")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 18
In vivo Characterization of the Opioid Receptor–Binding Profiles of Samidorphan and Naltrexone in Rats: Comparisons at Clinically Relevant Concentrations
Authors Tan LA, Gajipara N, Sun L , Bacolod M, Zhou Y, Namchuk M , Cunningham JI
Received 3 May 2022
Accepted for publication 7 October 2022
Published 1 November 2022 Volume 2022:18 Pages 2497—2506
DOI https://doi.org/10.2147/NDT.S373195
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Roger Pinder
Laura A Tan, Nileshkumar Gajipara, Lei Sun, Maria Bacolod, Ying Zhou, Mark Namchuk, Jacobi I Cunningham
Alkermes, Inc, Waltham, MA, USA
Correspondence: Laura A Tan, Alkermes, Inc, 852 Winter St, Waltham, MA, 02451, USA, Tel +1 781-609-6390, Email [email protected]
Introduction: The atypical antipsychotic olanzapine is approved for the treatment of schizophrenia and bipolar I disorder; however, weight gain and metabolic dysregulation associated with olanzapine therapy have limited its clinical utility. In clinical studies, treatment with the combination of olanzapine and the opioid receptor antagonist samidorphan (OLZ/SAM) mitigated olanzapine-associated weight gain while providing antipsychotic efficacy similar to that of olanzapine. Although samidorphan is structurally similar to the opioid receptor antagonist naltrexone, the two differ in their pharmacokinetics and in vitro binding affinities to mu, delta, and kappa opioid receptors (MOR, DOR, and KOR, respectively). The objective of this series of nonclinical studies was to compare the in vivo binding profiles of samidorphan and naltrexone and their receptor occupancies at MOR, DOR, and KOR in rat brains.
Methods: Male rats were injected with samidorphan or naltrexone to obtain total and unbound plasma and brain concentrations representing levels observed in humans at clinically relevant oral doses. Subsequently, samidorphan and naltrexone brain receptor occupancy at MOR, DOR, and KOR was measured using ultra-performance liquid chromatography and high-resolution accurate-mass mass spectrometry.
Results: A dose-dependent increase in samidorphan occupancy was observed at MOR, DOR, and KOR (EC50: 5.1, 54.7, and 42.9 nM, respectively). Occupancy of naltrexone at MOR (EC50: 15.5 nM) and KOR was dose dependent; minimal DOR occupancy was detected. At the clinically relevant unbound brain concentration of 23.1 nM, samidorphan bound to MOR, DOR, and KOR with 93.2%, 36.1%, and 41.9% occupancy, respectively. At 33.5 nM, naltrexone bound to MOR and KOR with 79.4% and 9.4% occupancy, respectively, with no binding at DOR.
Discussion: At clinically relevant concentrations, samidorphan occupied MOR, DOR, and KOR, whereas naltrexone occupied only MOR and KOR. The binding profile of samidorphan differs from that of naltrexone, with potential clinical implications.
Keywords: ALKS 3831, bipolar I disorder, Lybalvi, olanzapine, opioid receptor, schizophrenia
Plain Language Summary
Olanzapine is a medication for treating schizophrenia and bipolar I disorder that can cause substantial weight gain as an unwanted side effect. Treatment with a combination of olanzapine and another drug, samidorphan, results in less weight gain than treatment with olanzapine alone.
Samidorphan is an opioid receptor blocker that binds to receptors in the opioid receptor system, which is involved in the regulation of weight and metabolism. Samidorphan has a structure similar to that of naltrexone, another opioid receptor blocker. However, samidorphan and naltrexone differ in how they are processed in the body. They also differ in their binding to the mu, delta, and kappa opioid receptors (MOR, DOR, and KOR, respectively) in cell-based experiments.
In this study, we evaluated the differences in samidorphan and naltrexone binding to opioid receptors in a live organism as opposed to cells in a dish. Using rats as a whole-organism model of humans, we identified doses of samidorphan and naltrexone that mimic those associated with clinical efficacy in humans. We then used these doses to evaluate how each drug bound to MOR, DOR, and KOR in the brain.
Samidorphan bound to all three opioid receptors tested, while naltrexone bound only to MOR and KOR, but not DOR.
Based on these data, samidorphan has a unique binding profile at opioid receptors that distinguishes it from naltrexone and that can result in different clinical effects.
