")
Back to Journals » International Journal of Nanomedicine » Volume 14
In vitro and in vivo mechanism of hepatocellular carcinoma inhibition by beta-TCP nanoparticles
Authors Liu L, Dai H, Wu Y, Li B, Yi J, Xu C, Wu X
Received 2 November 2018
Accepted for publication 19 March 2019
Published 13 May 2019 Volume 2019:14 Pages 3491—3502
DOI https://doi.org/10.2147/IJN.S193192
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Langlang Liu, Honglian Dai, Yanzeng Wu, Binbin Li, Jiling Yi, Chao Xu, Xiaopei Wu
State Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, Wuhan, People’s Republic of China
Background: Studies have showed that nanoparticles have a certain anti-cancer activity and can inhibit many kinds of cancer cells. β-tricalcium phosphate nanoparticles (nano-β-TCP) displays better biodegradation, but the application and mechanism of nano-β-TCP in anti-cancer activity are still not clear.
Purpose: The objective of this study was to synthesize nano-β-TCP and investigate its inhibitory properties and mechanism on hepatocellular carcinoma (HepG2) cells in vitro and in vivo.
Methods: Nano-β-TCP was synthesized using ethanol-water system and characterized. The effects of nano-β-TCP on cell viability, cell uptake, intracellular oxidative stress (ROS), cell cycle and apoptosis were also investigated with HepG2 cells and human hepatocyte cells (L-02). Intratumoral injection of nano-β-TCP was performed on the xenograft liver cancer model to explore the inhibitory effect and mechanism of nano-β-TCP on liver tumors.
Results: In vitro results revealed that nano-β-TCP caused reduced cell viability of HepG2 cells in a time-and dose-dependent manner. Nano-β-TCP was internalized through endocytosis and degraded in cells, resulting in obvious increase of the intracellular Ca2+, and PO43-, ions. Nano-β-TCP induced cancer cells to produce ROS and induced apoptosis of tumor cells by an apoptotic signaling pathways both in extrinsic and intrinsic pathway. In addition, nano-β-TCP blocked cell cycle of HepG2 cells in G0/G1 phase and disturbed expression of some related cyclins. In vivo results showed that 40 mg/kg of nano-β-TCP had no significant toxic side effects, but could effectively suppress hepatocellular carcinoma growth.
Conclusion: These findings revealed the anticancer effect of nano-β-TCP and also clarified the mechanism of its inhibitory effect on hepatocellular carcinoma.
Keywords: nano-β-TCP, HepG2 cells, ROS, cell cycle, apoptosis, inhibition
Introduction
Published by the World Health Organization, “Global Cancer Report 2014” shows that China ranks first in the world in new cases of cancer. Among these cases, hepatocellular carcinoma, known as the king of cancers, has one of the highest incidences of malignant tumors, with the morbidity and mortality exceeding about 50%.1–4 Patients with hepatocellular carcinoma are often in the middle and late stage of diagnosis, losing the best chance of surgery and local treatment. The conventional treatment methods, including surgical treatment, drug intervention, chemotherapy, and radiotherapy, have poor healing outcomes. So far, the treatment of hepatocellular carcinoma is still an imminent problem that has not been satisfactorily solved.
In recent years, nanomaterials, due to unique physical and chemical properties, have been widely used in biomedicine, biotechnology, and other fields, including medical imaging,5 drug carrier,6 and cancer treatment.7 Many studies have shown that a variety of nanoparticles, such as metal nanoparticles (Ag, Au, Fe, Zinc),8,9 magnetic nanoparticles,10,11 and inorganic nanoparticles (Hydroxyapatite, Single-walled carbon nanotube),12–15have antitumor effects and restrain different tumors growth, inducing cancer cell apoptosis. For example, silicon dioxide nanoparticles show an obvious apoptosis-inducing effect on human lung adenocarcinoma A549 cells and activated intracellular reactive oxygen species (ROS) and glutathione (GSH).16 Titanium dioxide nanoparticles have resulted in the death of human lung cancer SPC-A1 cells by inducing oxidative stress.17,18 Activated carbon nanoparticles (ACNP) induced apoptosis of BGC-823 cells by overproducing of ROS and activating the mitochondrial signal transduction pathway.19 Fe3O4 nanoparticles were gradually ingested by hepatocellular carcinoma cells and inhibited the proliferation of cancer cells by causing a decrease in mitochondria membrane potential and inducing cell apoptosis.20,21 Among these anti-tumor nanomaterials, calcium phosphate nanomaterials have also attracted great attention. Numerous studies have reported that hydroxyapatite (HA) nanoparticles showed different inhibitory effects on human cancer cells (MGC80-3, HepG2, Hela, and MG63), but had no obvious toxicity on normal cells.22,23 In addition, particles in nanoscale exhibited stronger anti-tumor activity than that in micron grade and reduced the drug’s toxic side-effects when combined with other chemotherapy drugs.24–29
Moreover, as one of the calcium phosphate materials, nano-β-TCP displays better biodegradation than HA, but there are few reports on its application in cancer treatment. Hence, targeting the difficult problems of treatment in malignant tumors, the inhibitory effect and mechanism of nano-β-TCP were investigated with hepatocellular carcinoma as the model. The effects of nano-β-TCP on cell viability, cell uptake, intracellular ROS content, cell cycle, cell apoptosis, and tumor suppression in vitro and in vivo were investigated. The mechanism of the inhibitory effect of nano-β-TCP on hepatocellular carcinoma is shown in Figure 1.
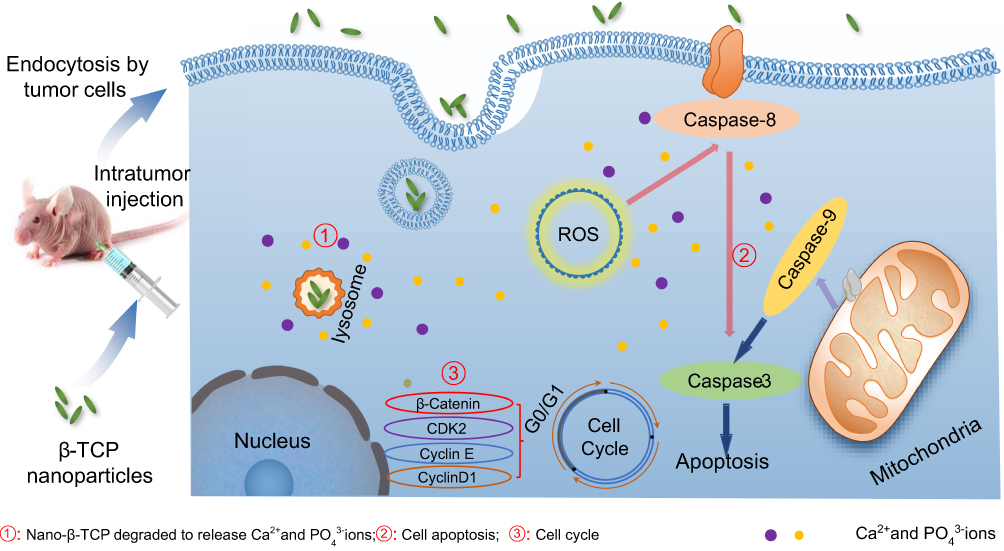 | Figure 1 Schematic representation of inhibitory mechanism of nano-β-TCP on hepatocellular carcinoma. Intratumor injection of nano-β-TCP was performed on the xenograft hepatocellular carcinoma model. Nano-β-TCP was internalized into tumor cells by nonspecific endocytosis and rapidly degraded in the acidic lysosome to release Ca2+ and PO43- ions (①). Cell apoptosis was activated by extrinsic and intrinsic apoptosis pathways synergistically orchestrated with the ROS generation (②). In addition, the expression of some related cyclins (CyclinD1, CDK2, CyclinE, and β-catenin) was inhibited, eventually leading to cell cycle blocking in G0/G1 phase (③). |
Materials and methods
Materials
Ca(NO3)2•4H2O, (NH4)2HPO4, anhydrous ethanol, and ammonia solution were purchased from Sinopharm Chemical Reagent Co. (Beijing, China). HepG2 and human hepatocyte (L-02) cells were provided by China Center for Type Culture Collection. DMEM medium, RPMI-1640, phosphate buffered saline (PBS), penicillin-streptomycin, and trypsin-EDTA were purchased from HyClone. Fetal bovine serum (FBS) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were provided by Tianhang Biotechnology Co. (Zhejiang, China). 2‘,7‘-dichlorofluorescein diacetate (DCFH/DA), propidium iodide (PI), Hoechst33342, HRP-conjugated IgG, Caspase-3, 8, and 9 fluorescence assay kits were purchased from Beyotime Institute of Biotechnology (Haimen, China).
