")
Back to Journals » OncoTargets and Therapy » Volume 12
In vitro mitochondrial-targeted antioxidant peptide induces apoptosis in cancer cells
Authors Zhan W, Liao X, Li L, Chen Z, Tian T, Yu L, Chen Z
Received 5 March 2019
Accepted for publication 18 July 2019
Published 6 September 2019 Volume 2019:12 Pages 7297—7306
DOI https://doi.org/10.2147/OTT.S207640
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sanjeev K. Srivastava
Wei Zhan*,1, Xin Liao*,2, Lianghe Li3, Zhongsheng Chen3, Tian Tian4, Lei Yu5, Zupeng Chen6
1Department of Colorectal Surgery, The Affiliated Hospital of Guizhou Medical University, Guiyang 550004, People’s Republic of China; 2Department of Imaging, The Affiliated Hospital of Guizhou Medical University, Guiyang 550004, People’s Republic of China; 3Department of Surgery, Clinical Medical College, Guizhou Medical University, Guiyang 550004, People’s Republic of China; 4Department of Pathophysiology, Basic Medical College, Guizhou Medical University, Guiyang 550004, People’s Republic of China; 5Department of Pathology, Guiyang Maternal and Child Health Hospital, Guiyang 550004, People’s Republic of China; 6Department of Neurosurgery, The First Affiliated Hospital of Zhejiang Chinese Medical University, Hangzhou 310006, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zupeng Chen
Department of Neurosurgery, The First Affiliated Hospital of Zhejiang Chinese Medical University, No. 54, Youdian Road, Hangzhou 310006, People’s Republic of China
Tel +86 05 718 707 2175
Email [email protected]
Introduction: Reactive oxygen species (ROS) are major contributors to cancer and involved in numerous tumor proliferation signaling pathways. Mitochondria are the major ROS-producing organelles, and ROS are produced from the synthesis of adenosine triphosphate and cell metabolism.
Methods: A novel mitochondria-targeted peptide, namely KRSH, was synthesized and characterized. KRSH consists of four amino acids; lysine and arginine contain positively charged groups that help KRSH target the mitochondria, while tyrosine and cysteine neutralize excessive endogenous ROS, thereby inhibiting tumorigenesis.
Results: The results indicated that KRSH is specifically inhibiting the growth of HeLa and MCF-7 cancer cell lines. However, MCF10A cells can resist the effects of KRSH even in a relative higher concentration. The dichloro-dihydro-fluorescein diacetate and MitoSOXTM Red assay suggested that KRSH drastically decreased the level of ROS in cancer cells. The mitochondrial depolarization assay indicated that treatment with KRSH at a dose of 50 nM may decrease the mitochondrial membrane potential leading to apoptosis of HeLa and MCF-7 cells.
Conclusion: In other studies, investigating rat liver mitochondria, the uptake of KRSH may reach 80% compared with that for mitoquinone. Therefore, KRSH was designed as a superior peptide antioxidant and a mitochondria-targeting anticancer agent.
Keywords: reactive oxygen species, ROS, oxidative stress, mitochondrial targeted, anticancer
Introduction
Mitochondria play multiple essential roles in eukaryotic cells. Primarily, they are responsible for the production of adenosine triphosphate (ATP) to support the cell metabolism. Different types of carbon sources, such as glycolysis, fatty acid, and amino acid, are transferred to ATP. All organic carbon sources will be metabolized to pyruvate and enter the Krebs cycle in the mitochondrial matrix through a reaction with nicotinamide adenine dinucleotide and flavin adenine dinucleotide. This reaction carries high energy electrons to the electron transport chain (ETC) in the inner mitochondrial membrane.1 This oxidative phosphorylation (OXPHOS) pathway is responsible for the production of most of the ATP yield. Except for the production of ATP, mitochondria are involved in apoptotic pathways by regulating the release of cytochrome c, which promotes cell death.2,3 In addition, mitochondria are involved in other forms of cell death, including necrosis, autophagy, and necroptosis.4
Regarding the energy engineer, mitochondria produce a large amount of cellular ROS, largely contributing to the development of cancer. The majority of mitochondrial ROS is leaking electrons from the ETC in the process of OXPHOS. A small amount of the mitochondrial ROS is produced through cell glycolysis and the tricarboxylic acid cycle.5 In addition, several enzymatic reactions generate non-mitochondrial ROS, such as peroxisomes and cytochrome P-450, as a byproduct. Nicotinamide adenine dinucleotide phosphate oxidase is identified as a typical ROS generator, contributing a large amount of extra-mitochondrial ROS.6 However, the production of mitochondrial ROS in normal tissues and cells is strictly controlled by endogenous antioxidants, such as glutathione, superoxide dismutase, and catalase. Following mitochondrial dysfunction, elevated levels of ROS will contribute to tumorigenesis signaling pathways. Increased ROS may also damage the DNA,7 proteins, and lipids, and ultimately lead to the development of cancer.