Introduction
Treatment with atypical antipsychotics can result in weight gain and metabolic dysregulation, which often leads to discontinuation and/or hospitalization.1 Olanzapine, one of the most efficacious atypical antipsychotics, is associated with a high risk for extreme weight gain, which has limited its clinical utility.2 A combination of olanzapine and the opioid receptor antagonist samidorphan (OLZ/SAM; Lybalvi, Alkermes, Inc.) was approved in the United States in May 2021 for the treatment of schizophrenia and bipolar I disorder in adults.3 Although samidorphan alone is not a weight-loss agent,4 in clinical trials, treatment with OLZ/SAM mitigated olanzapine-associated weight gain while maintaining the antipsychotic efficacy of olanzapine.1,5,6 OLZ/SAM therefore may provide a treatment option for patients who would benefit from olanzapine, with a reduced risk of significant weight gain.7,8
The opioid receptor system consists of mu, delta, and kappa receptors (MOR, DOR, and KOR, respectively), and each plays a distinct role in the regulation of weight and metabolism.9–12 In studies examining the effects of OLZ/SAM in rats and nonhuman primates, the addition of samidorphan to olanzapine attenuated olanzapine-associated weight gain, increases in adiposity, and insulin insensitivity.5 Additionally, in a double-blind, Phase 3 study of OLZ/SAM versus olanzapine in patients with schizophrenia, treatment with OLZ/SAM resulted in a significant mitigation of olanzapine-associated weight gain.1 Treatment with OLZ/SAM was also associated with smaller increases in waist circumference compared with olanzapine. Waist circumference increases as a marker for central adiposity is consistently associated with an increased risk of developing diabetes mellitus and coronary heart disease.1,13
Samidorphan was synthesized as a 3-carboxamido-4-hydroxy analog of naltrexone,14 and the two opioid receptor antagonists are structurally similar.15 However, they are differentiated by their distinct receptor-binding characteristics (Table 1)15 and pharmacokinetic profiles (Table 2).14,16,17 In vitro, samidorphan binds to human MOR, DOR, and KOR and is an antagonist at MOR and a partial agonist at DOR and KOR.16 Samidorphan binds with higher affinity to MOR and DOR than naltrexone, and functionally is a more potent antagonist at MOR.16 In vitro, the half-maximal inhibitory concentration (IC50) of samidorphan at human MOR is 5-fold lower than naltrexone and is approximately 20-fold lower at DOR and 3-fold lower at KOR (Table 3).14,16
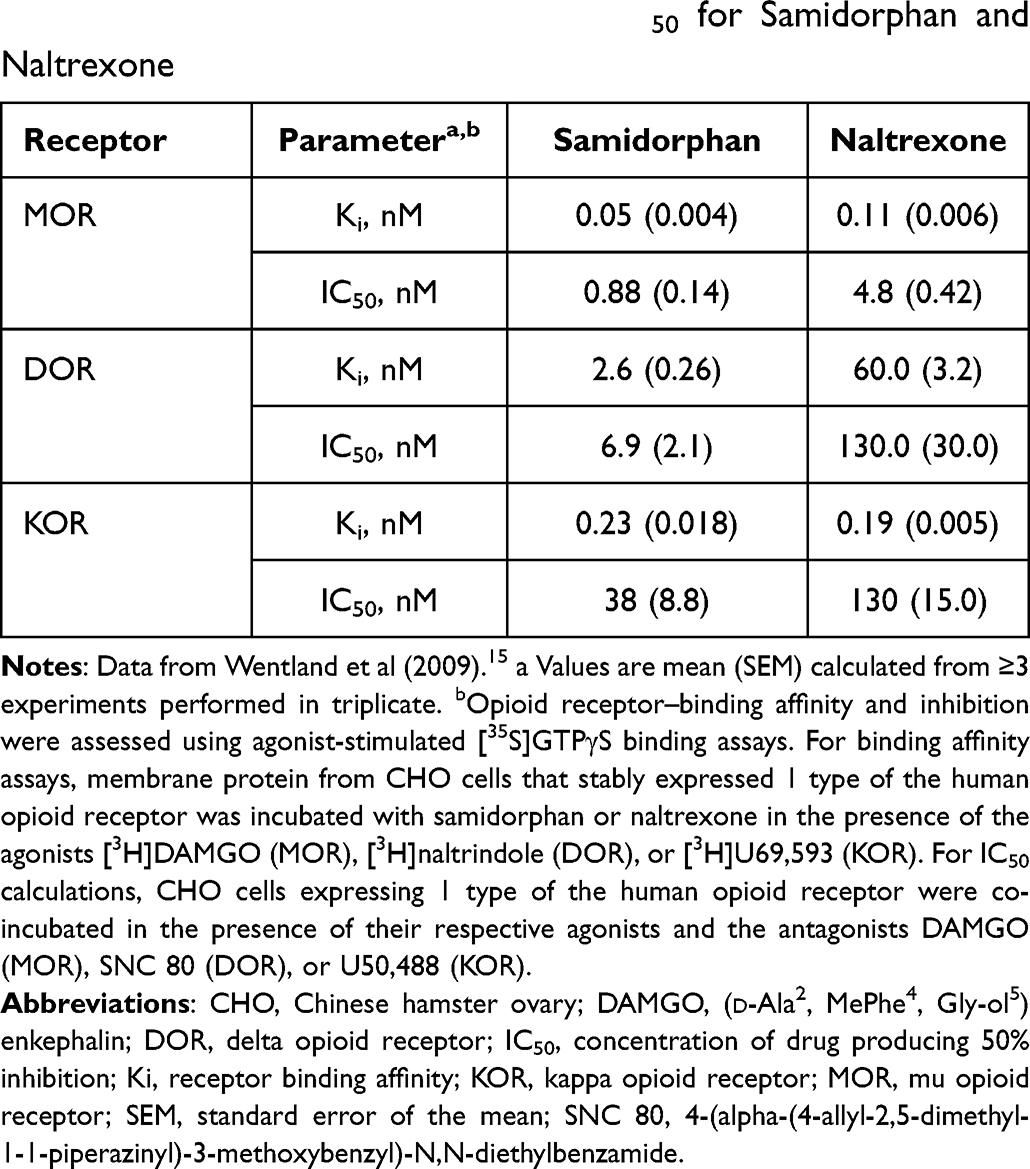 |
Table 1 In vitro Binding Affinity and IC50 for Samidorphan and Naltrexone |
 |
Table 2 Comparison of the Human Pharmacokinetics of Samidorphan and Naltrexone |
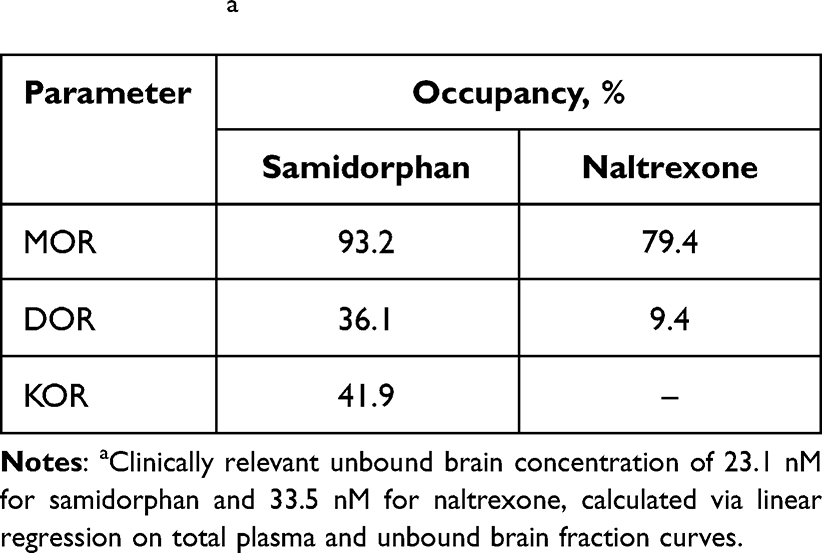 |
Table 3 In vivo Target Occupancies for Samidorphan and Naltrexone at Clinically Relevant Unbound Brain Concentrationsa |
Naltrexone is used for the treatment of patients with alcohol and opioid dependence and has been assessed, albeit in a limited capacity, for its potential to attenuate olanzapine-associated weight gain.18 Neither naltrexone nor any other pharmacologic treatment is approved to prevent and/or treat antipsychotic-associated weight gain or metabolic dysfunction. Although samidorphan and naltrexone have differentiated in vitro binding affinities and pharmacokinetic properties, their in vivo binding characteristics, especially at clinically relevant concentrations, have not been assessed. Here, we present receptor occupancy studies designed to determine the relationship between compound dose, plasma and brain concentrations, and fractional occupancy at receptor targets for samidorphan and naltrexone.