Preparation and characterization of nano-β-TCP
Nano-β-TCP used in the experiment was synthesized by an ethanol-water system in our laboratory. Briefly, a stoichiometric amount of Ca(NO3)2•4H2O and (NH4)2HPO4 were dissolved in anhydrous ethanol and deionized water, respectively. Then the two solutions were dropwise mixed under stirring thoroughly at 40°C and a constant pH of 7.0 with ammonia solution. After that, the mixed solution was placed in an oven at 30°C. The precipitate was centrifuged, washed with deionized water and anhydrous ethanol several times to remove NH4+ and NO3− ions, and then dried in a vacuum oven at 80°C for 12 hours. Finally, the dried powder was calcined at 800°C for 2 hours in a muffle furnace, employing a heating rate of 15°C/minute. Nano-β-TCP was sterilized by autoclaving at 121°C for 20 minutes and sonicated prior to cell experiments.
The phase and crystallization of the sample were characterized by X-ray diffraction (XRD) (D/MAX-RBRU-200B, Japan). The characteristic groups of the samples were studied using Fourier transform infrared spectroscopy (FTIR) (Nicolet6700, USA). The morphology was observed by field-emission transmission electron microscopy (TEM) (JEM2100F, Japan).
Cell viability (MTT assay)
HepG2 and L-02 cells were, respectively, cultured in DMEM and RPMI-1640 medium supplemented with 10% FBS and 1% penicillin-streptomycin at 37°C under a humidified atmosphere containing 5% carbon dioxide. Cells were trypsinized with 0.25% trypsin-EDTA and passaged upon attaining 70% confluence in a cell culture flask.
The effects of nano-β-TCP on the viabilities of HepG2 and L-02 cells were determined by MTT assay.30 In brief, the exponentially growing HepG2 and L-02 cells were seeded into a 96-well plate at a density of 1×104 cells/well and allowed to attach for 24 hours. Then, the culture medium was replaced by fresh DMEM and RPMI-1640 medium containing different concentrations of nano-β-TCP (0, 50, 100, 200, 400 μg/mL), respectively. The cells were incubated at 37°C with 5% CO2 for 24 and 48 hours. Afterwards, 20 μL of filtered MTT working solution (5 mg/mL) was added to each well, and the cells were further incubated for 4 hours to allow the yellow dye to be transformed into blue crystals. Thereafter, the unreacted dye solution was removed, and 200 μL of DMSO solution was added. The absorbance value was measured at 490 nm using a full-wavelength microplate reader. Cell viability was calculated according to the following formula: Cell viability (%)=[Atest]/[Acontrol]×100%.
Cellular uptake of nano-β-TCP
HepG2 and L-02 cells were seeded on culture dishes for 24 hours. Then, the cells were treated with nano-β-TCP (400 μg/mL) for 48 hours. At the end of the incubation time, cells in both groups were collected in centrifuge tubes and fixed in 2.5% glutaraldehyde, post-fixed with 1% osmium tetroxide, dehydrated in an ascending series of ethanol, embedded in epoxy resin, and sectioned. The sections were double stained with lead citrate and uranyl acetate and, thereafter, observed with a biological transmission electron microscope.
HepG2 and L-02 cells were treated with nano-β-TCP (0, 400 μg/mL) for 48 hours. Then the culture medium was discarded and washed three times by PBS. The cell lysing reagent (RIPA Lysis Buffer) is added to allow the cells to fully lyse. After centrifugation, the supernatant is collected and placed in a clean centrifuge tube. The concentration of intracellular calcium and phosphorus ions were measured by inductively coupled plasma atomic emission spectroscopy (ICP-OES, USA).
ROS generation
The level of intracellular ROS generation was estimated by using 2,7-dichlorofluorescein diacetate (DCFH/DA). HepG2 and L-02 cells were plated on 6-well plates and incubated with nano-β-TCP (400 μg/mL) for 48 hours. Then, the culture medium was discarded and washed three times by PBS. DCFH/DA (10 μM) with serum free medium was added to each well and incubated at 37°C for 20 minutes, the fluorescence was observed under the inverted fluorescence microscope. Rosup (50 mg/mL) is a compound mixture which was used as a positive control.
Analysis of cell cycle
HepG2 and L-02 cells were incubated with nano-β-TCP (400 μg/mL) for 48 hours. After incubation, cells in each group were harvested, washed with PBS, and fixed in ice-cold 70% ethanol at 4°C for 2 hours or more . Fixed cells were centrifuged at 1,000 rpm for 5 minutes at 4°C and incubated with PI mixture (50 mg/mL) for 30 minutes in darkness. The cell cycle distribution was measured by flow cytometry at 488 nm.
Cell apoptosis
Annexin V-FITC/PI and Hochest33342 stain assay were applied to examine cell apoptosis induced by nano-β-TCP. Briefly, HepG2 and L-02 cells were plated on culture dishes and treated with nano-β-TCP (400 μg/mL) in an incubator for 48 hours. Then, both cell types were carefully harvested, washed with PBS, and centrifuged at 1,000 rpm for 5 minutes. Annexin V-FITC/PI was added to the cell suspension and incubated for 10 minutes in the dark at room temperature. Flow cytometry analysis was carried out to assess the percentage of apoptotic cells. As to nuclear morphology, the cell nucleus was then stained with Hochest33342 for 30 minutes at 4°C and checked under a fluorescence microscope.
Western blotting
Cells were plated on culture dishes and exposed to the concentration of nano-β-TCP at 400 μg/mL for 48 hours, followed by centrifugation at 1,000 rpm for 5 minutes and harvest. Cell lysis buffer (containing protease inhibitors) was added to the pellet for 30 minutes on ice. After that, cells were centrifuged for 5 minutes and the supernatant was transferred into another new tube to measure the concentration of protein in the sample using BCA protein assay kit. For each sample, protein was loaded to the SDS-polyacrylamide gel and separated at 75 V for 2 hours, then transferred to a nitrocellulose membrane. The membrane was blocked by 5% skim milk in TBST on a rocking platform for 1 hour at room temperature. After treatment with primary antibodies (1:500 or 1:1,000 dilution) at 4°C overnight, the membrane was washed three times using TBST, then followed incubation with HRP-conjugated secondary antibodies (1:3,000 dilution) for 30 minutes at room temperature. The immunoreactive blots of apoptosis indexes (BAX, Caspase-3, Caspase-8, and Caspase-9)and cyclins (CyclinD1, CDK2, CyclinE, β-catenin) were measured using the ECL kit.
