")
Back to Journals » International Journal of Nanomedicine » Volume 11
In vitro inflammatory effects of hard metal (WC–Co) nanoparticle exposure
Authors Armstead AL, Li B
Received 31 August 2016
Accepted for publication 2 October 2016
Published 21 November 2016 Volume 2016:11 Pages 6195—6206
DOI https://doi.org/10.2147/IJN.S121141
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Carlos Rinaldi
Andrea L Armstead,1,2 Bingyun Li1–3
1Department of Orthopaedics, School of Medicine, 2School of Pharmacy, West Virginia University, 3Mary Babb Randolph Cancer Center, Morgantown, WV, USA
Abstract: Identifying the toxicity of nanoparticles (NPs) is an important area of research as the number of nanomaterial-based consumer and industrial products continually rises. In addition, the potential inflammatory effects resulting from pulmonary NP exposure are emerging as an important aspect of nanotoxicity. In this study, the toxicity and inflammatory state resulting from tungsten carbide–cobalt (WC–Co) NP exposure in macrophages and a coculture (CC) of lung epithelial cells (BEAS-2B) and macrophages (THP-1) at a 3:1 ratio were examined. It was found that the toxicity of nano-WC–Co was cell dependent; significantly less toxicity was observed in THP-1 cells compared to BEAS-2B cells. It was demonstrated that nano-WC–Co caused reduced toxicity in the CC model compared to lung epithelial cell monoculture, which suggested that macrophages may play a protective role against nano-WC–Co-mediated toxicity in CCs. Nano-WC–Co exposure in macrophages resulted in increased levels of interleukin (IL)-1β and IL-12 secretion and decreased levels of tumor necrosis factor alpha (TNFα). In addition, the polarizing effects of nano-WC–Co exposure toward the M1 (pro-inflammatory) and M2 (anti-inflammatory) macrophage phenotypes were investigated. The results of this study indicated that nano-WC–Co exposure stimulated the M1 phenotype, marked by high expression of CD40 M1 macrophage surface markers.
Keywords: nanoparticle, nanotoxicity, inflammation, macrophage, lung disease
Introduction
The majority of nanomaterial or nanoparticle (NP) exposure occurs via the pulmonary route1,2 and, upon inhalation, macrophages are recruited to the area as a part of the body’s defense mechanism to promote particle clearance from the lungs.3,4 Phagocytic by nature, macrophages quickly identify and engulf the NPs as a part of their inherent physiological response mechanism.4,5 However, during this process, the macrophage accumulates a large quantity of NPs. Depending on the NPs, this accumulation within the cell may cause toxicity or induce a secondary immune response and stimulate a local or systemic inflammatory process.5–7 Therefore, along with addressing the direct toxic effects of the multitude of NPs currently used in consumer products, it is important to consider the inflammatory response that these materials may generate upon exposure.5
Macrophage-mediated inflammation typically occurs in response to bacterial or viral infections and tissue injuries as a part of the normal healing process, but it can also occur as a result of foreign particle inhalation.5,8 Depending on the stimulus, macrophages differentiate into either a “classically” activated M1 or an “alternatively” activated M2 phenotype, which have signature characteristics that distinguish them from one another.9–14 Classical M1 macrophages play important roles in infection clearance, typically in response to lipopolysaccharide (LPS), which stimulates a high-level secretion of pro-inflammatory cytokines such as tumor necrosis factor alpha (TNFα) and interleukin (IL) 12p70 (IL-12) and promotes a type 1 T helper (Th1) immune response.9–11,14 Macrophages activated by other factors, such as endogenous IL-4 and glucocorticoid hormones, fall under the alternatively activated M2 phenotype. M2 macrophages generally secrete high levels of IL-10 and IL-1β to promote the type 2 T helper (Th2) immune response.9–11,14 In addition, emerging evidence indicates that macrophage activation/polarization toward the M1 or M2 phenotype may play critical roles in tumor growth and cancer progression.8,9,15,16 It is believed that M1 macrophages promote tumor resistance due to their high-level secretion of pro-inflammatory cytokines and potent effector cell functions.10,11 In contrast, M2 macrophages are believed to possess tumor-promoting functions due to their promotion of angiogenesis, tissue remodeling and repair mechanisms.10,11 Further, the tumor microenvironment can influence tumor-associated macrophages to undergo a “phenotypic switching” from M1 to M2, and thus tumor-promoting, phenotype.10,15
Given this body of literature, it has been suggested that inflammation resulting from pulmonary NP exposure may play a role in the toxicity associated with exposure,5–7,17–21 and that the inflammatory state resulting from particle exposure may play a role in cancer progression.5,8 This is especially concerning in occupational settings,22,23 where workers undergo pulmonary exposure to NPs on a daily basis.17,24 In particular, exposure to tungsten carbide–cobalt (WC–Co) NPs in occupational settings is known to cause hard metal lung disease (HMLD), characterized by progressive inflammation and fibrosis of the lung,25–29 which is further associated with a twofold increased risk of lung cancer.30–32 While the toxicity of WC–Co particles has been studied,33–50 the relationship between WC–Co toxicity, inflammation and lung cancer remains poorly understood.
In addition, there is limited information regarding the interaction of multiple cell types during WC–Co NP exposure (ie, immune cells and epithelial cells), as the majority of previous in vitro studies employed single-cell (monoculture) models in their examination of WC–Co toxicity.33,34,36,40–42,45,47,48 This design is a disadvantage, as coculture (CC) models more closely represent the in vivo environment during a WC–Co NP exposure. To address this gap in understanding, the current study examined the effects of nano-WC–Co particle exposure in vitro using monoculture and CC cell models composed of macrophages (THP-1) and lung epithelial cells (BEAS-2B) to assess the toxicity, stimulation of inflammatory cytokine secretion and M1/M2 macrophage polarization. We hypothesized that exposure to nano-WC–Co stimulates inflammation in macrophages and may promote polarization toward the M1 pro-inflammatory macrophage phenotype.
Materials and methods
Materials and reagents
THP-1 human monocyte cell line (TIB-202) and BEAS-2B cells were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). Nano-WC–Co composite particles were purchased from Inframat Advanced Materials (Manchester, CT, USA). RPMI-1640 medium for THP-1 cell culture was purchased from ATCC. Phosphate-buffered saline (PBS), Dulbecco’s Modified Eagle’s Medium (DMEM), 0.25% trypsin/ethylenediaminetetraacetic acid (EDTA), versene (EDTA-based cell detachment reagent), penicillin/streptomycin, beta-mercaptoethanol and fetal bovine serum (FBS) were purchased from Lonza (Allendale, NJ, USA). Isopropanol, hydrochloric acid, Triton-X-100, thiazolyl blue tetrazolium bromide (MTT reagent), phorbol-12-mystirate-13-acetate (PMA), LPS and enzyme-linked immunosorbent assay (ELISA) kits for human IL-12 (#RAB0252), IL-10 (#RAB0244), IL-1β (#RAB0273) and TNFα (#RAB0476) were purchased from Sigma-Aldrich (St Louis, MO, USA). Flow cytometry staining buffer (containing 0.2% bovine serum albumin and sodium azide), recombinant human IL-4, human immunoglobulin G (IgG), antihuman CD40-APC and antihuman CD206-FITC antibodies were purchased from BD Biosciences (Franklin Lakes, NJ, USA).
Nano-WC–Co particle preparation
Nano-WC–Co stock particle suspensions (5 mg/mL) were prepared in sterile PBS (containing 10% FBS to prevent agglomeration) by sonication under 120 W power output, frequency 20 kHz, for 1 min with an Omni International Sonic Ruptor (Omni International, Kennesaw, GA, USA). Sonication was performed in 30 mL plastic vials immobilized in an ice bath to prevent heating during particle dispersion. Dilute nano-WC–Co particle suspensions (1–1,000 μg/mL) were prepared on the day of each experiment in DMEM containing 10% FBS from the 5 mg/mL stock particle suspension.
