")
Back to Journals » Pediatric Health, Medicine and Therapeutics » Volume 9
Improving outcomes for patients with distal renal tubular acidosis: recent advances and challenges ahead
Authors Watanabe T
Received 16 October 2018
Accepted for publication 19 November 2018
Published 12 December 2018 Volume 2018:9 Pages 181—190
DOI https://doi.org/10.2147/PHMT.S174459
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Roosy Aulakh
Toru Watanabe
Department of Pediatrics, Niigata City General Hospital, Niigata City 950-1197, Japan
Abstract: Primary distal renal tubular acidosis (dRTA) is a rare genetic disorder caused by impaired distal acidification due to a failure of type A intercalated cells (A-ICs) in the collecting tubule. dRTA is characterized by persistent hyperchloremia, a normal plasma anion gap, and the inability to maximally lower urinary pH in the presence of systemic metabolic acidosis. Common clinical features of dRTA include vomiting, failure to thrive, polyuria, hypercalciuria, hypocitraturia, nephrocalcinosis, nephrolithiasis, growth delay, and rickets. Mutations in genes encoding three distinct transport proteins in A-ICs have been identified as causes of dRTA, including the B1/ATP6V1B1 and a4/ATP6V0A4 subunits of the vacuolar-type H+-ATPase (H+-ATPase) and the chloride–bicarbonate exchanger AE1/SLC4A1. Homozygous or compound heterozygous mutations in ATP6V1B1 and ATP6V0A4 lead to autosomal recessive (AR) dRTA. dRTA caused by SLC4A1 mutations can occur with either autosomal dominant or AR transmission. Red blood cell abnormalities have been associated with AR dRTA due to SLC4A1 mutations, including hereditary spherocytosis, Southeast Asia ovalocytosis, and others. Some patients with dRTA exhibit atypical clinical features, including transient and reversible proximal tubular dysfunction and hyperammonemia. Incomplete dRTA presents with inadequate urinary acidification, but without spontaneous metabolic acidosis and recurrent urinary stones. Heterozygous mutations in the AE1 or H+-ATPase genes have recently been reported in patients with incomplete dRTA. Early and sufficient doses of alkali treatment are needed for patients with dRTA. Normalized serum bicarbonate, urinary calcium excretion, urinary low-molecular-weight protein levels, and growth rate are good markers of adherence to and/or efficacy of treatment. The prognosis of dRTA is generally good in patients with appropriate treatment. However, recent studies showed an increased frequency of chronic kidney disease (CKD) in patients with dRTA during long-term follow-up. The precise pathogenic mechanisms of CKD in patients with dRTA are unknown.
Keywords: urinary acidification, clinical features, treatment, prognosis, gene, pathogenesis
Introduction
Primary distal (type 1) renal tubular acidosis (dRTA) is a rare genetic disorder caused by impaired distal acidification due to a failure of type A intercalated cells (A-ICs) of the collecting tubule.1 dRTA is characterized by persistent hyperchloremia, a normal plasma anion gap, and the inability to maximally lower urinary pH in the presence of systemic metabolic acidosis.2 Common clinical features of dRTA include vomiting, failure to thrive, polyuria, hypercalciuria, hypocitraturia, nephrocalcinosis, nephrolithiasis, growth delay, and rickets.1,2 The clinical variant of dRTA that presents with inadequate urinary acidification without spontaneous metabolic acidosis is termed incomplete dRTA (idRTA).2,3
Mutations in genes encoding three distinct transport proteins have been identified as causes of dRTA: the B1/ATP6V1B1 and a4/ATP6V0A4 subunits of the vacuolar-type H+-ATPase (H+-ATPase) and the chloride–bicarbonate exchanger AE1/SLC4A1.3 However, because a genetic cause is determined in only 70%–80% of patients with dRTA,4,5 mutations in additional genes are likely to cause dRTA.3,6
The clinical manifestations of patients with dRTA depend on the underlying gene mutations.3 For example, the majority of patients with dRTA caused by mutations in genes encoding for H+-ATPase develop sensorineural hearing loss (SNHL).6 Autosomal dominant (AD) dRTA with heterozygous AE1 gene mutations causes less severe clinical manifestations.7 However, a recent study showed that clinical features are not specific indicators of the underlying causal gene.5
Herein, we review the recent advances in the pathogenesis, underlying gene mutations, atypical clinical features, incomplete type, kidney stone formation, treatment, and long-term outcome of dRTA.
Mechanisms of renal acid excretion
The western diet plus endogenous metabolism generates 1 mmol/kg body weight per day of nonvolatile acids in adults.8 An additional 1–2 mmol/kg body weight per day of nonvolatile acids is produced from the formation of hydroxyapatite from growth bone in children.7 The kidneys maintain acid–base homeostasis by reabsorbing filtered HCO3−, excreting acid in the form of NH4+ and titratable acids, and producing new HCO3− to replenish that lost to metabolism.3 The distal nephron is responsible for the secretion of H+ that is then buffered by NH3 and titratable acids, leading to urinary acidification.6,7
The distal convoluted tubules and cortical collecting ducts consist of principal cells, which reabsorb sodium via the epithelial sodium channels, acid-secreting A-ICs, and base-secreting type B intercalated cells (B-ICs). A-ICs mainly contribute to urinary acidification by generating new HCO3− and excreting NH3 into the urine (Figure 1).3,9,10 CO2 is hydrated by cytosolic carbonic anhydrase type II (CAII) that is present in all intercalated cells (ICs) of the collecting tubule and forms H+ and HCO3−. A-ICs secrete H+ by apical H+-ATPase with an additional contribution from H+–K+-ATPase. A-ICs release HCO3− into the blood in exchange for Cl− via the basolateral anion exchanger AE1. Cl− exits the cell by the potassium chloride co-transporter (KCC4) or the chloride channel (ClC-Kb).7 B-ICs express an apical Cl−–HCO3− exchanger, pendrin, and a basolateral H+-ATPase, responsible for net base secretion. Chronic acidosis converts B-ICs to A-ICs, which is mediated by an extracellular matrix protein hensin/DMBT1, and increases net acid secretion.11,12 Although the mechanism by which ICs sense a change in blood pH is unclear, Schwartz et al recently showed that principal cells respond to acid by producing SFD1 which regulates ICs subtype distribution.13
