")
Back to Archived Journals » Current Biomarker Findings » Volume 7
Improved monitoring dynamics through use of a tethered enzyme biosensor to detect and quantify neuron-specific enolase activity levels in biofluids
Authors Cohen R, Nelson JL, Mukai C, Travis AJ
Received 23 February 2017
Accepted for publication 30 May 2017
Published 22 September 2017 Volume 2017:7 Pages 1—7
DOI https://doi.org/10.2147/CBF.S135368
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Hung Khong
Roy Cohen, Jacquelyn L Nelson, Chinatsu Mukai, Alexander J Travis
Baker Institute for Animal Health, College of Veterinary Medicine, Cornell University, Ithaca, NY, USA
Purpose: Neuron-specific enolase (NSE) is an isoform of the glycolytic enzyme enolase. For decades, changes in NSE levels in biofluids have been studied for their clinical value in diagnosis, prognosis and monitoring of a plethora of pathologies and conditions, including brain injuries, various kinds of cancers and tumors, and other diseases (eg, Guillain-Barré syndrome and Creutzfeldt-Jakob). However, because of its long 24–30 hours half-life, NSE’s diagnostic and prognostic power, specifically for brain injuries, was found to be limiting in respect to other biomarkers with faster dynamics (eg, S100β). Thus, we hypothesize that measuring NSEs fast decaying enzymatic activity, rather than only amounts of protein, will provide improved resolution of dynamic temporal changes.
Methods: Our previously reported tethered enzyme-based biosensor was used here to provide rapid measurement of changes in NSE activity as measured in vitro following incubation in canine and human serum or plasma. NSE activity was then compared with total protein amounts to assess the relative changes of these two parameters.
Results: We find that in serum, recovered plasma, and fresh plasma at 37°C, NSE protein levels (NSEP) are highly stable, whereas the enzymatic activity (NSEA) decays at a considerably faster rate.
Conclusion: Because the decay rate of NSEA is significantly higher as measured in vitro than its reported half-life in biofluids, and taking into account physiological clearance rates, measuring its enzymatic activity can provide better assessment of dynamic changes in the levels of this biomarker. A convenient and rapid technology for measurement of NSE enzymatic activity would improve understanding of NSE temporal dynamics, in comparison to more commonly used diagnostic methods.
Keywords: biomarker, enzyme activity, tethered enzymes, biosensor, brain injuries, tumor markers
Introduction
Neuron-specific enolase (NSE) is expressed predominantly in neurons and peripheral neuroendocrine cells.1 Remarkably, in some neurons NSE accounts for 3–4% of the total soluble protein, which led to common use of NSE as a clinical marker for neuronal and neuroendocrine cells.2
Levels of NSE in biofluids have been used extensively in diagnostics of various pathologies and conditions. Specifically, NSE has been one of the most extensively studied biomarkers for the diagnosis and prognosis of brain injuries,3 including ischemic stroke,4 seizures,5 comatose after cardiopulmonary resuscitation for cardiac arrest,6 neurological outcome after cardiac arrest,7 severe or mild traumatic brain injury (mTBI, concussion8), and Creutzfeldt–Jakob disease.3
NSE has also been broadly suggested as a diagnostic marker for various kinds of cancers, including small cell lung cancer9,10 and neuroblastoma,11 where its concentration correlates with tumor burden, number of metastatic sites, and response to treatment.3
The overwhelming majority of NSE biomarker studies have utilized antibody-based methods to detect and quantify protein amounts in biological samples (primarily blood and cerebrospinal fluid [CSF]). NSE’s clinical value as a biomarker is predominantly determined from its total circulating protein amounts (NSE protein, NSEP), using protein analytic methods including various solid-phase immunoassays,12,13 sandwich-type enzyme immunoassay14 (using polyclonal or monoclonal antibodies15), electrophoretic determination,16 and analysis by liquid chromatography-selected reaction monitoring/mass spectrometry (LC-SRM-MS).17 With these protein-centered approaches, it has been shown that the biological half-life for NSEP in human body fluids is between 24 and 30 hours.16,18 Importantly, the length of half-life influences both a biomarker’s predictive capabilities and optimal timing of sampling.19
The biological function of enolase (2-phospho-d-glycerate hydrolyase; EC 4.2.1.11) has long been known. These metal-activated metalloenzymes catalyze the dehydration of 2-phospho-d-glycerate (2-PG) to phosphoenolpyruvate (PEP) in the glycolytic pathway and the reverse reaction, the hydration of PEP to 2-PG, in gluconeogenesis.1 However, only a few attempts were made to detect NSE in biofluids by its enzymatic activity (ie, NSE activity, NSEA), either by directly measuring kinetics20 or by an immunocapture step followed by enolase activity bioluminescence assay.21 Interestingly, these reported methods have not been employed for mainstream NSE detection tests.