Cell culture and THP-1 macrophage (M0) activation
BEAS-2B cells were maintained and passaged upon confluence in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. THP-1 cells were maintained and passaged upon confluence in RPMI-1640 supplemented with 10% FBS, 1% penicillin/streptomycin and 0.5 mM beta-mercaptoethanol. All cells were maintained in an incubator at 37°C and 5% CO2. Differentiation of THP-1 cells to macrophages (M0) was achieved through the addition of 10 ng/mL PMA. After 48 h, THP-1 to M0 differentiation was confirmed via examination of cell morphology using a light microscope,13,51 where M0 cells underwent a signature change in morphology and became adherent to the culture dish. For CC, M0 cells were rinsed once with PBS and incubated with versene (EDTA-based cell dissociation reagent) for 15 min. M0 cells were then pipetted gently to ensure detachment from the culture dish and resuspended in DMEM at the desired concentration for CC seeding.
Macrophage and epithelial cell CC
To establish the CC at a ratio of 3:1 (lung epithelial cell: macrophage), BEAS-2B cells were trypsinized, resuspended in DMEM and seeded in a 96-well plate at a density of 1.5×105 cells/mL and allowed to adhere for 1 h. BEAS-2B attachment was confirmed using light microscopy prior to the addition of M0 cells. M0 cells were seeded directly on top of the BEAS-2B monolayer at a density of 5×104 cells/mL in DMEM and cocultured for 24 h to allow attachment and cellular interaction prior to nano-WC–Co exposure.
Nano-WC–Co particle exposure
Nano-WC–Co particle exposure was achieved by aspirating the medium from each well and replacing immediately with an equivalent volume of nano-WC–Co particle suspension at a concentration of 1, 10, 100 or 1,000 μg/mL and incubated at 37°C and 5% CO2 for exposure periods of 2, 6, 12, 24 and 48 h; controls were treated with LPS (100 ng/mL) or IL-4 (20 ng/mL). For the M1/M2 flow cytometry experiments, M0 cells were treated with either control LPS, IL-4 or 0, 1, 10 and 100 μg/mL nano-WC–Co for exposure periods of 1, 2 and 5 days.
Cell viability after nano-WC–Co exposure
Following nano-WC–Co exposure, the medium containing nano-WC–Co was aspirated from each well and cells were rinsed once with PBS to remove excess particles. Then, 100 μL un-supplemented DMEM was added to each well, followed by the addition of 10 μL MTT reagent to achieve a final concentration of 0.5 mg/mL MTT per well. Cells were incubated for 2 h at 37°C and 5% CO2 to allow conversion of the soluble tetrazolium salt (yellow) to formazan crystals (purple). Crystal formation was confirmed using light microscopy. Next, 100 μL of solubilization solution (0.1 M HCl in isopropanol containing 10% Triton-X) was added to each well to dissolve the formazan crystals, and the absorbance of each well was recorded immediately at 570 nm using a Bio-Tek μQuant microplate reader (Bio-Tek, Winooski, VT, USA). Blank values were subtracted from all absorbance readings. Cell viability was calculated by dividing the absorbance of nano-WC–Co-treated cells by the absorbance of control cells receiving media treatment only and converted to a percentage. The presence of WC–Co particles did not interfere with the MTT assays in our studies as we described earlier.49
Macrophage polarization assay
THP-1 cells were seeded at a density of 5×105 cells/mL in RPMI-1640 containing 10 ng/mL PMA in a 12-well culture dish and incubated for 48 h to stimulate M0 cell differentiation. Media were then aspirated from each well and replaced with DMEM containing 100 ng/mL LPS (M1 stimulus/positive control), 20 ng/mL IL-4 (M2 stimulus/positive control) and 0, 1, 10 or 100 μg/mL nano-WC–Co particles. M0 cells were incubated with the M1, M2 or nano-WC–Co stimulus for 1, 2 and 5 days at 37°C and 5% CO2.
Flow cytometry
Following M1/M2/WC–Co exposure, M0 cells were rinsed once with PBS, detached with versene (described earlier) and transferred to 15 mL polystyrene tubes. Cells were centrifuged at 1,200 rpm for 7 min to pellet and resuspended in 1 mL ice-cold flow cytometry staining buffer. M0 cells were counted on a hemocytometer and a total of 3×105 cells per sample were transferred to 5 mL polystyrene tubes for subsequent staining. After centrifugation, cell pellets were resuspended in 100 μL staining buffer containing 10 μg human IgG and incubated for 30 min over ice to block macrophage Fc receptors and minimize nonspecific antibody binding. Next, cells were rinsed with 1 mL cold staining buffer to remove excess IgG and resuspended in 80 μL buffer. Then, 10 μL of each antibody (anti-CD40-APC or anti-CD206-FITC) was added to each appropriate tube, including positive/negative and single-stain controls and incubated for 1 h over ice. After staining, cells were rinsed three times with 1 mL cold staining buffer to remove unbound antibodies. After the final rinse, cell pellets were resuspended in 100 μL 0.4% paraformaldehyde to fix and stored overnight at 4°C. The next day, fixed cells were centrifuged, resuspended in 300 μL staining buffer and analyzed immediately on a BD LSR Fortessa flow cytometer. Instrument settings were defined at the beginning of each experiment using the cells only (no stain) and single-stain controls (CD40-APC only and CD206-FITC only) and applied for all subsequent experimental samples. Representative flow cytometry dot plots of macrophage staining controls are shown in Figure S1.
Inflammatory cytokine secretion in response to nano-WC–Co exposure
The concentrations of IL-12, IL-10, IL-1β and TNFα in cell culture supernatants were determined using ELISA kits. Following nano-WC–Co exposure, cell culture supernatants were collected after 6, 12, 24 and 48 h in 150 μL aliquots in a 96-well plate and preserved immediately at −80°C for later analysis. Once all supernatant samples had been collected, the 96-well plates were quickly thawed at room temperature and centrifuged briefly (500 rpm for 5 min) to pellet any cell debris or nano-WC–Co particles, which may interfere with the assay. The ELISA assays were then carried out according to the manufacturer’s instructions. Briefly, 100 μL supernatant or cytokine standard (prepared according to the instructions) was transferred to the appropriate well(s) of the 96-well ELISA plate and incubated at room temperature for 2.5 h with gentle shaking. Next, the solutions were discarded and each well was rinsed four times with prepared 1× wash buffer according to the instructions. Following the rinse step, 100 μL of biotinylated antibody was added to each well and incubated for 1 h with gentle shaking. Next, the solutions were discarded and the rinse step was repeated, followed by addition of 100 μL streptavidin solution to each well and incubation for 45 min with gentle shaking. Then, the solutions were discarded, the rinse step was repeated and 100 μL of tetramethylbenzidine (TMB) one-step substrate reagent was added to each well. Plates were incubated in the dark for an additional 30 min with gentle shaking, followed by the addition of 50 μL stop solution to each well. Plate absorbance was read immediately at 450 nm. Standard curves were prepared in duplicate for each ELISA plate. The concentration of cytokine(s) in each supernatant sample was then calculated based on the sample absorbance at 450 nm and the slope of the standard curve, according to the instructions.
Statistical analyses
All experiments were performed in triplicate, and data are presented as mean ± standard deviation. Statistical analysis was carried out by two-way analysis of variance (ANOVA) using GraphPad Prism Software (GraphPad Software, Inc., La Jolla, CA, USA). P-values <0.05 were considered significant.
Results
Nano-WC–Co characterization
Nano-WC–Co particles were prepared and characterized as described earlier.49 Briefly, nano-WC–Co particles averaged 98 nm in diameter in suspension determined by dynamic light scattering and transmission electron microscopy. Compositional analysis by energy-dispersive X-ray (EDX) determined that nano-WC–Co contained 72.13% tungsten, 13.42% cobalt, 7.63% carbon and 6.81% oxygen.
CC viability
Macrophages (THP-1), lung epithelial cells (BEAS-2B) and a 3:1 CC of BEAS-2B and THP-1 cells were exposed to nano-WC–Co at concentrations of 1, 10, 100 and 1,000 μg/mL for durations of 2, 6, 12, 24 and 48 h. In macrophage monoculture (THP-1), nano-WC–Co exposure did not induce significant changes in cell viability (compared to control) at 1 μg/mL but did induce a significant reduction in cell viability at 10 μg/mL after 48 h, at 100 μg/mL after 6, 12, 24 and 48 h and at 1,000 μg/mL after 2, 6, 12, 24 and 48 h of exposure (Figure 1). Consistent with our previous report,49 nano-WC–Co exposure caused a significant reduction in cell viability in lung epithelial cells (BEAS-2B) at 1 μg/mL after 24 and 48 h and at ≥10 μg/mL after 2, 6, 12, 24 and 48 h of exposure (Figure 1). The viability in THP-1 cells was significantly higher compared to BEAS-2B monoculture at 1 μg/mL after 2, 6 and 48 h and at 10, 100 and 1,000 μg/mL after 2, 6, 12, 24 and 48 h nano-WC–Co exposure (Figure 1).
