")
Back to Journals » International Journal of Nanomedicine » Volume 11
Improvement of intratumor microdistribution of PEGylated liposome via tumor priming by metronomic S-1 dosing
Authors Doi Y, Abu Lila AS , Matsumoto H, Okada T, Shimizu T, Ishida T
Received 5 August 2016
Accepted for publication 27 August 2016
Published 25 October 2016 Volume 2016:11 Pages 5573—5582
DOI https://doi.org/10.2147/IJN.S119069
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Yusuke Doi,1 Amr S Abu Lila,1–3 Haruna Matsumoto,1 Tomoko Okada,1 Taro Shimizu,1 Tatsuhiro Ishida1
1Department of Pharmacokinetics and Biopharmaceutics, Institute of Biomedical Sciences, Tokushima University, Tokushima, Japan; 2Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, Zagazig University, Zagazig, Egypt; 3Department of Pharmaceutics, Faculty of Pharmacy, Hail University, Hail, Saudi Arabia
Abstract: The efficient delivery of nanocarrier-based cancer therapeutics into tumor tissue is problematic. Structural abnormalities, tumor vasculature heterogeneity, and elevated intratumor pressure impose barriers against the preferential accumulation of nanocarrier-based cancer therapeutics within tumor tissues and, consequently, compromise their therapeutic efficacy. Recently, we have reported that metronomic S-1, orally available tegafur formulation, dosing synergistically augmented the therapeutic efficacy of oxaliplatin (l-OHP)-containing PEGylated liposome without increasing the toxicity in animal model. However, the exact mechanism behind such synergistic effect was not fully elucidated. In this study, therefore, we tried to shed the light on the contributions of metronomic S-1 dosing to the enhanced accumulation and/or spatial distribution of PEGylated liposome within tumor tissue. Tumor priming with metronomic S-1 treatment induced a potent apoptotic response against both angiogenic endothelial cells and tumor cells adjacent to tumor blood vessels, resulting in enhanced tumor blood flow via transient normalization of tumor vasculature, along with alleviation of intratumor pressure. Such a change in the tumor microenvironment imparted by S-1 treatment allows efficient delivery of PEGylated liposome to tumor tissue and permits their deep penetration/distribution into the tumor mass. Such a priming effect of S-1 dosing can be exploited as a promising strategy to enhance the therapeutic efficacy of nanocarrier-based cancer therapeutics suffering from inadequate/heterogeneous delivery to tumor tissues.
Keywords: tumor microenvironment, metronomic chemotherapy, S-1, liposome, nanocarrier, EPR effect
Introduction
Nanocarriers have been widely explored as promising vehicles for cancer therapy due to their higher loading efficiency, ability to protect the encapsulated payload, and ease of their surface functionalization.1–3 In addition, the propensity of nanocarriers to be selectively accumulated within tumor tissue, through the leaky tumor vasculature, via the enhanced permeability and retention (EPR) effect, maximized their applicability in many experimental and/or clinical settings.4 However, despite the fact that nanocarrier-based chemotherapeutics have shown efficacy in animal tumor models, the response rates vary among tumor types and within individual tumors.5,6 Several factors, including permeability of tumor vasculature, regional blood flow to the tumor, structural barriers inflicted by perivascular tumor cells, and/or extracellular matrix and tumor interstitial pressure, are reported to hinder the homogenous accumulation and/or the broad distribution of nanocarrier-based chemotherapeutics within the tumor tissue.7,8
Ideally, nanocarrier-based chemotherapeutics should be able to reach tumor site without systemic loss, extravasate through the tumor vasculature into tumor interstitium, penetrate all the way to reach the core of the tumor mass, release the entrapped payload, and completely eradicate the tumors. However, due to the aforementioned delivery barriers, the effect of an anticancer therapeutic is often limited to the periphery of the tumor mass close to the vasculature9,10 while more centralized regions of the tumor remain unaffected.11 This raises the potential for tumor relapse and/or metastasis.
Recently, various approaches have been dedicated to ensure the homogenous distribution and/or deep penetration of nanocarriers within the tumor tissue, reaching the tumor core. These approaches include the co-administration of certain agents, such as vascular endothelial growth factor (VEGF) inhibitors,12 angiotensin-II,13 tumor necrosis factor α (TNF-α),14 tumor matrix modifying agents,15 and low dose transforming growth factor-beta type I receptor inhibitor,16 in order to induce alteration in cancer-associated neovasculature or interstitium.
Tumor “priming” with a low dose of chemotherapeutic agent has been reported as an alternative strategy to augment the intratumor delivery/penetration of subsequently administered nanocarrier-based chemotherapeutics. Lu et al17 investigated the effect of tumor priming by a low dose of paclitaxel on the delivery and efficacy of subsequently administered nanomedicines. They confirmed that paclitaxel accumulation and tumor apoptosis were significantly higher when the tumors were pretreated “primed” with a small dose of paclitaxel. In the same context, tumor priming with cyclophosphamide promoted the increased accumulation and subsequent wide distribution of doxorubicin-containing PEGylated liposomes in the solid tumor. This effect was attributed to the transient increase in the density of CD31+-microvessels, which show high permeability for PEGylated liposomes.18 Interestingly, we have recently reported that priming the tumor tissue with a single injection of l-OHP-containing PEGylated liposomes enhanced the intratumor accumulation/distribution of subsequently injected doses.19,20
S-1, an oral 5-fluorouracil (5-FU)-based cytotoxic agent-containing tegafur (a prodrug of 5-FU),21 has been approved for clinical use in Japan and Europe. Recently, we demonstrated that metronomic S-1 dosing increased tumor accumulation of l-OHP-containing PEGylated liposomes and, consequently, promoted a synergistic therapeutic effect of S-1 and l-OHP-containing PEGylated liposomes without exerting severe toxicity.22 However, the underlying mechanism behind the enhanced tumor accumulation and broader intratumor distribution of PEGylated liposomes by tumor priming with S-1 dosing has not been fully elucidated. In the present study, therefore, we tried to reveal the effect of tumor priming with S-1 (metronomic S-1 dosing) on tumor microenvironment (tumor vasculature and perfusion) and spatial tumor distribution of PEGylated liposomes.