Here, we utilized our tethered enzyme biosensor technology (tethered enzyme technology [TET]), where pyruvate kinase (PK) and luciferase (Luc) are immobilized onto nanoparticles22 to detect NSE via its enzyme activity and determine the rates at which NSE protein or its activity are changing in serum and plasma. We have previously shown, both as a broad observation for glycolytic enzymes,23 and specifically for NSE detection,22 that solid-state enzyme reactions using oriented protein immobilization conferred more efficient coupled activity, and thus higher assay sensitivity, than non-tethered enzymes. This novel method enabled us to detect NSE in a rat model for brain injury and in human subjects with a rapid (<10 minutes), sensitive, and convenient assay.22 Here, we hypothesize that through its advantages and improved temporal resolution, it will offer a better approach to utilizing the clinical value of using NSEA as a biomarker.
Methods
Reagents
2-PG, PEP, ADP, ATP, inactivated human serum, and luciferin were purchased from Sigma (St Louis, MO, USA). Silica nanoparticles (Si-NPs) (500 nm) were purchased from Spherotech Inc (Lake Forest, IL, USA). Recombinant human NSE was purchased from MyBioSource (San Diego, CA, USA). Recovered human plasma of healthy adults was purchased from Zen-Bio Inc (Research Triangle Park, NC, USA), and samples were shipped overnight at 4°C.
Preparation of tethered enzyme assay
NP-PK and NP-Luc were generated and prepared as previously described.22 Briefly, His-Si4-PK and His-Si4-Luc were purified using Ni-NTA beads, and their enzymatic activity was tested in sodium phosphate buffer (50 mM) supplemented with MgCl2 (5 mM) and KCl (20 mM). The forward reaction for PK is PEP + ADP → pyruvate + ATP. The forward reaction for Luc is ATP + luciferin + Mg2+ → hv.
Collection of canine plasma
All experimental protocols were reviewed and approved by the Institutional Animal Care and Use Committee of Cornell University (protocol #2011–0004). A total of 3 mL of blood was collected from 3 adult dogs using Na-heparin 6 mL tubes. Tubes were spun at 1000× g for 5 minutes, and then plasma was aspirated and placed at 37°C for the duration of the experiments.
Measuring NSEA in plasma or serum samples
To measure NSE activity using the TET enzyme assay, reaction mixtures included a 1:1 ratio of NP-PK and NP-Luc, based on mass amounts of each enzyme bound to the NPs, resulting in roughly a 2:1 molar ratio of Luc to PK and a 2:1 activity ratio.22,31 NP-PK and NP-Luc were then supplemented with ADP, 2-PG, luciferin, Mg2+, and K+. A total of 10 µL samples of serum or plasma were added to individual wells in a 96-well plate, and the TET reaction mix (10 μL) was added just before luminescence measurements were initiated. To minimize variation among samples, a single batch of premixed TET reaction enzymes and substrates was used for all time points per each experiment. Although the TET reaction mixture was kept on ice throughout the experiments, some decay with a linear fashion was observed due to rundown of ADP and luciferin. Thus, the data presented in Figures 1–5 were corrected according to the measured decay rate as calculated from control reactions. The readout luminescence signal was integrated for 0.4 seconds and read continuously for 25 minutes using a TECAN Safire plate reader (TECAN US Inc., Morrisville, NC, USA). For calculation of NSE activity levels, the linear regression slope for the initial activity was calculated per well. Then, control wells (2-PG) were subtracted from the test channels, and the results normalized to the positive control wells (with 2-PG and enolase). At the last time point of each experiments, samples were brought up to 50–55°C (for plasma, due to heat-dependent coagulation) or 80°C (for serum), to inactivate NSE as a final negative control.