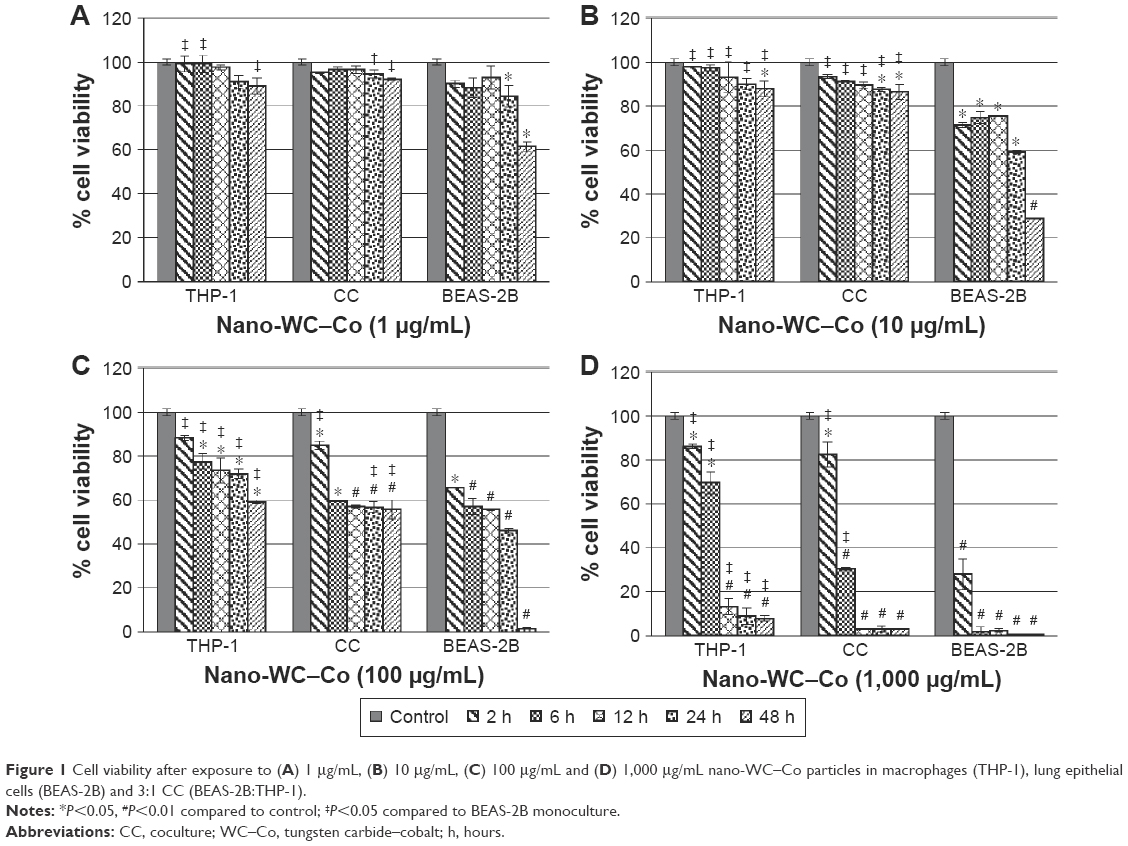 | Figure 1 Cell viability after exposure to (A) 1 μg/mL, (B) 10 μg/mL, (C) 100 μg/mL and (D) 1,000 μg/mL nano-WC–Co particles in macrophages (THP-1), lung epithelial cells (BEAS-2B) and 3:1 CC (BEAS-2B:THP-1). |
In the CC of BEAS-2B and THP-1 cells, nano-WC–Co exposure did not lead to significant changes in cell viability at 1 μg/mL but caused a significant reduction in cell viability (compared to control) at 10 μg/mL after 24 and 48 h and at 100 and 1,000 μg/mL after 2, 6, 12, 24 and 48 h of exposure (Figure 1). Compared to BEAS-2B monoculture, an increased cell viability was observed in the CC model at 1 μg/mL after 24 and 48 h, at 10 μg/mL after 2, 6, 12, 24 and 48 h, at 100 μg/mL after 2, 24 and 48 h and at 1,000 μg/mL after 2 and 6 h of exposure (Figure 1).
Macrophage polarization
Macrophage polarization toward the M1 and M2 phenotypes was examined after exposure to LPS (M1 positive control), IL-4 (M2 positive control) and 0, 1, 10 or 100 μg/mL nano-WC–Co for 1, 2 and 5 days. Cells staining positive for CD40-APC were considered M1-type macrophages and cells staining positive for CD206-FITC were considered M2-type macrophages. A summary of M1 and M2 flow cytometry staining is presented graphically in Figure 2. Representative dot plots depicting M1/M2 macrophage staining after exposure to nano-WC–Co for 1, 2 and 5 days are shown in Figure 3 (1 μg/mL), Figure 4 (10 μg/mL) and Figure 5 (100 μg/mL). Compared to the control group (0 μg/mL nano-WC–Co), for the staining control samples used to set the instrument gating parameters, a significant increase in CD40+ stained cells was observed following LPS exposure for 1, 2 and 5 days; IL-4 exposure caused a significant increase in CD206+ cells after 2- and 5-day exposure (Figure S2).