Materials and methods
Materials
Hydrogenated soy phosphatidylcholine (HSPC) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-n-[methoxy(polyethylene glycol)-2000] (mPEG2000-DSPE) were generously donated by NOF (Tokyo, Japan). Cholesterol (CHOL) was purchased from Wako Pure Chemical (Osaka, Japan). S-1, an oral fluorouracil antitumor drug that contains three pharmacological agents (tegafur, 5-chloro-2,4-dihydroxypyridine, and potassium oxonate) was generously donated by Taiho Pharmaceutical (Tokyo, Japan). 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) and 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine perchlorate (DiD) were purchased from Invitrogen (Paisley, UK). Fluorescein isothiocyanate (FITC)-labeled rabbit anti-rat CD31 IgG heavy and light chain polyclonal antibody was purchased from Abcam (Cambridge, UK). Hoechst 33342 and 3,3′- diheptyloxacarbocyanine iodide (DiOC7) were purchased from AnaSpec (Freemont, CA, USA). FITC-dextran (M.W. 150,000) and Drabkin’s reagent was purchased from Sigma-Aldrich (St Louis, MO, USA). In Situ Cell Death Detection Kit (TMR red) was purchased from Roche Diagnostics (Indianapolis, IN, USA). All other reagents were of analytical grade.
Animals and tumor cells
Five-week-old male BALB/c mice and BALB/c nu/nu mice were purchased from Japan SLC (Shizuoka, Japan). The experimental animals were allowed free access to water and mouse chow and were housed under controlled environmental conditions (constant temperature, humidity, and 12-hour dark–light cycle). All animal experiments were evaluated and approved by the Animal and Ethics Review Committee of the Tokushima University and conducted in accordance with the guidelines of animal welfare and rights in Japan.
Colon 26 (C26) murine colorectal carcinoma cell line was purchased from Cell Resource Center for Biomedical Research (Institute of Development, Aging and Cancer, Tohoku University, Sendai, Japan). A human colon carcinoma cell line, DLD-1 and DLD-1/FU (5-FU resistance), was kindly provided by Taiho Pharmaceutical.23 Each cell line was maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) (Nissui Pharmaceutical, Tokyo, Japan) and RPMI-1640 medium (Wako Pure Chemical) supplemented with 10% heat-inactivated fetal bovine serum (Japan Bioserum, Hiroshima, Japan), 100 units/mL penicillin, and 100 μg/mL streptomycin (ICN Biomedical, Aurora, OH, USA) in a 5% CO2/air incubator at 37°C, respectively.
Preparation of PEGylated liposomes
PEGylated liposomes were composed of HSPC/CHOL/mPEG2000-DSPE (2/1/0.2 molar ratio). Liposomes were prepared using the thin-film hydration technique.24 Briefly, the lipids were dissolved in chloroform. To follow accumulation and distribution in the solid tumor, 1 mol% of hydrophobic fluorescent dye (DiI or DiD) was incorporated into the lipid mixture. After evaporation of the organic solvent, the resulting lipid film was hydrated with 5% dextrose. The liposomes were sized by subsequent extrusion through polycarbonate membrane filters (Nuclepore, Pleasanton, CA, USA) with pore sizes of 400, 200, and 100 nm. The mean diameter of the liposomes was ~100 nm, as determined using a NICOMP 370 HPL submicron particle analyzer (Particle Sizing System, San Diego, CA, USA). The phospholipid concentration was determined by a colorimetric assay.25
Tumor model and S-1 treatment
BALB/c mice were inoculated subcutaneously in the back with 2×106 C26 cells suspended in 200 μL of complete DMEM medium. BALB/c nu/nu mice were inoculated subcutaneously in the back with 2×106 DLD-1 or DLD-1/FU cells suspended in 200 μL of complete RPMI 1640 medium.
Treatments were begun when the tumor volumes reached 40 to 60 mm3. S-1 was dissolved in distilled water at a concentration of 0.69 mg tegafur/mL and administered orally at the sub-maximum tolerated dose26 of 6.9 mg tegafur/kg/day for 7 days.
Tumor accumulation and distribution study of PEGylated liposome
In order to assess the effect of tumor priming with metronomic S-1 dosing on tumor accumulation and distribution of PEGylated liposomes, fluorescence (DiI or DiD)-labeled PEGylated liposomes (25 mg phospholipids/kg) were intravenously injected to the tumor-bearing mice on the day of final S-1 administration (Day 7).
For in vivo imaging study, mice received intravenous administration of DiD-labeled PEGylated liposomes (25 mg phospholipids/kg) and images of the mice were recorded at selected time points with a Fluorescence Image Analyzer LAS-4000 IR (Fujifilm, Tokyo, Japan). The fluorescence images were acquired with a 1/100 second exposure time.
For liposome-intratumoral distribution study, DiI-labeled PEGylated liposomes (25 mg phospholipids/kg) were intravenously injected into S-1 treated tumor-bearing mice. At 24 hours after injection, mice were euthanized. For fluorescence angiography, 5 minutes before scarification, FITC-dextran (5 mg/mouse) was intravenously administered into the mice. The tumors were harvested and snap-frozen in an optimal cutting temperature (OCT) compound (Sakura Fintechnical, Tokyo, Japan) by dry-iced acetone. Frozen samples were cut into sections of 5 μm thickness in cryostat (Leica Microsystems, Solms, Germany), mounted on a glass slide, and dried in air. Samples were directly observed by using a fluorescence microscopy (BZ-9000, Keyence, Osaka, Japan) to evaluate the intratumoral distribution of PEGylated liposomes. Three tumors per each group were subjected. At least ten randomly selected sections per tumor and 30 images were analyzed using analyze software (BZ analyzer, Keyence).
Immunostaining
Immunohistochemical staining for endothelial cells and apoptotic cells was done using anti-CD31 Ab and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining, respectively. Tumor of mice, which is treated with saline or S-1 (6.9 mg tegafur/kg/day) for 7 days, was harvested at 24 hours after final S-1 treatment. The removed tumors were snap-frozen in an OCT compound by dry-iced acetone. Frozen samples were sectioned at 5 μm thickness in a cryostat, mounted on a glass slide, and dried by air. The samples were sequentially fixed by incubation in 4% PFA for 15 minutes, washed with phosphate-buffered saline (PBS) (51 mM Na2HPO4, 12 mM NaH2PO4, 77 mM NaCl, pH 7.4), and blocked with 5% bovine serum albumin (BSA) in PBS for 30 minutes at room temperature. After washing with PBS, samples were incubated overnight with FITC-labeled rat anti-mouse CD31 (1:500) at 4°C. TUNEL staining was operated in accordance with recommended procedure. For nucleus staining, the samples were incubated with Hoechst 33342 (1 μg/mL) for 5 minutes at room temperature and washed with PBS. Then, samples were directly observed under a fluorescent microscopy BZ-9000.