Considering the increasing problems of traditional therapeutic methods against cancer, targeted drugs which are more tumor-specific and less toxic (eg, antibodies,8,9 anti-angiogenesis agents,10,11 and other small molecules) appear more reliable.12 Several mitochondria-targeted small molecules have been assessed in anticancer research. A previous study indicated that 5 nmol/kg SkQ1 per day in drinking water reduced the level of ROS prevented the formation of a tumor.13 In addition, MitoTEMPOL, mitoquinone (MitoQ), and mitochromanol-acetate, likely displayed anticancer activity through mechanisms involving mitochondrial membrane depolarization and/or surges in the levels of mitochondrial superoxide.14
Peptides consist of several amino acids (ie, 2–50), with a specific domain demonstrating better targeting and delivery.15 Cell-penetrating peptides have been applied in numerous therapeutic areas, such as diabetes, cardiovascular disease, Alzheimer’s disease, and cancer.16,17 The most common application of such peptides is mitochondrial targeting, easily crossing the mitochondrial membrane to repair mitochondrial dysfunction.18
Materials and methods
Peptides synthesis
The KRSH and KRA peptides were synthesized using Liberty Blue peptide synthesizer (CEM corporation, USA) on Rink amide AM resin (146 mg with a resin loading of 0.684 mmol/g) using microwave aided Fmoc-d-Ala-OH, Fmoc-d-Lys(Boc)-OH, Fmoc-d-Arg(Pbf)-OH, Fmoc-d-Tyr(tBu)-OH, and Fmoc-d-Cys(Trt)-OH were used to assemble the peptide using standard Fmoc solid phase peptide synthesis (SPPS) protocols. As shown in Figure 1, the peptides were synthesized of 0.1 mmol and peptide couplings were performed by using Fmoc amino acid (5.0 eq) (Sigma, USA), N, N′-Diisopropylcarbodiimide (DIC) (Sigma, USA) (5.0 eq) and Oxyma (10.0 eq) (Sigma, USA) in DMF (total volume 4 ml) with the microwave irradiated for 4 min. Upon completion of SPPS, the N-terminal was acetylated on resin using capping solution (acetic anhydride 0.5 mL and DiPEA 0.87 mL in 5 mL DMF) for 1 hr at room temperature under nitrogen bubbling. Then resin was then flushed with DMF (3×5 mL) and DCM (2×5 mL). The peptides were deprotected and cleaved using cleavage cocktail (TFA/TIPS/EDT/H2O =92.5:2.5:2.5:2.5) for 1 hrs. Precipitation in MTBE/Hexane (1:1) yielded the peptide, which was purified by preparative RP-HPLC. The purity and identity were confirmed via analytical HPLC and high-resolution mass spectrometry.
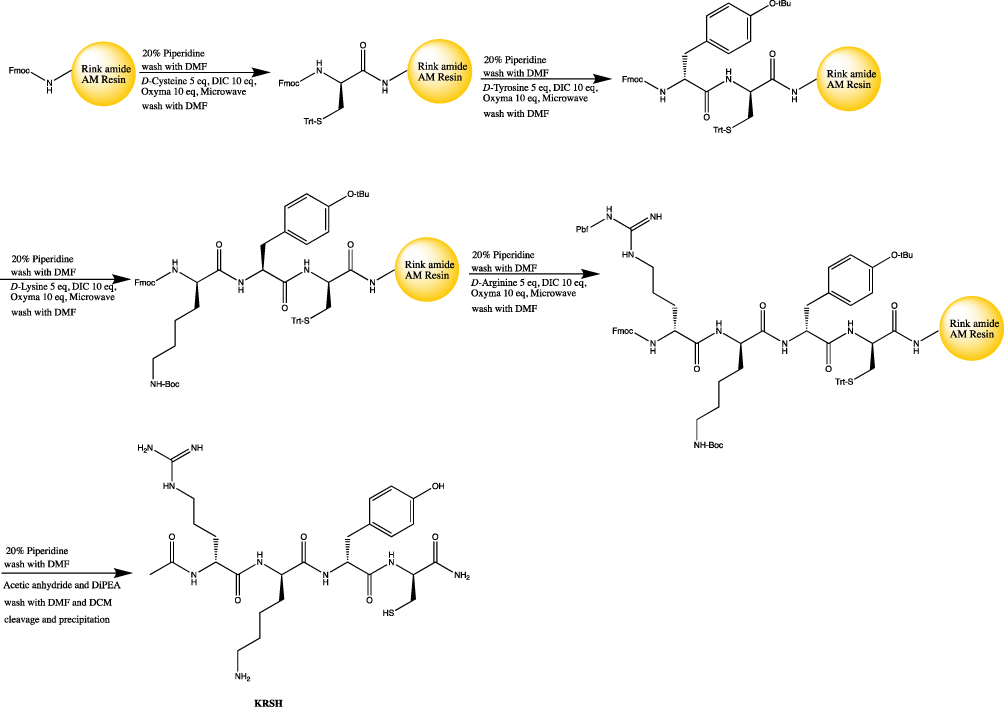 |
Figure 1 The scheme of solid phase peptide synthesis with microwave-aided. |
Cell culture and treatment
The MCF-7, HeLa, and MCF10A cells lines were cultured at 37 °C under a humidified atmosphere with 5% CO2 in Dulbecco’s Modified Eagle’s Medium basic media (C11995500BT; Gibco, USA), supplemented with 10% fetal bovine serum (16,000-044; Gibco, USA), containing penicillin (100 units/mL) and streptomycin (100 mg/mL; Gibco, USA) at 37 °C in a humidified atmosphere with 5% carbon dioxide. Cells were grown in six- or 96-well culture plates. The culture medium was replaced with fresh medium containing KRA and KRSH (50 nM) peptide solution and the cells were further incubated for 24 h. All cell lines were purchased from the National Infrastructure of Cell Line Resource platform (Beijing, China).
Neutral red (NR) assay
Cell survival was measured using the NR assay (Sigma-Aldrich, USA). Cells were seeded into 96-well culture plates with culture medium (with KRA and KRSH) and incubated for 24 h. For the cell survival experiment, different concentrations of KRSH solution were added and cells were further grown for 24 h. After washing with phosphate-buffered saline (PBS), 100 μl NR medium (50 μg/mL in Dulbecco’s Modified Eagle’s Medium) was added to all wells. Plates were incubated at 37 °C under a humidified atmosphere with 5% carbon dioxide for 2 h. The NR medium was removed, and wells were gently washed with PBS. Subsequently, 150 μl desorb solution (glacial acid: ethanol: H2O =1%: 50%: 49%) was used to extract NR from living cells. Plates were gently agitated for 5 min and the optical absorbance was recorded at 540 nm using an Infinite F-2000 microplate reader (Tecan, Switzerland).