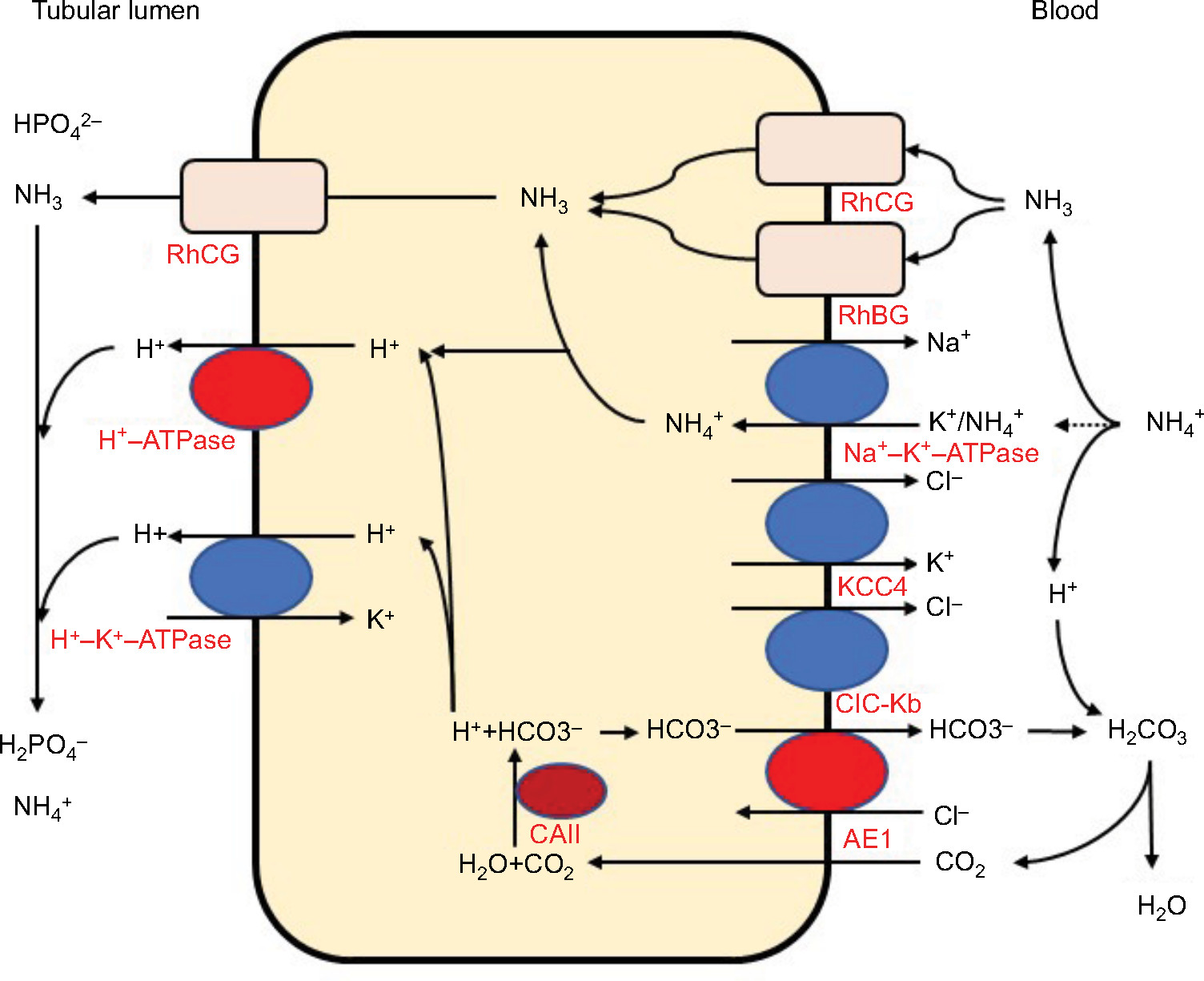 | Figure 1 Acid and ammonia secretion in type A intercalated cells in the distal nephron. Abbreviations: AE1, anion exchanger 1; CAII, carbonic anhydrase type II; ClC-Kb, chloride channel Kb; H+-ATPase, vacuolar H+-ATPase; KCC4, potassium chloride co-transporter 4; RhBG, rhesus blood group type B glycoprotein; RhCG, rhesus blood group type C glycoprotein. |
Because the capacity of the distal nephron to excrete acid as free H+ is limited, the distal nephron excretes the majority of acid coupled to urine buffers, ammonia (NH3), and titratable acids (mainly HPO42−). Therefore, the net acid secretion in urine is the sum of the ammonium (NH4+) charge plus the titratable acid (H2PO4−) charge minus the HCO3− charge.8 Under normal conditions, the kidneys excrete approximately one-third to one-half of the net acids as titratable acids and one-half to two-thirds as NH4+. Under conditions of chronic acidosis or acid load, NH4+ excretion can increase severalfold to tenfold, while titratable acid excretion shows only a small increase.8 Therefore, the excretion of NH4+ is the main mechanism for urine acidification under conditions of chronic acidosis.
In contrast to most urinary solutes, the majority of urinary NH3 is generated in the kidney.14 NH4+ is generated from glutamine in the proximal tubular cells and is secreted into the urine in the proximal tubule lumen, which is coupled with HCO3− recovery resulting from glutamine metabolism. NH4+ is primarily reabsorbed by the Na–K–2Cl co-transporter NKCC2 in the thick ascending limb of the loop of Henle and is accumulated in the interstitium. Interstitial NH3/NH4+ is then secreted into the cortical and medullary collecting duct lumen by several mechanisms: diffusion of medullary NH3 across the basolateral and apical membranes into the lumen; basolateral Na+–K+-ATPase that transports NH4+ into the cell by substituting NH4+ for K+ in the inner medullary collecting duct; NH3 uptake across the basolateral membrane via the rhesus blood group glycoproteins, RhBG and RhCG; and NH3 secretion across ICs via RhCG. Collecting duct NH3 secretion occurs by parallel NH3 and H+ transport, and apical H+ secretion involves both H+-ATPase and H+–K+-ATPase (Figure 1).8,14,15
Pathogenic mechanisms of dRTA
dRTA is caused by the failure of the kidney A-ICs to acidify the urine normally, which results from a dysfunction in any of the transporters involved in this process. Mutations in three transporter genes expressed in A-ICs have been identified as causes of dRTA, including the B1 (ATP6V1B1) and a4 (ATP6V0A4) subunits of H+-ATPase and the AE1/SLC4A1.16 However, because mutations in these genes are identified in only 70%–80% of patients with dRTA,4,5 dRTA is also likely to be caused by mutations in other genes.3,17 In addition to its genetic component, dRTA can also be acquired.7
Vacuolar H+-ATPase gene (ATP6V1B1 and ATP6V0A4) mutations
H+-ATPases are multisubunit enzymatic proton pumps, which consist of two domains, the V1 cytoplasmic domain (subunits A–H) and the V0 membrane domain composed of subunits a, d c″, c, and e.18 The V0 domain mediates proton transfer and requires ATP hydrolysis by the V1 domain.18,19 The B1 subunit is expressed in the kidney, inner ear, epididymis, and ciliary body of the eye. The a4 subunit is only expressed in the kidney, inner ear, and epididymis. In the kidney, the B1 subunit is expressed only in ICs of the late distal tubule, connecting segment, and collecting duct, while the a4 subunit is expressed in the proximal tubule, loop of Henle, and ICs of the late distal tubule, connecting segment, and collecting duct.20 Homozygous or compound heterozygous mutations in B1 and a4 lead to autosomal recessive (AR) dRTA.
Karet et al were the first to identify homozygous mutations in ATP6V1B1 in AR dRTA patients with early SNHL.21 Subsequently, Smith et al detected homozygous mutations in ATP6V0A4 in AR dRTA patients without SNHL.22 However, it became later obvious that a number of patients with AR dRTA with homozygous or compound heterozygous mutations in ATP6V0A4 developed SNHL in early childhood23 or in young adulthood.24 Compound heterozygous mutations in ATP6V1B1 were also found in some patients with dRTA.23,25,26
Premature termination codons, frameshift mutations, or splice-site mutations, which are predicted to disrupt the encoded protein, are found in most patients with ATP6V1B1 or ATP6V0A4 mutations, while missense mutations have been described in only a few patients.21–24 Experimental studies using rat inner medullary collecting duct cell culture or yeast models showed that dysfunction or impaired assembly of the B1 subunit with other protein complex subunits is the most common outcome of missense mutations in ATP6V1B1.27,28 Su et al recreated previously reported missense mutations of the a4 subunit G820R22 and R807Q24 in yeast to examine the effects on protein expression. They found that the G820R mutation caused a loss of required phosphofructokinase-1 (PFK) binding to the a4 subunit, while the R807Q mutation resulted in a loss of pump protein.29