Evaluation of tumor blood perfusion
To study the effect of S-1 dosing on tumor blood perfusion, the double-fluorescent dye technique27 based on the perfusion markers, Hoechst 33342 and DiOC7, was carried out. The different fluorescence excitation and emission properties of the two dyes allow for detection of temporal and spatial fluctuations in tumor perfusion. Briefly, the fluorescent dyes were intravenously administered into the C26 tumor-bearing mice treated with saline or S-1 (6.9 mg tegafur/kg/day) for 7 days with a 20-minute interval (1st: Hoechst 33342 (15 mg/kg), 2nd: DiOC7 (1 mg/kg)). The tumors were harvested at 5 minutes after the DiOC7 injection. Cryosections (10 μm thick) of the tumor were observed using laser scanning confocal microscope (LSM 510, Zeiss, Oberkochen, Germany).
To quantify the tumor blood flow, the amount of hemoglobin (Hb) and injected FITC-dextran in the tumor tissues was determined. FITC-dextran (5 mg/mouse) was intravenously administered into the C26 tumor-bearing mice, which is treated with saline or S-1 (6.9 mg tegafur/kg/day) for 7 days. At 5 minutes after injection, mice were euthanized and the tumor was removed. The tumor was homogenized on ice, and then the homogenates were centrifuged at 14,000× g for 15 minutes at 4°C. The supernatant was subjected to measure. Hb derived from erythrocytes in the sample was determined as an index of total blood volume in the tumor according to the method previously reported.28 The absorbance of samples at 540 nm was measured by a spectrophotometer. Fluorescence intensity of FITC of FITC-labeled dextran in the sample was determined as a functional vessel index in the tumor using a fluorescent spectrophotometer (Hitachi, Tokyo, Japan) at ex/em =495/520 nm. Relative perfusion index was calculated according to a formula as follows:
|
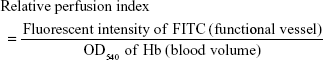
Statistics
All values are expressed as the mean ± standard deviation. Statistical analysis was performed with a two-tailed unpaired t-test using GraphPad InStat software (GraphPad Software, La Jolla, CA, USA). The level of significance was set at P<0.05.
Results
Effect of tumor priming with S-1 dosing on tumor accumulation and intratumor distribution of PEGylated liposome
DiI-labeled test PEGylated liposomes were injected intravenously into the lateral tail vein of S-1 treated mice to assess intratumor distribution of PEGylated liposome (Figure 1). Significant accumulation and wider distribution of the liposome were observed in the tumor sections (Figure 1A). In nontreated control tumor, the distribution pattern was entirely heterogeneous and the liposomes were mainly clustered in the edge of tumor section. On the other hand, in the tumor treated by S-1, the distribution pattern was still heterogeneous, but it became much wider and more uniform through the tumor tissue. The number of red spots does not necessarily reflect the amount of accumulated test PEGylated liposomes, but it reflects their accumulation region and area in the tumors. The number of red spots, relating to test PEGylated liposome, was relatively increased in the S-1 treated tumor sections (Figure 1B). This indicates that tumor priming with S-1 improves intratumor distribution of PEGylated liposome, which is consistent to our previous observation.22
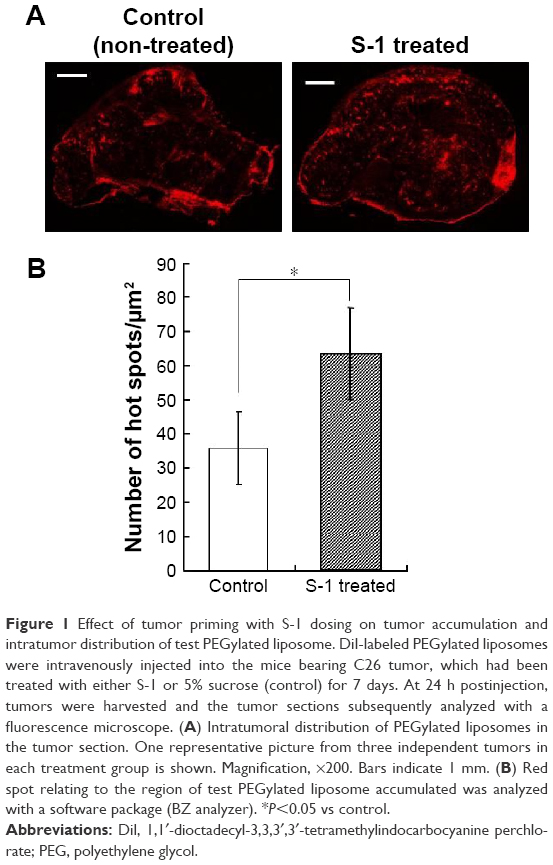 | Figure 1 Effect of tumor priming with S-1 dosing on tumor accumulation and intratumor distribution of test PEGylated liposome. DiI-labeled PEGylated liposomes were intravenously injected into the mice bearing C26 tumor, which had been treated with either S-1 or 5% sucrose (control) for 7 days. At 24 h postinjection, tumors were harvested and the tumor sections subsequently analyzed with a fluorescence microscope. (A) Intratumoral distribution of PEGylated liposomes in the tumor section. One representative picture from three independent tumors in each treatment group is shown. Magnification, ×200. Bars indicate 1 mm. (B) Red spot relating to the region of test PEGylated liposome accumulated was analyzed with a software package (BZ analyzer). *P<0.05 vs control. |
Induction of apoptosis on tumor cells by S-1 treatment
In order to clarify the mechanism of enhanced tumor accumulation of PEGylated liposome observed in Figure 1, the effect of S-1 priming on cellular apoptosis in the tumor was investigated using TUNEL staining (Figure 2A). In the control tumor, apoptotic cells were hardly detected. To the contrary, in the tumor sections treated with S-1, apoptosis were highly induced. Interestingly, S-1 treatment promoted apoptosis on not only endothelial cells (CD31 positive) but also tumor cells close to CD31 positive endothelial cells. This finding indicates that the frequent treatment with cytotoxic agent induces apoptosis against endothelial cells as well as tumor cells surrounding blood vessels.