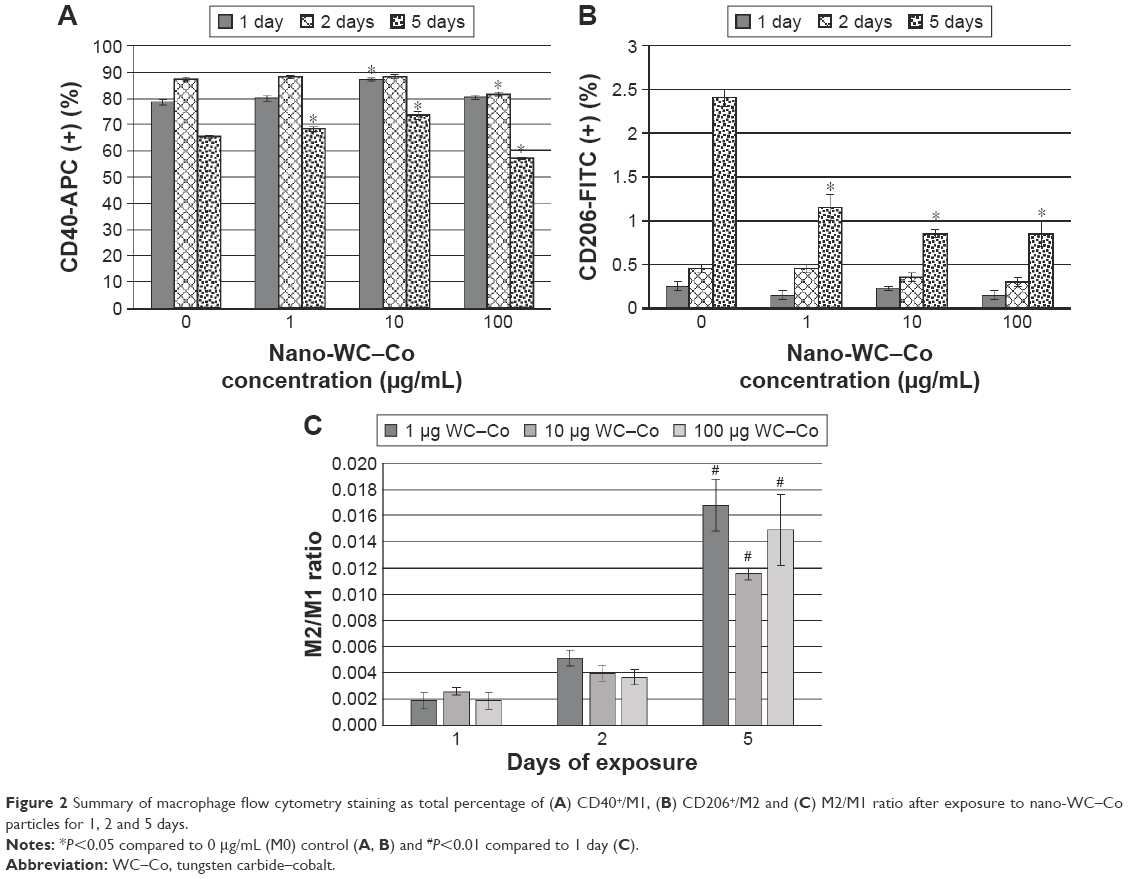 | Figure 2 Summary of macrophage flow cytometry staining as total percentage of (A) CD40+/M1, (B) CD206+/M2 and (C) M2/M1 ratio after exposure to nano-WC–Co particles for 1, 2 and 5 days. |
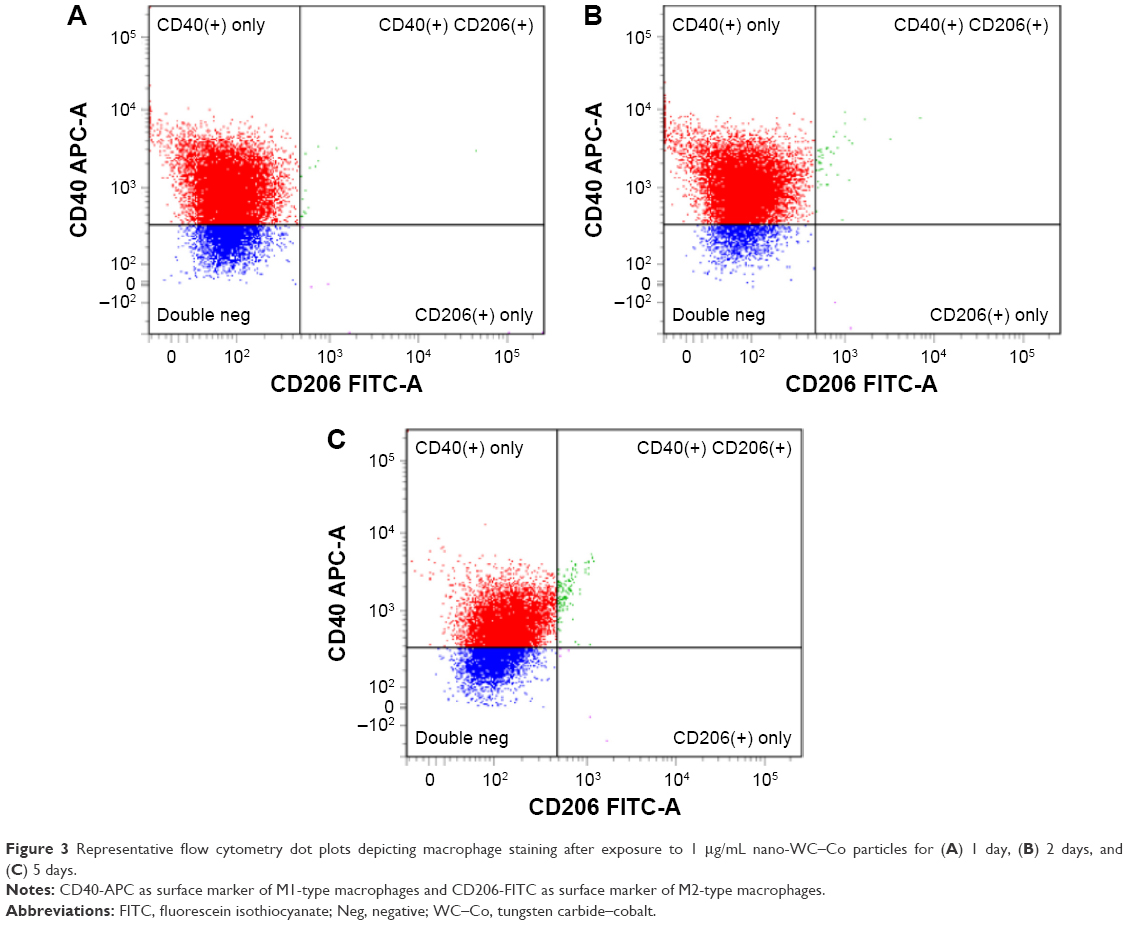 | Figure 3 Representative flow cytometry dot plots depicting macrophage staining after exposure to 1 μg/mL nano-WC–Co particles for (A) 1 day, (B) 2 days, and (C) 5 days. |
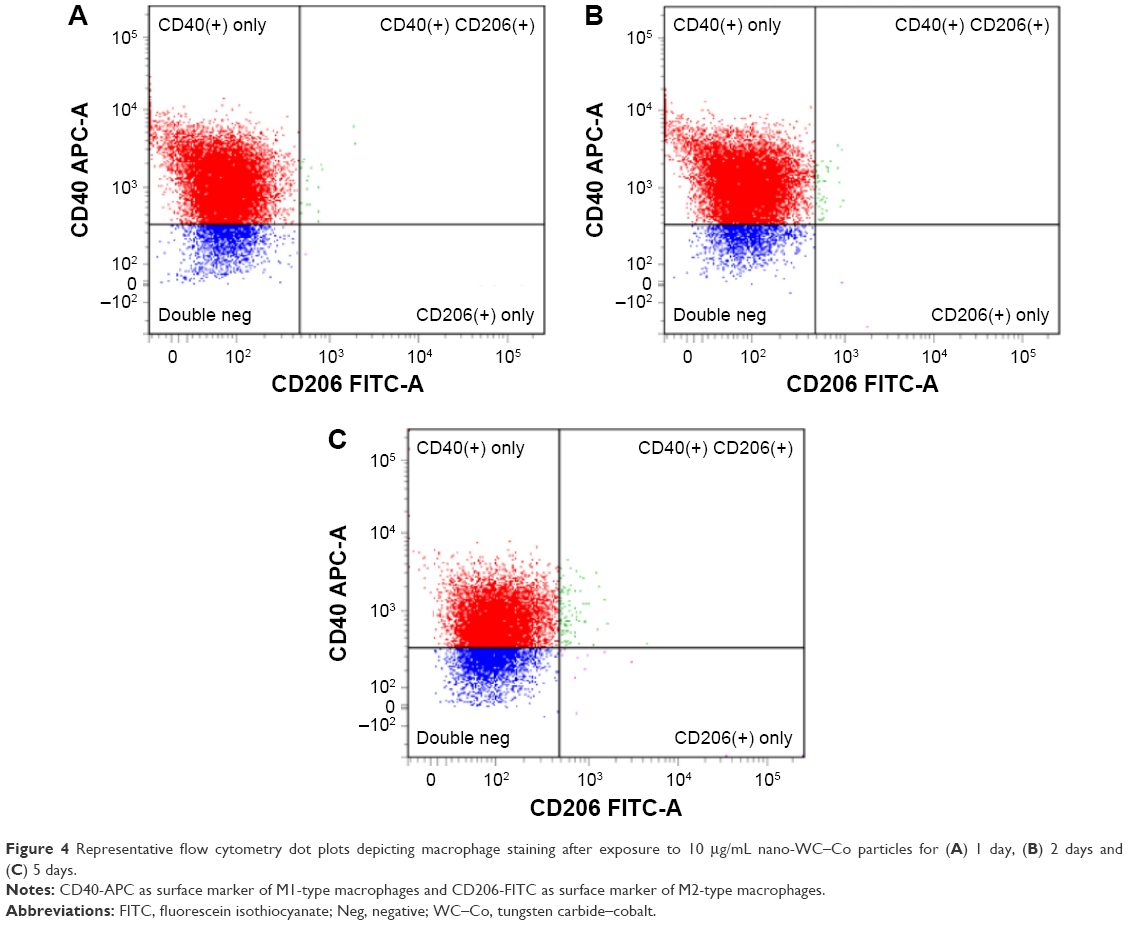 | Figure 4 Representative flow cytometry dot plots depicting macrophage staining after exposure to 10 μg/mL nano-WC–Co particles for (A) 1 day, (B) 2 days and (C) 5 days. |
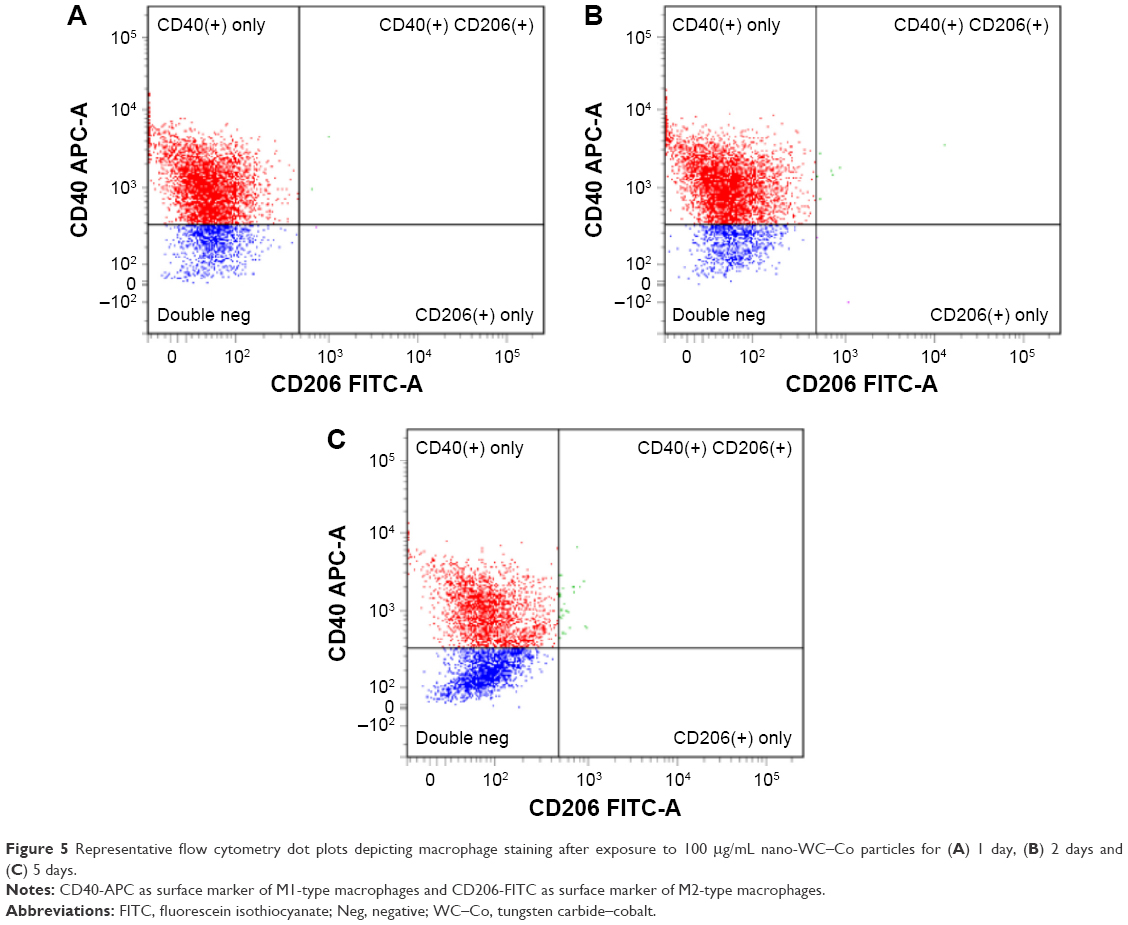 | Figure 5 Representative flow cytometry dot plots depicting macrophage staining after exposure to 100 μg/mL nano-WC–Co particles for (A) 1 day, (B) 2 days and (C) 5 days. |
In general, CD40+ staining was dominant in all the samples at the exposure periods studied (Figure 2). Compared to the control group (0 μg/mL WC–Co), cells exposed to 1 and 10 μg/mL nano-WC–Co had significantly higher CD40+ staining after 5 days of exposure. Exposure to 10 μg/mL nano-WC–Co also caused a significant increase in CD40+ cells after 1 day and cells exposed to 100 μg/mL demonstrated significantly lower numbers of CD40+ cells after 2 and 5 days of exposure (Figure 2A). Further, cells exposed to 1, 10 and 100 μg/mL nano-WC–Co had significantly less CD206+ cells after 5 days of exposure compared to the control group (0 μg/mL WC–Co; Figure 2B). With increasing nano-WC–Co exposure time from day 1 to day 2 and day 5, an increase in the ratio of M2/M1 was found; the ratio of M2/M1 was significantly higher after 5 days compared to 1 day for 1, 10 and 100 μg/mL nano-WC–Co (Figure 2C).