In vivo toxicity and antitumor activity
All animal experiments were conducted under an approved protocol of the Institutional Animal Care and Use Committee of Wuhan University of Technology. This committee approved the research and experiments. Female nude mice (BALB/c nu/nu, 6 weeks old) were provided by Hubei Provincial Center for Disease Control and Prevention. For the assessment of toxicity in vivo, the nude mice were randomly divided into three groups (three rats/group) and intraperitoneally injected with 0.2 mL/mouse of physiological saline or different concentrations of nano-β-TCP (0, 20, 40 mg/kg) three times a week. The whole period is about 24 days; before each injection, the weight of mice in each group was measured. On day 24, nude mice were anesthetized with phenobarbital and sacrificed. The major organs (liver and kidney) were collected, fixed with paraformaldehyde, embedded, and sectioned for hematoxylin and eosin (H&E) staining.
For antitumor activity, the solid HepG2 tumors established in nude mice were used to construct a hepatocellular carcinoma xenograft animal model. When tumor volume reached ∼250 mm3 (about 15 days), the nude mice with similar tumor size were randomly divided into three groups: negative control group, positive control group, and experimental group. Each mouse in these groups was intratumor injected with 0.2 mL of saline, doxorubicin hydrochloride (DOX, 5 mg/kg), and nano-β-TCP (40 mg/kg) three times a week, respectively. After the day of the treatment, nude mice were sacrificed to measure tumor size and weight, the formula is the following: Volume (mm3)=0.5×length×width2. Thereafter, the tumor was fixed with paraformaldehyde, embedded, and sectioned for histological examination.
Statistical analysis
Statistical analysis was performed on Software SPSS17.0. All data were presented as means±standard deviation (SD). Statistical differences were evaluated using the one-way ANOVA and considered to be significant when *P<0.05, **P<0.01.
Results and discussion
Characterization of nano-β-TCP
The representative XRD pattern, FTIR, and TEM image of nano-β-TCP prepared by ethanol-water system are shown in Figure 2. The XRD pattern exhibited a typical phase composition of β-TCP nanoparticles. The diffraction peak of the product was consistent with that of the β-TCP standard card (JCPDS NO.09–0169), indicating a high purity product with narrow diffraction peak and high strength (Figure 2A). The absorption spectra of PO43- ions (1044, 1078, and 972 cm−1) and P-O(H) stretching in HPO42- ions (945 cm−1) were observed in the FTIR spectra of nano-β-TCP (Figure 2B). The peaks of the single molecule of adsorbed water were also discerned at 1,618 and 3,437 cm−1. The 1,385 cm−1 band corresponded to the absorption peak of CO32-, which might be due to the fact that CO2 in the air was dissolved in the lattice of nano-β-TCP.31 In a typical TEM image of nano-β-TCP powder obtained by calcination at 800°C, as presented in Figure 2C, certain agglomeration of nano-β-TCP was observed, due to the large surface area and energy associated to these nanoparticles,32 indicating nanoparticles with a diameter of about 55 nm, 120 nm in length.
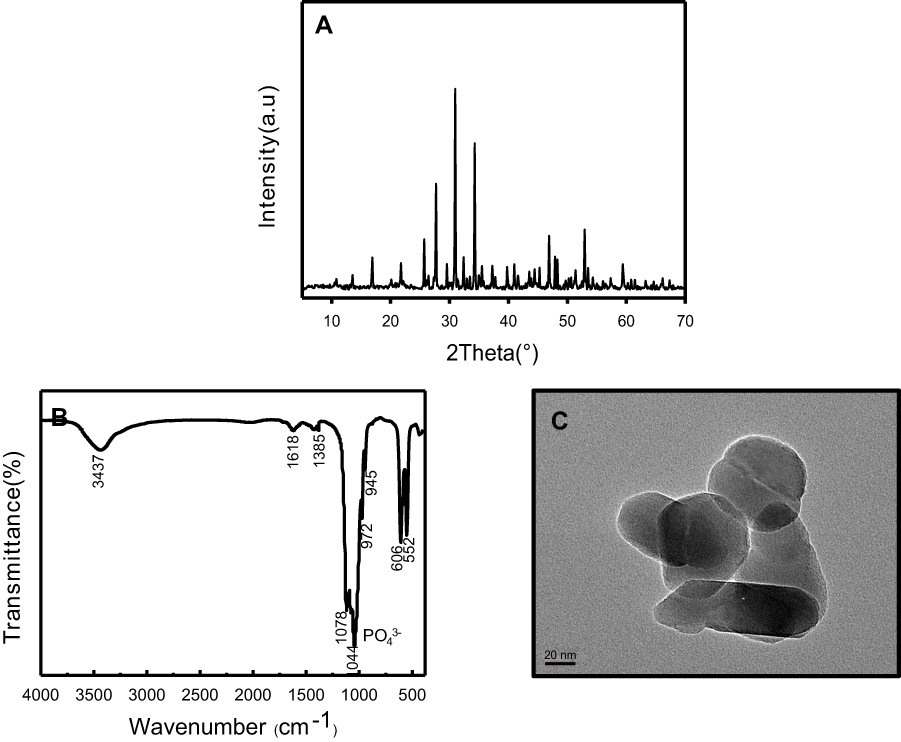 | Figure 2 Characterization of nano-β-TCP synthesized by ethanol-water system. (A) XRD pattern, (B) FTIR, and (C) TEM images of nano-β-TCP. (Scale bar: 20 nm).Abbreviations: FTIR, Fourier transform infrared spectroscopy; TEM, transmission electron microscopy; XRD, X-ray diffraction. |
Cell viability and cellular uptake
Based on the deposition feature of high concentration of nano-β-TCP, so here 0–400 μg/mL of nano-β-TCP was chosen in this study.23 After 24 hours, when the concentration of nano-β-TCP increased to 400 μg/mL, the viabilities of L-02 and HepG2 cells decreased to 69.52% and 50.11%, respectively. After 48 hours, with the concentration of nano-β-TCP increased to 400 μg/mL, the viabilities of L-02 and HepG2 cells decreased to 59.40% and 39.76%, respectively. These results indicate that nano-β-TCP can inhibit the viability of both cell types, with stronger inhibition for HepG2 cells. The viability of HepG2 cells showed a decreasing trend with the increase of the concentration of nano-β-TCP and incubation time. For instance, when the concentration of nano-β-TCP was 200 μg/mL, the viability of HepG2 cells decreased to 58.14% and 48.91% for 24 and 48 hours, respectively. The MTT assay revealed that nano-β-TCP reduced the viability of HepG2 cells in a time- and dose-dependent manner (Figure 3A).