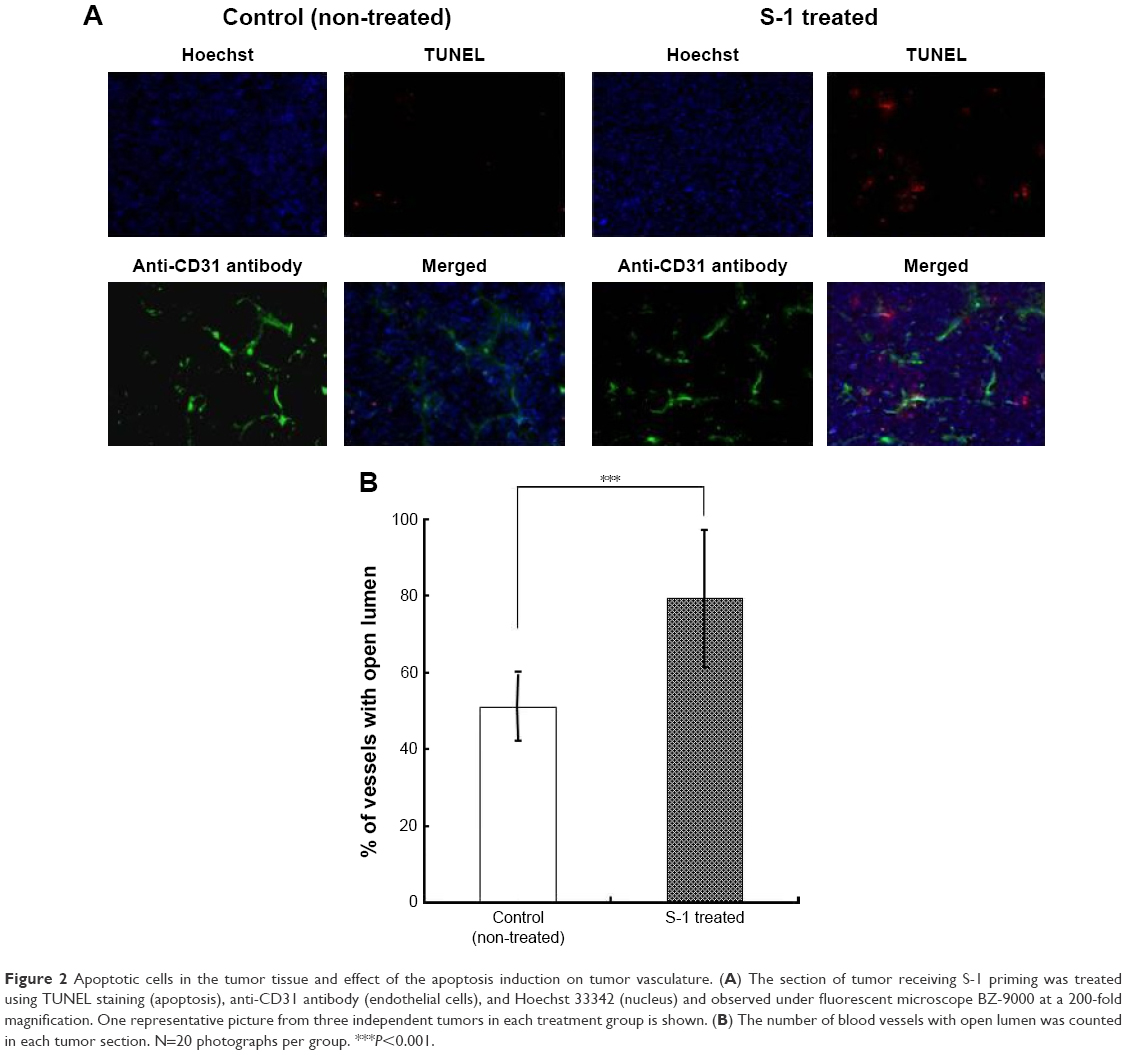 | Figure 2 Apoptotic cells in the tumor tissue and effect of the apoptosis induction on tumor vasculature. (A) The section of tumor receiving S-1 priming was treated using TUNEL staining (apoptosis), anti-CD31 antibody (endothelial cells), and Hoechst 33342 (nucleus) and observed under fluorescent microscope BZ-9000 at a 200-fold magnification. One representative picture from three independent tumors in each treatment group is shown. (B) The number of blood vessels with open lumen was counted in each tumor section. N=20 photographs per group. ***P<0.001. |
Next, the effect of apoptotic induction on tumor vasculature was investigated by counting the number of vessels with open lumen based on a previously reported method.29 In the S-1 treated tumor sections, a larger number of blood vessels with open lumen existed compared to those in control tumor sections (Figure 2B). It is known that tumor cells surrounding blood vessels are highly proliferative. Such tumor cells generate compressive mechanical force and then cause compression of tumor blood vessels. Apoptosis on the vessel-surrounding tumor cells caused by S-1 might relieve the mechanical force and consequently increase the number of blood vessels with open lumen.
Improved tumor blood flow by S-1 treatment
We then investigated the effect of S-1 priming on fluctuations in blood flow within tumor tissue using the double-fluorescent dye method described above. In this method, the dyes were administered separately with some interval into the same animal. Under this condition, it is assumed that the vessels with intermittent blood flow contain either dye, Hoechst 33342 or DiOC7, and the vessels with continuous blood flow contain both dyes. As shown in Figure 3A, in the control tumor, less dye distribution was observed and the regions stained by each dye were not merged. This indicates that blood flow in the control tumor has spatial and temporal heterogeneity. To the contrary, in the tumors treated with S-1, uniform merged distribution of both dyes was observed. It appears that the S-1 priming promotes homogeneous blood flow in the tumor.
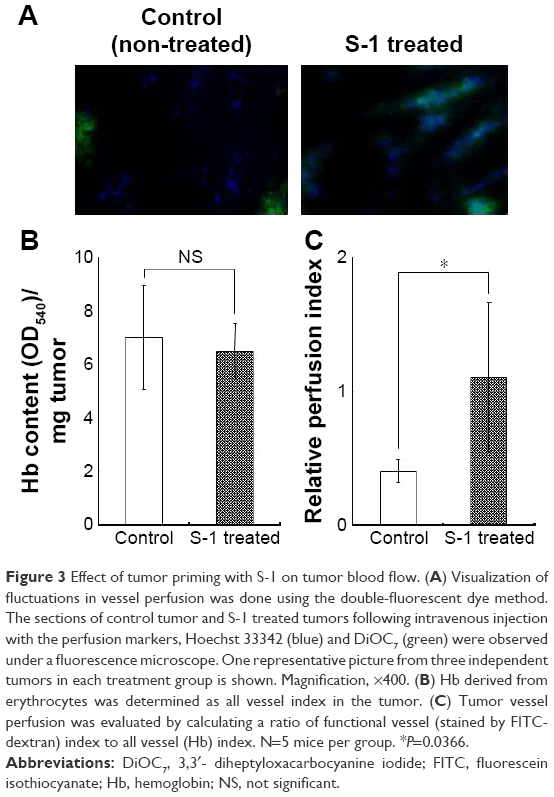 | Figure 3 Effect of tumor priming with S-1 on tumor blood flow. (A) Visualization of fluctuations in vessel perfusion was done using the double-fluorescent dye method. The sections of control tumor and S-1 treated tumors following intravenous injection with the perfusion markers, Hoechst 33342 (blue) and DiOC7 (green) were observed under a fluorescence microscope. One representative picture from three independent tumors in each treatment group is shown. Magnification, ×400. (B) Hb derived from erythrocytes was determined as all vessel index in the tumor. (C) Tumor vessel perfusion was evaluated by calculating a ratio of functional vessel (stained by FITC-dextran) index to all vessel (Hb) index. N=5 mice per group. *P=0.0366. |
Then, to evaluate the total blood volume in tumor and the flow continuity, the amount of Hb and relative perfusion index in tumor were evaluated. Hb-derived optical density (OD) per tumor weight represents tumor blood volume and the normalized value (relative perfusion index) of the injected FITC-dextran-based fluorescence by the blood volume reflects the perfused tumor blood volume. From the results of Hb quantification, control and S-1 treated tumor have the same amount of Hb, indicating that both tumors were equally vascularized (Figure 3B). On the other hand, relative perfusion index, which shows the ratio of perfused blood vessels in tumor, was significantly higher in S-1 treated tumor than control tumor (Figure 3C). These indicate that S-1 treatment does not affect total blood volume, but affects the ratio of functional (perfused) blood vessels in tumor.
