")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 11 » Issue 1
In vitro modeling of COPD inflammation and limitation of p38 inhibitor — SB203580
Authors Meng A, Zhang X, Wu S, Wu M, Li J, Yan X, Kopec-Harding K, Wu J
Received 3 November 2015
Accepted for publication 2 February 2016
Published 29 April 2016 Volume 2016:11(1) Pages 909—917
DOI https://doi.org/10.2147/COPD.S99810
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Richard Russell
Video abstract presented by Dr Aihong Meng.
Views: 301
Aihong Meng,1 Xiaopeng Zhang,2 Siyu Wu,1 Mingxia Wu,1 Jing Li,1 Xixin Yan,1 Kamilla Kopec-Harding,3 Jiakai Wu4
1Respiratory Division, The Second Hospital of Hebei Medical University, 2Department of Thoracic Surgery, Hebei Province General Hospital, Shijiazhuang, Hebei, Peoples’ Republic of China; 3Centre for Musculoskeletal Research, 4Centre for Respiratory and Allergy, Institute of Inflammation and Repair, University of Manchester, Manchester, UK
Background: Systemic inflammation and steroid resistance are the hallmarks of COPD. We examined the impact of p38 inhibitor (SB203580) in in vitro assays of systemic inflammation using pulmonary cells and patients’ sera.
Objective and methods: Data from 66 COPD patients and 15 age-/sex-matched healthy controls were compared. Interleukin-10 (IL-10), tumor necrosis factor-α (TNF-α), and CCL5 were measured in serum samples and culture media from peripheral blood mononuclear cells. The impact of sera on IL-10 and CCL5 expression in alveolar macrophage cell line (MH-S) was examined. The in vitro effects of SB203580 on lipopolysaccharide-induced inflammation were investigated.
Results: Peripheral blood mononuclear cells from Global Initiative for Chronic Obstructive Lung Disease (GOLD) D patients produced more CCL5 and TNF-α, and less IL-10 compared to GOLD A–C patients. SB203580 treatment suppressed CCL5 and TNF-α and stimulated IL-10 production; however, the effect of SB203580 on IL-10 was lower in the COPD group. Culture of MH-S cells with COPD serum showed a significant increase in CCL5 and a significant decrease in IL-10 compared to healthy serum. This effect was not suppressed with SB203580 treatment.
Conclusion: COPD serum has a potent proinflammatory effect on pulmonary cells. Inhibition of p38 phoshorylation had a limited effect in restoring impaired lymphocyte function and suppressing inflammation induced by COPD serum, implying important p38-independent inflammatory mechanisms in COPD.
Keywords: COPD, p38MAPK inhibitor, macrophage, CCL5, TNF-α, IL-10
Introduction
Coexistence of pulmonary as well as systemic inflammation is a hallmark of COPD.1,2 Although our understanding of the role of lung inflammation has been significantly advanced by studying lung histology, bronchoalveolar lavage (BAL) fluid, and induced sputum from patients,3–6 our knowledge of systemic inflammation in COPD remains sparse.
Elevated systemic inflammation is associated with increased disease severity and weight loss.1,7,8 Increased albumin levels in sputum samples of COPD patients are negatively associated with lung function. This may imply that plasma extravasation contributes to inflammation and disease progression.9 However, functional evidence of the role of systemic inflammation in disease pathogenesis is lacking.
Despite the extent of inflammation present in COPD patients, inhaled corticosteroids (ICSs) have little effect on either disease progression or mortality in most patients.10 This steroid resistance means that alternative anti-inflammatory medication is needed. The p38 mitogen-activated protein kinase (MAPK) signaling pathway is a major proinflammatory mechanism in COPD11 and has recently been the focus of much research effort in the field, culminating in several clinical studies.12,13 Lung immunohistochemistry showed increased p38 MAPK activation in COPD patients14 with increased phosphorylation in alveolar macrophages and in epithelial, CD20+, and CD8+ cells.15 In COPD alveolar macrophages, p38 MAPK activation is corticosteroid insensitive,16 which may be important in a particular subset of COPD macrophages. Several additional in vitro studies have observed decreased cytokine production by treating COPD pulmonary cells with p38 inhibitors.15 This evidence suggested that inhibition of p38 phosphorylation could be a rational therapeutic strategy in COPD.
To investigate the influence of blood exudates on pulmonary cells, in this study, we cultured MH-S alveolar macrophage cell line with serum from Global initiative for chronic Obstructive Lung Disease (GOLD) C/D patients and healthy controls. The effectiveness of p38 inhibitor (SB203580) in this serum-induced cell model was also investigated. In addition, pooled peripheral blood mononuclear cells (PBMCs) from patients of different GOLD groups under the treatment of SB203580 were assessed to identify the potential sources of proinflammatory mediators present in the blood and the cellular responses from PBMCs.
Methods
Study population
This study was approved by the ethical committee of the Second Hospital of Hebei Medical University. All subjects gave written informed consent.
COPD patients (n=66) were recruited according to the following criteria: forced expiratory volume in 1 second (FEV1)/forced vital capacity (FVC) ratio <0.7 and one or more of the following key indicators: 1) progressive and/or persistent dyspnea, 2) chronic cough, 3) chronic sputum production, and 4) history of exposure to risk factors (tobacco smoke).