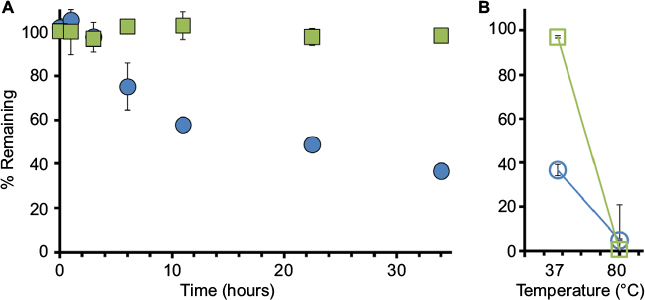 | Figure 1 Human NSE activity (NSEA) versus total protein (NSEP) as measured in inactivated human serum. Notes: (A) Commercially available inactivated human serum was spiked with recombinant human NSE and incubated at 37°C. Samples were collected at different time points as indicated and tested for NSE activity (via TET assay) or for total protein (samples sent to a commercial diagnostic laboratory, ARUP Labs, Salt Lake City, UT, USA). Data points for NSEA (blue circles) and NSEP (green squares) are presented as percentages of their initial values at time point zero. (B) At the last time point, samples were placed at 80°C for 30 minutes to achieve full inactivation and protein breakdown. Values are provided as avg ± SD, n=3. Abbreviations: NSE, neuron-specific enolase; TET, tethered enzyme technology; avg, average. |
Measuring NSEP in plasma or serum samples
Serum or plasma samples were collected at the indicated time points and kept at −80°C until the end of the experiment. Then, samples were shipped frozen on dry ice to ARUP laboratories (Salt Lake City, UT, USA) for quantification of amounts of NSE protein (provider uses the CanAg NSE EIA assay).
Theoretical calculation of NSEA decay rate in circulation
Using the standard half-life decay rate formulation: N(t) = N0(1/2)t/t1/2, where N0 is the initial quantity of NSEA or NSEP, N(t) is the remaining quantity, and t1/2 is the reported half-time for NSE protein in vivo (24 hours).16,18
Statistical analysis
Experiments were performed 3 times, and all samples were measured in triplicates. Data processing and analysis were carried out using Excel (Microsoft) and Origin (OriginLab, Northampton, MA, USA). Variability for PK and Luc activity between batches was addressed by preparing mixtures of the enzymes with equal activities. For Figure 4, a simple linear regression model was used to determine the decay rate of NSEA (using Excel function for linear regression). Data are presented as average ± standard deviation.
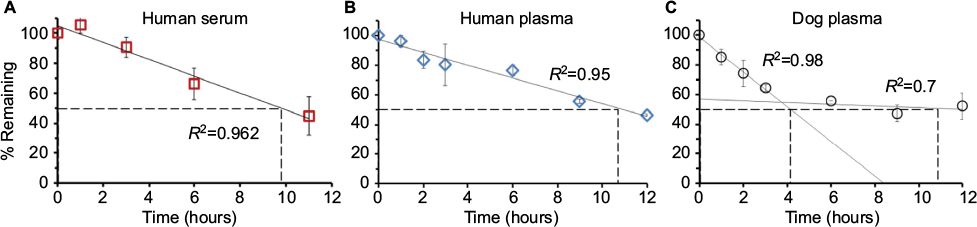 | Figure 4 Comparison of human NSE activity (NSEA) decay rates in biofluids at 37°C. Notes: The temporal profiles of NSEA during the first 12 hours as measured in experiments described for human serum (A), human plasma (B), and dog plasma (C) were evaluated. Within this time frame, NSEA in fresh dog plasma (C) demonstrated a biphasic profile with a rapid initial rate (t1/2=4.1 hours, 37°C) and a slower subsequent component. Slower decay rates with linear kinetics were observed for both inactivated serum (A) and human recovered plasma (B, t1/2=9.8 and t1/2=10.6 hours, respectively); however, all were significantly faster than the reported half-life for NSE protein amounts, which is ~24 hours. For (A) and (B), a simple linear regression model was used to determine the decay rate of NSEA (R2>0.95). For consistency with (A) and (B), the 2 phases of decay in (C) were fitted independently. Abbreviations: NSE, neuron-specific enolase; t1/2, half-life. |
Results
To determine the rate of protein degradation versus the loss of enzymatic activity, we conducted a comparative examination utilizing a “traditional” detection method (ie, CanAg NSE EIA assay) in parallel with the TET assay.