In mouse models, mice deficient in the B1 subunit show more alkaline urine without systemic acidosis when fed a standard diet. These mice also develop more severe metabolic acidosis after an acid load compared to normal mice, indicating a failure of normal urinary acidification.30 The B1-deficient mice do not develop hearing impairment or nephrocalcinosis. In contrast, mice lacking the a4 subunit demonstrate dRTA with severe metabolic acidosis, hypokalemia, early nephrocalcinosis, and severe hearing impairment with enlarged cochlear and endolymphatic ducts in the inner ear.31,32 In addition, these mice develop proximal renal tubular dysfunction with defective endocytic trafficking, proteinuria, and phosphaturia, which has not been reported in humans.33 This may be due to compensatory changes in the a1, a2, and a3 subunits in the proximal tubules of dRTA patients with the ATP6V0A4 mutation.17,34
Chloride–bicarbonate exchanger AE1 gene (SLC4A1) mutations
The Cl−–HCO3− exchanger AE1, encoded by SLC4A1, is expressed in the red blood cells (RBCs; eAE1, Band 3) and in the A-ICs of kidney (kAE1). SLC4A1 mutations can cause dRTA or RBC abnormalities including hereditary spherocytosis (HS), Southeast Asia ovalocytosis (SAO), hereditary stomatocytosis, and hereditary xerocytosis.35 Most mutations cause either dRTA or RBC abnormalities, whereas only a few mutations lead to abnormalities in both.3 dRTA caused by AE1 gene mutations can occur with either an AD or AR transmission (Table 1).1,6 AE1 mutations are rare and usually have an AD transmission in Caucasians, while AR dRTA is common in Asians.17 The clinical symptoms are more severe and the age of onset is earlier in patients with AR dRTA compared to patients with AD dRTA.7 Patients with AD dRTA can present with complete dRTA or idRTA, whereas patients with AR dRTA always present with complete dRTA.16 Although RBC abnormalities have been associated with AR dRTA, hemolytic anemia is extremely rare in AD dRTA, and only one family with AD idRTA and HS due to a heterozygous splicing mutation (c.1432–1G>A, Band 3PRIBRAM) has been reported.36
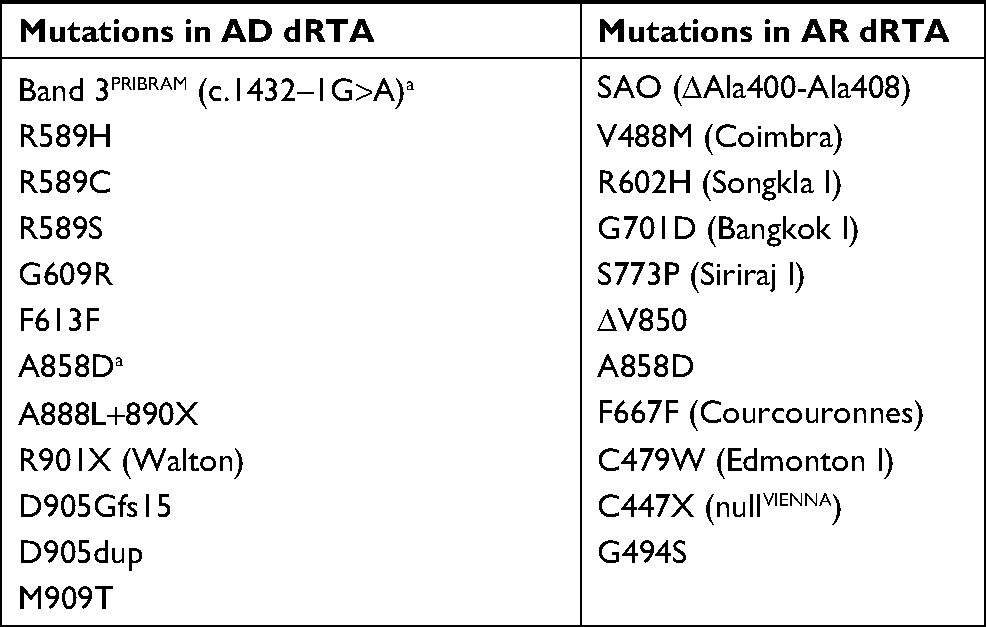 | Table 1 SLC4A1 mutations in dRTA Note: aHeterozygous Band 3PRIBRAM and A858D mutations cause incomplete dRTA. Abbreviations: AD, autosomal dominant; AR, autosomal recessive; dRTA, distal renal tubular acidosis; SAO, Southeast Asia ovalocytosis. |
AD AE1 gene (SLC4A1) mutations
Wrong et al reported a family with AD dRTA caused by a heterozygous mutation of R589H in AE1.37 Subsequent studies described AD dRTA with AE1 gene mutations including R589H,38–40 R589C,38 R589S,39 S613F,38 R901X,39 A858D,41 A888L+889X,42 G609R,43 D905Gfs15,44 D905dup,45 and M909T.46
Experimental studies using transfected cell models showed that AE1 gene mutations caused normal or modestly reduced anion transport activity and impaired trafficking with retention in the endoplasmic reticulum (ER). Heterodimers with the wild-type AE1 polypeptide caused a dominant negative trafficking phenotype (R589H and S613F)35,47–49 or mistargeting to the apical membranes or to both the apical and basolateral membranes (G609R, R901X, and M909T).43,46,49–51
In in vivo studies, mice lacking Ae1 exhibited complete dRTA with hemolytic anemia, while mice heterozygous for Ae1 showed no apparent defect.52,53 Mumtaz et al recently generated an Ae1 R607H knockin (KI) mouse, corresponding to the most common AD dRTA mutation in human AE1, R589H.54 They found that both homozygous and heterozygous R607H KI mice exhibited idRTA without RBC abnormalities.54 Mutant mice exhibited decreased levels of Ae1 in A-ICs, but preserved basolateral targeting of the mutant protein, and reduced expression of H+-ATPase due to impaired targeting to the apical membranes.9,54
AR AE1 gene (SLC4A1) mutations
Tanphaichitr et al first reported AR dRTA with a homozygous AE1 gene loss-of-function mutation, G701D, in two siblings in Thailand who had xerocytic hemolytic anemia with normal RBC AE1 activity.55 AE1 G701D interacts with an RBC AE1 chaperon, glycophorin A (GPA), which rescues the mutant protein in RBCs. GPA is not expressed in renal ICs, which explains the normal erythroid AE1 expression in these patients.35,55 Subsequently, Yenchitsomanus et al reported a homozygous AE1 G701D mutation in five out of eight Thai families with AR dRTA with mild RBC morphological abnormalities without hemolytic anemia and suggested that AE1 G701D was a common molecular defect in Thailand.56
SAO is a common hereditary condition in South East and Melanesia. SAO is caused by the AE1 (∆Ala400-Ala408) mutation, which causes ovalocytic erythrocytes to be resistant to invasion by the malarial parasite.57 Vasuvattakul et al reported two patients from Northeast Thailand with AR dRTA and SAO resulting from compound heterozygous AE1 G701D/SAO mutations.58 Bruce et al described AE1 gene mutations associated with AR dRTA and SAO in families from Malaysia and Papua New Guinea. The mutations included compound heterozygous AE1 G701D/SAO and ∆V850/SAO and homozygous AE1 ∆V850/∆V850.41 Patients with compound heterozygous AE1 A858D/SAO mutations in this study exhibited dRTA and SAO with AD transmission. Subsequently, other mutations were identified, including R602H/SAO from South Thailand,59 Q759H/SAO from Malaysia,60 and G701/SAO from the Philippines.61 Although homozygous SAO was long thought to be lethal,62 Picard et al recently reported a child with homozygous SAO mutations who had AR dRTA with severe dyserythropoietic and hemolytic anemia with SAO red cells.63
Approximately 20% of HS cases are caused by heterozygous AE1 gene mutations.64 Hence, HS can be associated with AR dRTA. Ribeiro et al reported a child with severe HS and AR dRTA caused by homozygous AE1 V488M (Band 3 Coimbra).65 Toye et al reported that homozygous S667F (Band 3 Courcouronnes) causes HS and idRTA.66 Subsequently, it was found that compound heterozygous E522K/G701D67 and C479W (Band 3 Edmonton I)/G701D68 and homozygous A858D69 cause AR dRTA with HS.