In vivo receptor occupancy studies have used liquid chromatography/mass spectrometry (LC-MS) to quantify low doses of MOR, DOR, and KOR antagonist tracers to measure receptor occupancy of those antagonists in rodent brain tissue19–21 and to determine their in vivo target engagement profiles. Receptor occupancy studies may also be used to estimate and compare receptor occupancy of drugs at clinically relevant concentrations. To explore the in vivo binding profile of samidorphan and naltrexone in greater detail, a series of nonclinical studies in rats was designed to determine the unbound brain concentrations of samidorphan and naltrexone at clinically relevant plasma concentrations observed in humans. The samidorphan concentration of 46.1 ng/mL targeted for these studies corresponds to the maximum plasma concentration (Cmax) of samidorphan at its therapeutic oral dose of 10 mg in OLZ/SAM.22 Likewise, the targeted naltrexone concentration of 16.1 ng/mL for these studies corresponds to the Cmax of naltrexone at its therapeutic oral dose of 50 mg.23 Calculated unbound brain concentrations were used to estimate receptor occupancies at MOR, DOR, and KOR in the brain to characterize the binding profiles of samidorphan and naltrexone at those concentrations.
Materials and Methods
Animals
Male Sprague Dawley rats aged approximately 10 weeks (Charles River Laboratories; Kingston, NY, USA) were used for all studies, which were conducted at Alkermes, Inc.
All rats used in these studies were housed, managed, and cared for in accordance with the Guide for the Care and Use of Laboratory Animals,24 and experiment protocols were approved by the Alkermes Institutional Animal Care and Use Committee. Rats were housed 3 per cage and were maintained on a 12:12-hour light/dark cycle (lights off at 18:00 hours) in a temperature- and humidity-controlled environment (22° ± 2°C; 45% ± 10% relative humidity). Rats were fed standard chow (Rodent Diet 5001; Lab Diets, St Louis, MO, USA) and tap water, and were allowed to eat and drink ad libitum.
Test Compounds
Samidorphan was synthesized as a salt (
Determination of Total and Unbound Samidorphan and Naltrexone Concentrations in Rat Plasma and Brain Tissues
Pharmacokinetic studies were conducted to relate the plasma and unbound brain concentrations in rats to plasma concentrations in humans. Doses were chosen to bracket clinically relevant plasma concentrations of samidorphan or naltrexone as described above. Briefly, 10 groups of rats were used: 5 groups of rats (n=6 per group) were injected subcutaneously with samidorphan at 0.03, 0.1, 0.3, 1.0, or 3.0 mg/kg at a dose volume of 1 mL/kg, whereas 5 groups of rats (n=6 per group) were injected subcutaneously with naltrexone at 0.01, 0.03, 0.1, 0.3, or 1.0 mg/kg at a dose volume of 1 mL/kg. Thirty minutes after dosing (coinciding with the Cmax of these compounds),22 blood was collected via direct cardiac puncture following anesthesia using carbon dioxide. Blood samples were centrifuged (3500 rpm, 10 minutes at 5°C) within 30 minutes of collection to obtain plasma. Brains were collected, rinsed, weighed, and homogenized. Plasma and brain samples were stored at −80°C until analysis. Aliquots of plasma and homogenized brain samples were transferred into 96-well plates and extracted with 4 volumes of acetonitrile containing an internal standard (naltrexone-D3 20 ng/mL; Cerilliant, Round Rock, TX, USA). After centrifugation, supernatants were transferred into a 96-well plate and analyzed. The total concentration of samidorphan and naltrexone in plasma and brain samples was measured using a liquid chromatography with tandem mass spectrometry (LC-MS/MS) system consisting of a Shimadzu SIL 30 AC autosampler (Columbia, MD, USA), two Shimadzu LC-30AD pumps, and a Shimadzu DGU-20A degasser (Columbia, MD, USA) connected to a SCIEX API 5500 mass spectrometer (Framingham, MA, USA). Positive electrospray ionization was used with the mass spectrometer in multiple reaction monitoring acquisition mode. The m/z mass transitions used for samidorphan, naltrexone, and internal standard (naltrexone-D3) were 371.00 >336.10, 342.00 >324.00, and 345.20 >327.20, respectively. The columns used were Phenomenex, Kinetix, C18, column (2.1 × 30 mm, 2.7 µm particle size) and Restek, Raptor FluoroPhenyl column (2.1 × 50 mm, 2.7 µm particle size) for samidorphan and naltrexone, respectively. Samples were run on a linear AB gradient (A: 0.1% formic acid in water; B: 0.1% formic acid in acetonitrile) at a flow rate of 1.0 mL/min. A calibration curve was constructed by plotting the peak area ratio of analyte to internal standard versus nominal concentration for all standards to quantify the concentration of samidorphan and naltrexone. Analyst 1.4.2 software (SCIEX) was used to fit a curve to the plot using a weighted (1/X2) linear least squares regression. The unbound concentrations of samidorphan and naltrexone in plasma and brain homogenate were obtained by normalizing with in vitro plasma and brain binding measured using a rapid equilibrium dialysis.