Detection of ROS
The levels of intracellular and mitochondrial ROS in MCF-7 and HeLa cells were estimated using the fluorescence-labeled probe 2′,7′-Dichlorofluorescin diacetate (DCFH-DA) (Beyotime, Shanghai, China) and the mitochondrial superoxide indicator MitoSOXTM Red (Thermo Fisher Scientific, USA), respectively. The intensity of fluorescence was measured through flow cytometry (BD Biosciences, USA). Nuclei were counterstained with Hoechst 33342 (Beyotime, Shanghai, China). Images of cells were obtained using an AMG EVOS fluorescence microscope (Thermo Fisher Scientific, USA).
Colony formation assay
MCF-7 and HeLa cells were seeded in six-well plates (500 cells per well) with 1 mL culture medium. Cells were treated with KRA and KRSH for 24 h. After 1 week, colonies were fixed with 2 mL methanol for 5 min and stained for 30 min with Giemsa stain (Solarbio, Beijing, China). Colonies with >50 cells were scored as survivors and the Image Pro Plus 6.0 software was used to count the colonies in each picture.
5-ethynyl-2´-deoxyuridine (EdU) assay and release of cytochrome c
MCF-7 and HeLa cells were seeded in six-well plates and incubated overnight. Subsequently, the cells were incubated with KRA and KRSH for 24 h. Cell proliferation was tested using an EdU assay kit (KTA2030; Abbkine, USA) according to the instructions provided by the manufacturer. The release of cytochrome c was detected using an enzyme-linked immunosorbent assay (ELISA) kit (CSB-E08530h; Cusabio, USA) according to the instructions provided by the manufacturer.
Apoptosis assay
Cell apoptosis was assessed using FITC-Annexin V and propidium iodide (PI) staining (556,547; BD Pharmingen). HeLa and MCF-7 cells were grown in six-well plates for 24 h to achieve complete attachment prior to treatment with 50 nM KRA and KRSH peptide. After 24 h, the cells were trypsinized and washed twice with PBS. Subsequently, the number of cells was adjusted to 1×106 cells/mL using binding buffer. Cell suspension (100 μl) was mixed with 5 μl FITC-Annexin V in the dark at room temperature for 5 min, followed by the addition of 5 μl PI dye, and incubated for another 10 min. The cell samples were tested within 30 min using a flow cytometer (BD Biosciences, USA).
Mitochondrial membrane potential assay
The JC-1 probe, which forms monomers or aggregates under different mitochondrial membrane potentials, was employed to measure the mitochondrial membrane potential in cells. In intact polarized mitochondria, the JC-1 probe is presented as a JC-1 aggregate emitting a red signal, whereas depolarized mitochondria induce the formation of JC-1 monomers emitting a green signal. Cells were cultured in six-well plates after the indicated treatments, incubated with 1 mL pre-warmed JC-1 staining solution (5 μg/mL) (Solarbio, Beijing, China) at 37 °C for 20 min, and washed thrice with PBS. The cells were collected, and the intensity of fluorescence was measured through flow cytometry (BD Biosciences, USA). Mitochondrial membrane depolarization was indicated by an increase in the green/red fluorescence intensity ratio.
Concentration of KRSH peptide in mitochondria
Rat liver mitochondria were prepared as previously described to investigate the accumulation of Mito-N in mitochondria.19 The mitochondria (250 μg of protein per mL) were incubated for 5 min at 37 °C in 250 mM sucrose, 5 mM Hepes, and 1 mM ethyleneglycoltetraacetic acid (pH 7.4) supplemented with 500 nM KRSH, MitoQ, or dihydronicotinamide. The mitochondria were subsequently pelleted by centrifugation (16,000 g for 5 min), and the residual supernatant was removed. Buffer B (acetonitrile: water: trifluoroacetic acid =95: 5: 0.1) (250 μl) was added, and the mixture was immediately and vigorously vortexed. The samples were centrifuged, and the supernatant was transferred to fresh tubes and diluted using an equal volume of buffer A (acetonitrile: water: trifluoroacetic acid =5: 95: 0.1). The samples were analyzed immediately through high-performance liquid chromatography. The mitochondrial uptake of KRSH was determined as the ratio to the uptake of MitoQ.20
Statistical analysis
All data were analyzed using analysis of variance in the SPSS version 13 software and presented as the mean ± standard deviations. A p<0.05 denoted statistical significance.
Results
The KRSH peptide was synthesized and the purity was verified by analytical HPLC
As shown in Figure 2A, the KRSH is consist of the D-amino acid and analytical trace indicated that the purity of KRSH is good enough for the biological testing. HRMS (m/z): [M+H]+ calculated for C26H44N9O6S+ 610.3130, found 610.3097.
