")
Back to Journals » Advances and Applications in Bioinformatics and Chemistry » Volume 16
In silico Screening of Potential SARS-CoV-2 Main Protease Inhibitors from Thymus schimperi
Authors Mengist HM , Khalid Z, Adane F
Received 30 October 2022
Accepted for publication 11 January 2023
Published 18 January 2023 Volume 2023:16 Pages 1—13
DOI https://doi.org/10.2147/AABC.S393084
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Maria A. Miteva
Hylemariam Mihiretie Mengist,1,* Zunera Khalid,2,* Fentahun Adane3
1Department of Medical Laboratory Science, College of Medical and Health Sciences, Debre Markos University, Debre Markos, Ethiopia; 2School of Basic Medical Sciences, Division of Life Sciences and Medicine, University of Science & Technology of China, Langfang, People’s Republic of China; 3Department of Biomedical Sciences, College of Medical and Health Sciences, Debre Markos University, Debre Markos, Ethiopia
*These authors contributed equally to this work
Correspondence: Hylemariam Mihiretie Mengist, Email [email protected]
Background: COVID-19 is still instigating significant social and economic chaos worldwide; however, there is no approved antiviral drug yet. Here, we used in silico analysis to screen potential SARS-CoV-2 main protease (Mpro) inhibitors extracted from the essential oil of Thymus schimperi which could contribute to the discovery of potent anti-SARS-CoV-2 phytochemicals.
Methods: The absorption, distribution, metabolism, excretion, and toxicity (ADMET) profiles of compounds were determined through SwissADME and ProToxII servers. AutoDock tools were used for molecular docking analysis studies, while Chimera, DS studio, and LigPlot were used for post-docking studies. Molecular dynamic simulations were performed for 200 ns under constant pressure.
Results: All compounds exhibited a bioavailability score of ≥ 0.55 entailing that at least 55% of the drugs can be absorbed unchanged. Only five (9%), nine (16%) and two (3.6%) of the compounds showed active hepatotoxicity, carcinogenicity, and immunotoxicity, respectively. Except for flourazophore P, which showed a little mutagenicity, all other compounds did not show mutagenic properties. On the other hand, only pinene beta was found to have a little cytotoxicity. Five compounds demonstrated effective binding to the catalytic dyad of the SARS-CoV-2 Mpro substrate binding pocket, while two of them (geranylisobutanoate and 3-octane) are found to be the best hits that formed hydrogen bonds with Glu166 and Ser144 of SARS-CoV-2 Mpro.
Conclusion: Based on our in silico analysis, top hits from Thymus schimperi may serve as potential anti-SARS-CoV-2 compounds. Further in vitro and in vivo studies are recommended to characterize these compounds for clinical applications.
Keywords: structural analysis, SARS-CoV-2, main protease, inhibitors, Thymus schimperi
Introduction
Coronavirus disease 19 (COVID-19) is caused by the novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Since its emergence in December 2019, it exerts a big economic and social impact globally. Data from worldometer (https://www.worldometers.info/coronavirus/) showed that COVID-19 cases surpass 655 million with a death toll of over 6.6 million as of December 14, 2022. It is unknown when the challenge posed by the pandemic will last. However, its impact could be longer due to various means of transmission, large asymptomatic carriers, absence of point-of-care cost-effective tests, absence of therapeutics and resistance of variants to vaccines.1–6 One of the challenges in tackling the prevention and control activities is the presence of various modes of transmission.1
Studies have been discovering promising inhibitors of SARS-CoV-2. Previously, the FDA approved remdesivir to use in case of emergency7; however, it has limited clinical outcomes in non-mechanically ventilated severely ill patients,8 indicating that further efforts are required to identify therapeutically effective antivirals.9 Owing to their key role in the viral cycle and relatively conserved nature, targeting viral enzymes like the main protease or 3C-like protease (Mpro or 3CLpro), papain-like protease (PLpro), non-structural protein 12 (nsp12), and RNA-dependent RNA polymerase (RdRP) could be promising.10
The main protease (Mpro) of SARS-CoV-2 plays key roles in viral replication and assembly11,12 and human protease enzymes with similar specificity have not been reported so far,13,14 making it the ultimate potential drug target. The Mpro is a cysteine protease where two protomers (each containing Domains I, II, and III) form a homodimer. Residues 8–101 and 102–184 form Domains I and II, respectively. A Cys-His catalytic dyad, which is proteolytically active, is located between Domains I and II.15–19
Viral protease enzymes have a relatively conserved sequence, and they also have crucial roles in viral maturation and assembly which make them potential therapeutic targets. So far, the FDA approved several antiviral drugs targeting viral proteases including HIV-1 protease inhibitors20 and hepatitis C virus (HCV) NS3/4A protease inhibitors.21 Since it is a key viral enzyme, the main protease of coronaviruses is a potential drug target that several studies are focusing on.11 The Mpro digests polyproteins to release enzymes essential for replication and it also has NTPase and RNA helicase activity.22,23 In this regard, studies looking for drugs against COVID-19 have been targeting SARS-CoV-2 Mpro. Therefore, it is essential to design antivirals targeting SARS-CoV-2 Mpro as it could have a potential clinical application.24
Repurposed broad-spectrum drugs, new drugs, medicinal plants, and known antivirals effectively inhibited SARS-CoV-2 Mpro with potential antiviral effects.9 Among others, peptidomimetic alpha ketoamide inhibitors,25,26 Michael acceptor compounds,27 carmofur,27,28 ebselen,27,29 aldehyde-based compounds,30 and 6e,31 clinically approved drugs (lopinavir/ritonavir),32 antiplatelet drug dipyridamole,33,34 boceprevir (anti-Hepatitis C virus), GC-376, calpain inhibitors (II, XII) and GC-37335–37 exhibited effective anti-SARS-CoV-2 Mpro activities.
Molecular docking and molecular dynamic simulations are becoming famous means of in silico drug discovery. These methods determine the effective binding of lead compounds in the active site of the Mpro of SARS-CoV-2. Accordingly, a study identified 12 best hits from Super Natural II and Traditional Chinese Medicine databases.38 Besides, several potential compounds were identified from the traditional Chinese Medicine database interacting with active site residues of the Mpro (His41, Gly143, and Cys145).39 Qumar et al40 also recommended the top nine hits from several phytochemicals to consider as potential anti-SARS-CoV-2 Mpro lead molecules. Studies also showed that phytochemicals demonstrated strong pharmacokinetic and drug-likeness properties with acceptable toxicity profiles.41 Bioflavonoids42 also showed promising drug candidacy for SARS-CoV-2. In addition to targeting Mpro, phytochemicals have been demonstrating effective in silico drug potential for other core viral components including RNA polymerase.43
The structure–function-based designing of drugs is an important phase in drug discovery. In addition to drug repurposing, studying the in silico structural basis of prospective SARS-CoV-2 Mpro inhibitors from medicinal plants is considered pivotal to further study the antiviral activity of these compounds for clinical application. In this regard, in silico mutational analysis of the Mpro helps for better drug repurposing.44 Several studies have been investigating the in silico potential inhibitory activities of compounds against the Mpro of SARS-CoV-2. A study identified that three clinically approved drugs (glibenclamide, bedaquiline, and miconazole) effectively bind on the active site of SARS-CoV-2 Mpro with possible inhibitory activities.45
In another study, Kanhed et al46 identified ritonavir, nelfinavir, and saquinavir to be potent Mpro inhibitors. Ritonavir formed hydrogen bonds with Gly143 and Cys145 with its (thiazol-5-yl) methylcarbamate of oxygen, while the thiazolyl ring formed polar contacts with Thr25, Thr26 and Leu27 of the S1’ subsite. Nelfinavir stabilized its binding with Mpro via hydrogen bonding with Glu166, and with His41 and Tyr54 in the S2 subsite. Depending on the structure of the pocket, compounds containing oxirane rings are suggested to be good Mpro inhibitors.47 Arbutin, terbutaline, barnidipine, tipiracil, and aprepitant were identified as potential hits forming different hydrophilic, hydrophobic, and electrostatic interactions with Mpro.48 Thioflavonol is a synthetic flavonoid analog that showed a strong binding with the conserved residues in the S1 subsite.49 These and other studies illustrated the efficient binding of compounds on the substrate binding pocket (active) site of the Mpro indicating their potential anti-SARS-CoV-2 roles.