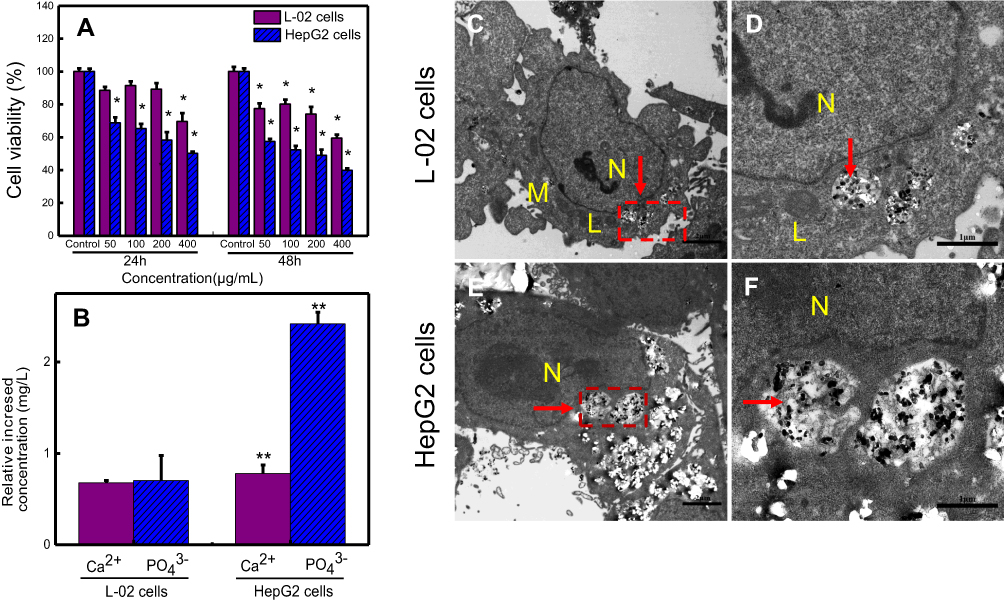 | Figure 3 (A) Comparison of the cell viabilities of HepG2 and L-02 cells treated with different concentrations of nano-β-TCP for 24 and 48 hours. (B) The relative increased intracellular content of Ca2+ and PO43- ions in L-02 and HepG2 cells treated with nano-β-TCP (400 μg/mL) for 48 hours (vs control group, *P<0.05, **P<0.01). TEM images of L-02 (C, D) and HepG2 (E, F) cultured with nano-β-TCP (400 μg/mL) for 48 h. (D and F are enlarged views of C and E in red pane, respectively). Abbreviations: N, nucleus; M, mitochondria; L, lysosomes; the particles labelled by red arrows are nano-β-TCP. |
Cells take up nanoparticles primarily through endocytosis, which is affected by the shape, size, and surface charge of nanomaterials.33 For example, rod-like nano-HA had an appreciable advantage over spherical-shaped nanoparticles with regard to cellular uptake.34 Even though there is repulsion between negatively charged particles and the electronegative cytomembrane, there is evidence that uptake of electronegative nanoparticles by cells can be achieved.35,36 After being co-cultured with nano-β-TCP for 48 hours, nano-β-TCP was penetrated into both kinds of cells by endocytosis. Nano-β-TCP was mainly found in vesicles in the cytoplasm close to the nucleus and didn’t enter the nucleus. This result is slightly different from that of HA in the HepG2 nucleus,23 the distribution of nanomaterials in cells is related to the properties of the material. There was relatively intact cell morphology, clearly visible mitochondria and lysosomes, and few vacuoles in L-02 cells (Figures 3C and D). However, HepG2 cells had irregular shape, invisible mitochondria, and lysosomes, and so many vesicles induced by nano-β-TCP (Figures 3E and F). Although there is repulsion between negatively charged particles and the electronegative cytomembrane, abundant negatively charged groups on the surface of cancer cells generate more negative charge of cancer cells than normal cells, which are mainly derived from the sialic acid residues protruding from the apical surface of the plasma membrane. Nano-β-TCP can be expected to have higher adhering capacity to HepG2 than L-02 cells on the basis of the electrostatic interactions between negatively charged sites on the cell membrane and the positive binding sites on the β-TCP surface. Since much more nano-β-TCP in HepG2 was observed than that in L-02 cells, this result agrees well with the finding of Daniels et al.37–39
The concentrations of intracellular Ca2+ and PO43- ions after co-culturing with nano-β-TCP (400 μg/mL) for 48 hours are shown in Figure 3B. Comparing with the control group, the concentrations of Ca2+ and PO43- ions in both cell types increased, especially in HepG2 cells (**P<0.01). The possible reasons are the following: (1) Nano-β-TCP is degraded in the acidic lysosome in both cell types after ingesting, leading to an increase in calcium and phosphorus ions;36 (2) HepG2 cells have a slightly stronger uptake capability of nano-β-TCP than L-02 cells, resulting in higher uptake of nano-β-TCP and higher intracellular Ca2+ and PO43-.
Measurement of intracellular ROS
Many nanomaterials can be used as anticancer drug carriers, because they can induce cancer cells to generate ROS when contacted with cells, resulting in cancer cell apoptosis. For example, HA induced human breast cancer cells (McF-7) to produce ROS and activate p53, causing McF-7 cells apoptosis.24 The release of Zn2+ ion from zinc oxide nanoparticles after intracellular degradation could produce ROS which, in turn, led to apoptosis of HepG2 cells.40 Can nano-β-TCP, similarly, induce HepG2 cells to produce ROS? To verify this hypothesis, the ROS Assay Kit was used to detect ROS levels in cells. DCFH-DA itself has no fluorescence and can freely penetrate the cell membrane and be hydrolyzed to generate DCFH. The DCFH does not permeabilize the cell membrane, making it easy for the probe to be loaded into the cell. ROS in cells can oxidize non-fluorescent DCFH to produce fluorescent DCF. The fluorescence of DCF can reflect the level of intracellular ROS, the higher green fluorescence intensity, the more ROS content. After being co-cultured with nano-β-TCP (400 μg/mL) for 48 hours, the green fluorescence intensity of L-02 cells, as expected, obviously increased (level=125.43) compared to the control group (level=86.28), but less than that in the Rosup treatment group (level=161.04), similarly, the green fluorescence intensity of HepG2 cells also increased (level=144.95) compared to the control group (level=116.33) (Figures 4A–G). Rosup, as a positive control group, is a mixture which can significantly improve the ROS content in cells within 30 minutes. In addition, green fluorescence intensity of HepG2 cells in the control group was slightly higher than that of L-02 cells, which is consistent with the literature report.41 The above findings showed that nano-β-TCP could induce HepG2 cells to produce ROS.
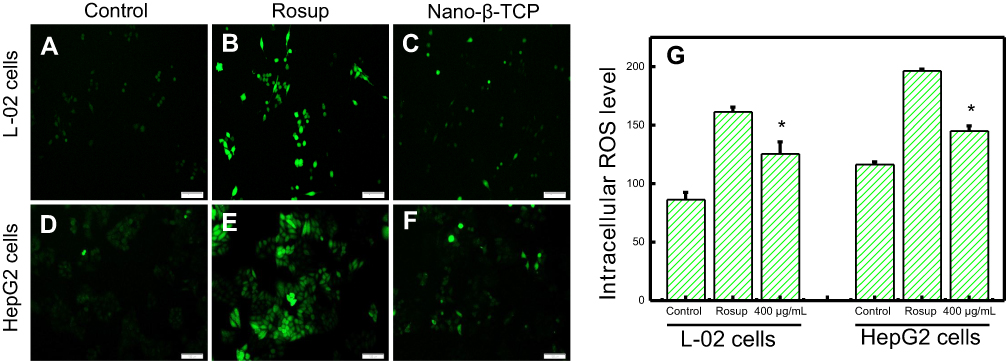 | Figure 4 Fluorescent of the intracellular ROS using DCFDA dye in L-02 (A–C) and HepG2 (D–F) cells for 48 hours. (A) and (D) Control group, (B) and (E) Treated with Rosup, (C) and (F) Treated with nano-β-TCP. (G) The intracellular ROS level in both cell types for 48 hours (vs control group, *P<0.05). |
Cell cycle and apoptosis of tumor cells
Cell cycle
During the cell proliferation, the cell cycle is divided into different phases: G0, G1, S, G2, M. Many substances, such as protein synthesis and DNA replication, occur mainly in G1 and S phases. According to differences in DNA content, the cell cycle can generally be divided into G0/G1, S, G2/M phase. Compared with the control group, the percentage of G0/G1 phase increased 1.21%, while the proportion of S phase and G2/M phase decreased by 0.54% and 0.66%, respectively, when L-02 cells were treated with nano-β-TCP (400 μg/mL). The percentage change of G0/G1 phase in HepG2 was slightly larger than that in L-02 cells. The proportion of G0/G1 phase increased 7.64%, while the proportion of S phase and G2/M phase decreased 3.28% and 4.36%, respectively. The result indicated that nano-β-TCP had a certain blocking effect on cell cycle, especially for HepG2 cells (Figures 5A–D).