Effect of S-1 priming on intratumoral microdiffusion of PEGylated liposome
Microdiffusion of PEGylated liposome in the tumor tissue was studied histologically at a higher magnification. Fluorescent angiography with FITC-dextran showed that DiI-labeled PEGylated liposomes were observed at outside of vessel contrasted by FITC-dextran in both S-1 treated and control tumors (Figure 4A). This obviously indicates that PEGylated liposomes extravasate from blood vessels due to the EPR effect. In the tumor treated with S-1, the PEGylated liposome diffusion area was enlarged (Figure 4A). In addition, the PEGylated liposome diffused much far from blood vessel (Figure 4B). In the control tumor, the PEGylated liposomes were accumulated in the region close to the vessels (Figure 4B). These indicate that S-1 priming results in enhancement of liposome diffusion from blood vessel to tumor interstitium.
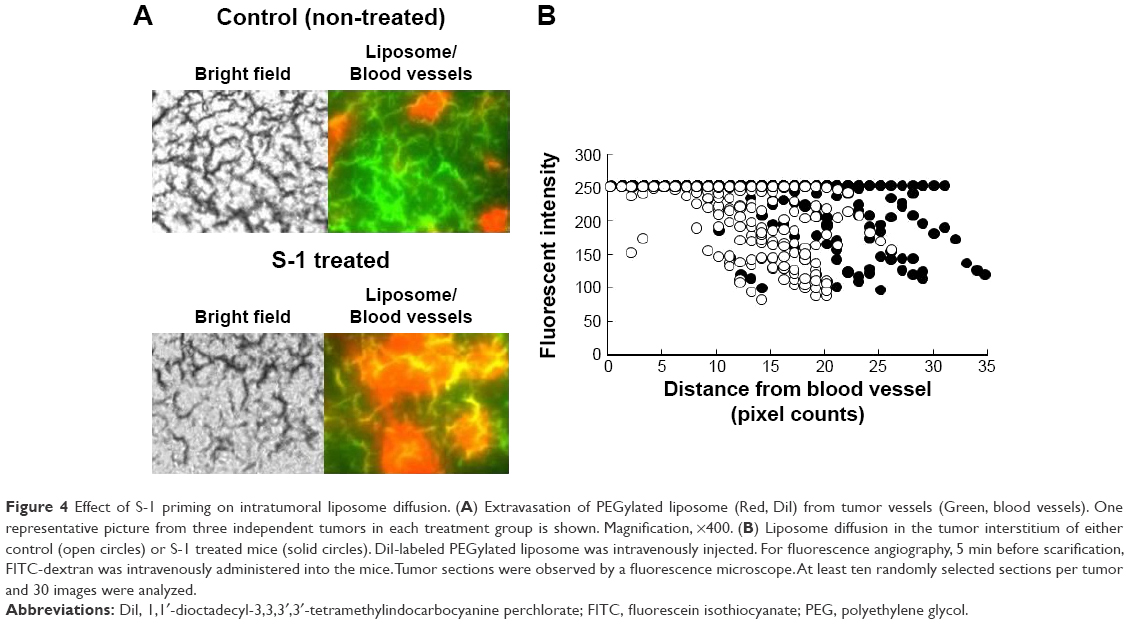 | Figure 4 Effect of S-1 priming on intratumoral liposome diffusion. (A) Extravasation of PEGylated liposome (Red, DiI) from tumor vessels (Green, blood vessels). One representative picture from three independent tumors in each treatment group is shown. Magnification, ×400. (B) Liposome diffusion in the tumor interstitium of either control (open circles) or S-1 treated mice (solid circles). DiI-labeled PEGylated liposome was intravenously injected. For fluorescence angiography, 5 min before scarification, FITC-dextran was intravenously administered into the mice. Tumor sections were observed by a fluorescence microscope. At least ten randomly selected sections per tumor and 30 images were analyzed. |
Effect of tumor priming with S-1 on accumulation of PEGylated liposome in 5-FU-resistant tumor
As described above, it is assumed that S-1 priming-induced apoptosis of tumor cells adjacent to blood vessels is one of the major elements to gain deeper penetration and diffusion of PEGylated liposome in tumor tissue (Figures 2 and 4). To obtain strong evidence, the accumulation of PEGylated liposome in 5-FU-resistant tumor (DLD-1/FU) was studied (Figure 5). In 5-FU sensitive DLD-1 tumor, S-1 priming increased tumor accumulation of PEGylated liposome, compared to non-S-1 treated (control) as expected (Figure 5A and C). The accumulation of PEGylated liposome reached to the maximum level 72 hours following intravenous injection and then gradually decreased. Even at 120 hours, sufficient PEGylated liposomes were still retained in the tumor. In the 5-FU-resistant tumor (DLD-1/FU), although tumor accumulation of PEGylated liposome was detected in the same manner as in the 5-FU sensitive DLD-1 tumor, S-1 priming did not enhance the tumor accumulation of PEGylated liposome (Figure 5B and C).