The genus Thymus contains about 350 species traditionally being used to treat different diseases worldwide.50 Members of the genus, mostly T. schimperi, locally known as “Tosign”, in Ethiopia are used to treat diseases. Its leaves are used as spices in various Ethiopian foods and traditional medicines to treat different diseases including colds.51 No significant in vivo and silico rat embryonic toxicity was observed from compounds derived from the essential oil of this plant,52 indicating the further application of this plant extract for other infectious diseases including COVID-19. Here, we used in silico analysis to identify potential SARS-CoV-2 Mpro inhibitors from Thymus schimperi which could provide structural insights to discover potent anti-SARS-CoV-2 phytochemicals.
Materials and Methods
ADMET Analysis of Compounds
Extraction of 56 compounds from the essential oil of Thymus schimperi was done by using GC-MS.52 Then, the PubChem CID number of each compound was obtained from PubChem53 and two-dimensional structures were built by Chemdraw (8.0).54 SMILES were created through the Swiss ADME web tool.55 Finally, the in silico absorption, distribution, metabolism, and excretion profiles were analyzed by using the online SwissADME server55 and ProToxII predicted the in silico toxicity profile of the ligands.56
Ligand and Protein Preparation
The ligands were prepared in PDB format first and then in PDBQT format suitable for molecular docking. Regarding protein preparation, the water molecules were removed, polar hydrogens were added, Kollam united atom force field was used to add charges and finally the structure was saved in a PDBQT format for further molecular docking analysis. To run the molecular docking, the crystal structure of SARS-CoV-2 Mpro (PDB ID: 6y2e)25 was prepared and refined similar to previous studies by using AutoDock tools 1.5.7.57–59 His41 was considered as the active site residue for molecular docking purposes. The protein we used (PDB ID: 6y2e) was solved by X-ray crystallography at a resolution of 1.75 Å.
Receptor Grid Preparation
The grid box for the Mpro was generated through AutoDock tools58 around the active site. The Grid Box center was set as 75, 75, and 75 for the X, Y, and Z centers, respectively. The substrate-binding site is in the cleft between domains I and II and the protomers, which bind each other through N-terminus residues 1–7, are located between domains II and III with roles in the formation of the substrate-binding site.18,25,40,60,61 Mpro is a cysteine protease digesting viral polyproteins. First, a proton is transferred from Cys145 to His41 with a simultaneous nucleophilic attack of the carbonyl carbon atom of the peptide bond by the sulfur atom of Cys145 resulting in a thiohemiketal intermediate with subsequent protease activity.62
Comparative Molecular Docking Analysis
Three docking tools (AutoDock Vina,58 GOLD (Genetic Optimization for Ligand Docking) and MOE (Molecular Operating Environment)) were used for the molecular docking study of the 56 compounds (Supplementary Table 1). The selected compounds were sketched and minimized by ChemDraw Ultra 3D and saved into Mol2 and PDB format for molecular docking purposes.
The molecular docking of selected compounds with the Mpro was performed by AutoDock Vina to assess the inhibitory action of ligands against the Mpro. The ligand and protein were converted into pdbqt files using MGL tools. Grid sizes were adjusted to 75 × 75×75 Å in the X, Y and Z axes, respectively, with the grid spacing value of 0.650 Å to cover the target receptor. The fitness score was determined to select the best docking pose of inhibitors in the binding pocket. The best binding affinity was selected in all the docking processes.
After importing all the ligands into MOE, MDB file conversion, 3D protonation and energy minimization was conducted to 0.01 gradient. The protein structure concerns of the Mpro PDB structure were resolved in MOE. Before energy minimization, hydrogen atoms were introduced to structures with their typical geometry, and all solvent molecules were removed. Adjustments were made to the scoring methodologies’ default values. After the docking processes were completed, the acquired poses were analyzed, and the poses with the lowest allowable rmsd refine values and the same binding mode as the native ligand were chosen.
Alkyl, hydrogen, and hydrophobic interactions between the ligand and receptor were investigated through Chimera v1.8.1 and LigPlot63 tools within the range of 5Å while BIOVIA Discovery Studio Visualizer v21.1 was used to identify the key interacting residues with maximum binding energies. Molecular dynamics (MD) simulations were conducted to validate docking results and assess the binding behavior and stability of prospective drugs by using the GROMACS 2016.4 Linux command line suite.25
Validation of the Molecular Docking Method
The molecular docking protocol was validated by using an N3 peptide inhibitor as a reference ligand.27 Then, a decoy set of ligands were used along with the active ligands. After the generation of the decoy dataset, all the ligands were prepared by MGL tool for docking.
Molecular Dynamics (MD) Simulation
The best-scored docking models of the most promising lead compound (3-octane) in complex with Mpro were selected as the starting coordinates for a 200-ns all-atom molecular dynamics simulation using the GROMACS-2016 program (GNU, General Public License; http://gromacs.org) and CHARMM36 force field. Solvation of the ligand–protein complex was done within a cubic box of the transferable intermolecular potential with a three-point (TIP3P) water model93,93 with a minimum distance of 10 between the protein and each side of the 3D boxes. CHARMM General Force Field (CGenFF) tool was used to define the CHARMM force field parameters for the examined ligand64 (ParamChem project; https://cgenff.umaryland.edu/). The protein residues were allocated to their standard ionization states under physiological conditions (pH 7.0), and the whole complexes were neutralized by adding enough K+ and Cl− ions via the Monte Carlo ion-placing method. Heavy atom retaining, and maintaining original protein folding was also considered using a 1000 kJ/mol.N m2 force constant in a three-step MD simulation. First, each system geometry was optimized with 5000 iterations (5 ps) of the steepest descent algorithm. In the succeeding step, the system was conditioned for 100,000 iterations (100 ps) at each stage of two-staged equilibration. The initial equilibration step was carried out using a constant number of particles, volume, and temperature (NVT) ensemble following the Berendsen temperature coupling method for controlling the temperature within the 3D box. Subsequently, the second equilibration stage was conducted in a constant number of particles, pressure, and temperature (NPT) ensemble at 1 atm and 303.15 K utilizing the Parrinello-Rahman barostat as a reference. Lastly, MD simulations were performed for 200 ns under constant pressure.
Results
ADME and Toxicity of the Compounds
Based on online server ADMET analysis, potential SARS-CoV-2 Mpro inhibitors from Thymus schimperi exhibited variable water solubility, GI absorption, blood–brain barrier (BBB) permeant, bioavailability score and synthetic accessibility. Among the screed compounds, 83.9% (47/56) of them are water-soluble, while the rest are either poorly soluble or moderately soluble. About 48% (27/56) of the compounds are predicted to be highly absorbable in the gastrointestinal (GI) system but 17.9% (10/56) of them are not able to penetrate the BBB. All compounds have a bioavailability score of ≥0.55 entailing that at least 55% of the drugs can be absorbed unchanged. Besides, almost all compounds have a synthetic accessibility (SA) score of below 5.00 while 35.7% (20/56) of them have a SA score of below 3.00 indicating their ease of synthetic accessibility (Supplementary Table 1).
The lethal dose (LD50) of the compounds ranges from 113 to 5700 mg/kg. Two compounds, alpha-phellandrene and myrcenol, showed non-toxic (with a toxicity class value of 6 and LD50 value >5000 mg/kg). Except for 6 (10.7%) compounds exhibiting a toxicity class of 3 indicating “toxic if swallowed”, other 48 (85.7%) compounds have a toxicity class of 4/5 indicating that these compounds are harmful/may be harmful if swallowed. Only five (9%), nine (16%) and two (3.6%) of the compounds exhibited active hepatotoxicity, carcinogenicity and immunotoxicity, respectively. Except for flourazophore P with a little mutagenicity, all other compounds did not show mutagenic properties. On the other hand, only pinene beta was found to have a little cytotoxicity (Supplementary Table 2).