First, to examine the extent and dynamics of passive changes in activity, recombinant human NSE was added to heat-inactivated human serum at a final concentration of 100 ng/mL. The spiked serum was then incubated at 37°C, while samples were collected at 0, 1, 2, 3, 6, 11, 22, and 34 hours (Figure 1A). For each time point, the collected sample was divided, where one aliquot was used for testing the NSE activity using the tethered enzyme assay and the second was frozen (−80°C) for protein amount analysis. As previously reported, the TET assay includes PK and Luc that are immobilized on to Si-NPs (NPs, NP-PK, and NP-Luc respectively), through a bio-affinity tag specific for silica (Si4).22 The NPs with tethered enzymes were supplemented with reagents necessary for the forward reaction as described in the “Methods” section. The enzymatic activity of NSE in each sample was calculated from the initial slopes of the luminescent signal coming out of the test reaction, in comparison with the negative control, in which the enolase substrate 2-PG was omitted (refer the “Methods” section and Cohen et al22 for details). Immediately following the collection of the last time point sample, the remaining serum was incubated at 80°C for 30 minutes for complete inactivation of NSE activity (Figure 1B).
Our analysis showed that in heat-inactivated serum, 60% of NSE’s activity was lost within 10 hours of start of the experiment, whereas minimal decrease was observed for the total amount of NSEP (Figure 1A). Heating the serum to 80°C not only completely inactivated the remaining enzymatic activity but also denatured the protein to a point where it is undetectable by the antibody-based assay (Figure 1B).
Because heat inactivation (by the provider) potentially minimizes any intrinsic protein-degrading activity, we next sought to determine the NSEA and NSEP changes in recovered human plasma (ie, commercially available plasma that is collected as a byproduct from patients who underwent blood collection for other purposes). Importantly, plasma, which is more representative of whole blood, might also contain factors that could affect both enzyme activity and protein stability (eg, through oxidation24 and proteolytic degradation25).
As described for the serum experiment, recombinant human NSE was then added to recovered human plasma at a final concentration of 100 ng/mL. The human plasma was collected by Zen-Bio Inc via Li-heparin tubes from healthy adult donors and shipped to us at 4°C overnight. Human plasma samples were tested with the tethered enzyme assay for NSEA at 0, 1, 2, 3, 6, 9, 12, 24, and 28 hour time points, and corresponding aliquots were saved at −80°C for protein analysis (NSEP). At the last time point, the plasma was heat treated to 55°C, resulting in 3-fold decrease in NSE activity and over 2-fold decrease in protein levels (using higher temperatures for inactivation, such as 80°C used for serum, caused coagulation, and thus could not be used in this case).
Our data showed that although minimal protein degradation was observed, NSEA decay dynamics were comparable to these found in human serum (Figure 2).