Definition of COPD severity
COPD severity was determined according to 2011 GOLD guidelines A–D, based on symptoms, airflow obstruction, and exacerbation history. Symptom burden was measured by either the modified Medical Research Council (mMRC) questionnaire or the COPD assessment test (CAT). Classification of COPD severity is as follows (Table 1).
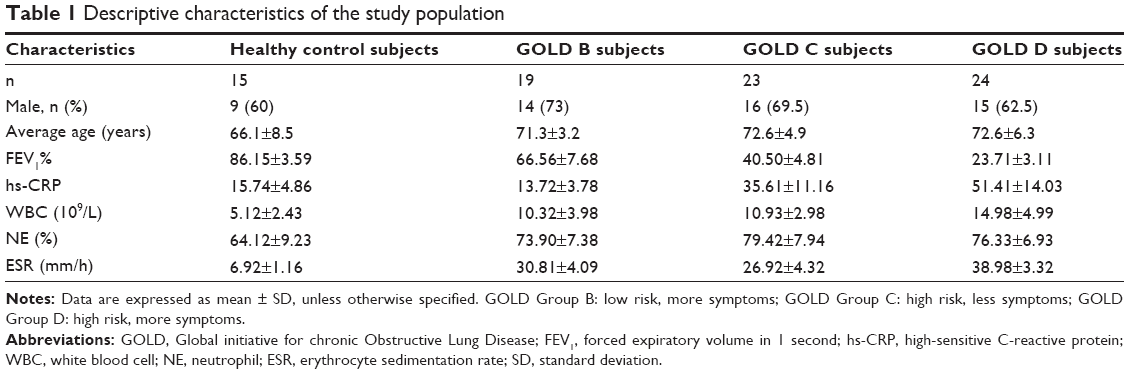 | Table 1 Descriptive characteristics of the study population |
GOLD A
Low symptom burden (mMRC of 0–1 or CAT <10), FEV1 of 50% or greater, and low exacerbation rate (0–1/year).
GOLD B
Higher symptom burden (mMRC ≥2 or CAT ≥10), FEV1 of 50% or greater, and low exacerbation rate (0–1/year).
GOLD C
Low symptom burden (mMRC of 0–1 or CAT <10), FEV1 <50%, and/or high exacerbation rate (≥2/year).
GOLD D
Higher symptom burden (mMRC ≥2 or CAT ≥10), FEV1 <50%, and/or high exacerbation rate (≥2/year).
Fifteen age- and sex-matched healthy volunteers were recruited as a control group. The exclusion criteria were severe cardiovascular disease, severe digestive disease, malignancies, kidney disease, diabetes, and other respiratory disease such as bronchiolitis, bronchiectasis, tuberculosis, and pulmonary embolism.
Procedures and sample collection
FEV1 was measured before discharge (MicroLoop 3535; Micro Medical Ltd, Rochester, UK). Blood was collected on the same day. All subjects fasted for at least 8 hours before collecting blood. Venous blood (3 mL) was collected in a tube without anticoagulant and centrifugated at 3,000 rpm for 10 minutes and then refrigerated at −80°C to detect cytokines; venous blood (5 mL) was collected in a similar manner for PBMC isolation. An aliquot of blood of each participant was sent to the blood testing department of the Second Hospital of Hebei Medical University to examine the white blood cell (WBC) count, percentage neutrophil (NE%), and erythrocyte sedimentation rate (ESR). Serum extraction and PBMC isolation were performed within 2 hours of blood withdrawal. Both serum and PBMC samples were maintained at −80°C until use.
Cell culture
Cell morphology appeared normal in all the conditions tested. After completion of the experiments, cell supernatants were collected and stored, cells were detached using trypsin methods and extracted for protein, and a small aliquot of cells was used for viability test; in all conditions tested, the viability was above 95% graph.
PBMC culture
Another 5 mL of blood was collected in a tube with anticoagulant ethylenediaminetetraacetic acid (EDTA) to separate PBMCs. PBMCs were isolated by density gradient sedimentation. PBMCs from the same severity group were pooled (1×106 cells/mL), and 1 mL of sample was added to each well in a 24-well plate and supplemented with 10% of inactivated serum. Pooled PBMC samples were cultured in either RPMI1640 culture media with 10% fetal calf serum or media supplemented with serially diluted SB203508 (5, 10, or 15 μM final concentration) for 1 hour before the addition of lipopolysaccharide (LPS, 1 μg/mL) or vehicle control. After 90 minutes, cell culture media were collected and stored at −80°C. Each condition was performed in triplicate.
Mouse alveolar macrophage cell line culture (MH-S cell line)
The murine alveolar macrophage MH-S cell line was purchased from ATCC (CRL-2019). MH-S cell lines were cultured in RPMI1640 medium with 10% fetal calf serum. Cells were plated into 24-well plates (5×106 cells/mL) by adding 1 mL per well. After 24 hours, culture medium was replaced with fresh RPMI1640 supplemented with 1%, 5%, or 10% pooled plasma, collected from COPD patients (pooled samples from 47 GOLD C or D patients) or healthy volunteers (pooled samples from 15 healthy volunteers). After 6 hours of culture, SB203580 or vehicle control was added for 20 minutes prior to stimulation for 6 hours with either LPS (1 μg/mL) or vehicle control. Each condition was performed in triplicate.