Molecular Docking
In molecular docking, five compounds from Thymus schimperi (amphotericin-gamma, geranylisobutanoate, 3-octane, vetivenene_beta and germacrene-D) were the top hits based on lowest binding energy and highest gold score (Table 1). These compounds form hydrogen bonds, van der Waals interactions and alkyl interactions with the amino acids of Mpro (Supplementary Figure 1). Among these, amphotericin-gamma, geranylisobutanoate, 3-octane and germacrene-D showed strong binding on the His41-Cys-145 catalytic dyad of the SARS-CoV-2 Mpro substrate binding pocket (Supplementary Figure 2). Except for beta-vetivenene, other compounds exhibited strong interaction with the Mpro residues through all Chimera, DS studio and LigPlot analyses. Further, according to DS studio and LigPlot analysis, geranylisobutanoate, and 3-octane form hydrogen bonds Glu166 and Ser144 indicating that these compounds are the best among others (Table 1). Based on the highest binding affinity (−5.0 kJ/Mol) and gold score (44.88), 3-octane–Mpro protein–ligand complex was taken for MD simulation studies for investigating the potentiality of a small drug-like molecule to occupy the catalytic dyad of Mpro cavity in a way that would disrupt the function of Mpro (Figure 1).
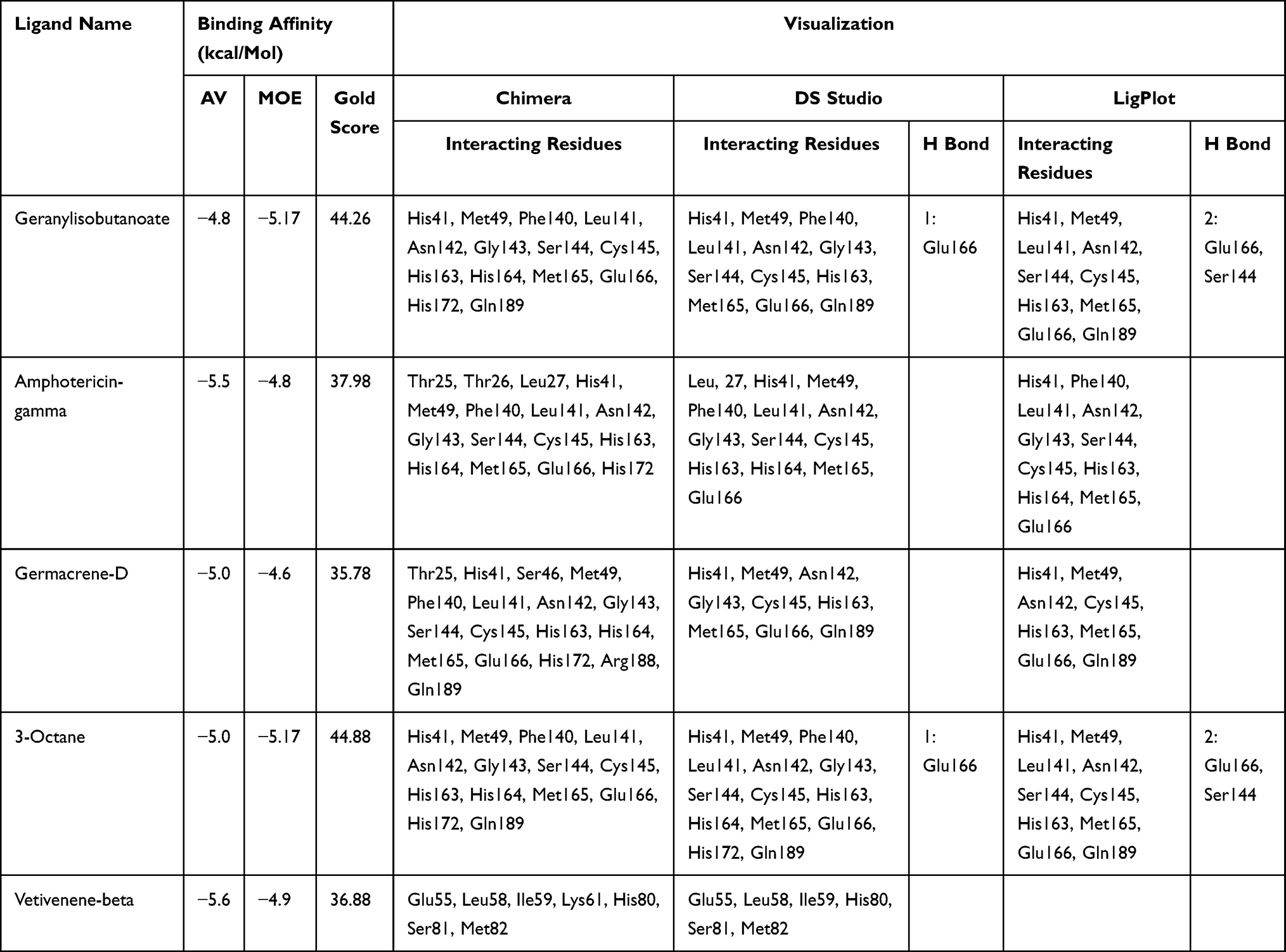 |
Table 1 Summary of Top Five Hits Screened Against SARS-CoV-2 Mpro |
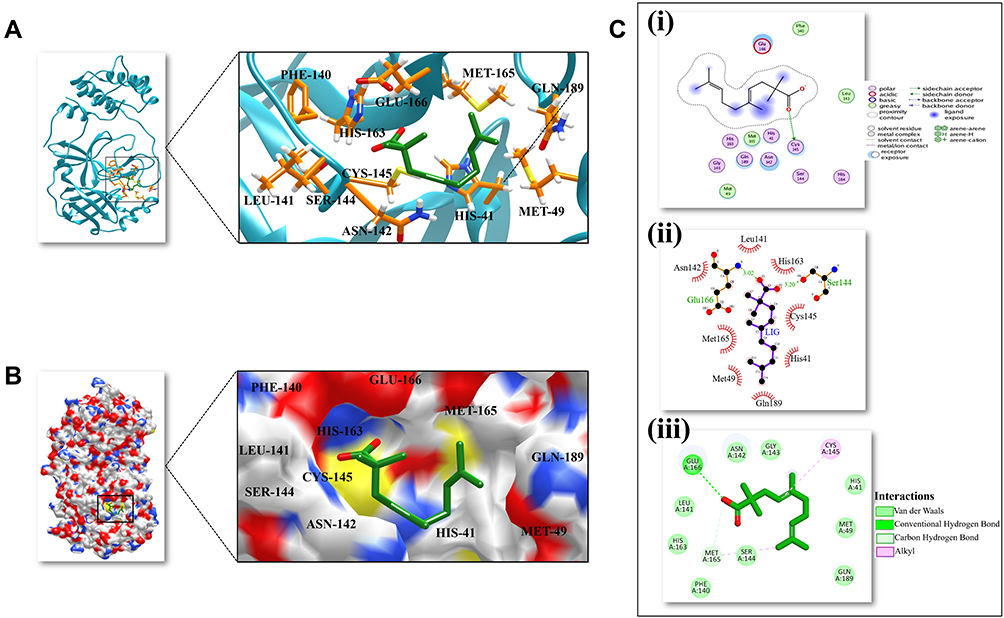 |
Figure 1 3D and 2D representation of the best docked pose of the compound 3-octane highlighting the critical binding site residues of Mpro. (A) Ribbon representation of the ligand 3-octane bound to the Mpro. The blue color represents the ribbon representation of Mpro while the ligand is represented in green color. The protein residues interacting with the ligand are represented in orange color. (B) The surface representations of the Mpro depicting the ligand are tightly bound within the binding groove of the protein. (C) The 2D representation of the docked pose protein–ligand complex. (i) 2D interaction diagram of 3-octane–Mpro complex generated by MOE. (ii) Ligplot representation of the protein–ligand complex drawn by LigPlot+. (iii) 2D depiction of the docked complex highlighting hydrogen bonds and cation-p interaction through DS Visualizer. |
Molecular Dynamics Simulation Studies
The stability of the projected docked 3-octane–Mpro complex was evaluated by an all-atom MD simulation study. Implementing this type of study would also give useful insights concerning the dynamic nature of both the ligand and Mpro, as well as an evaluation of the ligand’s most significant binding interactions with essential catalytic site residues.65 Enrolling of the predicted ligand–protein complex for 3-octane and Mpro protein was done within a 200 ns MD simulation run.