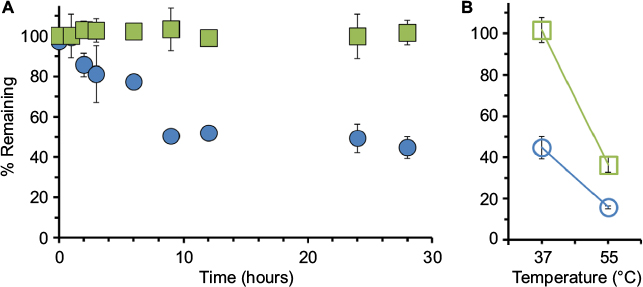 | Figure 2 Human NSE activity (NSEA) versus total protein (NSEP) as measured in human plasma. Notes: (A) Recovered human plasma was spiked with recombinant human NSE and incubated at 37°C. Samples were collected at different time points as indicated and tested for NSE activity (via TET assay) or for total protein (samples sent to external diagnostic laboratory service at ARUP Labs, Salt Lake City, UT, USA). Data points for NSEA (blue circles) and NSEP (green squares) are presented as percentage of initial values. (B) At the last time point, samples were placed at 55°C for 30 minutes to inactivate the enzyme activity. Values are provided as avg ± SD, n=3. Abbreviations: NSE, neuron-specific enolase; TET, tethered enzyme technology; avg, average. |
Finding only small differences in enzymatic activity decay rates between serum and the 24-hour-old recovered plasma encouraged us to test whether fresh plasma would have a more pronounced effect on NSEA. Thus, canine plasma was chosen for its availability in close proximity to the testing site, in addition to the fact that in laboratory-maintained dogs, drugs or confounding substances are not present and that no long-term, inducible changes in the composition of the plasma would be present from prior consumption or exposure to environmental factors.
As with previous experiments, recombinant human NSE was added to plasma collected from adult dogs maintained in a laboratory colony at a final concentration of 100 ng/mL. The spiked plasma was then incubated at 37°C, and samples were collected at 0, 1, 2, 3, 6, 9, 12, 24, and 30 hours (Figure 3). Similar to our findings in human serum and plasma, as indicated in Figure 3, our data show that NSEP was stable over the whole incubation period. Attempted enzyme inactivation by heat treatment of the plasma to 50°C for 30 minutes at the last time point resulted in only a marginal loss in NSEA and NSEP; however, higher temperatures had strong coagulation effect and could not be used (refer the “Discussion” section).
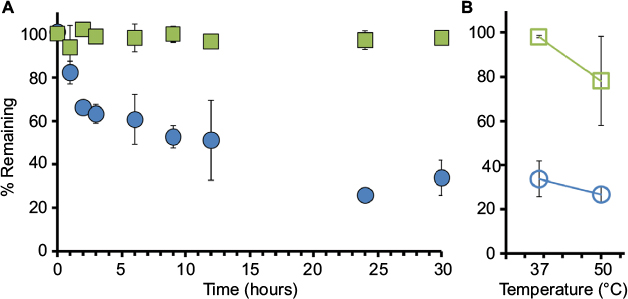 | Figure 3 Human NSE activity (NSEA) versus total protein (NSEP) as measured in fresh dog plasma. Notes: (A) Collected dog plasma was spiked with recombinant human NSE and incubated at 37°C. Samples were collected at different time points as indicated and tested for NSE activity (via TET assay) or for total protein (samples sent to a commercial diagnostic laboratory, ARUP Labs, Salt Lake City, UT, USA). Data points for NSEA (blue circles) and NSEP (green squares) are presented as percentage of initial values. (B) At the last time point, samples were placed at 50°C for 30 minutes to achieve NSE enzyme inactivation. Values are provided as avg ± SD, n=3. Abbreviations: NSE, neuron-specific enolase; TET, tethered enzyme technology; avg, average. |
Nonetheless, as shown in Figure 4C, our results exhibit biphasic decay dynamics in fresh dog plasma for NSEA, demonstrating an initial decay rate of t1/2=4 hours, which was reduced to t1/2=11 hours for the second component.
The different rates at which NSEA decayed during the first 12 hours of experiments in fresh plasma, as shown in Figure 4, revealed an initial 12% per hour NSEA decay rate, followed by a slower component of ~2% per hour. In contrast, over the same time frame, the loss of NSE enzymatic activity in recovered serum or plasma samples (Figure 4A and B) was mostly linear with rates of 4–6% per hour, respectively.