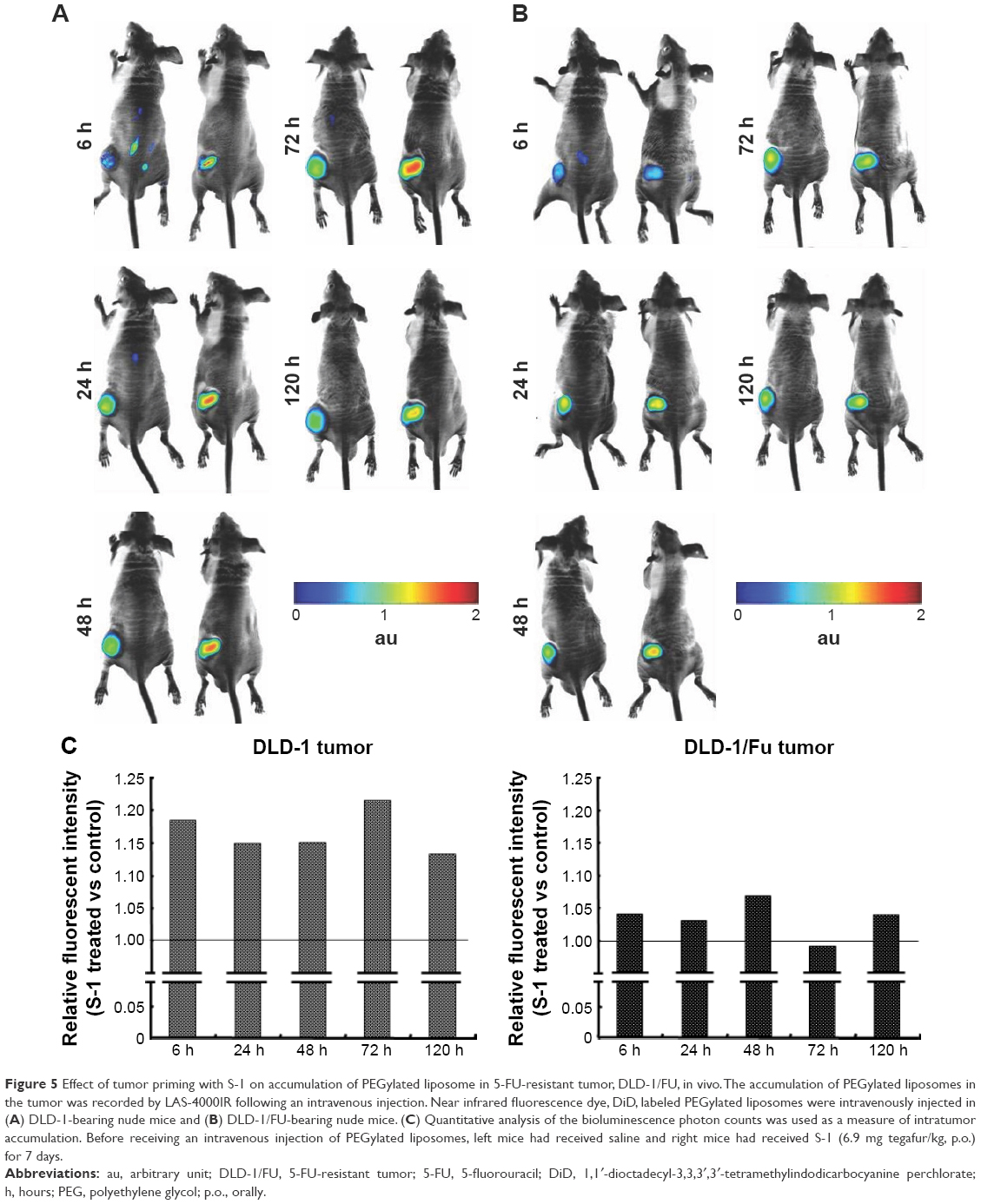 | Figure 5 Effect of tumor priming with S-1 on accumulation of PEGylated liposome in 5-FU-resistant tumor, DLD-1/FU, in vivo. The accumulation of PEGylated liposomes in the tumor was recorded by LAS-4000IR following an intravenous injection. Near infrared fluorescence dye, DiD, labeled PEGylated liposomes were intravenously injected in (A) DLD-1-bearing nude mice and (B) DLD-1/FU-bearing nude mice. (C) Quantitative analysis of the bioluminescence photon counts was used as a measure of intratumor accumulation. Before receiving an intravenous injection of PEGylated liposomes, left mice had received saline and right mice had received S-1 (6.9 mg tegafur/kg, p.o.) for 7 days. |
Discussion
Nanocarrier-based drug delivery is emerging as a powerful tool in several diseases, including cancer. Its long circulation characteristics, imparted by surface decoration, along with the ability to extravasate to tumor sites, via the EPR effect, significantly improved the safety and tolerability of nanocarrier-based chemotherapeutics.4,30 However, despite these improvements, the overall therapeutic efficacy of such nanocarriers-based systems remains modest; currently, these platforms offer only marginal improvements over conventional formulations.31,32 Several biological barriers, relating mainly to tumor microenvironment, including poor perfusion of inner regions of solid tumors, malformed vasculature, increased interstitial fluid pressure, and/or dense extracellular matrix, are found to impair drug delivery and impede uniform distribution of nanocarrier-based chemotherapeutics throughout tumor tissue. Accordingly, a vast amount of research has been directed toward the development of new approaches to overcome such tumor-related biological barriers and, thereby, enhance tumor accumulation and distribution of nanocarrier-based chemotherapeutics. In the present study, therefore, we emphasized the contribution of metronomic S-1 dosing on modulating the physiological state of tumors. Our results emphasized that tumor priming with S-1 dosing could induce superior accumulation and uniform microdistribution of PEGylated liposomes in tumor site via modulating tumor blood flow, vascular leakiness, and extracellular matrix penetration prior to administration of nanocarriers.
Metronomic chemotherapy, continuous administration of lower doses, relative to maximum tolerated dose, without drug-free breaks over extended periods, is currently well acknowledged for its potent anti-angiogenic effect and indirect role in inhibiting tumor growth.33 In addition, various studies have confirmed the potential of metronomic chemotherapy to directly affect tumor cells themselves.34 Accordingly, the cytotoxic effect imparted by metronomic S-1 dosing on both tumor endothelial cells of angiogenic vessels and tumor cells immediately adjacent to tumor vasculature (Figure 2) is anticipated to enhance the tumor accumulation and broad distribution of PEGylated liposomes via not only increasing the intratumor space available for PEGylated liposome accumulation but also by alleviating the mechanical compression exerted on tumor blood vessels by proliferating tumor cells and, consequently, improve tumor perfusion (Figure 3) and enhance the extravasation of PEGylated liposomes from tumor blood vessels resulting in deeper diffusion, broader distribution (Figure 4), and enhanced accumulation within the tumor tissue (Figure 5A).