Trajectory Analysis of 3-Octane–Mpro Complex
MD experiments conducted in the complex elucidated the plausible mechanism of inhibition. The shape of the protein influences the conformational dynamics, thus understanding the functional flexibility of a biological macromolecule is of utmost important.65 The average RMSD for the 3-octane–Mpro complex was found to be 0.25 Å when computed for 200 ns which indicated that the system was highly stable during the MD simulations (Figure 2A). Similar to previous RMSD analysis, the obtained Rg plot confirmed better protein–ligand complex stability with an average value of 0.25 Å (Figure 2B).
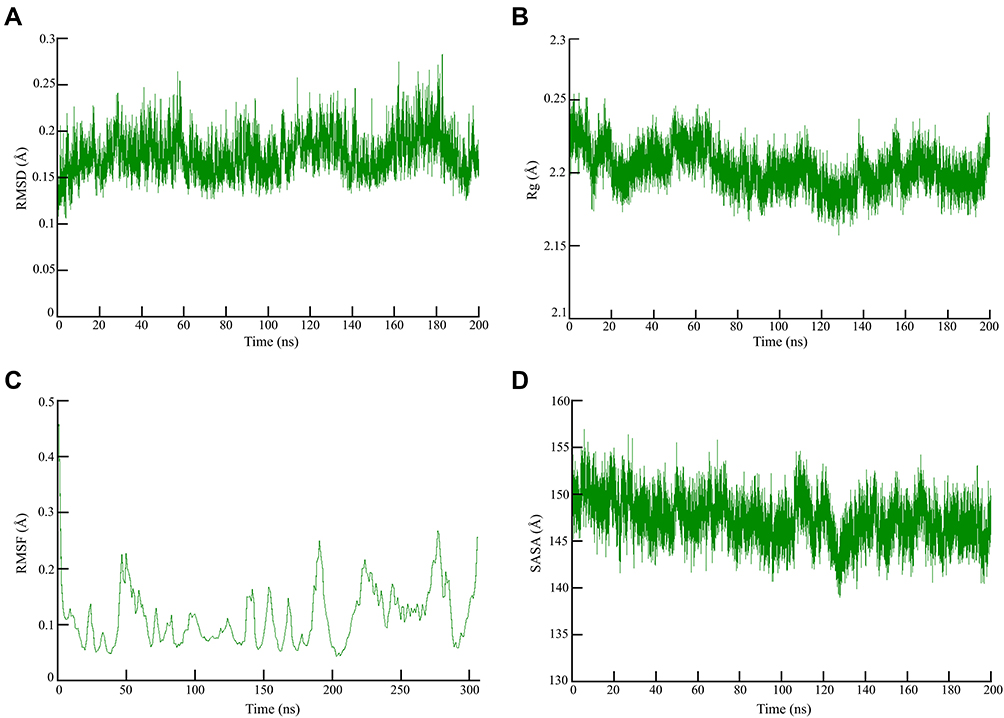 |
Figure 2 Global stability analysis of ligand-hACE2 protein complexes throughout 200 ns all-atom MD simulation. (A) Analysis of RMSD trajectories for the 3-octane–Mpro protein complex throughout 200 ns MD simulation deciphering the primary conformational switches. (B) The radius of gyration for the protein–ligand complex reflects the complex structure’s global stability. (C) RMSF of protein–ligand complex depicting fluctuations across protein residues during 200 ns of MD simulations. (D) Extent of Mpro binding site coverage via SASA analysis along with the time evolution 200 ns all-atom MD simulation. |
As shown in Figure 3C, RMSF values were 0.2 Å indicating the flexibility of residues forming the catalytic dyad. The 200 ns of MD run showed that the flexibility of the residues reached a peak upon binding of the ligand with the receptor as depicted by the RMSF. Significant fluctuations were observed at SER-46, LEU-50, THR-190, THR-224, and ASN-277 and PHE-305 with an average RMSF value of 0.2 Å (Figure 2C). SASA corresponds with the molecular surface area that can be assessed by solvent molecules, hence providing a quantitative assessment of the level of protein/solvent interaction. The study was performed on the atoms of residues bordering the Mpro binding site to estimate the solvent-exposed region (Figure 2D).
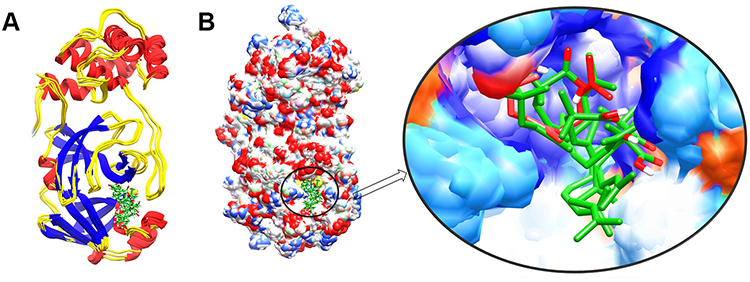 |
Figure 3 The structure superimposition of MD trajectories (0 ns, 50 ns, 100 ns, 150 ns, and 200 ns) over the pre-simulated 3-octane–Mpro complex. (A) The secondary structure representation of superimposed protein–ligand complexes. The ligand is represented in green color. (B) The surface representation of Mpro representing that ligand is bound within the same active site groove throughout the 200 ns of MD simulation run considering it stable active site conformation of the ligand. |
Conformational Analysis Across the Selected Trajectories
To detect the structural variability of the simulated system, the MD trajectories (0 ns, 50 ns, 100 ns, 150 ns and 200 ns) were superimposed on the pre-simulated 3-octane–Mpro docked complex (Figure 3) and produced the superimposed structure at 0.8 Å. The 3D and 2D interaction diagrams (Figure 4) of 0 ns, 50 ns, 100 ns, 150 ns and 200 ns trajectories presented that the interacting residues of Mpro were consistent with the course of the 200 ns MD simulation run. Interestingly, there is no significant orientation change for the ligand within the Mpro binding site between the time frames 0 and 200 ns. A slight shift caused a loss of the initial hydrogen bond with GLU-166. Stabilization of 3-octane within its new conformation/orientation was further mediated by several hydrophobic residues including His41, Leu141, Ser144, and Cys145.