Inflammatory cytokine secretion
The levels of inflammatory cytokines were quantified in the cell culture supernatant via ELISA after 6, 12, 24 and 48 h of exposure to 0, 1, 10 or 100 μg/mL nano-WC–Co (Figure 6) and either LPS (M1 stimulus) or IL-4 (M2 stimulus; Figures S3 and S4). Nano-WC–Co treatment caused varying effects on TNFα secretion (Figure 6A); 1 μg/mL nano-WC–Co caused a significant increase (P<0.05) in TNFα after 6 h of exposure, compared to control (0 μg/mL), but the levels were significantly lower after 12, 24 and 48 h of exposure. A decrease in TNFα, compared to control (0 μg/mL), was also observed at 10 μg/mL after 24 and 48 h and at 100 μg/mL after 12, 24 and 48 h of exposure to nano-WC–Co (Figure 6A).
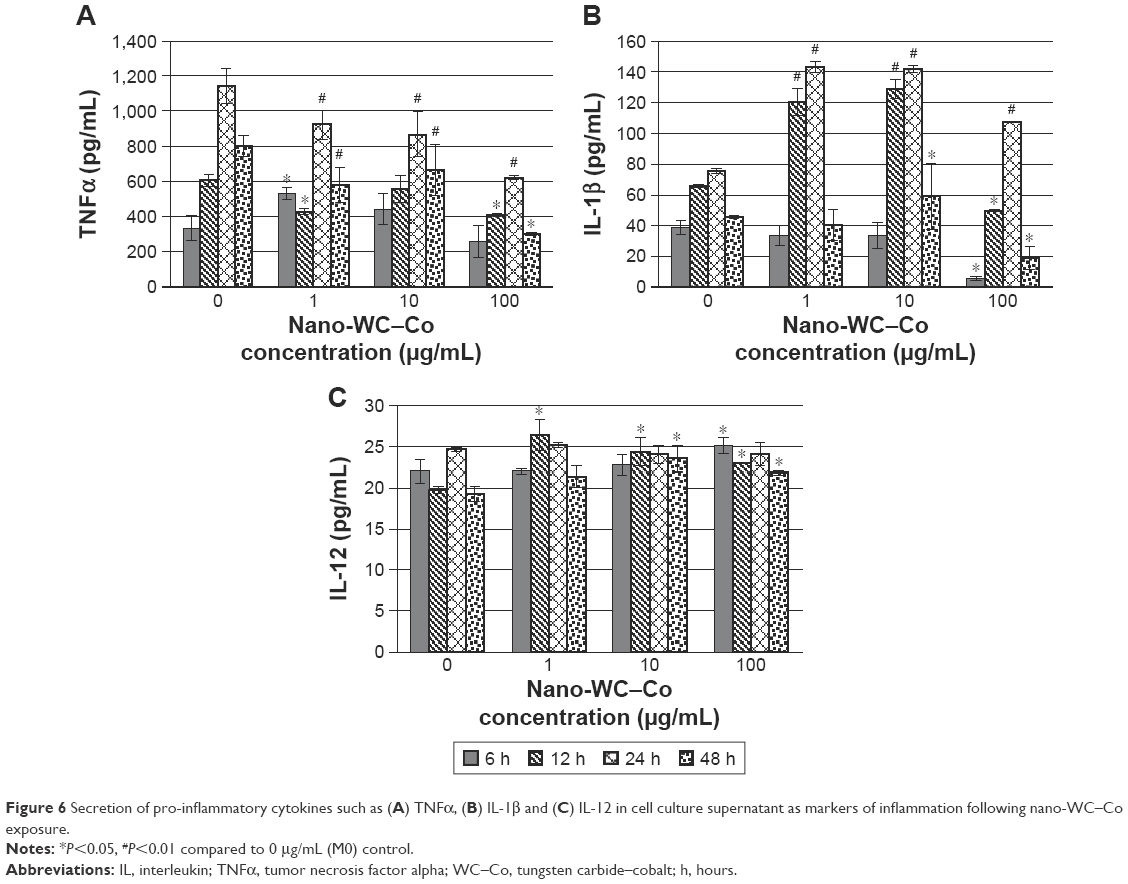 | Figure 6 Secretion of pro-inflammatory cytokines such as (A) TNFα, (B) IL-1β and (C) IL-12 in cell culture supernatant as markers of inflammation following nano-WC–Co exposure. |
In the IL-1β assay (Figure 6B), 1 μg/mL nano-WC–Co treatment caused a significant increase in IL-1β levels, compared to control (0 μg/mL), after 12 and 24 h of exposure. In the 10 μg/mL nano-WC–Co group, a significant increase in IL-1β was found at exposures of 12, 24 and 48 h. Compared to control (0 μg/mL), 100 μg/mL nano-WC–Co exposure led to a significant decrease in IL-1β after 6 and 12 h and a significant increase after 24 h followed by a significant decrease after 48 h of exposure (Figure 6B).