Another possibility of the enhanced tumor accumulation/distribution of PEGylated liposomes is the indirect effect of metronomic S-1 dosing on tumor vascular normalization. Jain has reported the efficacy of metronomic chemotherapy to induce a more structurally and functionally normal vasculature which, consequently, results in a more homogenous blood flow, improved perfusion, and more importantly efficient delivery and uniform distribution of systemically administered nanocarrier within tumor tissues.35 Our results (Figures 2B and 3) confirmed the potential of metronomic S-1 dosing to improve tumor perfusion and enhance the extravasation of PEGylated liposomes from tumor blood vessels allowing deeper diffusion and broader distribution within the tumor interstitium (Figures 4 and 5A).
It is worth noting that metronomic S-1 dosing could only enhance the tumor accumulation of PEGylated liposomes in 5-FU sensitive DLD-1, rather than 5-FU-resistant DLD-1/FU xenograft animal model (Figure 5). We assumed that, in 5-FU sensitive DLD-1 xenograft animal model, the cytotoxic effect of 5-FU, derived from S-1 treatment, on both tumor endothelial cells and tumor cells nearby blood vessels, as shown in Figure 2, could trigger the preferential accumulation of PEGylated liposomes within tumor tissues. On the other hand, in 5-FU-resistant DLD-1/FU xenograft animal model, 5-FU could exert its cytotoxic effect on only tumor endothelial cells, not on 5-FU-resistant tumor cells, and thus S-1 dosing failed, in part, to relieve the mechanical compression exerted on tumor blood vessels by proliferating tumor cells and consequently compromised tumor perfusion. As a result, S-1 priming failed to enhance the tumor accumulation of PEGylated liposomes. However, further studies are required to confirm our assumption.
Many therapeutic strategies have been adopted to modulate the tumor microenvironment and, thereby, enhance the accumulation/distribution of nanocarriers within tumor tissues. These strategies comprise the application of: 1) vascular permeability mediators, such as TNF-α,14 prostaglandin analogies,36 interleukin 2,37 VEGF,38 and nitric oxide donors,39 to enhance tumor vascular permeability via inducing vasodilatation and increased blood flow; 2) matrix modifying agents, such as matrix-degrading enzymes,40 to reduce extracellular matrix density, thus resulting in enhanced permeability of tumor interstitium, mitigation of mechanical stress, and therefore improvement of tumor perfusion; or 3) normalization of tumor vasculature, via using anti-VEGF or VEGF receptor antibodies,35,41 to make tumor vessels less leaky and thereby reduce interstitial fluid pressure, resulting in improved perfusion and penetration of nanocarriers. In the present study, tumor priming with repeated S-1 treatment was confirmed to modulate the tumor microenvironment, via more than one strategy, and thus promotes superior accumulation and broad distribution of PEGylated liposomes within tumor tissue.
Recently, we emphasized the synergistic potential of a combined therapy of metronomic S-1 dosing and l-OHP-containing PEGylated liposomes. Such a synergistic response was skewed mainly toward the potent cytotoxic priming effect of sequentially administered l-OHP-containing PEGylated liposomes, rather than the daily administered S-1 dosing.19 In the present study, however, we clarified the contribution(s) of S-1 dosing to the synergistic antitumor efficacy of the combined treatment we previously reported. Based on our findings, we postulate that tumor priming with small doses of free chemotherapeutic agent, rather than nanocarriers-based chemotherapeutics, which are likely to face greater difficulties in terms of penetrating tumors than free small-molecular weight drugs, might represent a promising strategy to overcome the biological barriers imparted by tumor microenvironment and hence permit the efficient intratumor accumulation and uniform distribution of the co-administered nanocarriers-based chemotherapeutics.
Conclusion
We showed that tumor priming with S-1 treatment results in wider and more homogeneous distribution of PEGylated liposome in the tumor tissue. Repeated S-1 dosing induced apoptosis in the tumor cells surrounding tumor blood vessel and consequently increased the functional blood vessels, expanded the interstitial space, and finally promoted the enhanced intratumor accumulation and interstitial distribution of PEGylated liposome. Tumor priming with S-1 treatment represents a potentially viable mean to promote tumor-efficient delivery of antitumor agent-containing PEGylated liposome. The current study may have a significant impact on enhancing the delivery and efficacy of nanocarrier-based chemotherapeutics.
Acknowledgments
The Authors thank Mr JL McDonald for his helpful advice in preparing this manuscript. This study was supported, in part, by Grant-in-Aid for Scientific Research (B) (15H04639), the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Disclosure
The authors report no conflicts of interest in this work.