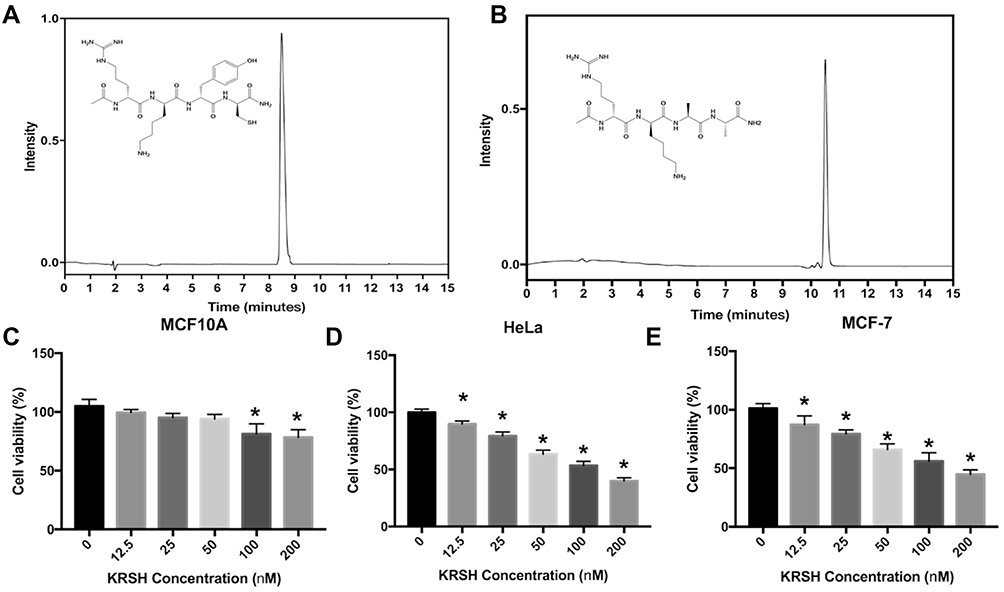 |
Figure 2 The analytical trace of the KRSH peptide and KRA (negative control) (A, B). Cell viability after treatment with the KRSH peptide at different doses (0–200 nM). The neutral red assay was employed to evaluate the survival rate of MCF10A, HeLa, and MCF-7 cells (C–E). *p<0.05 compared with control cells cultured in complete medium. |
The HPLC trace of negative control peptide, KRA, was shown in Figure 2B, the purity is good enough for next steps. HRMS (m/z): [M+H]+ calculated for C20H40N9O5+ 486.3147, found 486.3132.
KRSH peptide induces greater inhibition of growth in tumor cells versus normal cells
Cell toxicity was tested in three cell lines, namely MCF10A, HeLa, and MCF-7. As shown in Figure 2C–E, the KRSH peptide inhibited cell growth at high concentrations. However, its effect on tumor cells (HeLa and MCF-7) was more pronounced than that observed in normal cells (MCF10A). Notably, there was no significant difference between the treatment and control groups in MCF10A cells, even when the concentration of the KRSH peptide was increased to 50 nM. In contrast, notable cytotoxicity was found in tumor cell lines at a lower concentration (12.5 nM). The different effects of the KRSH peptide on tumor cells and normal cells may be attributed to disruption of the intracellular ROS balance, consequently interfering with cancer cell survival and metastasis. These finding could provide inspiration for future antitumor studies.21
KRSH peptide scavenges intracellular and mitochondrial ROS
DCFH-DA is an endogenous probe for the detection of ROS, which accumulates in cells and forms DCFH. It can react with ROS and is transferred to DCF with green fluorescence (Figure 3A and C). Tumor cells have higher levels of ROS than normal cells owing to the rapid proliferation rate. Therefore, chemotherapeutic drugs could be specifically designed to promote or suppress the levels of ROS and consequently antagonize tumor progression. As shown in Figure 3B and D, DCFH-DA and MitoSOXTM Red were employed to detect the levels of intracellular and mitochondrial ROS after treatment with KRA and the KRSH peptide. The results indicated that the KRSH peptide interfered with the accumulation of ROS in both HeLa and MCF-7 cells compared with the control and KRA-treated groups.
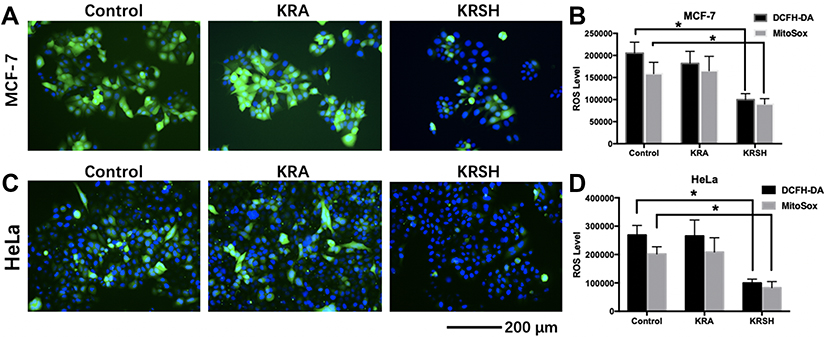 |
Figure 3 The levels of intracellular and mitochondrial ROS in different cell lines after treatment with KRA and the KRSH peptide for 24 h were detected using DCFH-DA and MitoSOXTM, and counterstained with Hoechst stain. The levels of ROS in MCF-7 cells (A, B) and HeLa cells (C, D) decreased significantly after treatment with the KRSH peptide. *p<0.05 compared with control cells cultured in complete medium.Abbreviations: ROS, reactive oxygen species; DCFH-DA, 2′,7′-dichlorofluorescin diacetate. |
Proliferation of HeLa and MCF-7 cells is inhibited by the KRSH peptide
The colony formation assay determines cell viability after treatment with cytotoxins. HeLa and MCF-7 cells were seeded into six-well plates and incubated with 50 nM KRA and KRSH peptide. At this concentration, the KRSH peptide drastically inhibited the proliferation of MCF-7 cells (Figure 4A and B) and HeLa cells (Figure 4A and C). Interestingly, there was no significant difference in the inhibition of cancer cell proliferation at 50 nM. Statistical analysis of the data (Figure 4B and C) suggested dose-dependent, distinct inhibitory effects on cell growth. This finding indicated that disruption in the levels of ROS may be an effective approach to killing tumor cells.