Because the total amounts of NSEP did not demonstrate any significant changes in serum or plasma over the incubation times, we hypothesize that a physiological clearance process must be the major mechanism driving the reported changes in protein half-life in vivo.16,18 Thus, as seen in Figure 5, combining the expected effects of physiological clearance (t1/2=24 hours) and enzymatic activity decay determined here, we calculated a theoretical in vivo half-life of ~4.5 hours for NSEA (Figure 5), which is more than 5 times faster than the reported t1/2 for NSEP.
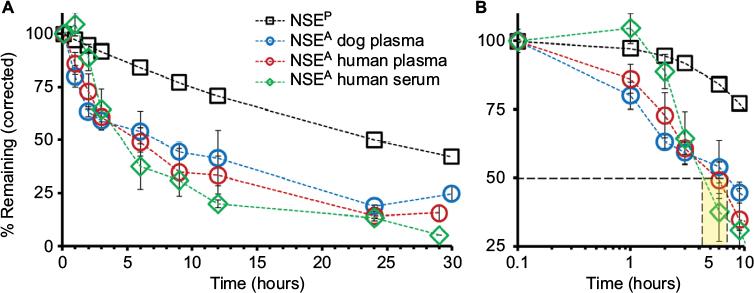 | Figure 5 Theoretical calculation of half-life (t1/2) for NSE activity (NSEA). Notes: (A) The reported half-life of NSE (black squares) was applied to our measured NSEA data points, using the standard half-life decay rate formulation (N(t) = N0(1/2)t/(t1/2), where N0 is the initial quantity of NSEA or NSEP, N(t) is the remaining quantity, and t1/2 is the reported half-time for NSE protein in vivo (24h). The corrected NSEA decay rates for human serum (green diamonds), human plasma (red circles), and dog plasma (blue circles) shown are to be expected when assuming a model where both physiological clearance (with the same rate as calculated for NSEP) and enzymatic inactivation are combined. (B) Our calculations provide a theoretical “in vivo” half-life of ~4.5 hours for NSEA in plasma (slower rates were calculated for serum or recovered plasma, as indicated in the highlighted area) versus the reported 24 hours for NSEP.” Abbreviations: NSE, neuron-specific enolase; t1/2, half-life; NSEP, NSE protein amount. |
Discussion
NSE is widely used as a diagnostic and prognostic marker for various diseases; however, because of its relatively long half-life (24–30 hours16), the predictive power of this biomarker is reduced.19 In addition, there is debate regarding the usefulness of NSE levels in measuring the extent of mTBI (concussion), thought to stem from the long half-life of NSE in blood.26 Here, we hypothesized that measuring the decay of NSE’s enzymatic activity, rather than its protein amount, would provide higher temporal resolution regarding the dynamics of its changes in biofluids.
Indeed, although some small variations in the dynamics of NSEA decay rates were found, our data revealed a dramatic difference between the loss of NSE activity and the stability of the NSE protein, when measured in serum, recovered plasma, and fresh plasma. The stability of NSEP observed here is in agreement with previous reports13 and suggests that the indicated half-life of NSE is predominantly regulated by physiological clearance, with little effect from proteolysis or degradation.
Our initial experiments in heat-inactivated serum were used to determine the baseline loss of NSEA and NSEP over time at 37°C. The observed faster decay rate for NSEA in fresh plasma versus serum or recovered plasma was not unexpected and could be largely attributed to oxidization and other processes that have the capacity to affect enzyme activity and conformational stability. Such processes might involve degradation by plasma thrombin,27 or proteases,25 and reactive oxygen species damage introduced by plasma enzymes including xanthine oxidase, NADPH oxidases, and oxygenase.24 Importantly, the inactivating effect of such components is expected to decay over time, as in the case of the 24-hour-old recovered plasma. We do not rule out any other effects mediated by clotting factors that would be more active in fresh plasma28 on NSEA. These processes could also explain the biphasic nature of the NSEA decay seen in fresh plasma, whereas NSEA decay in inactivated serum or recovered plasma was found to be linear. We speculate that the second slower decay rate in fresh plasma is the result of loss of enzymatic activity of the inactivating components that would be expected to decrease over time as well.