Several other AE1 mutations have been reported to cause AR dRTA with RBC abnormalities other than SAO or HS. These include compound heterozygous ∆V850/A858D (red cells of bizarre shapes and anemia)41 and G701D/A858D (ovalocytes and acanthocytes),70 homozygous A858D (hemolytic anemia with elliptocytosis, stomatocytosis, and acanthocytosis71 or acanthocytosis and echinocytosis),72 and nonsense mutation of S447X, Band 3 nullVIENNA (severe hemolytic anemia).73 In addition, AE1 mutations causing AR dRTA without RBC abnormalities have been reported, including homozygous ∆V85041 and compound heterozygous G494S/G701D44 and S773P/G701D.59
Experimental studies using transfected cell models have revealed that these mutations cause Golgi retention due to impaired trafficking with loss of function, which is rescued by GPA (G701D),48,55,74 misfolding and targeting for degradation (S773P),48,74 decreased AE1 function (∆V850);41 ER retention due to impaired trafficking with normal anion transport function, which is rescued by GPA (S667F),66 mild trafficking impairment (E522K);67 and ER retention due to impaired trafficking and misfolding (C479W).68
Other candidate genes for dRTA
Because ~20% of patients with dRTA do not have mutations in genes for H+-ATPase or AE1, additional candidate genes have been identified in mouse models of dRTA.3,6 These include HKα2 (colonic H+–K+-ATPase),75 Kcc4 (K+–Cl− co-transporter, Kcc4),76 genes for the H+-ATPase C, G, and d subunits,77 Foxi1 (the Forkhead transcription factor, Foxi1),78 Rhcg (the ammonia transporter, Rhcg),79,80 Slc26a7 (Cl−–HCO3− exchanger, Slc26a7, co-localized with AE1),81 DMBT1 (component of the pathway of acidosis-induced conversion of B-ICs into A-ICs, hensin),11 GPR4 (proton sensing G protein-coupled receptor, GPR4),82 NHE4 (Na+–H+ exchanger 4, NHE4),83 SLC4A5 (Na+–HCO3− co-transporter, NBCe2),84 Atp6ap2 (ATPase H+ transporting lysosomal accessory protein 2, atp6ap2),85 and Ncoa7 (nuclear receptor coactivator 7, Ncoa7: an H+-ATPase interacting protein), as shown in Table 2.86 Although most of these genes have not been previously identified in human disease, Enerbäck et al identified homozygous FOXI1mutations in two families with AR dRTA and early-onset SNHL.87 Furthermore, a recent study found homozygous or compound heterozygous WDR72 (tryptophan–aspartate repeat domain 72, WDR72: a protein possibly associated with intracellular endocytic vesicle trafficking) mutations in two families with AR dRTA (Table 2).88
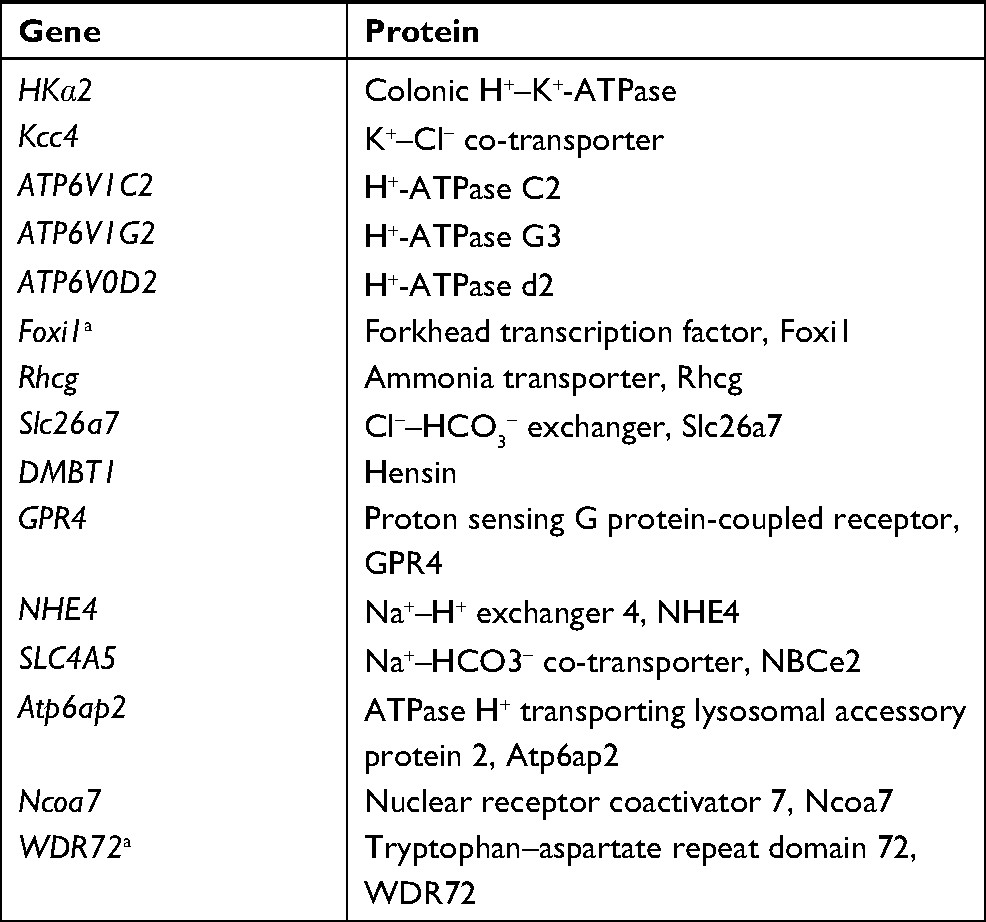 | Table 2 Candidate genes for distal renal tubular acidosis Note: aMutations in these genes were recently reported in humans. |
Causes of acquired dRTA
The main causes of acquired dRTA include medications and, most commonly, autoimmune diseases, with Sjögren syndrome being the most frequent cause. Although the precise pathogenic mechanisms of dRTA development in Sjögren syndrome are unclear, several studies suggest that autoantibodies against carbonic anhydrase or A-ICs transporters are involved in the pathogenesis.89 Systemic lupus erythematosus, thyroiditis, and renal transplantation have been reported as other autoimmune causes of dRTA.2
Medications including amphotericin B, foscarnet, analgesic abuse, lithium, melphalan, and amiloride also cause dRTA.2,7 In the case of amphotericin B and foscarnet, the mechanisms mediating these effects involved increased membrane permeability in the collecting duct and increased mitochondrial dysfunction in renal tubular cells, respectively.90 The mechanisms associated with other drugs are not clear.
Atypical clinical features of dRTA
Some patients with dRTA exhibit atypical clinical features, which include transient and reversible proximal tubular dysfunction and hyperammonemia.
Reversible proximal tubular dysfunction in patients with dRTA
Reversible proximal tubular dysfunction has been reported in patients with dRTA. This appears to involve defects in bicarbonate reabsorption,91,92 low-molecular-weight proteinuria,93 hypouricemia with uricosuria, phosphaturia, and generalized aminoaciduria.94,95 Besouw et al recently reported that 16 of 24 patients with dRTA showed transient and partial Fanconi syndrome that resembled Dent disease or Low syndrome. Proximal tubular dysfunction was only seen in children with mutations in subunits of the H+-ATPase and in those with unknown mutations.96
Although the exact mechanism underlying reversible proximal tubular dysfunction is unclear, it has been suggested to involve hypokalemic nephropathy32,94 and/or dysfunction of the receptor-mediated endosomal pathway in renal proximal tubule cells.3,97 The chloride transporter ClC-5 (2Cl−–H+ exchanger) is expressed in the apical endosomes of renal proximal tubules containing H+-ATPase. ClC-5 normally acts in the endosomal pathway by coupling with H+-ATPase. Mutations of the ClC-5 gene cause Dent disease.98 Picollo et al demonstrated that low extracellular pH or acidosis inhibits ClC-5 function by reducing the driving force for 2Cl−–H+ exchange.99 Therefore, inhibition of ClC-5 function due to systemic acidosis may lead to partial renal Fanconi syndrome in patients with dRTA.97
Hyperammonemia in patients with dRTA
Hyperammonemia, first described by Miller and Schwartz in dRTA, was not initially recognized as an important clinical feature of dRTA.100 Subsequently, Miura et al reported a high frequency of hyperammonemia in patients with dRTA (four of six patients with available data), suggesting that hyperammonemia is a common feature of dRTA.25 Hyperammonemia was reported in 15 patients with dRTA in a recent systematic review101 and in a case report.102 In these patients, a negative correlation was observed between blood ammonia and bicarbonate levels, and alkali therapy resulted in a rapid normalization of ammonia levels.101 Increased renal ammonia synthesis in response to acidosis, without appropriate ammonia excretion, may result in hyperammonemia in dRTA.100,101
Incomplete distal renal tubular acidosis
idRTA, first described in 1959, presents with inadequate urinary acidification without spontaneous metabolic acidosis.103 Failure to acidify urinary pH <5.3 in the NH4Cl load was considered diagnostic for idRTA.89 However, because urinary acidification capacity is a continuous trait, idRTA is not a distinct entity, and may be a variant of normal urinary acidification.89,104
Patients with idRTA commonly exhibit hypocitraturia, hypercalciuria, nephrocalcinosis, and nephrolithiasis.105,106 A wide range of idRTA prevalence has been reported in patients with recurrent urinary stones.104 Bone abnormalities also have been frequently reported in patients with idRTA, including rickets107 or growth failure108 in children and osteoporosis and osteopenia in adults.109–111 However, other studies found no association between idRTA and lower bone mass.1,65,112