Measurement of cytokines and hs-CRP from serum or culture media
Levels of heat-sensitive C-reactive protein (hs-CRP), CCL5, tumor necrosis factor-α (TNF-α) and interleukin-10 (IL-10) from either serum or cell culture media were measured by commercial ELISA kits (Boster, Wuhan, People’s Republic of China).
Statistical analysis
All the three serum cytokines were normally distributed; one-way analysis of variance (ANOVA) was used to analyze the group differences. Multiple analyses were adjusted by Bonferroni correction and P<0.05 was considered significant. Error bar represents 95% confidence interval. For PBMC and MH-S cell line culture, each cell culture condition was performed in triplicate. Data are expressed as mean ± standard deviation (SD). All analyses were conducted using SPSS 22.0 software (IBM Corporation, Armonk, NY, USA).
Results
Subjects characterization
Majority of the recruited COPD patients were male (Table 1). The levels of biomarkers for systemic inflammation (hs-CRP, WBC, and ESR) increased with increasing GOLD stage (Table 1).
Serum concentrations of IL-10, CCL5, and TNF-α during COPD exacerbation is dependent on GOLD stage
Healthy volunteers had significantly higher levels of serum IL-10 and significantly lower levels of CCL5 and TNF-α compared to all COPD patient groups (Figure 1A–C). Serum IL-10 decreased with increasing GOLD stage and was significantly lower in GOLD stage D patients compared to GOLD B patients. CCL5 and TNF-α levels increased significantly with each GOLD stage (Figure 1A–C).
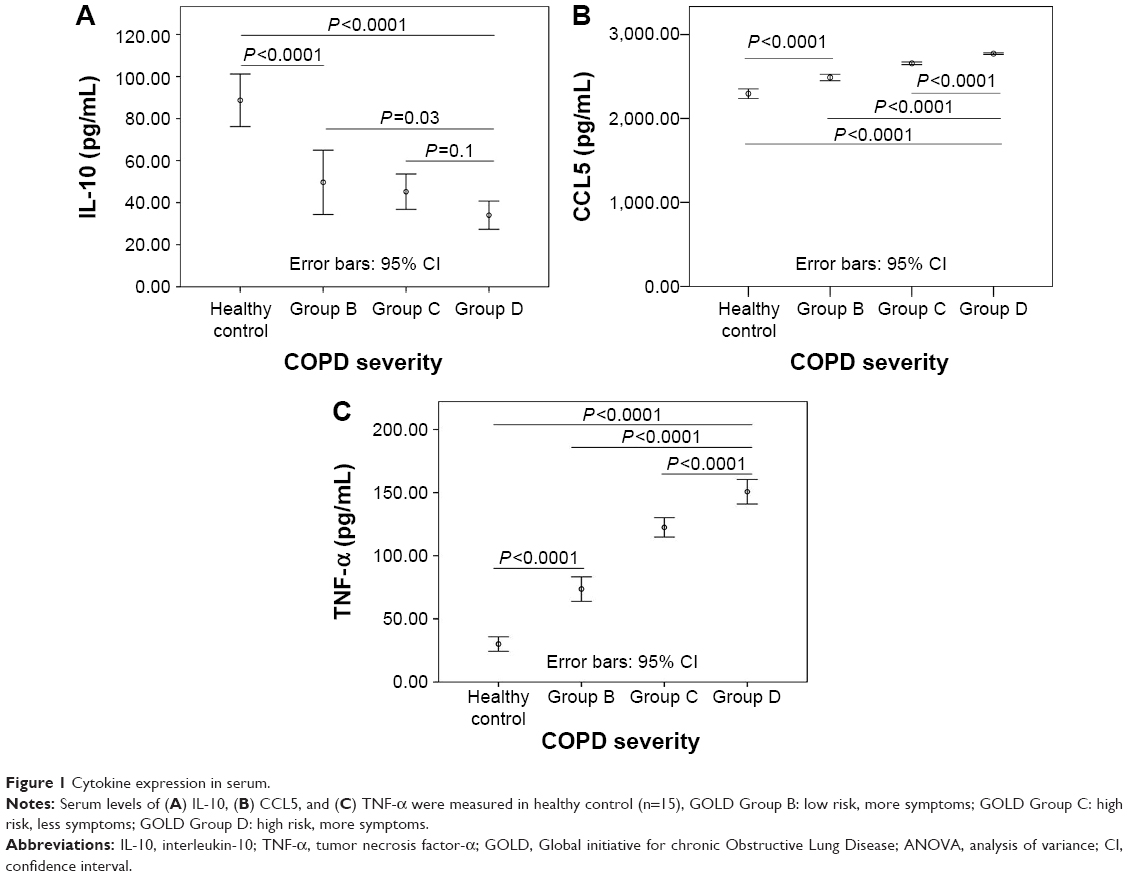 | Figure 1 Cytokine expression in serum. |
Cytokine production in pooled PBMC cultures
Pooled PBMC samples were used to assess the cellular cytokine response to an inflammatory stimulus (LPS) in the presence or absence of the p38 inhibitor SB203580.