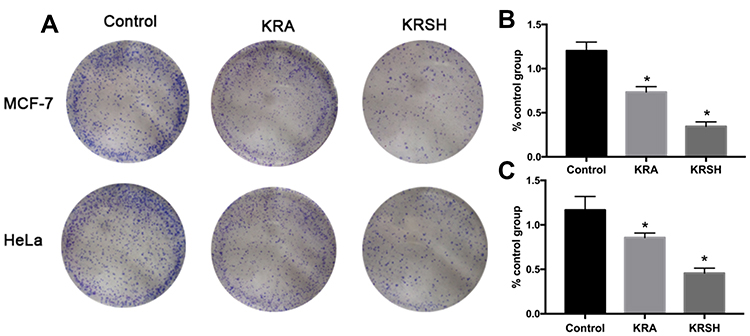 |
Figure 4 Colony formation assay was performed on (A, B) HeLa cells and (A, C) MCF-7 cells after treatment with 50 nM KRA and the KRSH peptide for 24 h using the Giemsa stain. The results are expressed as the mean ± SD from at least three independent experiments. *p<0.05 compared with control cells cultured in complete medium.Abbreviation: SD, standard deviation. |
The proliferation ability of cells was also investigated through EdU staining. As shown in Figure 5, DNA synthesis was inhibited in both cell lines treated with KRSH. The proportion of EdU-positive cell was decreased by approximately 50% after a 24-h incubation, suggesting the inhibition of tumor cell proliferation by KRSH.
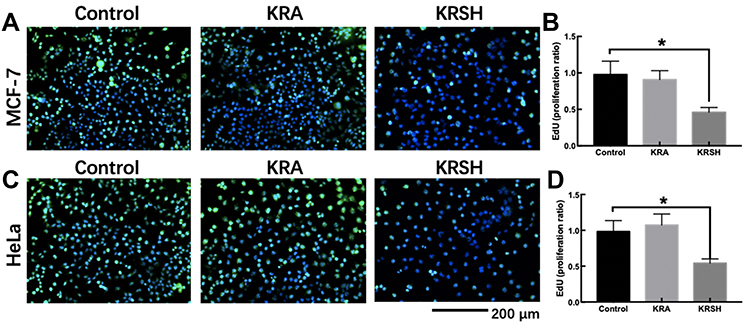 |
Figure 5 EdU staining to detect cell proliferation after treatment with KRSH. (A) MCF-7 cells were treated with EdU for 6 h prior to click reaction, and the data analysis is displayed in the graph (B). The analysis was performed to calculate the signal intensity in EdU-positive cells based on individual DAPI signal. (C) The proliferation of HeLa cells after treatment with the peptide was also detected using the EdU reaction system after a 6-h incubation. The statistical analysis is displayed in the graph (D) using the same calculation. *p<0.05, compared with control cells cultured in complete medium, one-way ANOVA.Abbreviations: EdU, 5-ethynyl-2’-deoxyuridine; DAPI, 4′,6-diamidino-2-phenylindole; ANOVA, analysis of variance. |
KRSH peptide induces apoptosis in HeLa and MCF-7 cells
Inhibited cells that are unable to perform mitosis activate the apoptotic pathways. Figure 6 shows the apoptotic population of HeLa and MCF-7 cells after treatment with KRA and the KRSH peptide. Late apoptosis increased to 12.5% and 17.8% in treated MCF-7 and HeLa cells respectively, in contrast to control cells (1.09% and 0.55%, respectively). Cell apoptosis was further investigated by determining the release of cytochrome c, which is involved in the apoptotic signaling triggered by mitochondria. Cell homogenates were tested after treatment with KRA and the KRSH peptide using an ELISA kit (Figure S2). The release of cytochrome c increased by approximately 10-fold in both cell lines, consistently with the results of Annexin V and PI staining.
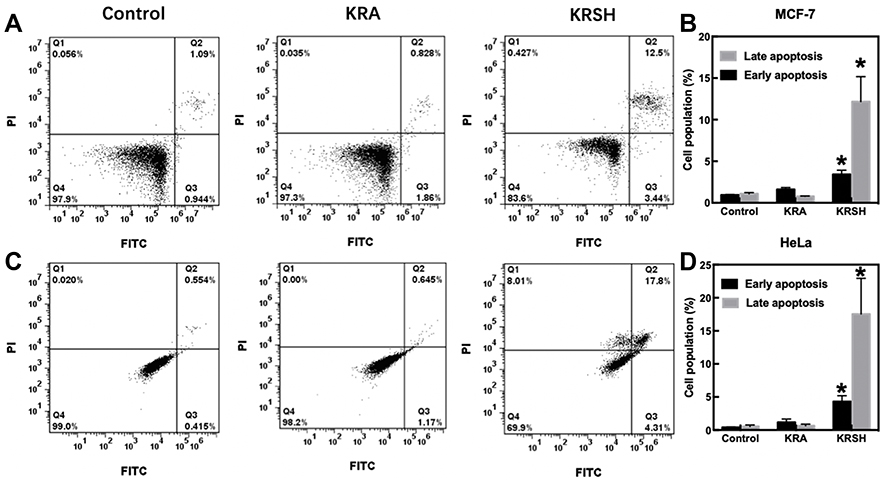 |
Figure 6 Cell apoptosis after incubation with 50 nM KRA and the KRSH peptide for 24 h. (A) Apoptosis in MCF-7 cells was distinguished through FITC-Annexin V coupled with PI staining. (B) The corresponding quantification of early and late apoptosis in MCF-7 cells. (C) Apoptosis in HeLa cells was determined using a flow cytometer, and the data analysis is displayed in (D). *p<0.05, compared with control cells cultured in complete medium, one-way ANOVA).Abbreviations: FITC, fluorescein isothiocyanate; ANOVA, analysis of variance. |
KRSH peptide induces mitochondrial depolarization
The protective effect of KRSH-induced apoptosis of cancer cells was further studied by examining mitochondrial depolarization. HeLa and MCF-7 mitochondria stained with JC-1 emitted red and/or green fluorescence. When JC-1 was accumulated into normal mitochondria, it appeared in the JC1 aggregate form (red fluorescence). The KRSH peptide decreased the mitochondrial potential and produced obvious green fluorescence compared with the control group. Therefore, the MCF-7 and HeLa cells showed a higher mitochondrial depolarization ratio compared with the control group (Figure 7).