In vivo Receptor Occupancy
The ability of samidorphan or naltrexone to engage MOR, DOR, or KOR in brain tissue was assessed in male Sprague Dawley rats using the in vivo receptor occupancy method19 at doses that result in plasma concentrations that bracketed clinically relevant plasma concentrations of samidorphan or naltrexone as described above. Briefly, 12 groups of rats were used: 6 groups of rats (n=6 per group) were injected subcutaneously with vehicle or samidorphan at 0.03, 0.1, 0.3, 1.0, or 3.0 mg/kg at a dose volume of 1 mL/kg, and 6 groups of rats (n=6 per group) were injected subcutaneously with vehicle or naltrexone at 0.01, 0.03, 0.1, 0.3, or 1.0 mg/kg at a dose volume of 1 mL/kg. For both cohorts of rats, receptor occupancy was measured 30 minutes after test compound administration by measuring the displacement of the non-radioactive tracers by ultra-performance liquid chromatography (UPLC) and high-resolution accurate-mass (HRAM) mass spectrometry. Low-dose antagonist tracers were naltrexone-D3 (MOR), naltriben (DOR), and GR103545 (KOR) at 10 µg/kg, 10 µg/kg, and 1.5 µg/kg, respectively, and were simultaneously administered as a single intravenous injection via the tail at a volume of 1 mL/kg. In previously published studies, non-labeled naltrexone was validated and utilized as a tracer for MOR.19–21 Based on in-house method validation studies, naltrexone-D3 binding was equivalent in the thalamus (specific binding) and cerebellum (nonspecific binding) (data not shown). In addition, HRAM mass spectrometry was able to resolve the masses of naltrexone and naltrexone-D3. As a result, naltrexone-D3 was selected as the tracer for MOR occupancy measurement. In pilot experiments, no difference was seen between ratios of total to nonspecific binding when tracers were administered individually or in a simultaneous injection (data not shown). Thirty minutes later, rats were sacrificed and brains were rapidly dissected. Tissues were stored at −80°C until analysis. To target areas of high neuroanatomical expression levels of MOR, DOR, and KOR in the brain, total binding to MOR was determined from thalamus tissue samples, whereas total binding for DOR and for KOR was determined from striatal tissue.25 Nonspecific binding was determined from tracer recovery in the cerebellum, which has very low expression of MOR, DOR, and KOR and does not have appreciable opioid ligand-receptor binding.19,25–27 Tissues were weighed and homogenized in 4 volumes of acetonitrile containing 0.1% (v/v) formic acid. The supernatant containing recovered tracers was diluted with an equal volume of internal standard (labetalol; Sigma Aldrich) in water containing 0.1% (v/v) formic acid. Analysis of tracers was performed on a Vanquish Ultimate 3000 LC system coupled to a Q Exactive Orbitrap mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). The separation was performed on an Acquity BEH shield RP 18 column (50 × 2.1 mm, 1.7 µm; Waters, Milford, MA, USA) maintained at 40°C. Samples were run on a linear AB gradient (A: ammonium acetate 10 mM in water; B: formic acid 0.1% in acetonitrile) from 10% to 98% B. The total UPLC run time was 3.1 minutes, with a 0.9-minute re-equilibration time at a flow rate of 0.2 mL/min. Analyses were performed on a Thermo Q Exactive mass spectrometer with an electrospray ion source, with capillary temperature at 250°C and voltage at 3.00 kV. Full mass range was set to m/z 100 to 1000, with positive polarity. For the targeted analytes (m/z 345.1888, 416.1856, and 414.1346 for naltrexone-D3, naltriben, and GR103545, respectively), the theoretical exact masses were used with mass accuracy of ± 5 ppm.
The resulting extracted ion chromatograms were integrated and the area under the curve was used for quantification. Calibration curves were generated by preparing a series of known quantities of analytes along with labetalol 50 ng/mL as an internal standard. Receptor occupancy was calculated using the following formula:19
% occupancy = 100×(1−[(Ratiot−1)/(Ratioc−1)])
in which “ratio” refers to the ratio of the test area (thalamus, striatum) relative to its nonspecific binding (cerebellum). Further, Ratiot refers to rats dosed with test compound, whereas Ratioc refers to the average ratio from rats dosed with vehicle.
Data Analysis
Data were compiled and analyzed in Prism 8 (GraphPad Software, La Jolla, CA, USA) and presented as means ± standard error. Linear regressions were performed on total plasma and unbound brain concentrations to interpolate unbound brain concentration relative to clinically relevant plasma concentrations of samidorphan (46.1 ng/mL) and naltrexone (16.1 ng/mL). Percent receptor occupancy was also expressed as a function of unbound brain concentration and as a 4-parameter nonlinear regression curve fit with upper and lower bounds not constrained. Hill slope for DOR and KOR curves were constrained to 1 as the curve did not reach linearity. The half-maximal effective concentration (EC50) values (where possible) and occupancy at the calculated clinically relevant brain concentrations were interpolated using these curve fits.