References
Ng KK, Lovell JF, Zheng G. Lipoprotein-inspired nanoparticles for cancer theranostics. Acc Chem Res. 2011;44(10):1105–1113. | ||
Patel S, Bhirde AA, Rusling JF, Chen X, Gutkind JS, Patel V. Nano delivers big: designing molecular missiles for cancer therapeutics. Pharmaceutics. 2011;3(1):34–52. | ||
Perche F, Torchilin VP. Recent trends in multifunctional liposomal nanocarriers for enhanced tumor targeting. J Drug Deliv. 2013;2013:705265. | ||
Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65(1–2):271–284. | ||
Van Elmpt W, Zegers CM, Reymen B, et al. Multiparametric imaging of patient and tumour heterogeneity in non-small-cell lung cancer: quantification of tumour hypoxia, metabolism and perfusion. Eur J Nucl Med Mol Imaging. 2016;43(2):240–248. | ||
Fokas E, McKenna WG, Muschel RJ. The impact of tumor microenvironment on cancer treatment and its modulation by direct and indirect antivascular strategies. Cancer Metastasis Rev. 2012;31(3–4):823–842. | ||
Lammers T, Kiessling F, Hennink WE, Storm G. Drug targeting to tumors: principles, pitfalls and (pre-) clinical progress. J Control Release. 2012;161(2):175–187. | ||
Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7(11):653–664. | ||
Kuh HJ, Jang SH, Wientjes MG, Weaver JR, Au JL. Determinants of paclitaxel penetration and accumulation in human solid tumor. J Pharmacol Exp Ther. 1999;290(2):871–880. | ||
Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer. 2006;6(8):583–592. | ||
Lankelma J, Dekker H, Luque FR, et al. Doxorubicin gradients in human breast cancer. Clin Cancer Res. 1999;5(7):1703–1707. | ||
Tong RT, Boucher Y, Kozin SV, Winkler F, Hicklin DJ, Jain RK. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. 2004;64(11):3731–3736. | ||
Iyer AK, Khaled G, Fang J, Maeda H. Exploiting the enhanced permeability and retention effect for tumor targeting. Drug Discov Today. 2006;11(17–18):812–818. | ||
Seki T, Carroll F, Illingworth S, et al. Tumour necrosis factor-alpha increases extravasation of virus particles into tumour tissue by activating the Rho A/Rho kinase pathway. J Control Release. 2011;156(3):381–389. | ||
Eikenes L, Tari M, Tufto I, Bruland OS, de Lange Davies C. Hyaluronidase induces a transcapillary pressure gradient and improves the distribution and uptake of liposomal doxorubicin (Caelyx) in human osteosarcoma xenografts. Br J Cancer. 2005;93(1):81–88. | ||
Kano MR, Bae Y, Iwata C, et al. Improvement of cancer-targeting therapy, using nanocarriers for intractable solid tumors by inhibition of TGF-beta signaling. Proc Natl Acad Sci U S A. 2007;104(9):3460–3465. | ||
Lu D, Wientjes MG, Lu Z, Au JL. Tumor priming enhances delivery and efficacy of nanomedicines. J Pharmacol Exp Ther. 2007;322(1):80–88. | ||
Ishida T, Shiraga E, Kiwada H. Synergistic antitumor activity of metronomic dosing of cyclophosphamide in combination with doxorubicin-containing PEGylated liposomes in a murine solid tumor model. J Control Release. 2009;134(3):194–200. | ||
Nakamura H, Doi Y, Abu Lila AS, Nagao A, Ishida T, Kiwada H. Sequential treatment of oxaliplatin-containing PEGylated liposome together with S-1 improves intratumor distribution of subsequent doses of oxaliplatin-containing PEGylated liposome. Eur J Pharm Biopharm. 2014;87(1):142–151. | ||
Abu Lila AS, Doi Y, Nakamura K, Ishida T, Kiwada H. Sequential administration with oxaliplatin-containing PEG-coated cationic liposomes promotes a significant delivery of subsequent dose into murine solid tumor. J Control Release. 2010;142(2):167–173. | ||
Sakata Y, Ohtsu A, Horikoshi N, Sugimachi K, Mitachi Y, Taguchi T. Late phase II study of novel oral fluoropyrimidine anticancer drug S-1 (1 M tegafur-0.4 M gimestat-1 M otastat potassium) in advanced gastric cancer patients. Eur J Cancer. 1998;34(11):1715–1720. | ||
Doi Y, Okada T, Matsumoto H, et al. Combination therapy of metronomic S-1 dosing with oxaliplatin-containing polyethylene glycol-coated liposome improves antitumor activity in a murine colorectal tumor model. Cancer Sci. 2010;101(11):2470–2475. | ||
Nakata E, Fukushima M, Takai Y, et al. S-1, an oral fluoropyrimidine, enhances radiation response of DLD-1/FU human colon cancer xenografts resistant to 5-FU. Oncol Rep. 2006;16(3):465–471. | ||
Ishida T, Maeda R, Ichihara M, et al. Accelerated clearance of PEGylated liposomes in rats after repeated injections. J Control Release. 2003;88(1):35–42. | ||
Bartlett GR. Colorimetric assay methods for free and phosphorylated glyceric acids. J Biol Chem. 1959;234(3):469–471. | ||
Ooyama A, Oka T, Zhao HY, Yamamoto M, Akiyama S, Fukushima M. Anti-angiogenic effect of 5-Fluorouracil-based drugs against human colon cancer xenografts. Cancer Lett. 2008;267(1):26–36. | ||
Bhattacharya A, Seshadri M, Oven SD, Tóth K, Vaughan MM, Rustum YM. Tumor vascular maturation and improved drug delivery induced by methylselenocysteine leads to therapeutic synergy with anticancer drugs. Clin Cancer Res. 2008;14(12):3926–3932. | ||
Balasubramaniam P, Malathi A, Viswanathan C. A simple technique for hemoglobin estimation to screen for anaemia. Indian J Physiol Pharmacol. 1992;36(3):213–214. | ||
Padera TP, Stoll BR, Tooredman JB, Capen D, di Tomaso E, Jain RK. Pathology: cancer cells compress intratumour vessels. Nature. 2004; 427(6976):695. | ||
Safra T, Muggia F, Jeffers S, et al. PEGylated liposomal doxorubicin (doxil): reduced clinical cardiotoxicity in patients reaching or exceeding cumulative doses of 500 mg/m2. Ann Oncol. 2000;11(8):1029–1033. | ||
O’Brien ME, Wigler N, Inbar M, et al. Reduced cardiotoxicity and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (CAELYX/Doxil) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Ann Oncol. 2004;15(3):440–449. | ||
Gradishar WJ, Tjulandin S, Davidson N, et al. Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil-based paclitaxel in women with breast cancer. J Clin Oncol. 2005; 23(31):7794–7803. | ||
Lien K, Georgsdottir S, Sivanathan L, Chan K, Emmenegger U. Low-dose metronomic chemotherapy: a systematic literature analysis. Eur J Cancer. 2013;49:3387–3395. | ||
Scharovsky OG, Mainetti LE, Rozados VR. Metronomic chemotherapy: changing the paradigm that more is better. Curr Oncol. 2009;16(2):7–15. | ||
Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307(5706):58–62. | ||
Tanaka S, Akaike T, Wu J, et al. Modulation of tumor-selective vascular blood flow and extravasation by the stable prostaglandin 12 analogue beraprost sodium. J Drug Target. 2003;11(1):45–52. | ||
Epstein AL, Mizokami MM, Li J, Hu P, Khawli LA. Identification of a protein fragment of interleukin 2 responsible for vasopermeability. J Natl Cancer Inst. 2003;95(10):741–749. | ||
Monsky WL, Fukumura D, Gohongi T, et al. Augmentation of transvascular transport of macromolecules and nanoparticles in tumors using vascular endothelial growth factor. Cancer Res. 1999;59(16): 4129–4135. | ||
Seki T, Fang J, Maeda H. Enhanced delivery of macromolecular antitumor drugs to tumors by nitroglycerin application. Cancer Sci. 2009;100(12):2426–2430. | ||
McKee TD, Grandi P, Mok W, et al. Degradation of fibrillar collagen in a human melanoma xenograft improves the efficacy of an oncolytic herpes simplex virus vector. Cancer Res. 2006;66(5):2509–2513. | ||
Goel S, Duda DG, Xu L, et al. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev. 2011;91(3): 1071–1121. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.