IL-10
Unstimulated PBMCs from healthy donors produced significantly higher levels of IL-10 compared to PBMCs from COPD patient groups. PBMCs from COPD patients produced less IL-10 at baseline corresponding to increasing GOLD stage (Figure 2A). LPS stimulation had no significant effect on IL-10 production from PBMCs except those from GOLD B patients, where IL-10 levels were significantly lower (P=0.001). SB203580 pretreatment showed a profound dosage-dependent induction of IL-10 in combination with LPS, but the levels of IL-10 expression in healthy PBMCs were highly significant than that in COPD patients.
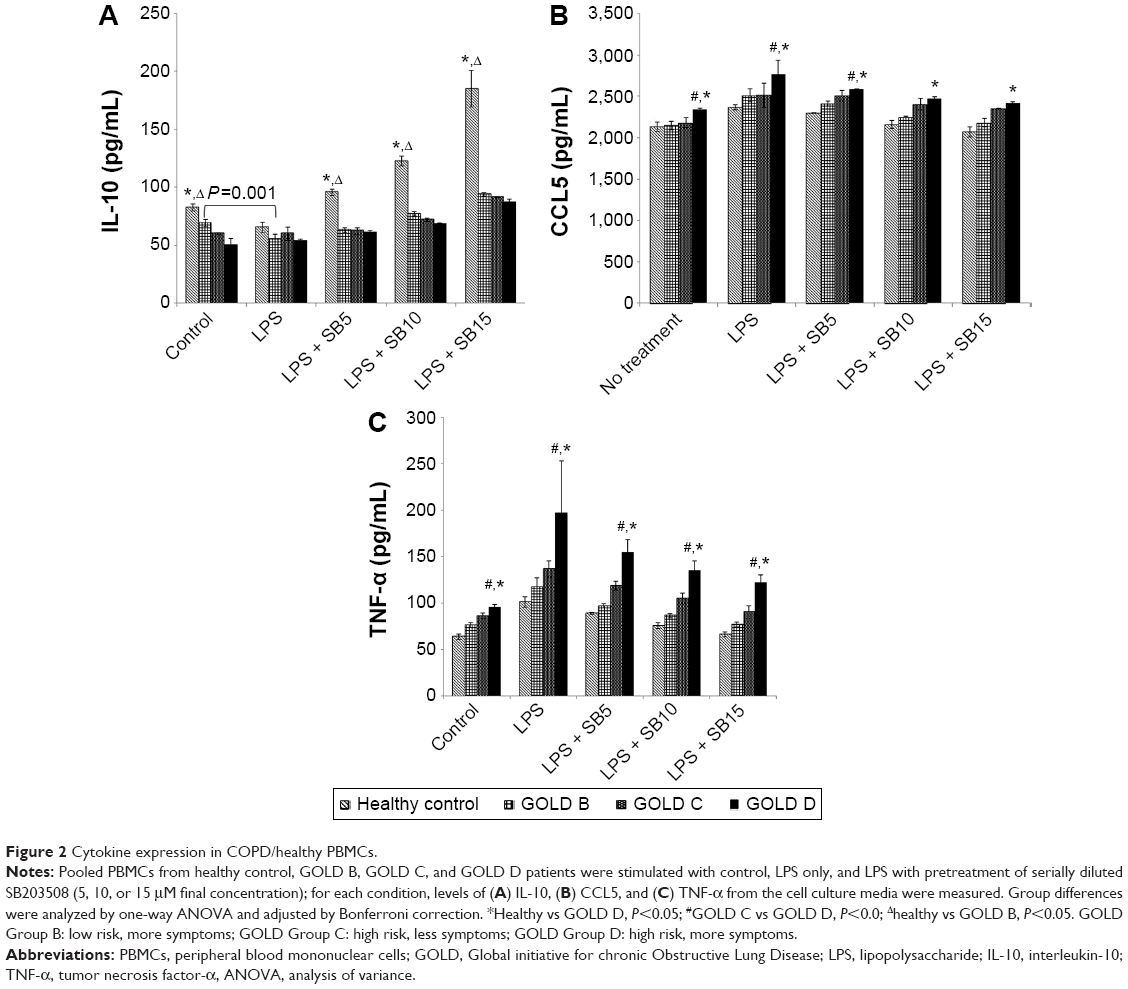 | Figure 2 Cytokine expression in COPD/healthy PBMCs. |
CCL5
Unstimulated PBMCs from GOLD D patients produced significantly higher levels (P<0.02) of CCL5 than PBMCs from less severe patients or healthy controls (Figure 2B). Levels of CCL5 were not significantly different between healthy controls and GOLD B and C patients (Figure 2B, P=1). LPS stimulation increased CCL5 in all the four groups; however, GOLD D PBMC still produced the highest levels of CCL5. SB203580 treatment suppressed CCL5 production in a dosage-dependent manner, except in PBMCs from GOLD C patients, where SB203580 had no significant effect on CCL5 (Figure 2B, P=0.38).