Results
Total and Unbound Plasma and Brain Concentrations
Total and unbound plasma and brain concentrations of samidorphan and naltrexone in rats are shown in Figure 1A and B, respectively. Unbound plasma fractions were 53.1% and 65.6% for samidorphan and naltrexone, respectively, whereas unbound brain fractions were 26.1% and 22.3% for samidorphan and naltrexone, respectively. The doses used bracketed the targeted clinically relevant plasma concentrations for samidorphan (Cmax of 46.1 ng/mL) and naltrexone (Cmax of 16.1 ng/mL). To determine the unbound brain concentration corresponding to these clinically relevant plasma concentrations, linear regressions were performed on total plasma and unbound brain fraction curves, resulting in calculated unbound brain concentrations of 23.1 nM and 33.5 nM for samidorphan and naltrexone, respectively.
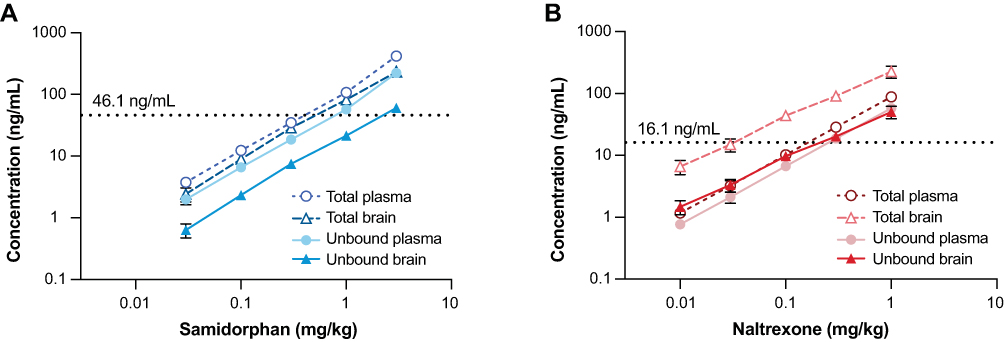 |
Figure 1 Total and unbound plasma and brain concentration of (A) samidorphan and (B) naltrexone in rats. Dose-concentration curves in plasma and brain of male Sprague Dawley rats 30 minutes after a single subcutaneous injection of samidorphan or naltrexone are depicted in log-log plots. In panel A, the horizontal dotted line indicates the total plasma Cmax (46.1 ng/mL) after oral administration of OLZ/SAM 20 mg/10 mg in humans. In panel B, the horizontal dotted line indicates the total plasma Cmax (16.1 ng/mL) after a 50-mg oral dose of naltrexone in humans. Data are expressed as means ± standard deviation, n=6 per group. Abbreviations: Cmax, maximum plasma concentration; OLZ/SAM, combination of olanzapine and samidorphan. |
In vivo Receptor Occupancy
For samidorphan, there was a dose-dependent increase in occupancy at MOR, DOR, and KOR (Figure 2A), saturating MOR at the highest doses. The EC50 values for samidorphan were 5.1, 54.7, and 42.9 nM for MOR, DOR, and KOR, respectively. For naltrexone, there was a dose-dependent increase in occupancy at MOR and KOR; minimal DOR occupancy was detected at the highest dose utilized (1 mg/kg) (Figure 2B). The EC50 for naltrexone at MOR was 15.5 nM. Receptor occupancies for samidorphan and naltrexone were plotted against unbound brain concentrations (Figure 3). At the clinically relevant unbound brain concentration of 23.1 nM, samidorphan bound to MOR, DOR, and KOR with 93.2%, 36.1%, and 41.9% occupancy, respectively. In comparison with samidorphan, at the clinically relevant unbound brain concentration of 33.5 nM, naltrexone occupied MOR (79.4%) and KOR (9.4%), with no binding at DOR (Table 3).
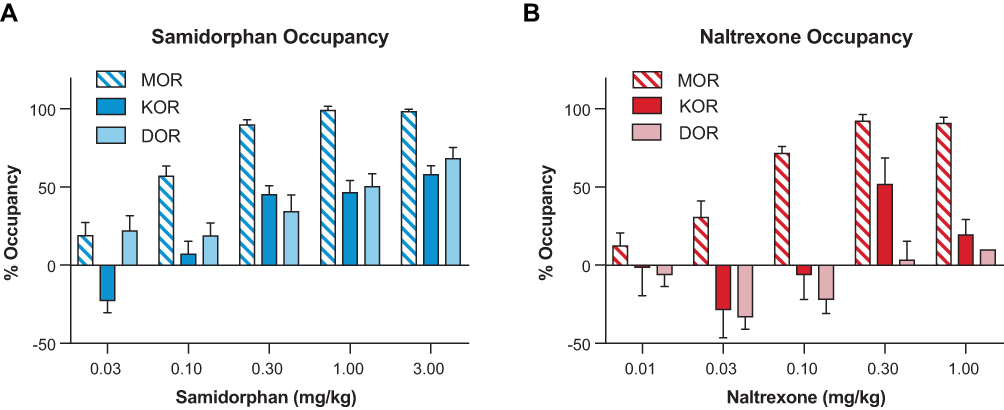 |
Figure 2 Dose-occupancy relationship for subcutaneously administered samidorphan (A) and naltrexone (B) at MOR, DOR, and KOR in male Sprague Dawley rats, as determined using an in vivo triple-tracer assay. (A) For samidorphan, there were dose-dependent increases in occupancy, saturating MOR at the higher doses (1–3 mg/kg). (B) For naltrexone, there were dose-dependent increases in occupancy, saturating MOR at the higher doses (0.3–1 mg/kg), whereas DOR occupancy was detected only at the highest doses. Data are expressed as means ± standard error of the mean. N=5–6 rats per group. Abbreviations: DOR, delta opioid receptor; KOR, kappa opioid receptor; MOR, mu opioid receptor. |
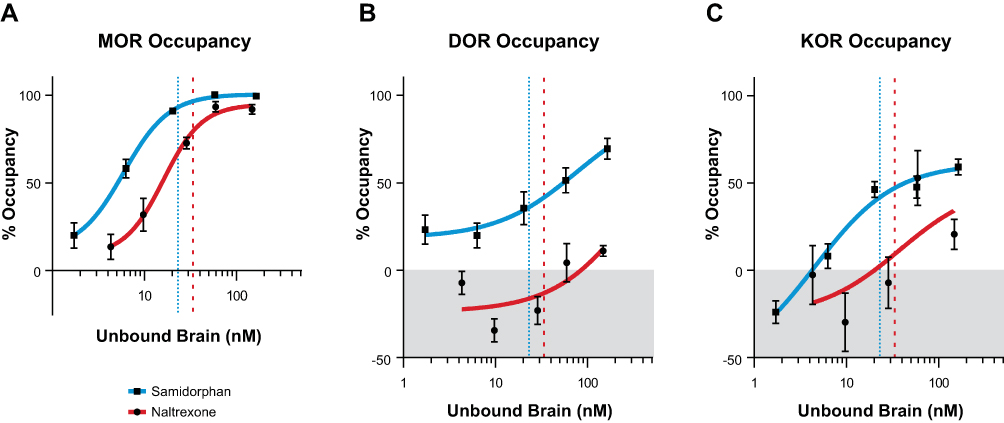 |
Figure 3 Comparison of in vivo receptor occupancies of samidorphan and naltrexone relative to unbound brain concentrations in Sprague Dawley rats at (A) MOR, (B) DOR, and (C) KOR. Blue dotted lines represent the clinically relevant unbound brain concentration for samidorphan at 23.1 nM for 46.1 ng/mL total plasma. Red dashed lines represent the clinically relevant unbound brain concentration for naltrexone at 33.5 nM for 16.1 ng/mL total plasma. At these concentrations, samidorphan occupied MOR (93.2%), DOR (36.1%), and KOR (41.9%), whereas naltrexone occupied only MOR (79.4%) and KOR (9.4%). Samidorphan had a leftward shift relative to naltrexone when comparing occupancy/brain concentration relationships, suggesting that samidorphan has higher affinity for these receptors. Abbreviations: DOR, delta opioid receptor; KOR, kappa opioid receptor; MOR, mu opioid receptor. |
