination. Mol Cancer Ther. 2006;5(10):2435–2443. | ||
Noorani L, Stenzel M, Liang R, Pourgholami MH, Morris DL. Albumin nanoparticles increase the anticancer efficacy of albendazole in ovarian cancer xenograft model. J Nanobiotechnology. 2015;13(1):25. | ||
Wang T, Kievit FM, Veiseh O, et al. Targeted cell uptake of a noninternalizing antibody through conjugation to iron oxide Nanoparticles in primary central nervous system lymphoma. World Neurosurg. 2013;80(1–2):134–141. | ||
Northwestern University; Office for Research. Center for Advanced Microscopy; Z Stack. Available from: https://cam.facilities.northwestern.edu/588–2/z-stack/. Accessed June 1, 2017. | ||
Confocal tutorial: Applications: 3D reconstruction. AMU Dept Pathol Univ Hel. Available from: http://www.hi.helsinki.fi/amu/AMU%20Cf_tut/cf_tut_part2-3.htm. Accessed June 1, 2017. | ||
Montero A, Fossella F, Hortobagyi G, Valero V. Docetaxel for treatment of solid tumours: a systematic review of clinical data. Lancet Oncol. 2005;6(4):229–239. | ||
Hernández-Vargas H, Palacios J, Moreno-Bueno G. Molecular profiling of docetaxel cytotoxicity in breast cancer cells: uncoupling of aberrant mitosis and apoptosis. Oncogene. 2007;26(20):2902–2913. | ||
Perelman A, Wachtel C, Cohen M, Haupt S, Shapiro H, Tzur A. JC-1: alternative excitation wavelengths facilitate mitochondrial membrane potential cytometry. Cell Death Dis. 2012;3(11):e430. | ||
Chu KS, Hasan W, Rawal S, et al. Plasma, tumor and tissue pharmacokinetics of Docetaxel delivered via nanoparticles of different sizes and shapes in mice bearing SKOV-3 human ovarian carcinoma xenograft. Nanomedicine Nanotechnology, Biol Med. 2013;9(5):686–693. | ||
Li X, Wang D, Zhang J, Pan W. Preparation and pharmacokinetics of docetaxel based on nanostructured lipid carriers. J Pharm Pharmacol. 2009;61(11):1485–1492. | ||
Wainberg ZA, Anghel A, Desai AJ, et al. Lapatinib, a dual EGFR and HER2 kinase inhibitor, selectively inhibits HER2-amplified human gastric cancer cells and is synergistic with trastuzumab in vitro and in vivo. Clin Cancer Res. 2010;16(5):1509–1519. | ||
Xu J, Gattacceca F, Amiji M. Biodistribution and pharmacokinetics of EGFR-targeted thiolated gelatin nanoparticles following systemic administration in pancreatic tumor-bearing mice. Mol Pharm. 2013;10(5):2031–2044. | ||
Nascimento AV, Singh A, Bousbaa H, Ferreira D, Sarmento B, Amiji MM. Mad2 checkpoint gene silencing using epidermal growth factor receptor-targeted chitosan nanoparticles in non-small cell lung cancer model. Mol Pharm. 2014;11(10):3515–3527. | ||
Nascimento AV, Gattacceca F, Singh A, et al. Biodistribution and pharmacokinetics of Mad2 siRNA-loaded EGFR-targeted chitosan nanoparticles in cisplatin sensitive and resistant lung cancer models. Nanomedicine. 2016;11(7):767–781. | ||
Nascimento AV, Singh A, Bousbaa H, Ferreira D, Sarmento B, Amiji MM. Combinatorial designed epidermal growth factor receptor targeted chitosan nanoparticles for encapsulation and delivery of lipid-modified platinum derivatives in wild type and resistant Non small cell lung cancer cells. Mol Pharm. 2015;12(12):4466–4477. | ||
Seshacharyulu P, Ponnusamy MP, Haridas D, Jain M, Ganti AK, Batra SK. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16(1):15–31. | ||