For IL-12 (Figure 6C), exposure to 1 μg/mL nano-WC–Co caused a significant increase compared to control (0 μg/mL) after 12 h of exposure. Then, 10 μg/mL nano-WC–Co caused a significant increase in IL-12 after 12 and 48 h of exposure and 100 μg/mL nano-WC–Co also caused a significant increase in IL-12 (compared to control) after 6, 12 and 48 h of exposure (Figure 6C).
Discussion
Due to the increased use of NPs in consumer and industrial applications,52 there is a critical need to clearly define the toxic and inflammatory effects of NPs which occur after exposure. We recently reported that commercially prepared nano-WC–Co particles induced a time- and dose-dependent toxicity in human lung epithelial cells (BEAS-2B) and were capable of being internalized, inducing oxidative stress and stimulating apoptotic cell death in vitro.49 Since macrophages play a critical role in promoting natural pulmonary particle clearance mechanisms, an important aspect of the current study was to define the toxic effects of nano-WC–Co particle exposure on macrophages in both monoculture and CC settings. To test the effects of nano-WC–Co particle exposure on macrophage-mediated inflammation and M1/M2 polarization, we selected monocyte-derived THP-1 cells as our macrophage model due to their prevalence in the literature and the ease with which THP-1 cells are differentiated toward a macrophage (M0) phenotype with PMA.13,14,51,53 In this study, a 3:1 CC ratio of BEAS-2B to THP-1 M0 was selected to represent the dynamic tissue environment within the lung during a particle inhalation scenario;54,55 the viability was compared between THP-1 and BEAS-2B monocultures and the 3:1 CC system.
Due to the inherent phagocytic nature of macrophages, we hypothesized that the presence of THP-1 M0 in the CC model would offer a “protective effect” against nano-WC–Co toxicity; therefore, increased viability would be observed in the CC system compared to BEAS-2B cells alone. In this case, the results of our cell viability study (Figure 1) are consistent with our hypothesis and we believe that macrophage engulfment of nano-WC–Co isolated the particles and prevented direct contact with the BEAS-2B cells, effectively attenuating nano-WC–Co toxicity to the extent reported previously in BEAS-2B cells.49 In fact, the toxicity of nano-WC–Co was found to be cell dependent and significantly less toxicity was observed in macrophages compared to BEAS-2B cells (Figure 2). While the 1,000 μg/mL dose is very high, we believe that it is relevant because the resulting lifetime accumulation of nano-WC–Co particles in the lung from occupational settings could be substantial. This idea is supported by histological findings in patients with HMLD, where large deposits of WC–Co are often visible in lung specimens.56–59
Macrophage polarization following the exposure to other metal NPs has been reported,60 so we explored the effects of nano-WC–Co exposure on macrophage polarization toward the M1 or M2 phenotype using flow cytometry by staining for two well-known macrophage cell membrane markers: CD40 as an M1 surface marker and CD206 (mannose receptor) as an M2 macrophage surface marker.12–14 Overall, high levels of CD40+/M1 staining were observed in nano-WC–Co-particle-exposed groups (Figure 2A) and in the LPS-stimulated M1 positive control group (Figure S2A). The prevalence of CD40+/M1 staining was much higher in nano-WC–Co-exposed groups than CD206+/M2 staining; while a slight increase in CD206+/M2 staining was observed at 5 days compared to 1 and 2 days after nano-WC–Co exposure; overall, the levels of CD206+/M2 macrophages were significantly lower than the control (0 μg/mL) group (Figure 2B). It seems that the ratio of M2/M1 increased with increasing exposure time (Figure 2C).
Therefore, at least in terms of surface marker expression, nano-WC–Co particles appear to induce strong CD40 expression, typical of an M1 classically activated phenotype, rather than increased levels of CD206 expression associated with M2 alternatively activated macrophages. However, there are a few potential limitations that may have contributed to the lack of CD206+ macrophages in our cell populations. Upon stimulation with PMA, THP-1 macrophages became extremely adherent to the culture dish and were difficult to detach for membrane surface staining; hence, it is possible that CD206 membrane receptors may have been damaged during the detachment process, causing low numbers of CD206+ cells overall. In addition, in our control (0 μg/mL) group, receiving PMA treatment only, a high level of CD40+/M1 staining was observed, which may indicate a predisposition toward the M1 surface markers in our control cells.
Next, we examined the secretion of pro-inflammatory cytokines including TNFα, IL-1β and IL-12 over an exposure period ranging from 6 to 48 h to determine the effects of nano-WC–Co exposure on cytokine expression over time (Figure 6). Given the strong CD40+/M1-type inflammatory macrophage surface marker expression in our flow cytometry assay, we expected to see complementary induction of pro-inflammatory cytokines in our ELISA assay. Typically, TNFα is secreted by activated macrophages and plays a primary role in the inflammatory immune response associated with infections due to bacterial or viral pathogens, such as promoting neutrophil chemotaxis and inducing acute phase proteins (C-reactive protein).61 A mixed TNFα response was observed in nano-WC-Co-treated macrophages compared to the control group, with higher levels after 6 h of exposure to low concentration (1 μg/mL) but similar or lower levels of TNFα compared to control at 10 and 100 μg/mL (Figure 6A).
IL-1β is a potent inflammatory cytokine that plays a critical role in the immune response to infection by promoting adhesion factors on endothelial cells, which allows for migration of macrophages and neutrophils to the site of infection.62 IL-1β is known to enhance systemic inflammation and mediate autoimmune disorders, such as rheumatoid arthritis.62,63 In contrast to TNFα, significantly higher levels of IL-1β, compared to control, were observed in all three nano-WC–Co treatment groups after 24 h of exposure and for the 1 and 10 μg/mL groups after 12 h (Figure 6B). Since IL-1β is produced largely by activated macrophages, the induction of high levels of IL-1β secretion may indicate a pro-inflammatory response and increased activation in macrophages exposed to nano-WC–Co particles.
IL-12 is known to induce phagocytic activation of macrophages, natural killer cells and cytotoxic T-lymphocytes as part of the innate immune response to bacterial, parasitic or intracellular infections.64–69 IL-12 also acts as an inducer of other cytokines, such as interferon-gamma (IFNγ). Like IL-1β, IL-12 may also contribute to chronic inflammation in rheumatoid arthritis, psoriasis and other immune disorders.64 In our THP-1 macrophage model, nano-WC–Co exposure also stimulated IL-12 secretion, since significantly higher IL-12 levels were found in nano-WC–Co-treated cells, compared to control, after 12 h of exposure (Figure 6C).