TNF-α
Baseline release of TNF-α increased in line with COPD severity (P<0.0001). LPS stimulation significantly potentiated TNF-α production, whereas SB203580 pretreatment inhibited it in all the groups in a dosage-dependent manner (Figure 2C). PBMC from GOLD D patients produced consistently more TNF-α in all conditions tested.
Cytokine expression by serum-stimulated MH-S cell line
The culture media were supplemented with various concentrations of pooled serum from healthy volunteers or GOLD C/D patients to observe the impact of serum on MH-S cell line and the influence of SB203580 treatment in terms of cytokine production.
IL-10
In the control (no stimulation) condition, 1% of serum supplement from GOLD C/D patients (COPD group) showed significant inhibition of IL-10 expression compared to cells cultured in 1% healthy serum; however, this effect did not seem to be dosage dependent as increased percentage of COPD serum concentration did not further suppress IL-10 expression (Figure 3A). LPS increased IL-10 production in all serum conditions tested, but the absolute amount was significantly higher in the healthy group (Figure 3A). There was no difference in IL-10 induction by LPS with varying serum concentrations in either group. SB203580 pretreatment suppressed IL-10 production, but the levels were still significantly higher than in the unstimulated cells (Figure 3A).
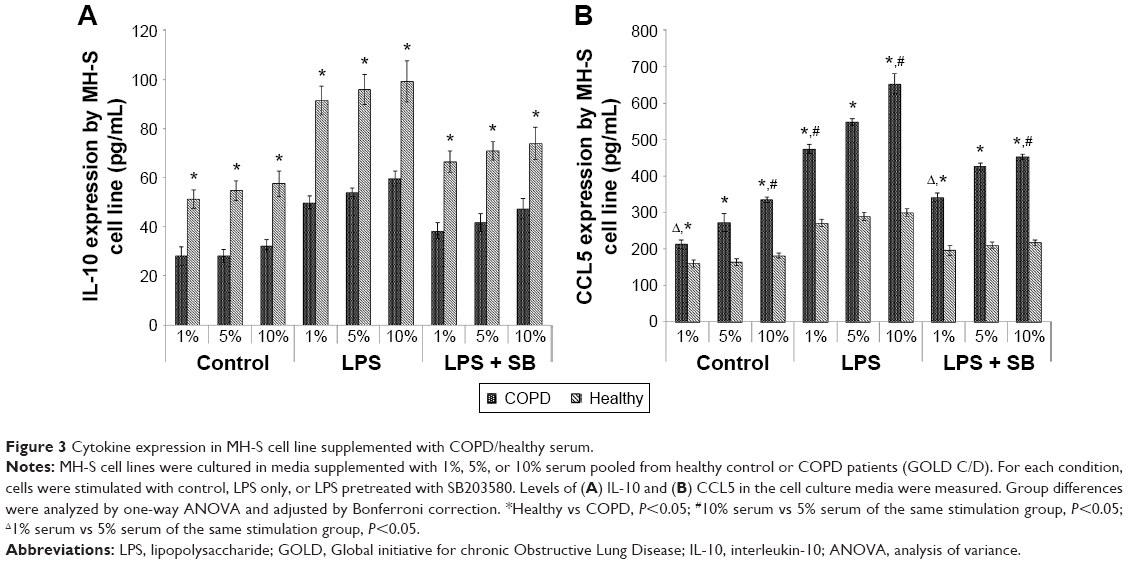 | Figure 3 Cytokine expression in MH-S cell line supplemented with COPD/healthy serum. |
CCL5
Unstimulated cells cultured in 1% COPD serum expressed significantly higher levels of CCL5 compared to healthy serum (Figure 3B). COPD serum showed a strong dosage-dependent stimulation of CCL5 production at baseline (Figure 3B). Ten percent COPD serum supplement enhanced CCL5 levels by 50% compared to cells cultured in 1% COPD serum. In contrast, there was no significant difference in CCL5 production with increasing amounts of healthy serum (Figure 3B). LPS stimulation increased CCL5 production in both COPD and healthy groups; however, this was significantly potentiated in the COPD group. SB203580 pretreatment significantly suppressed LPS-induced CCL5 production in all serum conditions, but the levels of CCL5 in the COPD group were still significantly higher than in the healthy group (Figure 3B).
Discussion
Main finding
Serum CCL5 and TNF-α levels significantly increased but IL-10 level significantly decreased with the progression of GOLD stage. In line with this observation, PBMCs from GOLD D patients produced higher levels of TNF-α and CCL5 but lower levels of IL-10 compared to less severe groups or healthy controls. LPS stimulation induced CCL5 and TNF-α expression in PBMCs but had little effect on IL-10 expression; SB203580 pretreatment prior to LPS stimulation induced IL-10 and suppressed TNF-α and CCL5 expression in a dosage-dependent manner. Of note, SB203580 pretreatment significantly induced IL-10 expression in healthy control group compared to patients of any GOLD groups. Using MH-S cell line, we demonstrated that 1% COPD serum supplement can suppress IL-10 expression and induce CCL5 expression. The induction of CCL5 by COPD serum was dosage dependent; however, the suppression of IL-10 by COPD serum remained constant, despite increase of serum percentage. The effect of LPS stimulation and SB203580 pretreatment on IL-10 expression in MH-S cell line culture was distinct from that of PBMC, where LPS stimulation increased IL-10 expression and SB203580 pretreatment suppressed IL-10 expression.