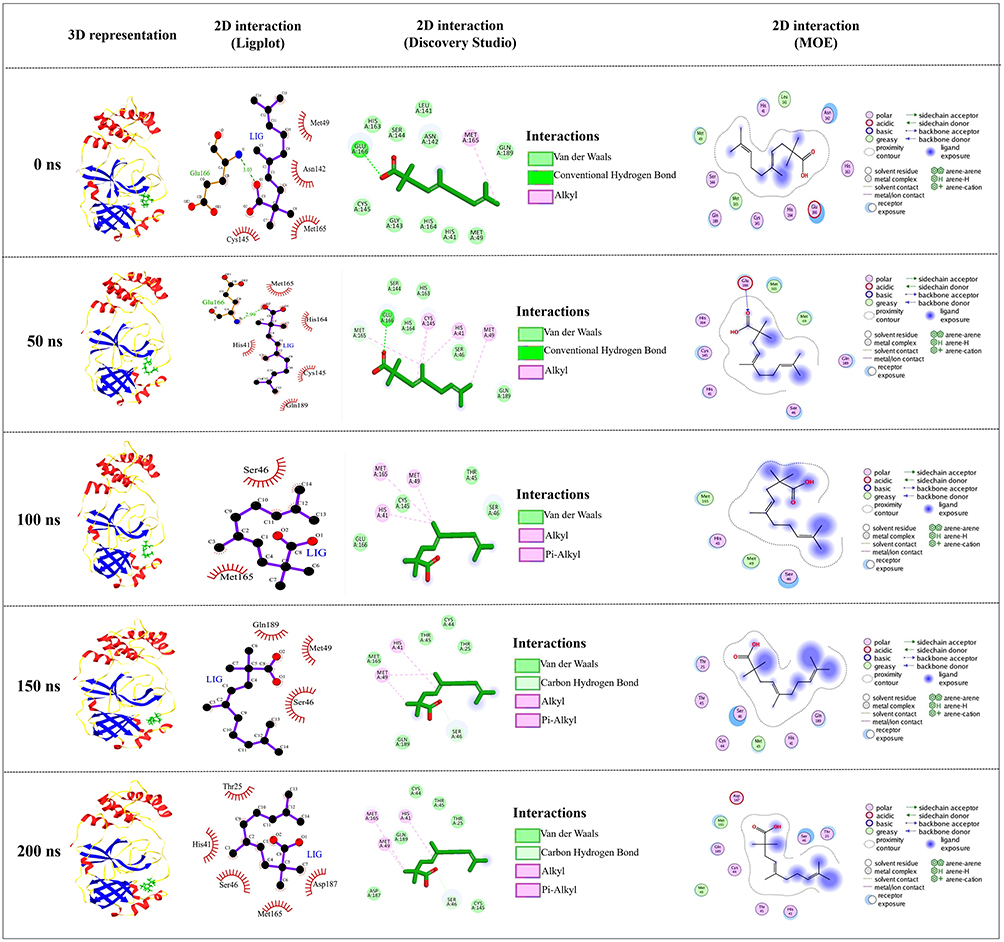 |
Figure 4 Conformations of the 3-octane complex at Mpro binding site through selected trajectories. |
Discussion
Computational drug design is quite an important tool to speed up drug discovery, especially in case of emergency.66 So far studies focused on both repurposing previously approved drugs and/or identifying new potent phytochemicals. Proposed plant metabolites are assumed to serve as potential anti-SARS-CoV-2 lead compounds to combat COVID-19.67 Medicinal plants especially those used in Chinese traditional medicine have been investigated for applications in COVID-19 treatment.68 Studies identified potential SARS-CoV-2 Mpro inhibitors from different plants and Traditional Chinese Medicine databases that effectively interact with the active site residues in the catalytic dyad of the Mpro38–40,69–73 with promising outcomes.
Here we screened 56 compounds from Thymus schimperi for their ADME, toxicity and binding profiles. Most of the screened compounds showed good water solubility, gastrointestinal absorption, and permeant to the BBB. According to bioavailability score definitions,74 our compounds could be well absorbed in humans since they have a good predicted bioavailability score of ≥0.55 which is similar to previous studies.75 Synthesizing natural compounds could be better to minimize the resource and time required to extract from their natural sources. In this regard, our molecules have an SA score below 5.00 indicating that these compounds are not difficult to synthesize,76 indicating their potential consideration for further optimization and drug development studies.
Five compounds from Thymus schimperi (amphotericin-gamma, geranylisobutanoate, 3-octane, vetivenene-beta and germacrene-D) were the top hits that formed hydrogen bonds, van der Waals interactions and alkyl interactions with the amino acids of Mpro. Specifically, geranylisobutanoate and 3-octane showed strong SARS-CoV-2 Mpro binding affinity with acceptable toxicity and ADME profiles except these compounds showed hepatotoxicity in in silico analysis. Interestingly, these compounds are under toxicity class 5 which is acceptable in medicine. The effective binding of these compounds to the Mpro and their good ADMET profiles is in line with previous studies.77–82 Thus, considering these compounds for further applications is of significance; however, in vitro, and in vivo chronic toxicity studies are pivotal.52
Previous studies also reported similar findings to our study. Numerous plant-derived phytochemicals such as curcumin, gartanin, robinetin,83 amentoflavone, gallocatechin gallate,84 chelidimerine, rutin, fumariline, catechin gallate, adlumidine, astragalin, somniferine,85 kaempferol, herbacetin, eugenol, 6-shogaol,86 triacontane, hexacosane, methyl linoleate, and methyl palmitoleate,87 cosmosiine, pelargoniding-3-O-glucose, and cleomiscosin88 formed similar binding patterns while docking with the main protease. Another study also reported that plant-derived phytochemicals including flavan 3-ols (catechins/procyanidins), complex oligomeric procyanidins (procyanidin A3, procyanidin A4, procyanidin A1, and procyanidin B3) exhibited very good binding characteristics like our compounds.89 Interestingly, the main protease of coronaviruses is relatively conserved.90–92 Thus, designing drugs against the main protease could broadly attack the virus variants. Additionally, the absence of human host-cell proteases with similar activity with viral main proteases make the prospective drugs to have less off-target actions.13,14
Bioinformatics analyses have demonstrated their value in the invention of innovative computer-assisted compounds against a variety of diseases, including neurological disorders, cancer, and pathogenic infections.93 To better comprehend the structure and function of Mpro, a crucial element in the design of medications, computational studies are necessary. Computational drug design methods also play a crucial part in determining which medicine is the best among others.24 MD simulation studies connect the theoretical and experimental studies in drug discovery by analyzing the protein–ligand complex. Further, these studies are crucial to determine the stability of the ligand-receptor complex.94 The MD simulation results in this study confirm the firm binding of 3-octane at 200 ns. These results present pieces of evidence that 3-octane could be a promising SARS-CoV-2 Mpro inhibitor, while in vitro and in vivo studies are yet required for further clinical application. The problem behind drug discovery is less probability of the anticipated compounds passing clinical trials. Therefore, continuous efforts and further investigation of compounds through in silico, in vitro and in vivo studies are required.
Compounds from food spices are reported to have potential anti-SARS-CoV-2 Mpro activities.95 Thymus schimperi is used as a food spice in Ethiopia. Studies have shown that essential oils from Thymus schimperi exhibited promising antimicrobial activities.96,97 In silico screening of lead compounds from food promote the therapeutic role of food in combating the COVID-19 pandemic.98 As the top hits in this study exhibited excellent binding and acceptable ADMET properties, further studies are recommended to optimize these compounds in the plant to design drugs tackling respiratory diseases. More importantly, considering geranylisobutanoate and 3-octane for future clinical applications could be of significance.
Many lessons have been learned from previous in silico and structure-based drug designing studies that would help prospective studies succeed fast in discovering effective antivirals to tackle COVID-19 and other related diseases.24 Future studies need to focus on the inhibitor enzyme complex which included atomic-level mechanisms of peptide cleavage, pharmacophore requirements of the Mpro, stability of the inhibitor-enzyme complex, and plasticity of the active site of Mpro. Besides, the occurrence of mutations at the domains and/or the active site affecting the pocket, the size and accommodation capacity of the subsites of the Mpro should be considered in designing new drugs or modifying previously known broad-spectrum drugs. Most studies solely report the binding affinity and energy of compounds towards the substrate-binding cleft of the Mpro, improvements considering the abovementioned points should be taken in the future.
Generally, structure–function analysis is a key factor in drug design and discovery where in silico studies are crucial to predict the drug ability of compounds in a short period in addition to its indispensable role in predicting the best drug candidates among others. The big challenge in computational drug designing is that the clinical use of these desired drugs is questionable corresponding to the possible limitations of passing clinical trials. Although our study characterizes a limited number of compounds, it provides a glimpse into the identification of lead compounds from food spices that could serve as therapeutic drugs in clinical medicine.