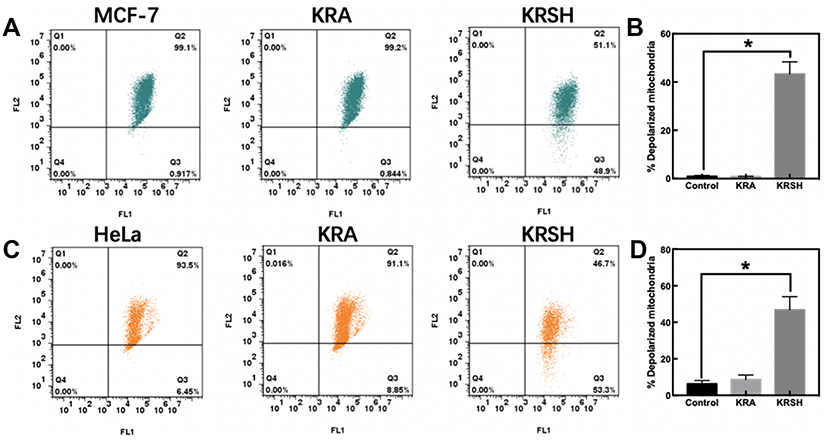 |
Figure 7 Mitochondrial membrane depolarization was determined using the JC-1 probe. (A) Depolarization in MCF-7 cells was detected after incubation with the peptide using a flow cytometer. (B) Quantitative analysis of the shift of mitochondrial red fluorescence to green fluorescence among groups, indicating the depolarization of the mitochondrial membrane. (C, D) HeLa cells were examined and analyzed using the same procedure. *p<0.05, compared with control cells cultured in complete medium, one-way ANOVA.Abbreviation: ANOVA, analysis of variance. |
KRSH peptide reaches a relatively high concentration in mitochondria
We measured the concentration of the KRSH peptide in rat liver mitochondria to investigate its distribution in the mitochondria. MitoQ is a well-investigated mitochondria-targeted small-molecule antioxidant, and its accumulation is measured using radio-labeled MitoQ. The accumulation of KRSH in mitochondria was approximately 80% compared with that measured for MitoQ (Figure S1).22
Discussion
Accumulating evidence suggests that the development of cancer requires an abundance of ATP for unlimited proliferation and/or survival. This means that mitochondrial metabolism is necessary for tumorigenesis. In the previous decade, the Warburg effect has been established as the prototypical model of cancer metabolism. This hypothesis suggested that “aerobic glycolysis” is the general way to produce sufficient energy for cancer cells, and may reflect impaired mitochondrial function.23 Recently, an increasing number of studies indicated that mitochondria in normal cells are structurally and functionally different from those present in malignant tumor cells.13,24 Mitochondria are the primary organelles involved in the production of intracellular ROS in the process of oxidative metabolism. However, endogenous antioxidants will scavenge ROS, thus controlling the levels of ROS. As a result of the highly effective OXPHOS, which occurs in cancer cells, a large amount of ROS is produced by the mitochondrial ETC. High levels of mitochondrial ROS (ie, superoxide anion and hydrogen peroxide) are required for signaling pathways to promote cancer cell proliferation and tumorigenesis.13,25 It has been reported that ROS produced from the ETC Complex III are involved in KRAS-mediated tumorigenesis through regulation of the ERK/MAPK signaling pathway.1 Another study indicated that overproduction of ROS and expression of Nox1 are associated with leukotriene B4 receptor 2-mediated survival of breast cancer cells.26
Mutations of mitochondrial DNA (mtDNA) are frequently observed in a wide range of cancer types.27 For instance, the most common mtDNA mutations in breast cancer are detected in the coding (dehydrogenase subunit 1 [ND1], ND4, ND5, and cytochrome b genes) or non-coding regions (D-loop) of the mitochondrial genome.28,29 Notably, a pathogenic mtDNA mutation in PC3 cells showed a drastic increase in the rate of tumor growth along with increased production of cellular ROS.30 In another study, ND6 mutations produced a deficiency in respiratory complex I activity, and were associated with overproduction of ROS.31
Although mitochondria are a promising target for the treatment of cancer, the delivery of antioxidants with high tumor specificity and lower toxicity into mitochondria remains a challenge. In the previous decades, Murphy et al developed the mitochondria-targeted antioxidant MitoQ, which contains a ubiquinone linked to the lipophilic penetrating cation triphenylphosphonium. With the assistance of the mitochondrial inner membrane potential (ΔΨ=140–180 mV), MitoQ can accumulate into the mitochondrial matrix by several hundred-fold.32 In addition to lipophilic cation targeting vectors, cell-penetrating peptides are commonly used and efficient vectors for the specific accumulation in mitochondria.33 Cell-penetrating peptides are a diverse family of peptides, consisting of 5–30 amino acids. These peptides can directly cross the mitochondrial membranes, and may rely on their small cationic group on the peptide side chains.34,35
According to the cell-permeable theory, we designed a peptide with high abundance of arginine and lysine, which can accumulate into mitochondria. Along the endocytic pathway, L-amino acids peptides are especially degraded by proteolytic enzymes, whereas D-amino acids peptide can decrase this degradation. Therefore, D-amino acid peptides are internalized through endocytosis at a higher level versus L-amino acids peptides.36 We synthesized the KRSH peptide using all the standard Fmoc D-amino acids to maintain a sufficient concentration of peptide. Similar to a study which tested all the amino acids, we found that antioxidant amino acids (ie, tryptophan, tyrosine, cysteine, and homocysteine) demonstrated good antioxidant activity.37 In addition, studies showed that N-acetyl cysteine exhibits strong potential for the inhibition of oxidative stress.38 N-acetyl cysteine may be a powerful antioxidant and precursor of glutathione,39 exerting a favorable effect on diabetes and Alzheimer’s disease.40,41 We selected cysteine and tyrosine as the antioxidants in KRSH to neutralize the overproduction of ROS, decrease the levels of ROS in cancer cells, and inhibit ROS-related pathways.