In addition, it is worth considering how the toxic effects of nano-WC–Co particles toward THP-1 macrophages (Figure 1) may have impacted the results of our inflammatory cytokine assay. In general, the 1 μg/mL nano-WC–Co dose was nontoxic up to 48 h in our viability assay, so higher observed levels of TNFα, IL-1β and IL-12 at this concentration are likely to reflect increased inflammatory cytokine secretion overall in nano-WC–Co-exposed macrophage populations. In contrast, significant toxicity was observed after 48 h of exposure to 10 μg/mL nano-WC–Co, so the lower levels of secreted inflammatory cytokines at this concentration seem consistent with increased nano-WC–Co toxicity at this time point. In addition, 100 μg/mL nano-WC–Co was toxic toward THP-1 macrophages at 6 h and beyond, and thus the secretion of increased levels of IL-12 (compared to control) at this concentration is especially significant, indicating that the remaining live macrophages (<80% compared to control) are stimulated to undergo a very strong IL-12-mediated inflammatory response. Overall, these results are consistent with our hypothesis and demonstrate that nano-WC–Co particles are capable of inducing a pro-inflammatory response in macrophages marked by high levels of IL-1β and IL-12 secretion and high expression of CD40 M1 surface markers.
Conclusion
This study examined the toxicity of nano-WC–Co in a CC, for the first time, of macrophages and lung epithelial cells and explored the effects of nano-WC–Co exposure on M1/M2 polarization and inflammatory cytokine secretion in THP-1 macrophages. The presence of THP-1 cells in the CC model was found to reduce the toxicity of nano-WC–Co compared to a monoculture of BEAS-2B cells, which suggested a protective role of macrophages against nano-WC–Co particle toxicity. In macrophages, nano-WC–Co exposure induced increased secretion of IL-1β and IL-12, which are indicators of a pro-inflammatory response. The M1/M2 polarization assay indicated a strong M1 phenotype (CD40+) in nano-WC–Co-treated macrophages after 1, 2 and 5 days of exposure. Overall, the outcomes of our cytokine ELISA and flow cytometry assay indicated that exposure to nano-WC–Co particles in vitro stimulates a pro-inflammatory cytokine response and polarization toward the M1 phenotype in macrophages.
Acknowledgments
The authors acknowledge the financial support from WV NASA EPSCoR, AO Foundation (Project S-13-15L), Osteosynthesis and Trauma Care Foundation, and Orthopaedic Research and Education Foundation, and also they acknowledge the WVU Flow Cytometry Core facility, operated by Kathy Brundage, which is supported by the National Institutes of Health equipment under grant number S10OD016165 and the Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant numbers P30GM103488 (CoBRE) and P20GM103434 (INBRE).
Disclosure
The authors report no conflicts of interest in this work.
References
Nurkiewicz TR, Porter DW, Hubbs AF, et al; HEI Health Review Committee. Pulmonary particulate matter and systemic microvascular dysfunction. Res Rep Health Eff Inst. 2011;164:3–48. | ||
Simeonova PP, Erdely A. Engineered nanoparticle respiratory exposure and potential risks for cardiovascular toxicity: predictive tests and biomarkers. Inhal Toxicol. 2009;21(suppl 1):68–73. | ||
Geiser M. Morphological aspects of particle uptake by lung phagocytes. Microsc Res Tech. 2002;57(6):512–522. | ||
Muhlfeld C, Rothen-Rutishauser B, Blank F, Vanhecke D, Ochs M, Gehr P. Interactions of nanoparticles with pulmonary structures and cellular responses. Am J Physiol Lung Cell Mol Physiol. 2008;294(5):L817–L829. | ||
Miyata R, van Eeden SF. The innate and adaptive immune response induced by alveolar macrophages exposed to ambient particulate matter. Toxicol Appl Pharmacol. 2011;257(2):209–226. | ||
Blum JL, Rosenblum LK, Grunig G, Beasley MB, Xiong JQ, Zelikoff JT. Short-term inhalation of cadmium oxide nanoparticles alters pulmonary dynamics associated with lung injury, inflammation, and repair in a mouse model. Inhal Toxicol. 2014;26(1):48–58. | ||
Gosens I, Post JA, de la Fonteyne LJ, et al. Impact of agglomeration state of nano- and submicron sized gold particles on pulmonary inflammation. Part Fibre Toxicol. 2010;7(1):37. | ||
Azad N, Rojanasakul Y, Vallyathan V. Inflammation and lung cancer: roles of reactive oxygen/nitrogen species. J Toxicol Environ Health B Crit Rev. 2008;11(1):1–15. | ||
Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122(3):787–795. | ||
Mosser DM. The many faces of macrophage activation. J Leukoc Biol. 2003;73(2):209–212. | ||
Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25(12):677–686. | ||
Aron-Wisnewsky J, Tordjman J, Poitou C, et al. Human adipose tissue macrophages: m1 and m2 cell surface markers in subcutaneous and omental depots and after weight loss. J Clin Endocrinol Metab. 2009;94(11):4619–4623. | ||
Daigneault M, Preston JA, Marriott HM, Whyte MK, Dockrell DH. The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages. PLoS One. 2010;5(1):e8668. | ||
Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol. 2006;177(10):7303–7311. | ||
Hao NB, Lu MH, Fan YH, Cao YL, Zhang ZR, Yang SM. Macrophages in tumor microenvironments and the progression of tumors. Clin Dev Immunol. 2012;2012:948098. | ||
Quatromoni JG, Eruslanov E. Tumor-associated macrophages: function, phenotype, and link to prognosis in human lung cancer. Am J Transl Res. 2012;4(4):376–389. | ||
Li JJ, Muralikrishnan S, Ng CT, Yung LY, Bay BH. Nanoparticle-induced pulmonary toxicity. Exp Biol Med (Maywood). 2010;235(9):1025–1033. | ||
Gustafsson A, Lindstedt E, Elfsmark LS, Bucht A. Lung exposure of titanium dioxide nanoparticles induces innate immune activation and long-lasting lymphocyte response in the Dark Agouti rat. J Immunotoxicol. 2011;8(2):111–121. | ||
Ma JY, Zhao H, Mercer RR, et al. Cerium oxide nanoparticle-induced pulmonary inflammation and alveolar macrophage functional change in rats. Nanotoxicology. 2011;5(3):312–325. | ||
Niwa Y, Hiura Y, Sawamura H, Iwai N. Inhalation exposure to carbon black induces inflammatory response in rats. Circ J. 2008;72(1):144–149. | ||
Nurkiewicz TR, Porter DW, Barger M, et al. Systemic microvascular dysfunction and inflammation after pulmonary particulate matter exposure. Environ Health Perspect. 2006;114(3):412–419. | ||
Madl AK, Unice K, Kreider M, Kovochich M, Bebenek IG, Abramson MM. Health risk ranking framework for the life cycle of nanomaterial-containing products: comparison of industrial versus consumer application settings. Int J Toxicol. 2013;32:65–69. | ||
Stefaniak AB, Day GA, Harvey CJ, et al. Characteristics of dusts encountered during the production of cemented tungsten carbides. Ind Health. 2007;45(6):793–803. | ||
Meyer-Bisch C, Pham QT, Mur JM, et al. Respiratory hazards in hard metal workers: a cross sectional study. Br J Ind Med. 1989;46(5):302–309. | ||
Chiappino G. Hard metal disease: clinical aspects. Sci Total Environ. 1994;150(1–3):65–68. | ||
Rivolta G, Nicoli E, Ferretti G, Tomasini M. Hard metal lung disorders: analysis of a group of exposed workers. Sci Total Environ. 1994;150(1–3):161–165. | ||
Wahbi ZK, Arnold AG, Taylor AJ. Hard metal lung disease and pneumothorax. Respir Med. 1997;91(2):103–105. | ||
Ruediger HW. Hard metal particles and lung disease: coincidence or causality? Respiration. 2000;67(2):137–138. | ||
Nemery B, Abraham JL. Hard metal lung disease: still hard to understand. Am J Respir Crit Care Med. 2007;176(1):2–3. | ||