Discussion
The endogenous opioid system is involved in the regulation of weight and metabolism,9–12,28 and opioid antagonism is a potential treatment pathway for mitigating antipsychotic-associated weight gain and metabolic dysregulation.18 Naltrexone, an opioid antagonist, was used in combination with olanzapine in a small clinical study in patients with schizophrenia or schizoaffective disorder. While the study noted small differences in body fat mass, there were no differences in body mass index or body weight after treatment with the naltrexone/olanzapine combination versus olanzapine plus placebo.18 To date, no further clinical trials with naltrexone have explored the combination to address olanzapine-associated weight gain. In contrast, samidorphan, a new opioid receptor antagonist, mitigated olanzapine-associated weight gain in both preclinical and clinical studies,1,5 and the combination of samidorphan and olanzapine is now approved in the United States for the treatment of schizophrenia and bipolar I disorder.3 Samidorphan and naltrexone, while structurally similar, have distinct in vitro binding properties at human opioid receptors. While the two antagonists exhibit similar activity for KOR in agonist-stimulated binding assays, samidorphan has a greater affinity for MOR and DOR compared with naltrexone.14–16
Results from the current study indicate an important distinction between samidorphan and naltrexone with respect to binding at DOR in vivo in that, at clinically relevant concentrations, samidorphan binds to MOR, DOR, and KOR, whereas naltrexone binds only to MOR and KOR. In addition to the differences in receptor binding profile, samidorphan has higher binding affinity in vivo for all three opioid receptors relative to naltrexone. Furthermore, although equally saturating MOR and reaching similar KOR binding in vitro (Table 1), increasing the dose of naltrexone to 10 times the therapeutic dose in vivo would still not achieve the same extent of binding at DOR as samidorphan at therapeutic doses.29
Evidence from opioid receptor gene deletion studies using murine knockout models indicates that MOR, DOR, and KOR each contribute to the regulation of weight and metabolism, with differing effects of deletion for each receptor type,30,31 and in humans, the MOR gene has been associated with dietary fat intake, adiposity,12 and susceptibility to type 2 diabetes mellitus.11 As samidorphan is differentiated from naltrexone by its DOR binding at clinically relevant concentrations, it is notable that central DOR mechanisms are involved in palatable food intake and reward32 and that DOR knockout mice have higher energy expenditure and greater thermogenesis in brown adipose tissue.10 Indeed, co-administration of samidorphan with olanzapine in rats at steady-state samidorphan concentrations similar to those used in this study (total plasma concentration of 46.1 ng/mL) attenuated olanzapine-associated weight gain and adiposity. In addition, samidorphan attenuated alterations in glucose utilization and insulin insensitivity that were observed in rats treated with olanzapine alone.5 However, these metabolic effects were not seen in rats treated with samidorphan alone, suggesting that the effects of samidorphan may manifest only in the context of olanzapine-induced metabolic sequelae.