Conclusion
Overall, our data indicated that KRSH is a promising antioxidant for the degradation of ROS in cancer cells and down-regulation of ROS-related pathways. KRSH did not induce any side effect in MCF10A cells, as the levels of ROS in normal cells are relatively lower compared with those measured in cancer cells (data not shown). The colony formation assay indicated that the KRSH peptide can effectively inhibit the proliferation of MCF-7 and HeLa cells by ≤50%. The mitochondrial depolarization assay indicated that treatment with the KRSH peptide increased mitochondrial depolarization through an unknown mechanism. This peptide will offer insights relevant to the design of future cell-permeable peptide-based agents. Such agents may be protease resistant and promote their activity inside endosomes. This important approach may help generate agents that are cell-permeable at sufficient concentrations and, thereby, more relevant to in-vivo applications.36
Abbreviations
ROS, Reactive Oxygen Species; ETC, Electron Transport Chain; OXPHOS, Oxidative Phosphorylation; SOD, Superoxide Dismutase; CAT, Catalase; SPPS, Solid Phase Peptide Synthesis; DIC, N,N′-Diisopropylcarbodiimide; DMF, Dimethylformamide; DCM, Dichloromethane; MTBE, tert-Butyl Methyl Ether; DCFH-DA, 2′,7′-Dichlorofluorescin diacetate.
Data sharing statement
The datasets used and/or analyzed during the current study are available from the all authors on reasonable request.
Acknowledgment
This work was supported by Technology Project on TCM in Zhejiang Province (Grant No. 2018ZB043); Science and Technology Fund Project of Guizhou Health and Family Planning Commission (Grant No. gzwjkj-2018-1-035).
Author contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Weinberg F, Hamanaka R, Wheaton WW, et al. Mitochondrial metabolism and ROS generat ion are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A. 2010;107(19):8788–8793. doi:10.1073/pnas.1003428107
2. Cai J, Yang J, Jones DP. Mitochondrial control of apoptosis: the role of cytochrome c. Biochim Biophys Acta. 1998;1366(1–2):139–149. doi:10.1016/s0005-2728(98)00109-1
3. Donovan M, Cotter TG. Control of mitochondrial integrity by Bcl-2 family members and caspase-independent cell death. Biochim Biophys Acta. 2004;1644(2–3):133–147. doi:10.1016/j.bbamcr.2003.08.011
4. Tait SW, Ichim G, Green DR. Die another way–non-apoptotic mechanisms of cell death. J Cell Sci. 2014;127(Pt 10):2135–2144. doi:10.1242/jcs.093575
5. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417(1):1–13. doi:10.1042/BJ20081386
6. Bedard K, Krause K-H. The NOX family of ROS-Generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87(1):245–313. doi:10.1152/physrev.00044.2005
7. Peng CA, Gaertner AAE, Henriquez SA, et al. Fluconazole induces ROS in Cryptococcus neoformans and contributes to DNA damage in vitro. PLoS One. 2018;13(12):e0208471–e0208471. doi:10.1371/journal.pone.0208471
8. Bax HJ, Josephs DH, Pellizzari G, Spicer JF, Montes A, Karagiannis SN. Therapeutic targets and new directions for antibodies developed for ovarian cancer. mAbs. 2016;8(8):1437–1455. doi:10.1080/19420862.2016.1219005
9. Gaborit N, Lindzen M, Yarden Y. Emerging anti-cancer antibodies and combination therapies targeting HER3/ERBB3. Hum Vaccin Immunother. 2015;12(3):576–592. doi:10.1080/21645515.2015.1102809
10. Slezak J, Kura B, Ravingerova T, Tribulova N, Okruhlicova L, Barancik M. Mechanisms of cardiac radiation injury and potential preventive approaches. Can J Physiol Pharmacol. 2015;93(9):737–753. doi:10.1139/cjpp-2015-0006
11. Zhao L, Zhao W, Liu Y, Chen X, Wang Y. Nano-hydroxyapatite-derived drug and gene co-delivery system for anti-angiogenesis therapy of breast cancer. Med Sci Monit. 2017;23:4723–4732. doi:10.12659/msm.902538
12. Urruticoechea A, Alemany R, Balart J, Villanueva A, Vinals F, Capella G. Recent advances in cancer therapy: an overview. Curr Pharm Des. 2010;16(1):3–10. doi:10.2174/138161210789941847
13. Yang Y, Karakhanova S, Hartwig W, et al. Mitochondria and mitochondrial ROS in cancer: novel targets for anticancer therapy. J Cell Physiol. 2016;231(12):2570–2581. doi:10.1002/jcp.25349
14. Pokrzywinski KL, Biel TG, Kryndushkin D, Rao VA. Therapeutic targeting of the mitochondria initiates excessive superoxide production and mitochondrial depolarization causing decreased mtDNA integrity. PLoS One. 2016;11(12):e0168283. doi:10.1371/journal.pone.0168283
15. Farsinejad S, Gheisary Z, Ebrahimi Samani S, Alizadeh AM. Mitochondrial targeted peptides for cancer therapy. Tumour Biol. 2015;36(8):5715–5725. doi:10.1007/s13277-015-3719-1