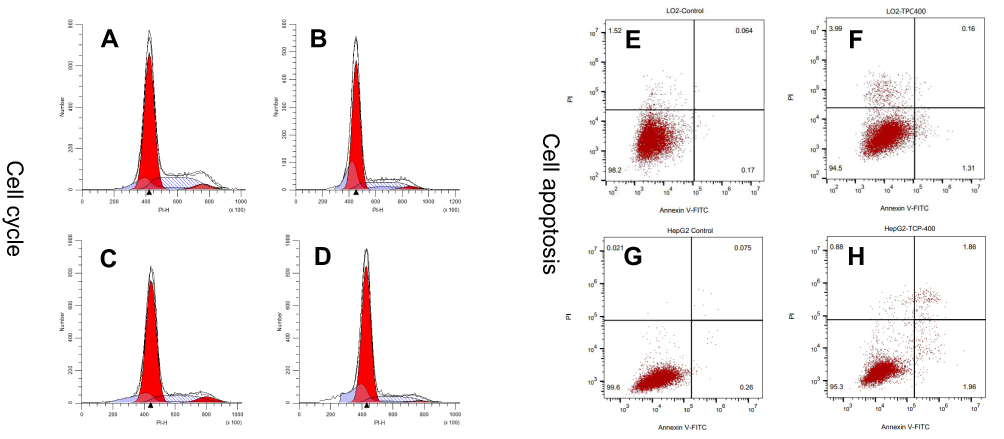 | Figure 5 The effects of nano-β-TCP on cell cycle (A–D) and cell apoptosis (E–H) for 48 hours. (A and E: L-02+0 μg/mL; B and F: L-02+400 μg/mL;C and G: HepG2+0 μg/mL, D and H: HepG2+0 μg/mL). |
The process of cell cycle can be blocked by the addition of exogenous substances.42,43 Nano-β-TCP could block the cell cycle in G0/G1 phase, probably involved in disturbing cell cycle signaling pathway. Therefore, some relative cyclins such as β-catenin, CDK2, CyclinE, and CyclinD1 in cells were tested (Figure 6E and Table 1). Compared with the control group, the expression of these proteins was lower in both cells treated with nano-β-TCP (400 μg/mL). The occurrence and development of tumor are closely related to abnormal cell cycle regulation. When the expression of β-catenin was inhibited, G0/G1 phase of HepG2 cells would be blocked and reducing the expression of CyclinE and CyclinD1, where molecules can induce cell cycle from G1 to S phase.44 In addition, the combination of cyclins with cyclin-dependent kinases (CDKs) plays an important role in regulation of cell cycle in a specific regulatory site. Therefore, as one of the CDKs, the expression of CDK2 in cells is also investigated. CDK2 mainly plays a role in the transformation of G1-S phase, and the decrease of its expression can inhibit the proliferation of HepG2 cell and induce cell apoptosis.
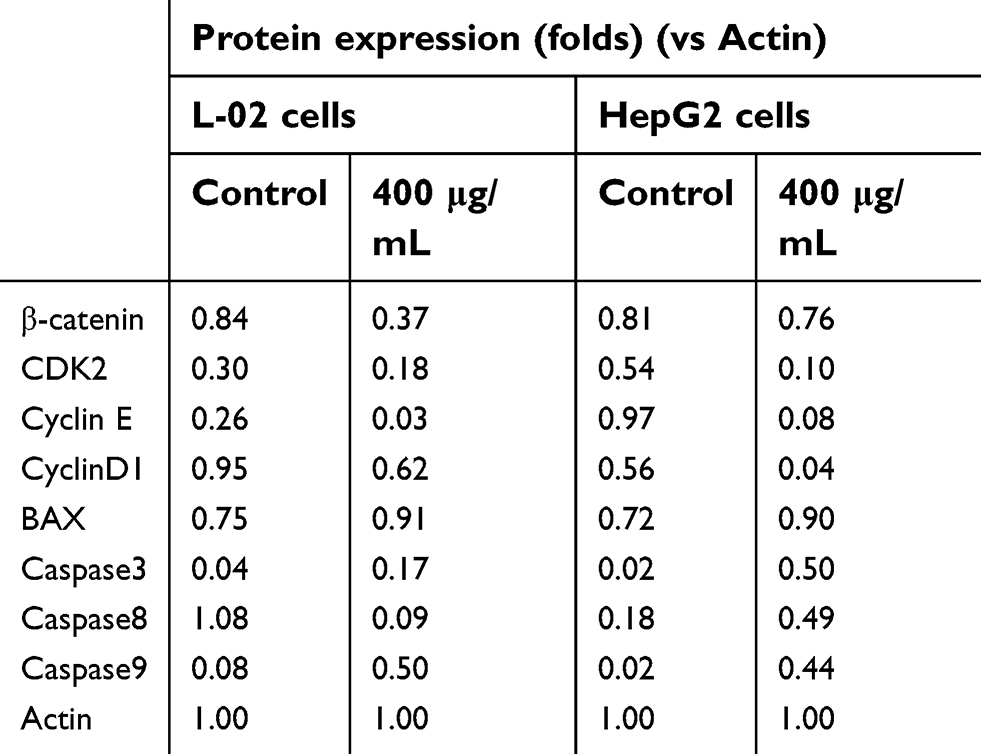 | Table 1 Effects of nano-β-TCP on cyclins (CyclinD1, CDK2, CyclinE, β-catenin) and apoptosis indexes (BAX, Caspase-3, 8, and 9)in L-02 and HepG2 cells |
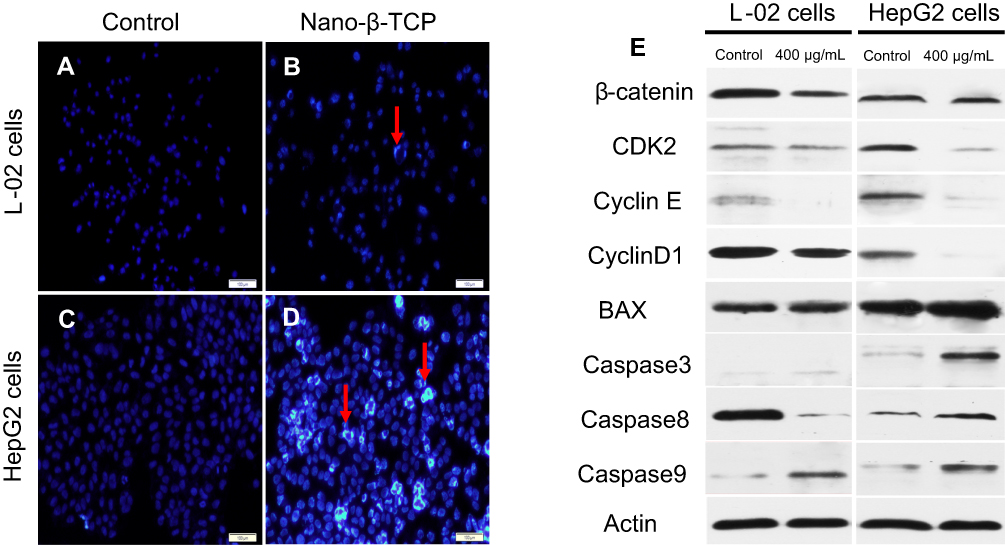 | Figure 6 (A–D) Fluorescent micrographs of cells before and after nano-β-TCP treatment for 48 hours. Cell nuclei were stained with Hochest33342. Apoptotic cells are labeled by red arrows. (E) Expression of some related cyclins (CyclinD1, CDK2, CyclinE, β-catenin) and apoptosis indexes (BAX, Caspase3, Caspase8, and Caspase9) in L-02 and HepG2 cells treated with nano-β-TCP for 48 hours. (Scale bar: 100 μm. Compared to the control group). |
Cell morphology and cell apoptosis
When cell apoptosis occurs, the nucleus morphology also changes. In order to examine change in the nucleus morphology, L-02 and HepG2 cells were incubated with Hochest33342, which can penetrate the cell membrane. The blue fluorescence intensity of apoptotic cells is more significantly enhanced than that of normal cells. Compared with the control group, there were no significant changes in fluorescence intensity, the nuclear morphology, and distribution of chromatin of L-02 cells in the experimental group. However, the fluorescence intensity was much brighter in HepG2 cells treated with nano-β-TCP than that in the untreated cells, indicating that there were many apoptotic cells. In addition, the nucleus of HepG2 cells treated with nano-β-TCP exhibited the classical morphology apoptotic characteristics: chromatin condensation and DNA fragmentation (Figures 6A–D). The above results showed that nano-β-TCP could also promote the apoptosis of L-02 and HepG2 cells.