Although the molecular basis for idRTA is unknown in most patients, heterozygous mutations in SLC4A1 (Band 3PPRIBRAM and A858D),36,41 ATP6V1B1 (F468fsX487),113 and ATP6V0A4 (S544L)114 have been reported. Furthermore, Dhayat et al reported that recurrent kidney stone formers with H+-ATPase B1 subunit p.E161K single-nucleotide polymorphism have idRTA with an increased prevalence of calcium phosphate kidney stones.115 A recent experimental study revealed that Ncoa7 (H+-ATPase interacting protein)-knockout mice have idRTA.86 Another study showed that haploinsufficiency of Atp6v1b in mice causes idRTA.116
Kidney stone formation in dRTA
The combination of hypercalciuria, hypocitraturia, and high urine pH contributes to the development of kidney stone formation and/or nephrocalcinosis in dRTA.6
Because bicarbonate is depleted from the extracellular fluid in dRTA, buffering of the retained nonvolatile acids promotes the release of calcium phosphate from bone, which increases urinary excretion of calcium and phosphate in dRTA.6,7 Moreover, metabolic acidosis decreases the function and expression of the TRPV5 calcium channel in the distal tubule, independent of parathyroid hormone and vitamin D, which also contributes to hypercalciuria in dRTA.117
Urinary citrate inhibits stone formation by complexing with calcium, inhibiting spontaneous nucleation, and preventing the growth of crystals.118 Metabolic acidosis increases citrate reabsorption in the proximal tubule via increased activity of sodium-dependent dicarboxylate transporter 1.17,117,118
A high urine pH increases the supersaturation of calcium phosphate in the tubular lumen, thereby increasing the risk of kidney stone formation.117
Treatment
The primary objectives of dRTA treatment are correction of metabolic acidosis and avoidance of disease-related complications, which include failure to thrive, growth retardation, rickets, osteoporosis, nephrolithiasis, and nephrocalcinosis.3,16,89 It is especially important to prevent nephrocalcinosis because progressive nephrocalcinosis may lead to chronic kidney disease (CKD) and end-stage renal disease in patients with dRTA.89
Alkali in the form of sodium and/or potassium bicarbonate or citrate salts should be administered to maintain a normal serum bicarbonate concentration of >20 mEq/L in infants and >22 mEq/L in children and adults.6,89 However, excessive sodium bicarbonate will increase the extracellular volume and decrease the reabsorption of proximal tubular HCO3−, which increases the need for alkali.6 In addition, because the increase in sodium intake after sodium citrate or sodium bicarbonate administration can result in increased urinary calcium excretion, potassium salts may be more effective than sodium salts for treatment of dRTA with kidney stones.119,120 As citrate salts can also correct hypocitraturia and prevent nephrolithiasis, potassium citrate is usually recommended.16,89
Young children with dRTA need higher doses of alkali because of the greater rate of acid production caused by the formation of hydroxyapatite associated with bone growth.7 The amount of alkali needed usually decreases with age from as much as 5–8 mEq/kg/day in infants to 3–4 mEq/kg/day in children after the age of 6 years to 1–2 mEq/kg/day in adults.6,89,92A recent study showed that children with mutations in ATP6V1B1 or ATP6V0A4 generally needed higher doses of alkali compared to those with SLC4A1 mutations.96 In patients with severe hypokalemia despite administration of potassium citrate, potassium supplementation may be required.96
Dietary modifications to increase urinary citrate excretion may benefit dRTA patients with kidney stones and hypocitraturia. Dietary modifications include increased intake of fluid and citrus fruits, normal intake of calcium, and restricted intake of sodium, oxalate, animal proteins, and fructose.118
Normalization of serum bicarbonate level, urinary calcium excretion, urinary low-molecular-weight protein levels (β2 microglobulin or α2 microglobulin), and growth rate are good markers for adherence to and/or adequacy of treatment.6,93,96 In addition, abdominal ultrasonography should be regularly checked to detect nephrocalcinosis and nephrolithiasis.
Long-term outcome
The prognosis of dRTA is generally good in patients treated with early and sufficient doses of alkali, but alkali administration does not improve hearing impairment in patients with dRTA and SNHL.16,96 Moreover, recent studies showed an increased frequency of CKD in patients with dRTA during long-term follow-up. Besouw et al reported 9 of 24 (37.5%) children with dRTA developed CKD stage 2.96 Palazzo et al also showed 16 of 51 (31.3%) patients with dRTA developed CKD during long-term follow-up and after puberty.5 Although the precise pathogenic mechanisms of CKD in patients with dRTA are unknown, the combination of nephrocalcinosis and hypokalemia, which results in tubulointerstitial damage, or kidney damage after repeated episodes of dehydration and acute kidney injury have been suggested as potential causal factors.5,17
Disclosure
The author reports no conflicts of interest in this work.
References
Fry AC, Karet FE. Inherited renal acidoses. Physiology. 2007;22(3):202–211. | ||
Rodríguez Soriano J. Renal tubular acidosis: the clinical entity. J Am Soc Nephrol. 2002;13(8):2160–2170. | ||
Mohebbi N, Wagner CA. Pathophysiology, diagnosis and treatment of inherited distal renal tubular acidosis. J Nephrol. 2018;31(4):511–522. | ||
Besouw MTP, Bienias M, Walsh P, et al. Clinical and molecular aspects of distal renal tubular acidosis in children. Pediatr Nephrol. 2017;32(6):987–996. | ||
Palazzo V, Provenzano A, Becherucci F, et al. The genetic and clinical spectrum of a large cohort of patients with distal renal tubular acidosis. Kidney Int. 2017;91(5):1243–1255. | ||
Gil-Peña H, Mejía N, Santos F. Renal tubular acidosis. J Pediatr. 2014;164(4):691–698. | ||
Quigley R, Wolf MT. Renal tubular acidosis in children. In: Avner ED, Harmon WE, Niaudet P, Yoshikawa N, Emma F, Goldstein SL. editors. Pediatric Nephrology. 7th ed. New York: Springer-Verlag Berlin Heidelberg; 2016:1273–1306. | ||
Hamm LL, Nakhoul N, Hering-Smith KS. Acid–base homeostasis. Clin J Am Soc Nephrol. 2015;10(12):2232–2242. | ||
Trepiccione F, Prosperi F, de La Motte LR, et al. New findings on the pathogenesis of distal renal tubular acidosis. Kidney Dis. 2017;3(3):98–105. | ||
Corey HE, Eckstein D. Renal tubular acidosis. In: Ronco C, Bellomo R, Kellum JA, Ricci Z, editors. Critical Care Nephrology. 3rd ed. Philadelphia: Elsevier; 2019:405–408. | ||
Gao X, Eladari D, Leviel F, et al. Deletion of hensin/DMBT1 blocks conversion of beta- to alpha-intercalated cells and induces distal renal tubular acidosis. Proc Natl Acad Sci USA. 2010;107(50):21872–21877. | ||