Conclusion
We identified potential compounds from Thymus schimperi that could serve as drugs to inhibit SARS-CoV-2 Mpro. Among the top hits, geranylisobutanoate and 3-octane exhibited strong binding to the catalytic dyad of SARS-CoV-2 Mpro with acceptable ADMET profiles. Interestingly, our results entail that 3-octane could be the most promising anti-SARS-CoV-2 Mpro inhibitor. Further studies are required to determine the in vitro and in vivo characteristics of the identified potential compounds to employ them in clinical applications.
Ethical Approval
Our study is a pure computational study and does not include human data and thus no need of ethical approval.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that there are no conflicts of interest.
References
1. Patel KP, Vunnam SR, Patel PA, et al. Transmission of SARS-CoV-2: an update of current literature. Eur J Clin Microbiol Infect Dis. 2020;39(11):2005–2011. doi:10.1007/s10096-020-03961-1
2. Zhang W, Du RH, Li B, et al. Molecular and serological investigation of 2019-nCoV infected patients: implication of multiple shedding routes. Emerg Microbes Infect. 2020;9(1):386–389. doi:10.1080/22221751.2020.1729071
3. Chow N, Fleming-Dutra K, Gierke R, et al. Preliminary estimates of the prevalence of selected underlying health conditions among patients with coronavirus disease 2019—United States, February 12–March 28, 2020. Morb Mortal Wkly Rep. 2020;69(13):382. doi:10.15585/mmwr.mm6913e2
4. Mekonnen D, Mengist HM, Derbie A, et al. Diagnostic accuracy of serological tests and kinetics of severe acute respiratory syndrome coronavirus 2 antibody: a systematic review and meta-analysis. Rev Med Virol. 2021;31:e2181. doi:10.1002/rmv.2181
5. Wang Y, Wang Y, Chen Y, Qin Q. Unique epidemiological and clinical features of the emerging 2019 novel coronavirus pneumonia (COVID-19) implicate special control measures. J Med Virol. 2020;92(6):568–576. doi:10.1002/jmv.25748
6. Cascella M, Rajnik M, Cuomo A, Dulebohn SC, Di Napoli R. Features, Evaluation, and Treatment of Coronavirus. Statpearls. Treasure Island (FL): StatPearls PublishingCopyright © 2020, StatPearls Publishing LLC.; 2020.
7. Beigel JH, Tomashek KM, Dodd LE, et al. Remdesivir for the treatment of Covid-19 — final report. N Eng J Med. 2020;383(19):1813–1826. doi:10.1056/NEJMoa2007764
8. Elsawah HK, Elsokary MA, Abdallah MS, ElShafie AH. Efficacy and safety of remdesivir in hospitalized Covid-19 patients: systematic review and meta-analysis including network meta-analysis. Rev Med Virol. 2021;31:e2187. doi:10.1002/rmv.2187
9. Mengist HM, Mekonnen D, Mohammed A, Shi R, Jin T. Potency, safety, and pharmacokinetic profiles of potential inhibitors targeting SARS-CoV-2 main protease. Front Pharmacol. 2021;11:2495. doi:10.3389/fphar.2020.630500
10. Zumla A, Chan JF, Azhar EI, Hui DS, Yuen K-Y. Coronaviruses—drug discovery and therapeutic options. Nat Rev Drug Discov. 2016;15(5):327–347. doi:10.1038/nrd.2015.37
11. Ziebuhr J, Snijder EJ, Gorbalenya AE. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J Gen Virol. 2000;81(4):853–879. doi:10.1099/0022-1317-81-4-853
12. Jain R, Mujwar S. Repurposing metocurine as main protease inhibitor to develop novel antiviral therapy for COVID-19. Struct Chem. 2020;31(6):2487–2499. doi:10.1007/s11224-020-01605-w
13. Zhang L, Lin D, Kusov Y, et al. α-Ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: structure-based design, synthesis, and activity assessment. J Med Chem. 2020;63(9):4562–4578. doi:10.1021/acs.jmedchem.9b01828
14. Hilgenfeld R. From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014;281(18):4085–4096. doi:10.1111/febs.12936
15. Anand K, Palm GJ, Mesters JR, Siddell SG, Ziebuhr J, Hilgenfeld R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra α‐helical domain. EMBO J. 2002;21(13):3213–3224. doi:10.1093/emboj/cdf327
16. Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300(5626):1763–1767. doi:10.1126/science.1085658
17. Xue X, Yang H, Shen W, et al. Production of authentic SARS-CoV Mpro with enhanced activity: application as a novel tag-cleavage endopeptidase for protein overproduction. J Mol Biol. 2007;366(3):965–975. doi:10.1016/j.jmb.2006.11.073
18. Yang H, Yang M, Ding Y, et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc Nat Acad Sci. 2003;100(23):13190–13195. doi:10.1073/pnas.1835675100
19. Shi J, Wei Z, Song J. Dissection Study on the severe acute respiratory syndrome 3C-like protease reveals the critical role of the extra domain in dimerization of the enzyme defining the extra domain as a new target for design of highly specific protease inhibitors. J Biol Chem. 2004;279(23):24765–24773. doi:10.1074/jbc.M311744200
20. Lv Z, Chu Y, Wang Y. HIV protease inhibitors: a review of molecular selectivity and toxicity. HIV/AIDS. 2015;7:95–104.
21. de Leuw P, Stephan C. Protease inhibitors for the treatment of hepatitis C virus infection. GMS Infect Dis. 2017;5:Doc08–Doc. doi:10.3205/id000034
22. Thiel V, Ivanov KA, Putics A, et al. Mechanisms and enzymes involved in SARS coronavirus genome expression. J Gen Virol. 2003;84(9):2305–2315. doi:10.1099/vir.0.19424-0
23. Shu T, Huang M, Wu D, et al. SARS-coronavirus-2 Nsp13 possesses NTPase and RNA helicase activities that can be inhibited by bismuth salts. Virol Sin. 2020;35(3):321–329. doi:10.1007/s12250-020-00242-1
24. Mengist HM, Dilnessa T, Jin T. Structural basis of potential inhibitors targeting SARS-CoV-2 main protease. Front Chem. 2021;9:622898. doi:10.3389/fchem.2021.622898
25. Zhang L, Lin D, Sun X, et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368(6489):409–412. doi:10.1126/science.abb3405
26. Mengist HM, Fan X, Jin T. Designing of improved drugs for COVID-19: crystal structure of SARS-CoV-2 main protease Mpro. Signal Transduct Target Ther. 2020;5(1):67. doi:10.1038/s41392-020-0178-y
27. Jin Z, Du X, Xu Y. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582(7811):289–293. doi:10.1038/s41586-020-2223-y
28. Jin Z, Zhao Y, Sun Y. Structural basis for the inhibition of SARS-CoV-2 main protease by antineoplastic drug carmofur. Nat Struct Mol Biol. 2020;27(6):529–532. doi:10.1038/s41594-020-0440-6
29. Sies H, Parnham MJ. Potential therapeutic use of ebselen for COVID-19 and other respiratory viral infections. Free Radic Biol Med. 2020;156:107–112. doi:10.1016/j.freeradbiomed.2020.06.032
30. Dai W, Zhang B, Jiang X-M, et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368(6497):1331–1335. doi:10.1126/science.abb4489
31. Rathnayake AD, Zheng J, Kim Y. 3C-like protease inhibitors block coronavirus replication in vitro and improve survival in MERS-CoV-infected mice. Sci Transl Med. 2020;12(557):eabc5332. doi:10.1126/scitranslmed.abc5332
32. Liu X, Wang X-J. Potential inhibitors against 2019-nCoV coronavirus M protease from clinically approved medicines. J Genet Genomics. 2020;47(2):119–121. doi:10.1016/j.jgg.2020.02.001
33. Li Z, Li X, Huang -Y-Y, et al. Identify potent SARS-CoV-2 main protease inhibitors via accelerated free energy perturbation-based virtual screening of existing drugs. Proc Nat Acad Sci. 2020;117(44):27381–27387. doi:10.1073/pnas.2010470117
34. Liu X, Li Z, Liu S, et al. Potential therapeutic effects of dipyridamole in the severely ill patients with COVID-19. Acta Pharm Sin B. 2020;10(7):1205–1215. doi:10.1016/j.apsb.2020.04.008
35. Vuong W, Khan MB, Fischer C. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat Commun. 2020;11(1):4282. doi:10.1038/s41467-020-18096-2
36. Ma C, Sacco MD, Hurst B, et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. bioRxiv Preprint 20200420051581; 2020.