Annexin V-FITC/PI double stain assay was used to determine cell apoptosis. The apoptosis rates in L-02 and HepG2 cells induced by nano-β-TCP increased 0.64% and 3.02% (vs control group), respectively, especially in HepG2 cells after treating with nano-β-TCP at 400 μg/mL (**P<0.01) (Figures 5E–H). To further verify this conclusion, some related apoptosis indexes (BAX, Caspase-3, Caspase-8, and Caspase-9) were detected in both cells. As we know, a series of related proteins were activated in progress of cell apoptosis. For example, BAX is a member of BCL-2member proteins which can promote cell apoptosis.45 Caspase (Cysteine-requiring Aspartate Protease) is a family of proteases that plays an important role in cell apoptosis. As a member of caspase, Caspase-3 is a key enzyme in the process of apoptosis, which functions include chromatin condensation, DNA fragmentation, and so on. Caspase-3 is activated by Caspase-8 (initiator of external apoptotic signaling pathway) and Caspase-9 (initiator of internal apoptotic signaling pathway),46 thereby promotes subsequent apoptotic signaling and cell apoptosis. Compared with the control group, the expression of BAX, Caspase-3, and Caspase-9 also increased in L-02 cells treated with 400 μg/mL nano-β-TCP for 48 hours, but the expression of Caspase-8 did not seem significant, the possible reason is that L-02 cells were not as sensitive as HepG2 cells in the external signal stimulation of nano-β-TCP. As to HepG2 cells, the expression of the above protein in the experimental group increased relative to the untreated group (Figure 6e and Table 1). The above findings indicate that apoptosis of L-02 and HepG2 cells treated with nano-β-TCP was activated by extrinsic and intrinsic apoptosis pathways.
Nano-β-TCP inhibited tumor growth with no toxic side-effect in vivo
The drug carrier that enters the body should reduce the toxic side-effects as far as possible. Here, the toxicity of nano-β-TCP in vivo was investigated through the change in weight of nude mice and the influence of major organs (liver and kidney). The effect of nano-β-TCP on the weight of nude mice in different concentrations (0, 20, 40 mg/kg) is shown in Figure 7A. During the treatment, there was an increasing trend in weight of nude mice in either the experimental group or the control group. On day 24, nude mice were anesthetized with phenobarbital and sacrificed; the kidney and liver were then processed and subjected to H&E staining. Compared with the control group, nano-β-TCP treatment did not cause any obvious abnormality or organ damage, as shown by representative images in Figures 7B–E. These results, thus, demonstrated that nano-β-TCP (<400 mg/kg) had no visible toxicity.
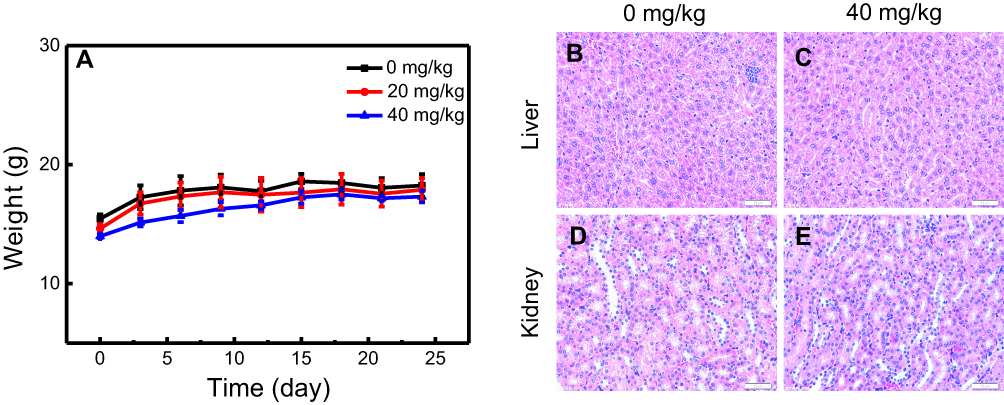 | Figure 7 In vivo toxicity of nano-β-TCP. (A) The effects of nano-β-TCP with different concentrations (0, 20, 40 mg/kg) on change in body weight. (B–E) H&E staining of liver and kidney sections from nude mice treated with nano-β-TCP. (B and D) Control; (C and E) 40 mg/kg. |
Based on the toxicity of nano-β-TCP in vivo, 40 mg/kg of nano-β-TCP was chosen to explore its inhibitory effect on hepatocellular carcinoma in nude mice bearing xenografted HepG2 cells. Adriamycin hydrochloride (DOX, 5 mg/kg) was used as a positive control group. In vivo evaluation of antitumor activity of nano-β-TCP was shown in Figure 8. When the nude mice were sacrificed, the volumes of liver tumor in different groups (0 mg/kg, DOX, 40 mg/kg) were 1800.48±484.84, 938.20±52.13, and 1047.35±118.49 mm3 (Figures 8A and B) (**P<0.01), respectively. The weights of Liver tumor treated with different groups (0 mg/kg, DOX, 40 mg/kg) were 2.34±0.85, 0.43±0.05, and 1.23±0.12 g (Figure 8C) (**P<0.01), respectively. The tumor volume and weight in the group treated with nano-β-TCP were lower than that in the negative control group and higher than that in the positive control group, because doxorubicin hydrochloride is an anticancer drug that can significantly inhibit the growth of liver tumors. These results indicate that nano-β-TCP can inhibit liver tumors in vivo (**P<0.01). To further verify this conclusion, Caspase-3, Caspase-8 and Caspase-9 in liver tumors were examined. Compared with the control group, the expression of the above proteins increased significantly in groups treated with DOX and nano-β-TCP (Figure 8D). The result indicates that nano-β-TCP can also promote the apoptosis of hepatocellular carcinoma cells in vivo, thereby promoting subsequent apoptotic signaling and cell apoptosis. which is consistent with that in vitro.