Interpretation
Severity of COPD is positively associated with systemic inflammation. The presence of inflammatory markers in circulation, sputum, and bronchoalveolar fluid suggests that systemic inflammation is one of the potential mechanisms responsible for both COPD and metabolic syndrome. Physical inactivity, skeletal muscle dysfunction, hypogonadism, and steroid use are also important causes of the metabolic syndrome in COPD.17 In our study, WBC count, ESR, and the levels of hs-CRP increased as the severity of COPD increased. This is consistent with a similar observation where elevated plasma inflammation-sensitive plasma protein levels, such as fibrinogen, ceruloplasmin, α1-antitrypsin, haptoglobin, and orosomucoid, were associated with an increased incidence of hospitalizations of patients with COPD.1
In line with that observation, elevated serum levels of proinflammatory cytokines (TNF-α and CCL5) and attenuated levels of anti-inflammatory cytokine (IL-10) were observed to be increased with COPD severity. Elevated levels of CCL5 were previously reported in serum,18 sputum,19 BAL fluid,20 and airway wall21 of COPD patients. The physiological impact of elevated CCL5 in serum is unclear. A recent study showed that in the lung, the gain-of-function allele in CCL5 is associated with severe emphysema in COPD and has been linked to small airways disease caused by repeated exposure to stimuli such as respiratory viruses like HRV and RSV; these viruses are detected in approximately half of the COPD exacerbations. In addition, infection with viruses was suggested to underlie the airway eosinophilia observed in some COPD patients.22 Sputum eosinophilia is an important predictor of time to exacerbation after ICS withdrawal (Liesker, 2011). High levels of TNF-α in sputum and serum were also reported; elevated levels of TNF-α in serum were associated with weight loss among COPD patients,7,8 and overexpression of TNF-α in the lung was suggested to be a driving factor for the inflammatory process.23–26
However, there were mixed reports regarding the relationship between blood IL-10 levels and COPD. Using immunotrapping assay, Moermans et al showed that IL-10 was significantly higher in the blood of patients with COPD compared to healthy controls;27 however, this results contradict with their sputum cell culture where cells from COPD patients released significantly less IL-10.27 In contrast, using ELISA, Zhang et al showed significantly higher levels of serum IL-10 from healthy nonsmokers compared to healthy smokers and COPD patients (GOLD stages I–IV), but IL-10 levels were similar between patients with different GOLD stages.28 Our serum IL-10 level was broadly similar to that reported by Zhang et al; however, we observed significantly lower serum IL-10 levels with increasing COPD severity. The difference between the two results might be due to the nature of the study design. In our study, blood was collected in the morning after at least 8 hours of fasting; however, the time of blood collection was not stated by Zhang et al.28
PBMCs from the GOLD D patients produced significantly higher levels of CCL5 and TNF-α but significantly lower level of IL-10 compared to the less severe counterparts and healthy control. This finding is consistent with the cytokine levels measured in serum. Altered peripheral lymphocyte functions in COPD patients have previously been reported.29 COPD PBMCs have fewer CD4+ cells that produce IL-10. It is unclear what drives the altered lymphocyte function; however, lymphocytes travel between the lung and circulatory compartments via lymph nodes.30 So, impaired lymphocyte function in peripheral blood may have a pathological impact on the lung. Furthermore, we observed a weakened SB203580 induction of IL-10 expression in the PBMCs of all the three GOLD groups. This observation may reflect an important limitation of SB203580 in the treatment of COPD, especially when trials intend to administer this drug orally.
Since blood exudation is a significant feature for COPD, we investigated the influence of blood exudates on pulmonary cells by culturing MH-S cells with human serum. Strikingly, MH-S cells cultured in 1% COPD serum produced significantly less IL-10 and significantly more CCL5 compared to serum from healthy volunteers. This indicates that COPD serum is a potent inducer of lung inflammation. Moreover, we observed a dosage-dependent effect of COPD serum on CCL5 production, but not for IL-10. We speculate that inhibition is already maximal at the lowest serum concentration. Interestingly, LPS stimulation after SB203580 pretreatment had the opposite effect on IL-10 production in MH-S cells compared to PBMCs. SB203580 suppressed LPS-induced IL-10 production in MH-S cells, whereas it potentiated LPS-induced IL-10 production in PBMCs. Of note, in spite of SB203580 pretreatment, cells cultured in COPD serum released significantly less IL-10 and significantly more CCL5 compared to healthy counterparts, suggesting that the effect of serum on cytokine expression maybe independent of the p38 pathway. Our previous study showed that LPS-mediated overproduction of TNF-α and IL-10 by MH-S is mediated at least partially by the MAPK pathway.31 Combining dexamethasone and SB203580 showed strong inhibition on the LPS-induced IL-10 secretion and STAT3 phosphorylation, which might reflect a very important drawback from the combined use of both anti-inflammatory agents.32 The current study showed that elevated p38 pathway is evident in COPD; however, recent clinical trials using p38 inhibitor showed mixed results,12,13 which implies that dysregulated p38 pathway may not be a generic pathway in every COPD case. One of the aims of this work was to explore the limitation of using p38 inhibitor as a potential anti-inflammatory drug in COPD. The result of this work demonstrated that non-diseased cells (MH-S) could be stimulated to become proinflammatory by exposing them to the serum of COPD patients, whereas immune cells (PBMCs) from COPD patients were proinflammatory even in the absence of serum. In both cases, p38 inhibitor showed limited effect in suppressing the proinflammatory cytokine production, which suggested the inflammatory pathway might only be partially dependent on p38 pathway.