16. Dinca A, Chien W-M, Chin MT. Intracellular delivery of proteins with cell-penetrating peptides for therapeutic uses in human disease. Int J Mol Sci. 2016;17(2):263. doi:10.3390/ijms17020263
17. Guidotti G, Brambilla L, Rossi D. Cell-penetrating peptides: from basic research to clinics. Trends Pharmacol Sci. 2017;38(4):406–424. doi:10.1016/j.tips.2017.01.003
18. Wu J, Li J, Wang H, Liu CB. Mitochondrial-targeted penetrating peptide delivery for cancer therapy. Expert Opin Drug Deliv. 2018;15(10):951–964. doi:10.1080/17425247.2018.1517750
19. Asin-Cayuela J, Manas A-RB, James AM, Smith RAJ, Murphy MP. Fine-tuning the hydrophobicity of a mitochondria-targeted antioxidant. FEBS Lett. 2004;571(1–3):9–16. doi:10.1016/j.febslet.2004.06.045
20. Kelso GF, Porteous CM, Coulter CV, et al. Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J Biol Chem. 2001;276(7):4588–4596. doi:10.1074/jbc.M009093200
21. Kumari S, Badana AK, Malla R. Reactive oxygen species: a key constituent in cancer survival. Biomark Insights. 2018;13:1177271918755391. doi:10.1177/1177271918771969
22. Ross Meredith F, Prime Tracy A, Abakumova I, et al. Rapid and extensive uptake and activation of hydrophobic triphenylphosphonium cations within cells. Biochem J. 2008;411(3):633. doi:10.1042/BJ20071369
23. Chen X, Qian Y, Wu S. The Warburg effect: evolving interpretations of an established concept. Free Radic Biol Med. 2015;79:253–263. doi:10.1016/j.freeradbiomed.2014.08.027
24. Modica-Napolitano JS, Weissig V. Treatment strategies that enhance the efficacy and selectivity of mitochondria-targeted anticancer agents. Int J Mol Sci. 2015;16(8):17394–17421. doi:10.3390/ijms160817394
25. Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol. 2015;11(1):9–15. doi:10.1038/nchembio.1712
26. Afanas’ev I. Reactive oxygen species signaling in cancer: comparison with aging. Aging Dis. 2011;2(3):219–230.
27. Yu M. Generation, function and diagnostic value of mitochondrial DNA copy number alterations in human cancers. Life Sci. 2011;89(3–4):65–71. doi:10.1016/j.lfs.2011.05.010
28. Tan DJ, Bai RK, Wong LJ. Comprehensive scanning of somatic mitochondrial DNA mutations in breast cancer. Cancer Res. 2002;62(4):972–976.
29. Parrella P, Xiao Y, Fliss M, et al. Detection of mitochondrial DNA mutations in primary breast cancer and fine-needle aspirates. Cancer Res. 2001;61(20):7623–7626.
30. Petros JA, Baumann AK, Ruiz-Pesini E, et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc Natl Acad Sci U S A. 2005;102(3):719–724. doi:10.1073/pnas.0408894102
31. Ishikawa K, Takenaga K, Akimoto M, et al. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science (New York, NY). 2008;320(5876):661–664. doi:10.1126/science.1156906
32. Murphy MP, Smith RA. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol. 2007;47:629–656. doi:10.1146/annurev.pharmtox.47.120505.105110
33. Szeto HH. Cell-permeable, mitochondrial-targeted, peptide antioxidants. Aaps J. 2006;8(2):E277–E283. doi:10.1007/bf02854898
34. Snyder EL, Dowdy SF. Cell penetrating peptides in drug delivery. Pharm Res. 2004;21(3):389–393. doi:10.1023/B:PHAM.0000019289.61978.f5
35. Chen ZP, Li M, Zhang LJ, et al. Mitochondria-targeted drug delivery system for cancer treatment. J Drug Target. 2016;24(6):492–502. doi:10.3109/1061186X.2015.1108325
36. Najjar K, Erazo-Oliveras A, Brock DJ, Wang TY, Pellois JP. An l- to d-amino acid conversion in an endosomolytic analog of the cell-penetrating peptide TAT influences proteolytic stability, endocytic uptake, and endosomal escape. J Biol Chem. 2017;292(3):847–861. doi:10.1074/jbc.M116.759837
37. Meucci E, Mele MC. Amino acids and plasma antioxidant capacity. Amino Acids. 1997;12(3):373–377. doi:10.1007/BF01373017
38. Dludla PV, Dias SC, Obonye N, Johnson R, Louw J, Nkambule BB. A systematic review on the protective effect of N-Acetyl Cysteine against diabetes-associated cardiovascular complications. Am J Cardiovasc Drugs. 2018;18(4):283–298. doi:10.1007/s40256-018-0275-2
39. Raffaele M, Barbagallo I, Licari M, et al. N-Acetylcysteine (NAC) ameliorates lipid-related metabolic dysfunction in bone marrow stromal cells-derived adipocytes. Evidence-based Complementary Altern Med. 2018;2018:5310961. doi:10.1155/2018/9567061
40. Shahidi S, Zargooshnia S, Asl SS, Komaki A, Sarihi A. Influence of N-acetyl cysteine on beta-amyloid-induced Alzheimer’s disease in a rat model: a behavioral and electrophysiological study. Brain Res Bull. 2017;131:142–149. doi:10.1016/j.brainresbull.2017.04.001
41. Wang B, Aw T Y, Stokes KY. N-acetylcysteine attenuates systemic platelet activation and cerebral vessel thrombosis in diabetes. Redox Biol. 2018;14:218–228. doi:10.1016/j.redox.2017.09.005
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.