Lasfargues G, Wild P, Moulin JJ, et al. Lung cancer mortality in a French cohort of hard-metal workers. Am J Ind Med. 1994;26(5):585–595. | ||
Ruokonen EL, Linnainmaa M, Seuri M, Juhakoski P, Soderstrom KO. A fatal case of hard-metal disease. Scand J Work Environ Health. 1996;22(1):62–65. | ||
Moulin JJ, Wild P, Romazini S, et al. Lung cancer risk in hard-metal workers. Am J Epidemiol. 1998;148(3):241–248. | ||
Lison D, Lauwerys R. In vitro cytotoxic effects of cobalt-containing dusts on mouse peritoneal and rat alveolar macrophages. Environ Res. 1990;52(2):187–198. | ||
Lison D, Lauwerys R. Study of the mechanism responsible for the elective toxicity of tungsten carbide-cobalt powder toward macrophages. Toxicol Lett. 1992;60(2):203–210. | ||
Lasfargues G, Lison D, Maldague P, Lauwerys R. Comparative study of the acute lung toxicity of pure cobalt powder and cobalt-tungsten carbide mixture in rat. Toxicol Appl Pharmacol. 1992;112(1):41–50. | ||
Huaux F, Lasfargues G, Lauwerys R, Lison D. Lung toxicity of hard metal particles and production of interleukin-1, tumor necrosis factor-alpha, fibronectin, and cystatin-c by lung phagocytes. Toxicol Appl Pharmacol. 1995;132(1):53–62. | ||
Lison D. Human toxicity of cobalt-containing dust and experimental studies on the mechanism of interstitial lung disease (hard metal disease). Crit Rev Toxicol. 1996;26(6):585–616. | ||
Adamis Z, Tatrai E, Honma K, Karpati J, Ungvary G. A study on lung toxicity of respirable hard metal dusts in rats. Ann Occup Hyg. 1997;41(5):515–526. | ||
De Boeck M, Hoet P, Lombaert N, Nemery B, Kirsch-Volders M, Lison D. In vivo genotoxicity of hard metal dust: induction of micronuclei in rat type II epithelial lung cells. Carcinogenesis. 2003;24(11):1793–1800. | ||
De Boeck M, Lombaert N, De Backer S, Finsy R, Lison D, Kirsch-Volders M. In vitro genotoxic effects of different combinations of cobalt and metallic carbide particles. Mutagenesis. 2003;18(2):177–186. | ||
Lombaert N, De Boeck M, Decordier I, Cundari E, Lison D, Kirsch-Volders M. Evaluation of the apoptogenic potential of hard metal dust (WC-Co), tungsten carbide and metallic cobalt. Toxicol Lett. 2004;154(1–2):23–34. | ||
Lombaert N, Lison D, Van Hummelen P, Kirsch-Volders M. In vitro expression of hard metal dust (WC-Co) – responsive genes in human peripheral blood mononucleated cells. Toxicol Appl Pharmacol. 2008;227(2):299–312. | ||
Kuhnel D, Busch W, Meissner T, et al. Agglomeration of tungsten carbide nanoparticles in exposure medium does not prevent uptake and toxicity toward a rainbow trout gill cell line. Aquat Toxicol. 2009;93(2–3):91–99. | ||
Ding M, Kisin ER, Zhao J, et al. Size-dependent effects of tungsten carbide-cobalt particles on oxygen radical production and activation of cell signaling pathways in murine epidermal cells. Toxicol Appl Pharmacol. 2009;241(3):260–268. | ||
Bastian S, Busch W, Kuhnel D, et al. Toxicity of tungsten carbide and cobalt-doped tungsten carbide nanoparticles in mammalian cells in vitro. Environ Health Perspect. 2009;117(4):530–536. | ||
Zhang XD, Zhao J, Bowman L, Shi X, Castranova V, Ding M. Tungsten carbide-cobalt particles activate Nrf2 and its downstream target genes in JB6 cells possibly by ROS generation. J Environ Pathol Toxicol Oncol. 2010;29(1):31–40. | ||
Busch W, Kuhnel D, Schirmer K, Scholz S. Tungsten carbide cobalt nanoparticles exert hypoxia-like effects on the gene expression level in human keratinocytes. BMC Genomics. 2010;11(1):65. | ||
Lombaert N, Castrucci E, Decordier I, et al. Hard-metal (WC-Co) particles trigger a signaling cascade involving p38 MAPK, HIF-1alpha, HMOX1, and p53 activation in human PBMC. Arch Toxicol. 2012;87:259–268. | ||
Armstead AL, Arena CB, Li B. Exploring the potential role of tungsten carbide cobalt (WC-Co) nanoparticle internalization in observed toxicity toward lung epithelial cells in vitro. Toxicol Appl Pharmacol. 2014;278(1):1–8. | ||
Armstead AL, Minarchick VC, Porter DW, Nurkiewicz TR, Li B. Acute inflammatory responses of nanoparticles in an intra-tracheal instillation rat model. PLoS One. 2015;10(3):e0118778. | ||
Takashiba S, Van Dyke TE, Amar S, Murayama Y, Soskolne AW, Shapira L. Differentiation of monocytes to macrophages primes cells for lipopolysaccharide stimulation via accumulation of cytoplasmic nuclear factor kappaB. Infect Immun. 1999;67(11):5573–5578. | ||
Adlakha-Hutcheon G, Khaydarov R, Korenstein R, et al. Nanomaterials, nanotechnology applications, consumer products, and benefits. Nanomater Risk Benefit. 2009:195–207. | ||
Maess MB, Wittig B, Cignarella A, Lorkowski S. Reduced PMA enhances the responsiveness of transfected THP-1 macrophages to polarizing stimuli. J Immunol Methods. 2014;402(1–2):76–81. | ||
Napierska D, Thomassen LC, Vanaudenaerde B, et al. Cytokine production by co-cultures exposed to monodisperse amorphous silica nanoparticles: the role of size and surface area. Toxicol Lett. 2012;211(2):98–104. | ||
Wottrich R, Diabate S, Krug HF. Biological effects of ultrafine model particles in human macrophages and epithelial cells in mono- and co-culture. Int J Hyg Environ Health. 2004;207(4):353–361. | ||
Naqvi AH, Hunt A, Burnett BR, Abraham JL. Pathologic spectrum and lung dust burden in giant cell interstitial pneumonia (hard metal disease/cobalt pneumonitis): review of 100 cases. Arch Environ Occup Health. 2008;63(2):51–70. | ||
Moriyama H, Kobayashi M, Takada T, et al. Two-dimensional analysis of elements and mononuclear cells in hard metal lung disease. Am J Respir Crit Care Med. 2007;176(1):70–77. | ||
Blanc PD. Is giant cell interstitial pneumonitis synonymous with hard metal lung disease? Am J Respir Crit Care Med. 2007;176(8):834; author reply 834–835. | ||
Dunlop P, Muller NL, Wilson J, Flint J, Churg A. Hard metal lung disease: high resolution CT and histologic correlation of the initial findings and demonstration of interval improvement. J Thorac Imaging. 2005;20(4):301–304. | ||
Antonios JK, Yao Z, Li C, Rao AJ, Goodman SB. Macrophage polarization in response to wear particles in vitro. Cell Mol Immunol. 2013;10(6):471–482. | ||
Pfeffer K. Biological functions of tumor necrosis factor cytokines and their receptors. Cytokine Growth Factor Rev. 2003;14(3–4):185–191. | ||
Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–550. | ||
Dinarello CA. Interleukin-1 beta and the autoinflammatory diseases. N Engl J Med. 2009;360(23):2467–2470. | ||
Gee K, Guzzo C, Che Mat NF, Ma W, Kumar A. The IL-12 family of cytokines in infection, inflammation and autoimmune disorders. Inflamm Allergy Drug Targets. 2009;8(1):40–52. | ||
Trinchieri G. Interleukin-12: a proinflammatory cytokine with immunoregulatory functions that bridge innate resistance and antigen-specific adaptive immunity. Annu Rev Immunol. 1995;13(1):251–276. | ||
Hamza T, Barnett JB, Li B. Interleukin 12 a key immunoregularoty cytokine in infection applications. Int J Mol Sci. 2010;11(3):789–806. | ||
Li B, Jiang B, Dietz MJ, Smith ES, Clovis NB, Rao KM. Evaluation of local MCP-1 and IL-12 nanocoatings for infection prevention in open fractures. J Orthop Res. 2010;28(1):48–54. | ||
Boyce BM, Lindsey BA, Clovis NB, et al. Additive effects of exogenous IL-12 supplementation and antibiotic treatment in infection prophylaxis. J Orthop Res. 2012;30(2):196–202. | ||
Li B, Jiang B, Boyce BM, Lindsey BA. Multilayer polypeptide nanoscale coatings incorporating IL-12 for the prevention of biomedical device-associated infections. Biomaterials. 2009;30:2552–2558. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.