Al-Awqati Q. Cell biology of the intercalated cell in the kidney. FEBS Lett. 2013;587(13):1911–1914. | ||
Schwartz GJ, Gao X, Tsuruoka S, et al. SDF1 induction by acidosis from principal cells regulates intercalated cell subtype distribution. J Clin Invest. 2015;125(12):4365–4374. | ||
Weiner ID, Verlander JW. Ammonia transporters and their role in acid–base balance. Physiol Rev. 2017;97(2):465–494. | ||
Weiner ID, Mitch WE, Sands JM. Urea and ammonia metabolism and the control of renal nitrogen excretion. Clin J Am Soc Nephrol. 2015;10(8):1444–1458. | ||
Batlle D, Haque SK. Genetic causes and mechanisms of distal renal tubular acidosis. Nephrol Dial Transplant. 2012;27(10):3691–3704. | ||
Kurtz I. Renal tubular acidosis: H+/base and ammonia transport abnormalities and clinical syndromes. Adv Chronic Kidney Dis. 2018;25(4):334–350. | ||
Blake-Palmer KG, Karet FE. Cellular physiology of the renal H+ATPase. Curr Opin Nephrol Hypertens. 2009;18(5):433–438. | ||
Breton S, Brown D. Regulation of luminal acidification by the V-ATPase. Physiology. 2013;28(5):318–329. | ||
Wagner CA, Finberg KE, Breton S, Marshansky V, Brown D, Geibel JP. Renal vacuolar H+-ATPase. Physiol Rev. 2004;84(4):1263–1314. | ||
Karet FE, Finberg KE, Nelson RD, et al. Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet. 1999;21(1):84–90. | ||
Smith AN, Skaug J, Choate KA, et al. Mutations in ATP6N1B, encoding a new kidney vacuolar proton pump 116-kD subunit, cause recessive distal renal tubular acidosis with preserved hearing. Nat Genet. 2000;26(1):71–75. | ||
Vargas-Poussou R, Houillier P, Le Pottier N, et al. Genetic investigation of autosomal recessive distal renal tubular acidosis: evidence for early sensorineural hearing loss associated with mutations in the ATP6V0A4 gene. J Am Soc Nephrol. 2006;17(5):1437–1443. | ||
Stover EH, Borthwick KJ, Bavalia C, et al. Novel ATP6V1B1 and ATP6V0A4 mutations in autosomal recessive distal renal tubular acidosis with new evidence for hearing loss. J Med Genet. 2002;39(11):796–803. | ||
Miura K, Sekine T, Takahashi K, et al. Mutational analyses of the ATP6V1B1 and ATP6V0A4 genes in patients with primary distal renal tubular acidosis. Nephrol Dial Transplant. 2013;28(8):2123–2130. | ||
Mohebbi N, Vargas-Poussou R, Hegemann SC, et al. Homozygous and compound heterozygous mutations in the ATP6V1B1 gene in patients with renal tubular acidosis and sensorineural hearing loss. Clin Genet. 2013;83(3):274–278. | ||
Yang Q, Li G, Singh SK, Alexander EA, Schwartz JH. Vacuolar H+-ATPase B1 subunit mutations that cause inherited distal renal tubular acidosis affect proton pump assembly and trafficking in inner medullary collecting duct cells. J Am Soc Nephrol. 2006;17(7):1858–1866. | ||
Fuster DG, Zhang J, Xie XS, Moe OW. The vacuolar-ATPase B1 subunit in distal tubular acidosis: novel mutations and mechanisms for dysfunction. Kidney Int. 2008;73(10):1151–1158. | ||
Su Y, Blake-Palmer KG, Sorrell S, et al. Human H+ATPase a4 subunit mutations causing renal tubular acidosis reveal a role for interaction with phosphofructokinase-1. Am J Physiol Renal Physiol. 2008;295(4):F950–F958. | ||
Finberg KE, Wagner CA, Bailey MA, et al. The B1-subunit of the H+ ATPase is required for maximal urinary acidification. Proc Natl Acad Sci USA. 2005;102(38):13616–13621. | ||
Norgett EE, Golder ZJ, Lorente-Cánovas B, Ingham N, Steel KP, Karet Frankl FE. Atp6v0a4 knockout mouse is a model of distal renal tubular acidosis with hearing loss, with additional extrarenal phenotype. Proc Natl Acad Sci USA. 2012;109(34):13775–13780. | ||
Lorente-Cánovas B, Ingham N, Norgett EE, Golder ZJ, Karet Frankl FE, Steel KP. Mice deficient in H+-ATPase a4 subunit have severe hearing impairment associated with enlarged endolymphatic compartments within the inner ear. Dis Model Mech. 2013;6(2):434–442. | ||
Hennings JC, Picard N, Huebner AK, et al. A mouse model for distal renal tubular acidosis reveals a previously unrecognized role of the V-ATPase a4 subunit in the proximal tubule. EMBO Mol Med. 2012;4(10):1057–1071. | ||
Schulz N, Dave MH, Stehberger PA, Chau T, Wagner CA. Differential localization of vacuolar H+-ATPases containing a1, a2, a3, or a4 (ATP6V0A1-4) subunit isoforms along the nephron. Cell Physiol Biochem. 2007;20(1-4):109–120. | ||
Alper SL. Molecular physiology and genetics of Na+-independent SLC4 anion exchangers. J Exp Biol. 2009;212(Pt 11):1672–1683. | ||
Rysavá R, Tesar V, Jirsa M, Brabec V, Jarolím P. Incomplete distal renal tubular acidosis coinherited with a mutation in the band 3 (AE1) gene. Nephrol Dial Transplant. 1997;12(9):1869–1873. | ||
Wrong O, Unwin R, Fine LG, Cohen E, Thakker R, Tanner M. Unravelling of the molecular mechanisms of kidney stones. Report of a Meeting of Physicians and Scientists. Lancet. 1996;348(9041):1561–1565. | ||
Bruce LJ, Cope DL, Jones GK, et al. Familial distal renal tubular acidosis is associated with mutations in the red cell anion exchanger (Band 3, AE1) gene. J Clin Invest. 1997;100(7):1693–1707. | ||
Karet FE, Gainza FJ, Györy AZ, et al. Mutations in the chloride–bicarbonate exchanger gene AE1 cause autosomal dominant but not autosomal recessive distal renal tubular acidosis. Proc Natl Acad Sci USA. 1998;95(11):6337–6342. | ||
Jarolim P, Shayakul C, Prabakaran D, et al. Autosomal dominant distal renal tubular acidosis is associated in three families with heterozygosity for the R589H mutation in the AE1 (band3) Cl−/HCO3−exchanger. J Biol Chem. 1998;273(11):6380–6388. | ||
Bruce LJ, Wrong O, Toye AM, et al. Band 3 mutations, renal tubular acidosis and South-East Asian ovalocytosis in Malaysia and Papua New Guinea: loss of up to 95% band 3 transport in red cells. Biochem J. 2000;350(1):41–51. | ||
Cheidde L, Vieira TC, Lima PR, Saad ST, Heilberg IP. A novel mutation in the anion exchanger 1 gene is associated with familial distal renal tubular acidosis and nephrocalcinosis. Pediatrics. 2003;112(6 Pt 1):1361–1367. | ||
Rungroj N, Devonald MA, Cuthbert AW, et al. A novel missense mutation in AE1 causing autosomal dominant distal renal tubular acidosis retains normal transport function but is mistargeted in polarized epithelial cells. J Biol Chem. 2004;279(14):13833–13838. | ||
Zhang Z, Liu KX, He JW, et al. Identification of two novel mutations in the SLC4A1 gene in two unrelated Chinese families with distal renal tubular acidosis. Arch Med Res. 2012;43(4):298–304. | ||
Shao L, Xu Y, Dong Q, Lang Y, Yue S, Miao Z. A novel SLC4A1 variant in an autosomal dominant distal renal tubular acidosis family with a severe phenotype. Endocrine. 2010;37(3):473–478. | ||
Fry AC, Su Y, Yiu V, Cuthbert AW, Trachtman H, Karet Frankl FE. Mutation conferring apical-targeting motif on AE1 exchanger causes autosomal dominant distal RTA. J Am Soc Nephrol. 2012;23(7):1238–1249. | ||
Quilty JA, Li J, Reithmeier RA. Impaired trafficking of distal renal tubular acidosis mutants of the human kidney anion exchanger kAE1. Am J Physiol Renal Physiol. 2002;282(5):F810–F820. | ||