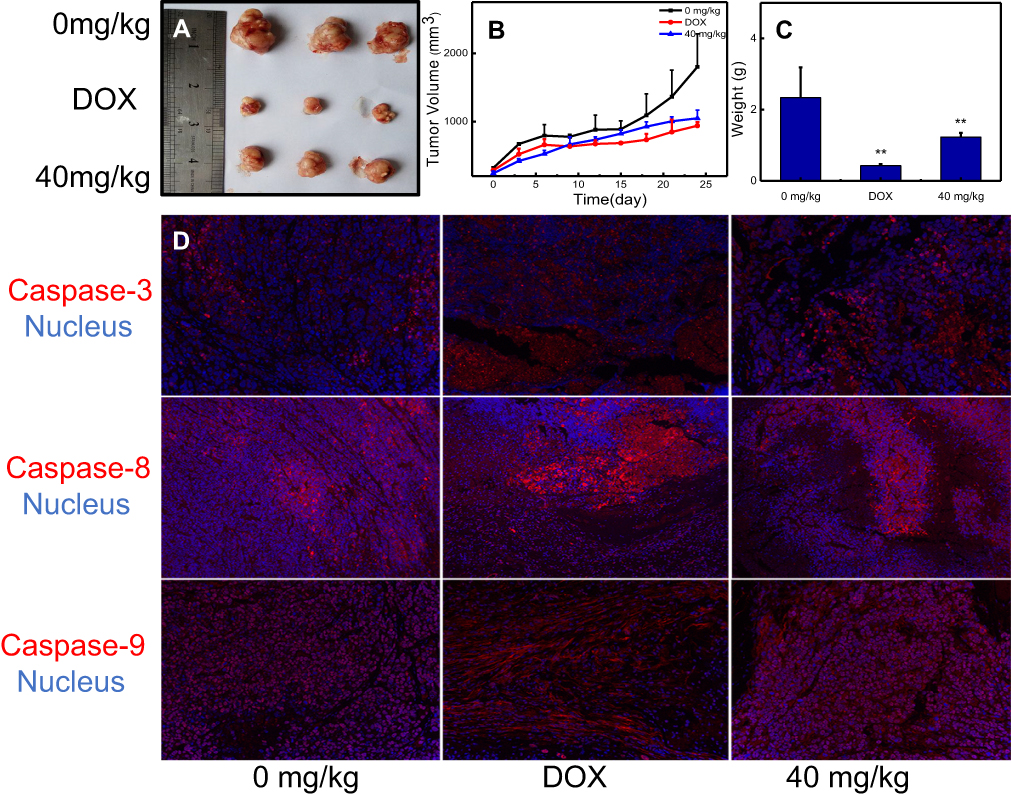 | Figure 8 In vivo evaluation of antitumor activity of nano-β-TCP. (A) Tumor size; (B) Tumor volume; (C) Tumor weight; (D) Nano-β-TCP-induced tumor cell apoptosis (Caspase-3, Caspase-8, and Caspase-9), indicating that the mechanisms of nano-β-TCP-induced caspase-dependent apoptosis were also adequate for the in vivo (vs control group); **P<0.01. |
Conclusions
In summary, we have successfully synthesized nano-β-TCP and investigated the mechanism of its antitumor activity. In vitro results revealed that nano-β-TCP caused reduced cell viability of HepG2 cells in a time- and dose-dependent manner, and induced an increment of intracellular ROS. Nanoparticles were internalized through endocytosis and degraded in cells, resulting in the intracellular calcium and phosphorous ions obvious improvement. Nano-β-TCP induced apoptosis of tumor cells by apoptotic signaling both in extrinsic and intrinsic pathways. In addition, nano-β-TCP could block the cell cycle of HepG2 cells in G0/G1 phase and disturb the expression of some cyclins. In vivo results showed that 40 mg/kg of nano-β-TCP had no significant toxic side-effects, but could effectively suppress liver tumor growth. Consequently, these findings revealed the anticancer effect of nano-β-TCP and also initially clarified the mechanism of its inhibitory effect on hepatocellular carcinoma.
Acknowledgments
This work was supported by the National Key Research and Development Program of China (No. 2016YFC1101605 and 2016YFB1101302), the National Natural Science Foundation of China (No. 51772233, 51772233, and 81190133), the Key Technology Research and Development Program of Hubei province (No. 2015BAA085), and the Excellent Dissertation Cultivation Funds of Wuhan University of Technology (No. 2016-YS-011).
Author contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Chen W, Zheng R, Zhang S, et al. Annual report on status of cancer in China, 2010. Chin J Cancer Res. 2014;26(1):48–58. doi:10.3978/j.issn.1000-9604.2014.01.08
2. Chen W, Zheng R, Zeng H, Zhang S, He J. Annual report on status of cancer in China, 2011. Chin J Cancer Res. 2015;27(1):2–12. doi:10.3978/j.issn.1000-9604.2015.01.06
3. Yau T, Tang VYF, Yao TJ, Fan ST, Lo CM, Poon RTP. Development of Hong Kong liver cancer staging system with treatment stratification for patients with hepatocellular carcinoma. Gastroenterology. 2014;146(7):1691–1700.e1693. doi:10.1053/j.gastro.2014.02.032
4. Chen W, Zheng R, Zhang S, et al. Report of incidence and mortality in China cancer registries, 2009. Chin J Cancer Res. 2013;25(1):10–21. doi:10.3978/j.issn.1000-9604.2012.12.04
5. Chapman S, Dobrovolskaia M, Farahani K, et al. Nanoparticles for cancer imaging: the good, the bad, and the promise. Nano Today. 2013;8(5):454–460. doi:10.1016/j.nantod.2013.06.001
6. Cao B, Yang M, Zhu Y, Qu X, Mao C. Stem cells loaded with nanoparticles as a drug carrier for in vivo breast cancer therapy. Adv Mater. 2014;26(27):4627–4631. doi:10.1002/adma.201401550
7. Fiorillo M, Verre AF, Iliut M, et al. Graphene oxide selectively targets cancer stem cells, across multiple tumor types: implications for non-toxic cancer treatment, via “differentiation-based nano-therapy”. Oncotarget. 2015;6(6):3553–3562. doi:10.18632/oncotarget.3348
8. Selim ME, Hendi AA. Gold nanoparticles induce apoptosis in MCF-7 human breast cancer cells. Asian Pac J Cancer Prev. 2012;13(4):1617–1620.
9. Zhang L, Wang L, Hu Y, et al. Selective metabolic effects of gold nanorods on normal and cancer cells and their application in anticancer drug screening. Biomaterials. 2013;34(29):7117–7126. doi:10.1016/j.biomaterials.2013.05.043
10. Gobbo OL, Sjaastad K, Radomski MW, Volkov Y, Prina-Mello A. Magnetic nanoparticles in cancer theranostics. Theranostics. 2015;5(11):1249–1263. doi:10.7150/thno.11544
11. Lima-Tenório MK, Gómez Pineda EA, Ahmad NM, Fessi H, Elaissari A. Magnetic nanoparticles: in vivo cancer diagnosis and therapy. Int J Pharm. 2015;493(1):313–327. doi:10.1016/j.ijpharm.2015.07.059
12. Zhang Q, Wang X, Li PZ, et al. Biocompatible, uniform, and redispersible mesoporous silica nanoparticles for cancer‐targeted drug delivery in vivo. Adv Funct Mater. 2014;24(17):2450–2461. doi:10.1002/adfm.201302988
13. Choi S, Coonrod S, Estroff L, Fischbach C. Chemical and physical properties of carbonated HA affect breast cancer cell behavior. Acta Biomater. 2015;24:333–342. doi:10.1016/j.actbio.2015.06.001
14. Chu SH, Karri S, Ma YB, Feng DF, Li ZQ. In vitro and in vivo radiosensitization induced by hydroxyapatite nanoparticles. Neuro-Oncology. 2013;15(7):880. doi:10.1093/neuonc/not030
15. Liang C, Diao S, Wang C, et al. Tumor metastasis inhibition by imaging‐guided photothermal therapy with single‐walled carbon nanotubes. Adv Mater. 2014;26(32):5646–5652. doi:10.1002/adma.201401825
16. Lin W, Huang Y-W, X-D Z, Ma Y. In vitro toxicity of silica nanoparticles in human lung cancer cells. Toxicol Appl Pharmacol. 2006;217(3):252–259. doi:10.1016/j.taap.2006.10.004
17. Liu S, Xu L, Zhang T, Ren G, Yang Z. Oxidative stress and apoptosis induced by nanosized titanium dioxide in PC12 cells. Toxicology. 2010;267(1):172–177. doi:10.1016/j.tox.2009.11.012
18. Xu M, Huang N, Xiao Z, Lu Z. Photoexcited TiO2 nanoparticles through •OH-radicals induced malignant cells to necrosis. Supramolecular Sci. 1998;5(5):449–451. doi:10.1016/S0968-5677(98)00048-0
19. Qiu-Lian QU, Zhang YG. Effects of three kinds of nanoparticles on the mitochondrial membrane potential and level of reactive oxygen species in human gastric carcinoma cell line BGC-823. Bull Acad Mil Med Sci. 2010;34(4):306–309.