37. Choy K-T, Wong AY-L, Kaewpreedee P, et al. Remdesivir, lopinavir, emetine, and homoharringtonine inhibit SARS-CoV-2 replication in vitro. Antiviral Res. 2020;178:104786. doi:10.1016/j.antiviral.2020.104786
38. Abel R, Chen Q, Paredes Ramos M, et al. Computational prediction of potential inhibitors of the main protease of SARS-CoV-2. Front Chem. 2020;8:1162. doi:10.3389/fchem.2020.590263
39. Selvaraj C, Panwar U, Dinesh DC, et al. Microsecond MD simulation and multiple-confirmation virtual screening to identify potential anti-COVID-19 inhibitors against SARS-CoV-2 main protease. Front Chem. 2020;8:1179.
40. Ul Qamar MT, Alqahtani SM, Alamri MA, Chen -L-L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J Pharm Anal. 2020;10(4):313–319. doi:10.1016/j.jpha.2020.03.009
41. More-Adate P, Lokhande KB, Swamy KV, Nagar S, Baheti A. GC-MS profiling of Bauhinia variegata major phytoconstituents with computational identification of potential lead inhibitors of SARS-CoV-2 M(pro). Comput Biol Med. 2022;147:105679. doi:10.1016/j.compbiomed.2022.105679
42. Lokhande K, Nawani N, Pawar S, Pawar S. Biflavonoids from Rhus succedanea as probable natural inhibitors against SARS-CoV-2: a molecular docking and molecular dynamics approach. J Biomol Struct Dyn. 2022;40(10):4376–4388. doi:10.1080/07391102.2020.1858165
43. Pandey K, Lokhande KB, Swamy KV, Nagar S, Dake M. In silico exploration of phytoconstituents from phyllanthus emblica and aegle marmelos as potential therapeutics against SARS-CoV-2 RdRp. Bioinform Biol Insights. 2021;15:11779322211027403. doi:10.1177/11779322211027403
44. Pulakuntla S, Lokhande KB, Padmavathi P, et al. Mutational analysis in international isolates and drug repurposing against SARS-CoV-2 spike protein: molecular docking and simulation approach. Virusdisease. 2021;32(4):690–702. doi:10.1007/s13337-021-00720-4
45. Ferraz WR, Gomes RA, Novaes AL, Goulart Trossini GH. Ligand and structure-based virtual screening applied to the SARS-CoV-2 main protease: an in silico repurposing study. Future Med Chem. 2020;12(20):1815–1828. doi:10.4155/fmc-2020-0165
46. Kanhed AM, Patel DV, Teli DM, Patel NR, Chhabria MT, Yadav MR. Identification of potential Mpro inhibitors for the treatment of COVID-19 by using systematic virtual screening approach. Mol Divers. 2020;25:383–401. doi:10.1007/s11030-020-10130-1
47. Palese LL The structural landscape of SARS-CoV-2 main protease: hints for inhibitor search. ChemRxiv Preprint; 2020.
48. Baby K, Maity S, Mehta CH, Suresh A, Nayak UY, Nayak Y. Targeting SARS-CoV-2 main protease: a computational drug repurposing study. Arch Med Res. 2020;52:38–47.
49. Batool F, Mughal EU, Zia K, et al. Synthetic flavonoids as potential antiviral agents against SARS-CoV-2 main protease. J Biomol Struct Dyn. 2020;5:1–12.
50. Belaqziz R, Harrak R, Romane A, Oufdou K, ElFels MAE. Antimicrobial and insecticidal activities of the endemic thymus broussonetti boiss. and thymus maroccanus ball. Rec Nat Prod. 2010;4(4):230.
51. Damtie D, Mekonnen Y. Thymus species in Ethiopia: distribution, medicinal value, economic benefit, current status and threatening factors. Ethiop Sci Technol. 2015;8(2):81–92. doi:10.4314/ejst.v8i2.3
52. Adane F, Asres K, Ergete W, et al. Composition of the essential oil Thymus schimperi and evaluation of its acute and subacute toxicity in Wistar albino rats: in silico toxicity studies. Evid Based Complement Altern Med. 2021;2021:1. doi:10.1155/2021/5521302
53. Kim S, Chen J, Cheng T, et al. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. 2019;47(D1):D1102–D9. doi:10.1093/nar/gky1033
54. Mendelsohn LD. ChemDraw 8 Ultra, Windows and Macintosh versions. J Chem Inf Comput Sci. 2004;44(6):2225–2226. doi:10.1021/ci040123t
55. Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7(1):1–13. doi:10.1038/srep42717
56. Banerjee P, Eckert AO, Schrey AK, Preissner R. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018;46(W1):W257–W63. doi:10.1093/nar/gky318
57. Sanner MF. Python: a programming language for software integration and development. J Mol Graph Model. 1999;17(1):57–61.
58. Huey R, Morris GM, Forli S. Using AutoDock 4 and AutoDock vina with AutoDockTools: a tutorial. Scripps Res Inst Mol Graphics Lab. 2012;10550:92037.
59. Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–2791. doi:10.1002/jcc.21256
60. Hsu M-F, Kuo C-J, Chang K-T, et al. Mechanism of the maturation process of SARS-CoV 3CL protease. J Biol Chem. 2005;280(35):31257–31266. doi:10.1074/jbc.M502577200
61. Chou C-Y, Chang H-C, Hsu W-C, Lin T-Z, Lin C-H, Chang -G-G. Quaternary structure of the severe acute respiratory syndrome (SARS) coronavirus main protease. Biochemistry. 2004;43(47):14958–14970. doi:10.1021/bi0490237
62. Swiderek K, Moliner V. Revealing the molecular mechanisms of proteolysis of SARS-CoV-2 Mpro from QM/MM computational methods. Chem Sci. 2020;11:10626–10630. doi:10.1039/D0SC02823A
63. Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng Des Sel. 1995;8(2):127–134. doi:10.1093/protein/8.2.127
64. Vanommeslaeghe K, Hatcher E, Acharya C, et al. CHARMM general force field: a force field for drug‐like molecules compatible with the CHARMM all‐atom additive biological force fields. J Comput Chem. 2010;31(4):671–690. doi:10.1002/jcc.21367
65. Karplus M, Petsko GAJN. Molecular dynamics simulations in biology. Nature. 1990;347(6294):631–639. doi:10.1038/347631a0
66. Galindez G, Matschinske J, Rose TD, et al. Lessons from the COVID-19 pandemic for advancing computational drug repurposing strategies. Nat Comput Sci. 2021;1(1):33–41. doi:10.1038/s43588-020-00007-6
67. Bhuiyan FR, Howlader S, Raihan T, Hasan M. Plants metabolites: possibility of natural therapeutics against the COVID-19 pandemic. Front Med. 2020;7:444. doi:10.3389/fmed.2020.00444
68. Qamar M, Maryam A, Muneer I, et al. Computational screening of medicinal plant phytochemicals to discover potent pan-serotype inhibitors against dengue virus. Sci Rep. 2019;9:1433. doi:10.1038/s41598-018-38450-1
69. Sinha SK, Prasad SK, Islam MA, et al. Identification of bioactive compounds from Glycyrrhiza glabra as possible inhibitor of SARS-CoV-2 spike glycoprotein and non-structural protein-15: a pharmacoinformatics study. J Biomol Struct Dyn. 2021;39(13):4686–4700. doi:10.1080/07391102.2020.1779132
70. Patel CN, Jani SP, Jaiswal DG, et al. Identification of antiviral phytochemicals as a potential SARS-CoV-2 main protease (M(pro)) inhibitor using docking and molecular dynamics simulations. Sci Rep. 2021;11(1):20295. doi:10.1038/s41598-021-99165-4
71. Srivastav AK, Gupta SK, Kumar U Computational studies towards identification of lead herbal compounds of medicinal importance for development of nutraceutical against COVID-19; 2021.