Cordat E, Kittanakom S, Yenchitsomanus PT, et al. Dominant and recessive distal renal tubular acidosis mutations of kidney anion exchanger 1 induce distinct trafficking defects in MDCK cells. Traffic. 2006;7(2):117–128. | ||
Toye AM, Banting G, Tanner MJ. Regions of human kidney anion exchanger 1 (kAE1) required for basolateral targeting of kAE1 in polarised kidney cells: mis-targeting explains dominant renal tubular acidosis (dRTA). J Cell Sci. 2004;117(Pt 8):1399–1410. | ||
Quilty JA, Cordat E, Reithmeier RA. Impaired trafficking of human kidney anion exchanger (kAE1) caused by hetero-oligomer formation with a truncated mutant associated with distal renal tubular acidosis. Biochem J. 2002;368(Pt 3):895–903. | ||
Devonald MA, Smith AN, Poon JP, Ihrke G, Karet FE. Non-polarized targeting of AE1 causes autosomal dominant distal renal tubular acidosis. Nat Genet. 2003;33(2):125–127. | ||
Stehberger PA, Shmukler BE, Stuart-Tilley AK, Peters LL, Alper SL, Wagner CA. Distal renal tubular acidosis in mice lacking the AE1 (band3) Cl–/HCO3– exchanger (slc4a1). J Am Soc Nephrol. 2007;18(5):1408–1418. | ||
Akel A, Wagner CA, Kovacikova J, et al. Enhanced suicidal death of erythrocytes from gene-targeted mice lacking the Cl–/HCO3– exchanger AE1. Am J Physiol Cell Physiol. 2007;292(5): C1759–1767. | ||
Mumtaz R, Trepiccione F, Hennings JC, et al. Intercalated cell depletion and vacuolar H+-ATPase mistargeting in an Ae1 R607H knockin model. J Am Soc Nephrol. 2017;28(5):1507–1520. | ||
Tanphaichitr VS, Sumboonnanonda A, Ideguchi H, et al. Novel AE1 mutations in recessive distal renal tubular acidosis. Loss-of-function is rescued by glycophorin A. J Clin Invest. 1998;102(12):2173–2179. | ||
Yenchitsomanus PT, Vasuvattakul S, Kirdpon S, et al. Autosomal recessive distal renal tubular acidosis caused by G701D mutation of anion exchanger 1 gene. Am J Kidney Dis. 2002;40(1):21–29. | ||
Jarolim P, Palek J, Amato D, et al. Deletion in erythrocyte band 3 gene in malaria-resistant Southeast Asian ovalocytosis. Proc Natl Acad Sci USA. 1991;88(24):11022–11026. | ||
Vasuvattakul S, Yenchitsomanus PT, Vachuanichsanong P, et al. Autosomal recessive distal renal tubular acidosis associated with Southeast Asian ovalocytosis. Kidney Int. 1999;56(5):1674–1682. | ||
Sritippayawan S, Sumboonnanonda A, Vasuvattakul S, et al. Novel compound heterozygous SLC4A1 mutations in Thai patients with autosomal recessive distal renal tubular acidosis. Am J Kidney Dis. 2004;44(1):64–70. | ||
Choo KE, Nicoli TK, Bruce LJ, Tanner MJ, Ruiz-Linares A, Wrong OM. Recessive distal renal tubular acidosis in Sarawak caused by AE1 mutations. Pediatr Nephrol. 2006;21(2):212–217. | ||
Anacleto FE, Bruce LJ, Clayton P, Hegde S, Resontoc LP, Wrong O. Distal renal tubular acidosis in Filipino children, caused by mutations of the anion-exchanger SLC4A1 (AE1, Band 3) gene. Nephron Physiol. 2010;114(2):19–24. | ||
Liu SC, Jarolim P, Rubin HL, et al. The homozygous state for the band 3 protein mutation in Southeast Asian ovalocytosis may be lethal. Blood. 1994;84(10):3590–3591. | ||
Picard V, Proust A, Eveillard M, et al. Homozygous Southeast Asian ovalocytosis is a severe dyserythropoietic anemia associated with distal renal tubular acidosis. Blood. 2014;123(12):1963–1965. | ||
Jarolim P, Murray JL, Rubin HL, et al. Characterization of 13 novel band 3 gene defects in hereditary spherocytosis with band 3 deficiency. Blood. 1996;88(11):4366–4374. | ||
Ribeiro ML, Alloisio N, Almeida H, et al. Severe hereditary spherocytosis and distal renal tubular acidosis associated with the total absence of band 3. Blood. 2000;96(4):1602–1604. | ||
Toye AM, Williamson RC, Khanfar M, et al. Band 3 Courcouronnes (Ser667Phe): a trafficking mutant differentially rescued by wild-type band 3 and glycophorin A. Blood. 2008;111(11):5380–5389. | ||
Chang YH, Shaw CF, Jian SH, Hsieh KH, Chiou YH, Lu PJ. Compound mutations in human anion exchanger 1 are associated with complete distal renal tubular acidosis and hereditary spherocytosis. Kidney Int. 2009;76(7):774–783. | ||
Chu C, Woods N, Sawasdee N, et al. Band 3 Edmonton I, a novel mutant of the anion exchanger 1 causing spherocytosis and distal renal tubular acidosis. Biochem J. 2010;426(3):379–388. | ||
Sinha R, Agarwal I, Bawazir WM, Bruce LJ. Distal renal tubular acidosis with hereditary spherocytosis. Indian Pediatr. 2013;50(7):693–695. | ||
Khositseth S, Sirikanerat A, Wongbenjarat K, et al. Distal renal tubular acidosis associated with anion exchanger 1 mutations in children in Thailand. Am J Kidney Dis. 2007;49(6):841–850. | ||
Shmukler BE, Kedar PS, Warang P, et al. Hemolytic anemia and distal renal tubular acidosis in two Indian patients homozygous for SLC4A1/AE1 mutation A858D. Am J Hematol. 2010;85(10):824–828. | ||
Fawaz NA, Beshlawi IO, Al Zadjali S, et al. dRTA and hemolytic anemia: first detailed description of SLC4A1 A858D mutation in homozygous state. Eur J Haematol. 2012;88(4):350–355. | ||
Kager L, Bruce LJ, Zeitlhofer P, et al. Band 3 nullVIENNA, a novel homozygous SLC4A1 p.Ser477X variant causing severe hemolytic anemia, dyserythropoiesis and complete distal renal tubular acidosis. Pediatr Blood Cancer. 2017;64(3):e26227. | ||
Kittanakom S, Cordat E, Akkarapatumwong V, Yenchitsomanus PT, Reithmeier RA. Trafficking defects of a novel autosomal recessive distal renal tubular acidosis mutant (S773P) of the human kidney anion exchanger (kAE1). J Biol Chem. 2004;279(39):40960–40971. | ||
Simpson AM, Schwartz GJ. Distal renal tubular acidosis with severe hypokalaemia, probably caused by colonic H+-K+-ATPase deficiency. Arch Dis Child. 2001;84(6):504–507. | ||
Boettger T, Hübner CA, Maier H, Rust MB, Beck FX, Jentsch TJ. Deafness and renal tubular acidosis in mice lacking the K-Cl co-transporter Kcc4. Nature. 2002;416(6883):874–878. | ||
Smith AN, Borthwick KJ, Karet FE. Molecular cloning and characterization of novel tissue-specific isoforms of the human vacuolar H(+)-ATPase C, G and d subunits, and their evaluation in autosomal recessive distal renal tubular acidosis. Gene. 2002;297(1-2):169–177. | ||
Blomqvist SR, Vidarsson H, Fitzgerald S, et al. Distal renal tubular acidosis in mice that lack the forkhead transcription factor Foxi1. J Clin Invest. 2004;113(11):1560–1570. | ||
Biver S, Belge H, Bourgeois S, et al. A role for Rhesus factor Rhcg in renal ammonium excretion and male fertility. Nature. 2008;456(7220):339–343. | ||
Bourgeois S, Bounoure L, Christensen EI, et al. Haploinsufficiency of the ammonia transporter Rhcg predisposes to chronic acidosis: Rhcg is critical for apical and basolateral ammonia transport in the mouse collecting duct. J Biol Chem. 2013;288(8):5518–5529. | ||
Xu J, Song P, Nakamura S, et al. Deletion of the chloride transporter slc26a7 causes distal renal tubular acidosis and impairs gastric acid secretion. J Biol Chem. 2009;284(43):29470–29479. | ||