20. Ma M. Preparation of magnetite nanoparticles and interaction with cancer cells. J Southeast Univ. 2003;33(2):205–207.
21. Laurent S, Saei AA, Behzadi S, Panahifar A, Mahmoudi M. Superparamagnetic iron oxide nanoparticles for delivery of therapeutic agents: opportunities and challenges. Expert Opin Drug Deliv. 2014;11(9):1449–1470. doi:10.1517/17425247.2014.924501
22. Tang W, Yuan Y, Liu C, Wu Y, Lu X, Qian J. Differential cytotoxicity and particle action of hydroxyapatite nanoparticles in human cancer cells. Nanomedicine. 2014;9(3):397. doi:10.2217/nnm.12.217
23. Yuan Y, Liu C, Qian J, Wang J, Zhang Y. Size-mediated cytotoxicity and apoptosis of hydroxyapatite nanoparticles in human hepatoma HepG2 cells. Biomaterials. 2010;31(4):730–740. doi:10.1016/j.biomaterials.2009.09.088
24. Meena R, Kesari KK, Rani M, Paulraj R. Effects of hydroxyapatite nanoparticles on proliferation and apoptosis of human breast cancer cells (MCF-7). J Nanopart Res. 2012;14(2):712. doi:10.1007/s11051-011-0712-5
25. Olton DYE, Close JM, Sfeir CS, Kumta PN. Intracellular trafficking pathways involved in the gene transfer of nano-structured calcium phosphate-DNA particles. Biomaterials. 2011;32(30):7662–7670. doi:10.1016/j.biomaterials.2011.01.043
26. Sykes EA, Chen J, Zheng G, Chan WCW. Investigating the impact of nanoparticle size on active and passive tumor targeting efficiency. ACS Nano. 2014;8(6):5696–5706. doi:10.1021/nn500299p
27. Bañobre-López M, Teijeiro A, Rivas J. Magnetic nanoparticle-based hyperthermia for cancer treatment. Rep Pract Oncol Radiother. 2013;18(6):397–400. doi:10.1016/j.rpor.2013.09.011
28. Jiang W, Kim BYS, Rutka JT, Chan WCW. Nanoparticle-mediated cellular response is size-dependent. Nat Nanotechnol. 2008;3:145. doi:10.1038/nnano.2008.30
29. Xu J, Xu P, Li Z, Huang J, Yang Z. Oxidative stress and apoptosis induced by hydroxyapatite nanoparticles in C6 cells. J Biomed Mater Res A. 2012;100A(3):738–745. doi:10.1002/jbm.a.v100a.3
30. Elshafie H, Armentano M, Carmosino M, Bufo S, De Feo V, Camele I. Cytotoxic activity of origanum vulgare l. on hepatocellular carcinoma cell line hepg2 and evaluation of its biological activity. Molecules. 2017;22(9):1435. doi:10.3390/molecules22091435
31. Sanosh KP, Chu M-C, Balakrishnan A, Kim TN, Cho S-J. Sol–gel synthesis of pure nano sized β-tricalcium phosphate crystalline powders. Curr Appl Phys. 2010;10(1):68–71. doi:10.1016/j.cap.2009.04.014
32. Koshkaki MR, Ghassai H, Khavandi A, Seyfoori A, Molazemhosseini A. Effects of formaldehyde solution and nanoparticles on mechanical properties and biodegradation of gelatin/nano β-TCP scaffolds. Iran Polym J. 2013;22(9):653–664. doi:10.1007/s13726-013-0164-0
33. Yin M, Yin Y, Han Y, Dai H, Li S. Effects of uptake of hydroxyapatite nanoparticles into hepatoma cells on cell adhesion and proliferation. J Nanomater. 2014;2014(2014):1–7.
34. Hu L, Mao Z, Gao C. Colloidal particles for cellular uptake and delivery. J Mater Chem. 2009;19(20):3108–3115. doi:10.1039/b815958k
35. Motskin M, Wright DM, Muller K, et al. Hydroxyapatite nano and microparticles: correlation of particle properties with cytotoxicity and biostability. Biomaterials. 2009;30(19):3307–3317. doi:10.1016/j.biomaterials.2009.02.044
36. Cai Y, Liu Y, Yan W, et al. Role of hydroxyapatite nanoparticle size in bone cell proliferation. J Mater Chem. 2007;17(36):3780–3787. doi:10.1039/b705129h
37. Han Y, Li S, Cao X, et al. Different inhibitory effect and mechanism of hydroxyapatite nanoparticles on normal cells and cancer cells in vitro and in vivo. Sci Rep. 2014;4(3):7134. doi:10.1038/srep07134
38. Daniels TR, Delgado T, Rodriguez JA, Helguera G, Penichet ML. The transferrin receptor part I: biology and targeting with cytotoxic antibodies for the treatment of cancer. Clin Immunol. 2006;121(2):144–158. doi:10.1016/j.clim.2006.06.010
39. Daniels TR, Delgado T, Helguera G, Penichet ML. The transferrin receptor part II: targeted delivery of therapeutic agents into cancer cells. Clin Immunol. 2006;121(2):159–176. doi:10.1016/j.clim.2006.06.006
40. Martin CL, Bouvard D, Delette G. Discrete element simulations of the compaction of aggregated ceramic powders. J Am Ceram Soc. 2006;89(11):3379–3387. doi:10.1111/jace.2006.89.issue-11
41. Huang H-L, Fang L-W, Lu S-P, Chou C-K, Luh T-Y, Lai M-Z. DNA-damaging reagents induce apoptosis through reactive oxygen species-dependent Fas aggregation. Oncogene. 2003;22:8168. doi:10.1038/sj.onc.1206979
42. Asharani PV, Mun GLK, Hande MP, Valiyaveettil S. Cytotoxicity and genotoxicity of silver nanoparticles in human cells. ACS Nano. 2009;3(2):279–290. doi:10.1021/nn800596w
43. Foldbjerg R, Dang DA, Autrup H. Cytotoxicity and genotoxicity of silver nanoparticles in the human lung cancer cell line, A549. Arch Toxicol. 2011;85(7):743–750. doi:10.1007/s00204-010-0545-5
44. Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398(6726):422–426. doi:10.1038/18884
45. Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2013;15:49. doi:10.1038/nrm3722
46. Wang Y, Wang J, Hao H, et al. In vitro and in vivo mechanism of bone tumor inhibition by selenium-doped bone mineral nanoparticles. ACS Nano. 2016;10(11):9927–9937. doi:10.1021/acsnano.6b03835
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.