Prewett MC, Hooper AT, Bassi R, Ellis LM, Waksal HW, Hicklin DJ. Enhanced antitumor activity of anti-epidermal growth factor receptor monoclonal antibody IMC-C225 in combination with irinotecan (CPT-11) against human colorectal tumor xenografts. Clin Cancer Res. 2002;8(5):994–1003. | ||
Saltz LB, Meropol NJ, Loehrer PJ, Needle MN, Kopit J, Mayer RJ. Phase II trial of cetuximab in patients with refractory colorectal cancer that expresses the epidermal growth factor receptor. J Clin Oncol. 2004;22(7):1201–1208. | ||
Gunn J, Park SI, Veiseh O, Press OW, Zhang M. A pretargeted nanoparticle system for tumor cell labeling. Mol Biosyst. 2011;7(3):742–748. | ||
Wartlick H, Michaelis K, Balthasar S, Strebhardt K, Kreuter J, Langer K. Highly specific HER2-mediated cellular uptake of antibody modified nanoparticles in tumour cells. J Drug Target. 2004;12(7):461–471. | ||
Thomas TP, Patri AK, Myc A, et al. In vitro targeting of synthesized antibody conjugated dendrimer nanoparticles †. Biomacromolecules. 2004;5(6):2269–2274. | ||
Luo Y, Ling Y, Guo W, et al. Docetaxel loaded oleic acid-coated hydroxyapatite nanoparticles enhance the docetaxel-induced apoptosis through activation of caspase-2 in androgen independent prostate cancer cells. J Control Release. 2010;147(2):278–288. | ||
Pienta KJ. Preclinical mechanisms of action of docetaxel and docetaxel combinations in prostate cancer. Semin Oncol. 2001;28(4 Suppl 15):3–7. | ||
Tsakalozou E, Eckman AM, Bae Y. Combination Effects of Docetaxel and Doxorubicin in Hormone-Refractory Prostate Cancer Cells. Biochem Res Int. 2012;2012:1–10. | ||
Esmaeili F, Ghahremani MH, Ostad SN, et al. Folate-receptor-targeted delivery of docetaxel nanoparticles prepared by PLGA-PEG-folate conjugate. J Drug Target. 2008;16(5):415–423. | ||
Raju A, Muthu MS, Feng SS. Trastuzumab-conjugated vitamin E TPGS liposomes for sustained and targeted delivery of docetaxel. Expert Opin Drug Deliv. 2013;10(6):747–760. | ||
Narayanan S, Mony U, Vijaykumar DK, Koyakutty M, Paul-Prasanth B, Menon D. Sequential release of epigallocatechin gallate and paclitaxel from PLGA-casein core/shell nanoparticles sensitizes drug-resistant breast cancer cells. Nanomedicine. 2015;11(6):1399–1406. | ||
Li C, Newman RA, Wu Q-P, et al. Biodistribution of paclitaxel and poly(l −glutamic acid)-paclitaxel conjugate in mice with ovarian OCa-1 tumor. Cancer Chemother Pharmacol. 2000;46(5):416–422. | ||
Yue C, Liu P, Zheng M, et al. IR-780 dye loaded tumor targeting theranostic nanoparticles for NIR imaging and photothermal therapy. Biomaterials. 2013;34(28):6853–6861. | ||
Thiel KW, Hernandez LI, Dassie JP, et al. Delivery of chemo-sensitizing siRNAs to HER2+-breast cancer cells using RNA aptamers. Nucleic Acids Res. 2012;40(13):6319–6337. | ||
Sun X, Rossin R, Turner JL, et al. An assessment of the effects of shell cross-linked nanoparticle size, core composition, and surface PEGylation on in vivo biodistribution. Biomacromolecules. 2005;6(5):2541–2554. | ||
Li C, Wang C, Yang H, Zhao X, Wei N, Cui J. Liposomal topotecan formulation with a low polyethylene glycol grafting density: pharmacokinetics and antitumour activity. J Pharm Pharmacol. 2012;64(3):372–382. | ||
Steiner P, Joynes C, Bassi R, et al. Tumor growth inhibition with cetuximab and chemotherapy in non-small cell lung cancer xenografts expressing wild-type and mutated epidermal growth factor receptor. Clin Cancer Res. 2007;13(5):1540–1551. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.