In addition to having higher occupancy for the three opioid receptors relative to naltrexone, samidorphan also has a distinct pharmacokinetic profile (Table 2). Samidorphan has high oral bioavailability (69%)17 and an elimination half-life of 7 to 9 hours,33 making it suitable for once-daily dosing in combination with olanzapine (which has an extended half-life of 30 hours and is also dosed once daily34). In comparison, naltrexone has lower and highly variable oral bioavailability (5% to 40%) and a shorter half-life (4 hours).35
It should be noted that the receptor occupancy results reported here were performed in rats to best correlate receptor occupancy with unbound brain concentration in humans, and that these studies only indicate target engagement (ie, binding) but not any subsequent functional downstream activity (if any) at these receptors. Although an analogous human imaging study has yet to be conducted, our data are consistent with the rank order potencies observed in previous in vitro studies on human opioid receptors, especially at clinically relevant concentrations.14,16 Finally, future research would be needed to better evaluate the impact on functional activity and/or pharmacodynamic effects (in vivo) of the binding profiles at MOR, DOR, and KOR.
Conclusions
While both samidorphan and naltrexone bind to MOR and KOR at clinically relevant doses, samidorphan binds to DOR whereas naltrexone does not. These data further support the hypothesis that samidorphan, in combination with olanzapine, likely functions as an MOR and DOR antagonist to mitigate olanzapine-associated weight gain.
Abbreviations
Cmax, maximum plasma concentration; DOR, delta opioid receptor; EC50, half-maximal effective concentration; HRAM, high-resolution accurate-mass; IC50, half-maximal inhibitory concentration; KOR, kappa opioid receptor; LC-MS, liquid chromatography/mass spectrometry; LC-MS/MS, tandem mass spectrometry; MOR, mu opioid receptor; OLZ/SAM, a combination of olanzapine and samidorphan; UPLC, ultra-performance liquid chromatography.
Acknowledgments
The authors thank Mark S. Todtenkopf, PhD, of Alkermes, Inc., for assistance in the preparation and critical review of this manuscript. They also thank Michaela Cullum-Doyle, Adjovi Rodriguez, Abigail Hanks, Issam Ayoub, and Praveen Srivistava of Alkermes, Inc., for their technical assistance. Editorial support was provided by Peloton Advantage, LLC, an OPEN Health company, and funded by Alkermes, Inc.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
This study was sponsored by Alkermes, Inc. L.A. Tan, N. Gajipara, M. Bacolod, and Y. Zhou are employees of Alkermes, Inc., and may own stock/options in the company. M. Namchuk, L. Sun, and J.I. Cunningham were employees of Alkermes, Inc., at the time of this study, and may own stock/options in the company. M. Namchuk reports personal fees from Arrakis Therapeutics and Sionna Therapeutics, personal fees from Axonis, and grants from AbbVie outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Correll CU, Newcomer JW, Silverman B, et al. Effects of olanzapine combined with samidorphan on weight gain in schizophrenia: a 24-week phase 3 study. Am J Psychiatry. 2020;177(12):1168–1178. doi:10.1176/appi.ajp.2020.19121279
2. Berkowitz RL, Patel U, Ni Q, Parks JJ, Docherty JP. The impact of the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) on prescribing practices: an analysis of data from a large midwestern state. J Clin Psychiatry. 2012;73(4):498–503. doi:10.4088/JCP.10m06497
3. Lybalvi [package insert]. Waltham, MA: Alkermes, Inc; 2021.
4. Silverman BL, Martin W, Memisoglu A, DiPetrillo L, Correll CU, Kane JM. A randomized, double-blind, placebo-controlled proof of concept study to evaluate samidorphan in the prevention of olanzapine-induced weight gain in healthy volunteers. Schizophr Res. 2018;195:245–251. doi:10.1016/j.schres.2017.10.014
5. Cunningham JI, Eyerman DJ, Todtenkopf MS, et al. Samidorphan mitigates olanzapine-induced weight gain and metabolic dysfunction in rats and nonhuman primates. J Psychopharmacol. 2019;33(10):1303–1316. doi:10.1177/0269881119856850
6. Potkin SG, Kunovac J, Silverman BL, et al. Efficacy and safety of a combination of olanzapine and samidorphan in adult patients with an acute exacerbation of schizophrenia: outcomes from the randomized, phase 3 ENLIGHTEN-1 study. J Clin Psychiatry. 2020;81(2):19m12769. doi:10.4088/JCP.19m12769
7. Citrome L, Graham C, Simmons A, et al. An evidence-based review of OLZ/SAM for treatment of adults with schizophrenia or bipolar I disorder. Neuropsychiatr Dis Treat. 2021;17:2885–2904. doi:10.2147/NDT.S313840
8. Faden J, Serdenes R, Citrome L. Olanzapine-samidorphan combination tablets for the treatment of schizophrenia and bipolar I disorder—what is it, and will it be used? Expert Rev Neurother. 2022;22(5):365–376. doi:10.1080/14737175.2022.2060742
9. Czyzyk TA, Nogueiras R, Lockwood JF, et al. kappa-Opioid receptors control the metabolic response to a high-energy diet in mice. FASEB J. 2010;24(4):1151–1159. doi:10.1096/fj.09-143610
10. Czyzyk TA, Romero-Pico A, Pintar J, et al. Mice lacking delta-opioid receptors resist the development of diet-induced obesity. FASEB J. 2012;26(8):3483–3492. doi:10.1096/fj.12-208041