Because our in vitro assays lack physiological clearance (see example for LDH clearance in rabbits29 and clearance of NSE in urine30), an even faster rate of NSEA decay would be expected to be observed in patients (Figure 5). Although a pharmacokinetic approach is required to determine the decay rate of NSEA in vivo, our findings suggest that measurements of NSEA dynamics, such as provided by methodologies such as TET, will improve the clinical utility of this biomarker. For example, one could distinguish between recent and previous injury, as in assessing mTBI in athletes or soldiers who can be subjected to trauma on a repetitive basis. Alternatively, knowledge of the half-life of activity in vivo might enable one to look for and calculate the amount of NSE production or release into the bloodstream, such as by ongoing injury or growth of a tumor. Yet another use could be in patients with several co-morbidities, in which case chronic production might mask an acute injury/event if only NSEP were being measured.
Conclusion
We report here that NSEA has a much shorter half-life in vitro than NSEP. In vivo studies are needed to determine how physiological/metabolic clearance will affect half-life in clinical situations. Our findings highlight the potential improvement in clinical utility of NSEA as an independent biomarker or as one that can be measured in comparison to NSEP. Use of a tethered enzyme biosensor enables detection of the faster kinetics of changes in enzymatic activity compared to protein amount, enabling repetitive sampling over shorter time frames than might be feasible with currently available commercial ELISA-based tests. This new technology could prove to be diagnostically and prognostically more powerful in scenarios in which rapid changes might occur (eg, stroke), when assessing treatment outcomes (eg, cancer) or when repetitive injuries are suspected as potentially confounding factors (eg, mTBI in athletes). In these circumstances, NSEA might better reflect acute versus prior insults, the extent and timing of the most recent damage, and whether release was transient or is still ongoing.
Disclosure
The authors report no conflicts of interest in this work.
References
Vizin T, Kos J. Gamma-enolase: a well-known tumour marker, with a less-known role in cancer. Radiol Oncol. 2015;49(3):217–226. | ||
Marangos PJ, Schmechel DE. Neuron specific enolase, a clinically useful marker for neurons and neuroendocrine cells. Annu Rev Neurosci 1987;10:269–295. | ||
Isgro MA, Bottoni P, Scatena R. Neuron-specific enolase as a biomarker: biochemical and clinical aspects. Adv Exp Med Biol 2015;867:125–143. | ||
Ahmad O, Wardlaw J, Whiteley WN. Correlation of levels of neuronal and glial markers with radiological measures of infarct volume in ischaemic stroke: a systematic review. Cerebrovasc Dis 2012;33(1):47–54. | ||
Lee SY, Choi YC, Kim JH, Kim WJ. Serum neuron-specific enolase level as a biomarker in differential diagnosis of seizure and syncope. J Neurol 2010;257(10):1708–1712. | ||
Wijdicks EF, Hijdra A, Young GB, Bassetti CL, Wiebe S; Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter: prediction of outcome in comatose survivors after cardiopulmonary resuscitation (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2006;67(2):203–210. | ||
Stammet P, Collignon O, Hassager C, et al. Neuron-specific enolase as a predictor of death or poor neurological outcome after out-of-hospital cardiac arrest and targeted temperature management at 33 degrees C and 36 degrees C. J Am Coll Cardiol 2015;65(19):2104–2114. | ||
Kawata K, Liu CY, Merkel SF, Ramirez SH, Tierney RT, Langford D. Blood biomarkers for brain injury: what are we measuring? Neurosci Biobehav Rev 2016;68:460–473. | ||
Wang H, Zhang X, Liu X, Liu K, Li Y, Xu H. Diagnostic value of bronchoalveolar lavage fluid and serum tumor markers for lung cancer. J Cancer Res Ther 2016;12(1):355–358. | ||