Limitation
Although the MH-S cell line showed a good functional resemblance as primary alveolar macrophages, further validation using primary cells is needed.
The use of pooled PBMCs and serum for cell culture experiments provided a simple overview between cytokine response, disease severity, and cellular response to SB203580 treatment; however, even within the same GOLD groups, the symptoms may be driven by different disease mechanisms.33 Therefore, a future study investigating cellular responses of individual patients is essential to provide a clearer linkage between cytokine profiles (by means of a machine-learning approach) and clinical outcomes.
COPD is a complex disease that can be influenced by multiple factors such as genetic predisposition (eg, α1-antitrypsin), cigarette smoking, and bacterial/viral infection.34 LPS-induced PBMCs and MH-S cell line culture were oversimplified models to reflect inflammation in COPD. A more comprehensive study using additional stimuli such as cigarette extract followed by downstream omic analysis (transcriptomics and proteomics) might provide further understanding of the complex interplay between stimuli and cellular response.35
Conclusion
Systemic inflammation is highly associated with COPD severity. Dysregulation of cytokine production is evident in COPD PBMC, which can only partially be restored by SB203580 treatment. Serum from GOLD C and D patients is a potent inducer of inflammation in the pulmonary cells (MH-S cell line) and this effect was not reversed by SB203580 treatment.
Acknowledgments
This work was supported by a research grant from Hebei Provincial Health and Family Planning Commission (protocol 07047), Hebei Provincial Department of Science and Technology (protocol 15967753D), and the Second Hospital of Hebei Medical University.
Disclosure
The authors report no conflicts of interest in this work.
References
Engstrom G, Segelstorm N, Ekberg-Aronsson M, Nilsson PM, Lindgarde F, Lofdahl CG. Plasma markers of inflammation and incidence of hospitalisations for COPD: results from a population-based cohort study. Thorax. 2009;64(3):211–2115. | ||
O’Donnell RA, Peebles C, Ward JA, et al. Relationship between peripheral airway dysfunction, airway obstruction, and neutrophilic inflammation in COPD. Thorax. 2004;59(10):837–842. | ||
Saetta M, Di Stefano A, Turato G, et al. CD8+ T-lymphocytes in peripheral airways of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998;157(3 Pt 1):822–826. | ||
Pesci A, Balbi B, Majori M, et al. Inflammatory cells and mediators in bronchial lavage of patients with chronic obstructive pulmonary disease. Eur Respir J. 1998;12(2):380–386. | ||
Peleman RA, Rytila PH, Kips JC, Joos GF, Pauwels RA. The cellular composition of induced sputum in chronic obstructive pulmonary disease. Eur Respir J. 1999;13(4):839–843. | ||
Finkelstein R, Fraser RS, Ghezzo H, Cosio MG. Alveolar inflammation and its relation to emphysema in smokers. Am J Respir Crit Care Med. 1995;152(5 Pt 1):1666–1672. | ||
Karadag F, Karul AB, Cildag O, Yilmaz M, Ozcan H. Biomarkers of systemic inflammation in stable and exacerbation phases of COPD. Lung. 2008;186(6):403–409. | ||
Di Francia M, Barbier D, Mege JL, Orehek J. Tumor necrosis factor-alpha levels and weight loss in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1994;150(5 Pt 1):1453–1455. | ||
Minakata Y, Nakanishi M, Hirano T, Matsunaga K, Yamagata T, Ichinose M. Microvascular hyperpermeability in COPD airways. Thorax. 2005;60(10):882. | ||
Yang IA, Clarke MS, Sim EH, Fong KM. Inhaled corticosteroids for stable chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2012;7:CD002991. | ||
Saklatvala J. The p38 MAP kinase pathway as a therapeutic target in inflammatory disease. Curr Opin Pharmacol. 2004;4(4):372–377. | ||
Lomas DA, Lipson DA, Miller BE, et al. An oral inhibitor of p38 MAP kinase reduces plasma fibrinogen in patients with chronic obstructive pulmonary disease. J Clin Pharmacol. 2012;52(3):416–424. | ||
Watz H, Barnacle H, Hartley BF, Chan R. Efficacy and safety of the p38 MAPK inhibitor losmapimod for patients with chronic obstructive pulmonary disease: a randomised, double-blind, placebo-controlled trial. Lancet Respir Med. 2014;2(1):63–72. | ||