72. Kumar A, Choudhir G, Shukla SK, et al. Identification of phytochemical inhibitors against main protease of COVID-19 using molecular modeling approaches. J Biomol Struct Dyn. 2021;39(10):3760–3770. doi:10.1080/07391102.2020.1772112
73. Ebada SS, Al-Jawabri NA, Youssef FS, et al. Anti-inflammatory, antiallergic and COVID-19 protease inhibitory activities of phytochemicals from the Jordanian hawksbeard: identification, structure–activity relationships, molecular modeling and impact on its folk medicinal uses. RSC Adv. 2020;10(62):38128–38141. doi:10.1039/D0RA04876C
74. Martin YC. A bioavailability score. J Med Chem. 2005;48(9):3164–3170. doi:10.1021/jm0492002
75. Abdul-Hammed M, Adedotun IO, Olajide M, et al. Virtual screening, ADMET profiling, PASS prediction, and bioactivity studies of potential inhibitory roles of alkaloids, phytosterols, and flavonoids against COVID-19 main protease (M(pro)). Nat Prod Res. 2022;36(12):3110–3116. doi:10.1080/14786419.2021.1935933
76. Ertl P, Schuffenhauer A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J Cheminform. 2009;1(1):8. doi:10.1186/1758-2946-1-8
77. Sisakht M, Mahmoodzadeh A, Darabian M. Plant-derived chemicals as potential inhibitors of SARS-CoV-2 main protease (6LU7), a virtual screening study. Phytother Res. 2021;35(6):3262–3274. doi:10.1002/ptr.7041
78. Ullah S, Munir B, Al-Sehemi AG, et al. Identification of phytochemical inhibitors of SARS-CoV-2 protease 3CLpro from selected medicinal plants as per molecular docking, bond energies and amino acid binding energies. Saudi J Biol Sci. 2022;29(6):103274. doi:10.1016/j.sjbs.2022.03.024
79. Mahmud S, Hasan MR, Biswas S, et al. Screening of Potent phytochemical inhibitors against SARS-CoV-2 main protease: an integrative computational approach. Front Bioinform. 2021;1:1. doi:10.3389/fbinf.2021.717141
80. Shree P, Mishra P, Selvaraj C, et al. Targeting COVID-19 (SARS-CoV-2) main protease through active phytochemicals of ayurvedic medicinal plants – withania somnifera (Ashwagandha), Tinospora cordifolia (Giloy) and Ocimum sanctum (Tulsi) – a molecular docking study. J Biomol Struct Dyn. 2022;40(1):190–203. doi:10.1080/07391102.2020.1810778
81. Dey D, Hossain R, Biswas P, et al. Amentoflavone derivatives significantly act towards the main protease (3CLPRO/MPRO) of SARS-CoV-2: in silico admet profiling, molecular docking, molecular dynamics simulation, network pharmacology. Mol Divers. 2022;2022:1.
82. Chatterjee S, Maity A, Chowdhury S, Islam MA, Muttinini RK, Sen D. In silico analysis and identification of promising hits against 2019 novel coronavirus 3C-like main protease enzyme. J Biomol Struct Dyn. 2021;39(14):5290–5303. doi:10.1080/07391102.2020.1787228
83. Mahmud S, Uddin MAR, Paul GK, et al. Virtual screening and molecular dynamics simulation study of plant-derived compounds to identify potential inhibitors of main protease from SARS-CoV-2. Brief Bioinform. 2021;22(2):1402–1414. doi:10.1093/bib/bbaa428
84. Swargiary A, Mahmud S, Saleh MA. Screening of phytochemicals as potent inhibitor of 3-chymotrypsin and papain-like proteases of SARS-CoV2: an in silico approach to combat COVID-19. J Biomol Struct Dyn. 2022;40(5):2067–2081. doi:10.1080/07391102.2020.1835729
85. Mousavi SS, Karami A, Haghighi TM, et al. In silico evaluation of Iranian medicinal plant phytoconstituents as inhibitors against main protease and the receptor-binding domain of SARS-CoV-2. Molecules. 2021;26(18):5724. doi:10.3390/molecules26185724
86. Tallei TE, Tumilaar SG, Niode NJ, et al. Potential of plant bioactive compounds as SARS-CoV-2 main protease (Mpro) and spike (S) glycoprotein inhibitors: a molecular docking study. Scientifica. 2020;2020:2. doi:10.1155/2020/6307457
87. Mahmud S, Paul GK, Afroze M, et al. Efficacy of phytochemicals derived from Avicennia officinalis for the management of COVID-19: a combined in silico and biochemical study. Molecules. 2021;26(8):2210. doi:10.3390/molecules26082210
88. Mahmud S, Afrose S, Biswas S, et al. Plant-derived compounds effectively inhibit the main protease of SARS-CoV-2: an in silico approach. PLoS One. 2022;17(8):e0273341. doi:10.1371/journal.pone.0273341
89. Teli DM, Shah MB, Chhabria MT. In silico screening of natural compounds as potential inhibitors of SARS-CoV-2 main protease and spike RBD: targets for COVID-19. Front Mol Biosci. 2021;7:4. doi:10.3389/fmolb.2020.599079
90. Hayden FG, Turner RB, Gwaltney JM, et al. Phase II, randomized, double-blind, placebo-controlled studies of ruprintrivir nasal spray 2-percent suspension for prevention and treatment of experimentally induced rhinovirus colds in healthy volunteers. Antimicrob Agents Chemother. 2003;47(12):3907–3916. doi:10.1128/AAC.47.12.3907-3916.2003
91. Kim Y, Liu H, Galasiti Kankanamalage AC, et al. Reversal of the progression of fatal coronavirus infection in cats by a broad-spectrum coronavirus protease inhibitor. PLoS Pathog. 2016;12(3):e1005531. doi:10.1371/journal.ppat.1005531
92. Yang H, Xie W, Xue X, et al. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005;3(10):e324. doi:10.1371/journal.pbio.0030324
93. Khalid Z, Mannan SJP, Biology A. Revealing potential drug targets against Proto-oncogene Wnt10B by comparative molecular docking. Pure Appl Biol. 2018;7(2):565–574. doi:10.19045/bspab.2018.70070
94. Mangrio GR, Maneengam A, Khalid Z, et al. RP-HPLC method development, validation, and drug repurposing of sofosbuvir pharmaceutical dosage form: a multidimensional study. Environ Res. 2022;212:113282. doi:10.1016/j.envres.2022.113282
95. Ibrahim MAA, Abdelrahman AHM, Hussien TA, et al. In silico drug discovery of major metabolites from spices as SARS-CoV-2 main protease inhibitors. Comput Biol Med. 2020;126:104046. doi:10.1016/j.compbiomed.2020.104046
96. Nasir M, Tafess K, Abate D. Antimicrobial potential of the Ethiopian Thymus schimperi essential oil in comparison with others against certain fungal and bacterial species. BMC Complement Altern Med. 2015;15(1):260. doi:10.1186/s12906-015-0784-3
97. Damtie D, Mekonnen Y. Antibacterial activity of essential oils from Ethiopian thyme (Thymus serrulatus and Thymus schimperi) against tooth decay bacteria. PLoS One. 2020;15(10):e0239775. doi:10.1371/journal.pone.0239775
98. Di Matteo G, Spano M, Grosso M, et al. Food and COVID-19: preventive/co-therapeutic strategies explored by current clinical trials and in silico studies. Foods. 2020;9(8):1036. doi:10.3390/foods9081036
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.