Hu Q, Xiao P, Li J, Yu P. A retrospective analysis of serum tumor markers found in non-small cell lung cancer. J Cancer Res Ther 2016;12(1):117–120. | ||
Riley RD, Heney D, Jones DR, et al. A systematic review of molecular and biological tumor markers in neuroblastoma. Clin Cancer Res 2004;10(1 Pt 1):4–12. | ||
Gerbitz KD, Summer J, Thallemer J. Brain-specific proteins: solid-phase immunobioluminescence assay for neuron-specific enolase in human plasma. Clin Chem 1984;30(3):382–386. | ||
Mercer DW, Virji MA, Barry GE, Piper ML. New solid-phase enzyme immunoassay of neuron-specific enolase in serum: effect of storage temperature, lipemia, icterus, and hemolysis. Clin Chem 1990;36(8 Pt 1):1519. | ||
Kimura S, Uchikawa H, Yamamoto R, Kato K. Practicable enzyme immunoassay for neuron-specific enolase in human serum. J Appl Biochem 1984;6(5–6):319–324. | ||
Kimura S, Uchikawa H, Yamamoto R, Kato K. Two-site column enzyme immunoassay for neuron-specific enolase (NSE) in human serum using monoclonal antibodies. J Immunol Methods 1986;94(1–2):51–55. | ||
Viallard JL, Ven Murthy MR, Dastugue B. Rapid electrophoretic determination of neuron-specific enolase isoenzymes in serum. Clin Chem 1986;32(4):593–597. | ||
Torsetnes SB, Lovbak SG, Claus C, et al. Immunocapture and LC-MS/MS for selective quantification and differentiation of the isozymes of the biomarker neuron-specific enolase in serum. J Chromatogr B Analyt Technol Biomed Life Sci 2013;929:125–132. | ||
Johnsson P, Blomquist S, Luhrs C, et al. Neuron-specific enolase increases in plasma during and immediately after extracorporeal circulation. Ann Thorac Surg 2000;69(3):750–754. | ||
Thelin EP, Jeppsson E, Frostell A, et al. Utility of neuron-specific enolase in traumatic brain injury; relations to S100B levels, outcome, and extracranial injury severity. Crit Care 2016;20:285. | ||
Wevers RA, Jacobs AA, Hommes OR. A bioluminescent assay for enolase (EC 4.2.1.11) activity in human serum and cerebrospinal fluid. Clin Chim Acta 1983;135(2):159–168. | ||
Viallard JL, Murthy MR, Betail G, Dastugue B. Determination of serum neuron-specific enolase by differential immunocapture. Clin Chim Acta 1986;161(1):1–10. | ||
Cohen R, Lata JP, Lee Y, et al. Use of tethered enzymes as a platform technology for rapid analyte detection. PLoS One 2015;10(11):e0142326. | ||
Mukai C, Gao L, Nelson JL, et al. Biomimicry promotes the efficiency of a 10-step sequential enzymatic reaction on nanoparticles, converting glucose to lactate. Angew Chem Int Ed Engl. 2017. 56(1):235–238 | ||
Griffiths HR, Dias IH, Willetts RS, Devitt A. Redox regulation of protein damage in plasma. Redox Biol. 2014;2:430–435. | ||
Kushnir MM. Are samples in your freezer still good for biomarker discovery? Am J Clin Pathol 2013;140(3):287–288. | ||
Ingebrigtsen T, Romner B. Biochemical serum markers for brain damage: a short review with emphasis on clinical utility in mild head injury. Restor Neurol Neurosci 2003;21(3–4):171–176. | ||
Takayama K, Taguchi A, Yakushiji F, Hayashi Y. Identification of a degrading enzyme in human serum that hydrolyzes a C-terminal core sequence of neuromedin U. Biopolymers 2016;106(4):440–445. | ||
Feng L, Zhao Y, Zhao H, Shao Z. Effects of storage time and temperature on coagulation tests and factors in fresh plasma. Sci Rep 2014;4:3868. | ||
Wilkinson JH, Qureshi AR. Catabolism of plasma enzymes, as studied with 125I-labeled lactate dehydrogenase-1 in the rabbit. Clin Chem 1976;22(8):1269–1276. | ||
Gotoh Y, Hashimoto K, Tada K. Urine neuron-specific enolase and its clinical implication in patients with neuroblastoma. Tohoku J Exp Med 1986;149(1):67–72. | ||
Lata JP, Gao L, Mukai C, et al. Effects of nanoparticle size on multilayer formation and kinetics of tethered enzymes. Bioconjug Chem 2015;26(9):1931–1938. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.