Sun X, Yang LV, Tiegs BC, et al. Deletion of the pH sensor GPR4 decreases renal acid excretion. J Am Soc Nephrol. 2010;21(10):1745–1755. | ||
Bourgeois S, Meer LV, Wootla B, et al. NHE4 is critical for the renal handling of ammonia in rodents. J Clin Invest. 2010;120(6):1895–1904. | ||
Wen D, Yuan Y, Cornelius RJ, et al. Deficient acid handling with distal RTA in the NBCe2 knockout mouse. Am J Physiol Renal Physiol. 2015;309(6):F523–F530. | ||
Trepiccione F, Gerber SD, Grahammer F, et al. Renal Atp6ap2/(Pro)renin receptor is required for normal vacuolar H+-ATPase function but not for the renin–angiotensin system. J Am Soc Nephrol. 2016;27(11):3320–3330. | ||
Merkulova M, Pa˘unescu TG, Nair AV, et al. Targeted deletion of the Ncoa7 gene results in incomplete distal renal tubular acidosis in mice. Am J Physiol Renal Physiol. 2018;315(1):F173–F185. | ||
Enerbäck S, Nilsson D, Edwards N, et al. Acidosis and deafness in patients with recessive mutations in FOXI1. J Am Soc Nephrol. 2018;29(3):1041–1048. | ||
Rungroj N, Nettuwakul C, Sawasdee N, et al. Distal renal tubular acidosis caused by tryptophan–aspartate repeat domain 72 (WDR72) mutations. Clin Genet. 2018;94(5):409–418. | ||
Vallés PG, Batlle D. Hypokalemic distal renal tubular acidosis. Adv Chronic Kidney Dis. 2018;25(4):303–320. | ||
Kitterer D, Schwab M, Alscher MD, Braun N, Latus J. Drug-induced acid–base disorders. Pediatr Nephrol. 2015;30(9):1407–1423. | ||
Simón H, Orive B, Zamora I, Mendizabal S. The acidification defect in the syndrome of renal tubular acidosis with nerve deafness. Acta Paediatr Scand. 1979;68(2):291–295. | ||
Rodriguez-Soriano J, Vallo A, Castillo G, Oliveros R. Natural history of primary distal renal tubular acidosis treated since infancy. J Pediatr. 1982;101(5):669–676. | ||
Igarashi T, Kawato H, Kamoshita S. Reversible low-molecular-weight proteinuria in patients with distal renal tubular acidosis. Pediatr Nephrol. 1990;4(6):593–596. | ||
Watanabe T. Proximal renal tubular dysfunction in primary distal renal tubular acidosis. Pediatr Nephrol. 2005;20(1):86–88. | ||
Tasic V, Korneti P, Gucev Z, Hoppe B, Blau N, Cheong HI. Atypical presentation of distal renal tubular acidosis in two siblings. Pediatr Nephrol. 2008;23(7):1177–1181. | ||
Besouw MTP, Bienias M, Walsh P, et al. Clinical and molecular aspects of distal renal tubular acidosis in children. Pediatr Nephrol. 2017;32(6):987–996. | ||
Watanabe T. Renal Fanconi syndrome in distal renal tubular acidosis. Pediatr Nephrol. 2017;32(6):1093. | ||
Satoh N, Suzuki M, Nakamura M, et al. Functional coupling of V-ATPase and CLC-5. World J Nephrol. 2017;6(1):14–20. | ||
Picollo A, Malvezzi M, Accardi A. Proton block of the CLC-5 Cl–/H+ exchanger. J Gen Physiol. 2010;135(6):653–659. | ||
Miller SG, Schwartz GJ. Hyperammonaemia with distal renal tubular acidosis. Arch Dis Child. 1997;77(5):441–444. | ||
Clericetti CM, Milani GP, Lava SAG, Bianchetti MG, Simonetti GD, Giannini O. Hyperammonemia associated with distal renal tubular acidosis or urinary tract infection: a systematic review. Pediatr Nephrol. 2018;33(3):485–491. | ||
Gökceog˘lu AU, Tas¸ar MA, Yalaki Z, Günes¸ A, Bakır A. An infant with hypercalcemia and hyperammonia: inborn error of metabolism or not? Questions. Pediatr Nephrol. 2018. | ||
Wrong O, Davies HE. The excretion of acid in renal disease. Q J Med. 1959;28(110):259–313. | ||
Fuster DG, Moe OW. Incomplete distal renal tubular acidosis and kidney stones. Adv Chronic Kidney Dis. 2018;25(4):366–374. | ||
Osther PJ, Bollerslev J, Hansen AB, Engel K, Kildeberg P. Pathophysiology of incomplete renal tubular acidosis in recurrent renal stone formers: evidence of disturbed calcium, bone and citrate metabolism. Urol Res. 1993;21(3):169–173. | ||
Arampatzis S, Röpke-Rieben B, Lippuner K, Hess B. Prevalence and densitometric characteristics of incomplete distal renal tubular acidosis in men with recurrent calcium nephrolithiasis. Urol Res. 2012;40(1):53–59. | ||
Sluysmans T, Vanoverschelde JP, Malvaux P. Growth failure associated with medullary sponge kidney, due to incomplete renal tubular acidosis type 1. Eur J Pediatr. 1987;146(1):78–80. | ||
Oduwole AO, Giwa OS, Arogundade RA. Relationship between rickets and incomplete distal renal tubular acidosis in children. Ital J Pediatr. 2010;36:54. | ||
Weger M, Deutschmann H, Weger W, Kotanko P, Skrabal F. Incomplete renal tubular acidosis in “primary” osteoporosis. Osteoporos Int. 1999;10(4):325–329. | ||
Weger W, Kotanko P, Weger M, Deutschmann H, Skrabal F. Prevalence and characterization of renal tubular acidosis in patients with osteopenia and osteoporosis and in non-porotic controls. Nephrol Dial Transplant. 2000;15(7):975–980. | ||
Sromicki JJ, Hess B. Abnormal distal renal tubular acidification in patients with low bone mass: prevalence and impact of alkali treatment. Urolithiasis. 2017;45(3):263–269. | ||
Pongchaiyakul C, Domrongkitchaiporn S, Stitchantrakul W, Chailurkit LO, Rajatanavin R. Incomplete renal tubular acidosis and bone mineral density: a population survey in an area of endemic renal tubular acidosis. Nephrol Dial Transplant. 2004;19(12):3029–3033. | ||
Zhang J, Fuster DG, Cameron MA, et al. Incomplete distal renal tubular acidosis from a heterozygous mutation of the V-ATPase B1 subunit. Am J Physiol Renal Physiol. 2014;307(9):F1063–F1071. | ||
Imai E, Kaneko S, Mori T, Okado T, Uchida S, Tsukamoto Y. A novel heterozygous mutation in the ATP6V0A4 gene encoding the V-ATPase a4 subunit in an adult patient with incomplete distal renal tubular acidosis. Clin Kidney J. 2016;9(3):424–428. | ||
Dhayat NA, Schaller A, Albano G, et al. The vacuolar H+-ATPase B1 subunit polymorphism p.E161K associates with impaired urinary acidification in recurrent stone formers. J Am Soc Nephrol. 2016;27(5):1544–1554. | ||
Bourgeois S, Bettoni C, Baron S, Wagner CA. Haploinsufficiency of the mouse Atp6v1b1 gene leads to a mild acid–base disturbance with implications for kidney stone disease. Cell Physiol Biochem. 2018;47(3):1095–1107. | ||
Alexander RT, Cordat E, Chambrey R, Dimke H, Eladari D. Acidosis and urinary calcium excretion: insights from genetic disorders. J Am Soc Nephrol. 2016;27(12):3511–3520. | ||
Zuckerman JM, Assimos DG. Hypocitraturia: pathophysiology and medical management. Rev Urol. 2009;11(3):134–144. | ||
Sakhaee K, Nicar M, Hill K, Pak CY. Contrasting effects of potassium citrate and sodium citrate therapies on urinary chemistries and crystallization of stone-forming salts. Kidney Int. 1983;24(3):348–352. | ||
Lemann J, Gray RW, Pleuss JA. Potassium bicarbonate, but not sodium bicarbonate, reduces urinary calcium excretion and improves calcium balance in healthy men. Kidney Int. 1989;35(2):688–695. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.