Renda T, Baraldo S, Pelaia G, et al. Increased activation of p38 MAPK in COPD. Eur Respir J. 2008;31(1):62–69. | ||
Gaffey K, Reynolds S, Plumb J, Kaur M, Singh D. Increased phosphorylated p38 mitogen-activated protein kinase in COPD lungs. Eur Respir J. 2013;42(1):28–41. | ||
Armstrong J, Harbron C, Lea S, et al. Synergistic effects of p38 mitogen-activated protein kinase inhibition with a corticosteroid in alveolar macrophages from patients with chronic obstructive pulmonary disease. J Pharmacol Exp Ther. 2011;338(3):732–740. | ||
Naik D, Joshi A, Paul TV, Thomas N. Chronic obstructive pulmonary disease and the metabolic syndrome: consequences of a dual threat. Indian J Endocrinol Metab. 2014;18 (5):608–616. | ||
Tsai JJ, Liao EC, Hsu JY, Lee WJ, Lai YK. The differences of eosinophil- and neutrophil-related inflammation in elderly allergic and non-allergic chronic obstructive pulmonary disease. J Asthma. 2010;47(9):1040–1044. | ||
Fujimoto K, Kubo K, Yamamoto H, Yamaguchi S, Matsuzawa Y. Eosinophilic inflammation in the airway is related to glucocorticoid reversibility in patients with pulmonary emphysema. Chest. 1999;115(3):697–702. | ||
Linden M, Rasmussen JB, Piitulainen E, et al. Airway inflammation in smokers with nonobstructive and obstructive chronic bronchitis. Am Rev Respir Dis. 1993;148(5):1226–1232. | ||
Panzner P, Lafitte JJ, Tsicopoulos A, Hamid Q, Tulic MK. Marked up-regulation of T lymphocytes and expression of interleukin-9 in bronchial biopsies from patients with chronic bronchitis with obstruction. Chest. 2003;124(5):1909–1915. | ||
Chung KF. Cytokines in chronic obstructive pulmonary disease. Eur Respir J Suppl. 2001;34:50s–59s. | ||
Rich EA, Panuska JR, Wallis RS, Wolf CB, Leonard ML, Ellner JJ. Dyscoordinate expression of tumor necrosis factor-alpha by human blood monocytes and alveolar macrophages. Am Rev Respir Dis. 1989;139(4):1010–1016. | ||
Levine SJ, Larivee P, Logun C, Angus CW, Ognibene FP, Shelhamer JH. Tumor necrosis factor-alpha induces mucin hypersecretion and MUC-2 gene expression by human airway epithelial cells. Am J Respir Cell Mol Biol. 1995;12(2):196–204. | ||
Tosi MF, Stark JM, Smith CW, Hamedani A, Gruenert DC, Infeld MD. Induction of ICAM-1 expression on human airway epithelial cells by inflammatory cytokines: effects on neutrophil-epithelial cell adhesion. Am J Respir Cell Mol Biol. 1992;7(2):214–221. | ||
Wu T, Ikezono T, Angus CW, Shelhamer JH. Tumor necrosis factor-alpha induces the 85-kDa cytosolic phospholipase A2 gene expression in human bronchial epithelial cells. Biochim Biophys Acta. 1996;1310(2):175–184. | ||
Moermans C, Heinen V, Nguyen M, et al. Local and systemic cellular inflammation and cytokine release in chronic obstructive pulmonary disease. Cytokine. 2011;56(2):298–304. | ||
Zhang L, Cheng Z, Liu W, Wu K. Expression of interleukin (IL)-10, IL-17A and IL-22 in serum and sputum of stable chronic obstructive pulmonary disease patients. COPD. 2013;10(4):459–465. | ||
Zhu X, Gadgil AS, Givelber R, et al. Peripheral T cell functions correlate with the severity of chronic obstructive pulmonary disease. J Immunol. 2009;182(5):3270–3277. | ||
Lehmann C, Wilkening A, Leiber D, et al. Lymphocytes in the bronchoalveolar space reenter the lung tissue by means of the alveolar epithelium, migrate to regional lymph nodes, and subsequently rejoin the systemic immune system. Anat Rec. 2001;264(3):229–236. | ||
Meng AH, Zhang XP, Shi YN. The role of p-38MAPK and STAT3 in LPS-stimulated mouse alveolar macrophage. Exp Ther Med. 2014;8(6):1772–1776. | ||
Meng AH, Wang B, Zhang XP, Qi N, Liu DC, Wu JK. Additive suppression of LPS-induced IL-10 and TNF-g XP,pre-treatment of dexamethasone and SB203580 in a murine alveolar macrophage cell Line (MH-S). Inflammation. 2015;38:(3):1260–1266. | ||
Marsh SE, Travers J, Weatherall M, et al. Proportional classifications of COPD phenotypes. Thorax. 2008;63(9):761–767. | ||
Sandford AJ, Silverman EK. Chronic obstructive pulmonary disease. 1: susceptibility factors for COPD the genotype-environment interaction. Thorax. 2002;57(8):736–741. | ||
Reynier F, de Vos AF, Hoogerwerf JJ, et al. Gene expression profiles in alveolar macrophages induced by lipopolysaccharide in humans. Mol Med. 2012;18:1303–1311. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.