11. Gallagher CJ, Gordon CJ, Langefeld CD, et al. Association of the mu-opioid receptor gene with type 2 diabetes mellitus in an African American population. Mol Genet Metab. 2006;87(1):54–60. doi:10.1016/j.ymgme.2005.07.013
12. Haghighi A, Melka MG, Bernard M, et al. Opioid receptor mu 1 gene, fat intake and obesity in adolescence. Mol Psychiatry. 2014;19(1):63–68. doi:10.1038/mp.2012.179
13. Klein S, Allison DB, Heymsfield SB, et al. Waist circumference and cardiometabolic risk: a consensus statement from shaping america’s health: association for weight management and obesity prevention; NAASO, the obesity society; the American Society for Nutrition; and the American Diabetes Association. Am J Clin Nutr. 2007;85(5):1197–1202. doi:10.1093/ajcn/85.5.1197
14. Wentland MP, Lu Q, Lou R, Bu Y, Knapp BI, Bidlack JM. Synthesis and opioid receptor binding properties of a highly potent 4-hydroxy analogue of naltrexone. Bioorg Med Chem Lett. 2005;15(8):2107–2110. doi:10.1016/j.bmcl.2005.02.032
15. Wentland MP, Lou R, Lu Q, et al. Syntheses of novel high affinity ligands for opioid receptors. Bioorg Med Chem Lett. 2009;19(8):2289–2294. doi:10.1016/j.bmcl.2009.02.078
16. Bidlack JM, Knapp BI, Deaver DR, et al. In vitro pharmacological characterization of buprenorphine, samidorphan, and combinations being developed as an adjunctive treatment of major depressive disorder. J Pharmacol Exp Ther. 2018;367(2):267–281. doi:10.1124/jpet.118.249839
17. Kumar V, Lu H, Hard M, von Moltke L. Characterization of the pharmacokinetics of samidorphan in healthy volunteers: absolute bioavailability and the effect of food and age. Drugs R D. 2019;9(3):277–287. doi:10.1007/s40268-019-00280-5
18. Taveira TH, Wu WC, Tschibelu E, et al. The effect of naltrexone on body fat mass in olanzapine-treated schizophrenic or schizoaffective patients: a randomized double-blind placebo-controlled pilot study. J Psychopharmacol. 2014;28(4):395–400. doi:10.1177/0269881113509904
19. Need AB, McKinzie JH, Mitch CH, Statnick MA, Phebus LA. In vivo rat brain opioid receptor binding of LY255582 assessed with a novel method using LC/MS/MS and the administration of three tracers simultaneously. Life Sci. 2007;81(17–18):1389–1396. doi:10.1016/j.lfs.2007.09.005
20. Rorick-Kehn LM, Witkin JM, Statnick MA, et al. LY2456302 is a novel, potent, orally-bioavailable small molecule kappa-selective antagonist with activity in animal models predictive of efficacy in mood and addictive disorders. Neuropharmacology. 2014;77:131–144. doi:10.1016/j.neuropharm.2013.09.021
21. Rorick-Kehn LM, Witcher JW, Lowe SL, et al. Determining pharmacological selectivity of the kappa opioid receptor antagonist LY2456302 using pupillometry as a translational biomarker in rat and human. Int J Neuropsychopharmacol. 2015;18(2):pyu036. doi:10.1093/ijnp/pyu036
22. Sun L, McDonnell D, von Moltke L. Pharmacokinetics and short-term safety of ALKS 3831, a fixed-dose combination of olanzapine and samidorphan, in adult subjects with schizophrenia. Clin Ther. 2018;40(11):1845–1854. doi:10.1016/j.clinthera.2018.09.002
23. Weerts EM, Kim YK, Wand GS, et al. Differences in delta- and mu-opioid receptor blockade measured by positron emission tomography in naltrexone-treated recently abstinent alcohol-dependent subjects. Neuropsychopharmacology. 2008;33(3):653–665. doi:10.1038/sj.npp.1301440
24. Guide for the Care and Use of Laboratory Animals. National Research Council Committee for the Update of the Guide for the Care and Use of Laboratory Animals.
25. Mansour A, Fox CA, Burke S, et al. Mu, delta, and kappa opioid receptor mRNA expression in the rat CNS: an in situ hybridization study. J Comp Neurol. 1994;350(3):412–438. doi:10.1002/cne.903500307
26. Mansour A, Khachaturian H, Lewis ME, Akil H, Watson SJ. Anatomy of CNS opioid receptors. Trends Neurosci. 1988;11(7):308–314. doi:10.1016/0166-2236(88)90093-8
27. Thomasy SM, Moeller BC, Stanley SD. Comparison of opioid receptor binding in horse, Guinea pig, and rat cerebral cortex and cerebellum. Vet Anaesth Analg. 2007;34(5):351–358. doi:10.1111/j.1467-2995.2006.00337.x
28. Raffan E, Dennis RJ, O’Donovan CJ, et al. A deletion in the canine POMC gene is associated with weight and appetite in obesity-prone labrador retriever dogs. Cell Metab. 2016;23(5):893–900. doi:10.1016/j.cmet.2016.04.012
29. Crabtree BL. Review of naltrexone, a long-acting opiate antagonist. Clin Pharm. 1984;3(3):273–280.
30. Xiao K, Gillissie ES, Lui LMW, et al. Immune response to vaccination in adults with mental disorders: a systematic review. J Affect Disord. 2022;304:66–77. doi:10.1016/j.jad.2022.02.025
31. McIntyre RS, Citrome L, Cummings H, et al. Opioid antagonism mitigates antipsychotic-associated weight gain: focus on olanzapine. CNS Spectr. 2022;2022:1–12 doi:10.1017/S1092852922000116.
32. Castro DC, Berridge KC. Opioid hedonic hotspot in nucleus accumbens shell: mu, delta, and kappa maps for enhancement of sweetness “liking” and “wanting”. J Neurosci. 2014;34(12):4239–4250. doi:10.1523/JNEUROSCI.4458-13.2014
33. Turncliff R, DiPetrillo L, Silverman B, Ehrich E. Single- and multiple-dose pharmacokinetics of samidorphan, a novel opioid antagonist, in healthy volunteers. Clin Ther. 2015;37(2):338–348. doi:10.1016/j.clinthera.2014.10.001
34. Zyprexa [package insert]. Indianapolis, IN: Eli Lilly and Company; 2021.
35. Revia [package insert]. Pomona, NY: Duramed Pharmaceuticals, Inc; 2013.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.