")
Back to Journals » Clinical and Experimental Gastroenterology » Volume 14
Importance of Mevalonate Pathway Lipids on the Growth and Survival of Primary and Metastatic Gastric Carcinoma Cells
Authors Ortiz N, Delgado-Carazo JC, Diaz C
Received 10 March 2021
Accepted for publication 7 May 2021
Published 31 May 2021 Volume 2021:14 Pages 217—228
DOI https://doi.org/10.2147/CEG.S310235
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Anastasios Koulaouzidis
Natalia Ortiz,1 Juan Carlos Delgado-Carazo,2 Cecilia Díaz1,3
1Departamento de Bioquímica, Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica; 2Escuela de Biología, Universidad de Costa Rica, San José, Costa Rica; 3Instituto Clodomiro Picado, Facultad de Microbiología, Universidad de Costa Rica, San José, Costa Rica
Correspondence: Cecilia Díaz
Instituto Clodomiro Picado, Facultad de Microbiología, Universidad de Costa Rica, San José, 11501, Costa Rica
Email [email protected]
Purpose: This preclinical study aims to determine the effect of drugs that alter isoprenoids and cholesterol metabolism in the homeostasis of gastric carcinoma cell lines in the search for new therapeutic targets for stomach cancer.
Materials and Methods: Primary (AGS) and metastatic (NCI-N87) gastric cancer cell lines were treated with simvastatin and terbinafine, two inhibitors of the mevalonate pathway, and avasimibe, an inhibitor of cholesterol esterification. Cell viability and growth were measured as well as cholesterol levels and the expression of the hydroxy methyl-glutaryl CoA reductase (HMGCR) and the LDL receptor (LDLR).
Results: Primary and metastatic gastric carcinoma cells show different sensitivity to drugs that affect isoprenoid synthesis and the metabolism and uptake of cholesterol. Isoprenoids are involved in the growth and viability of both types of cells, but the role of free and esterified cholesterol for metastatic gastric cell survival is not as evident as for primary gastric cancer cells. Differential expression of LDLR due to mevalonate pathway inhibition suggests variations in the regulation of cholesterol uptake between primary and metastatic cancer cells.
Conclusion: These results indicate that at least for primary gastric cancer, statins and avasimibe are promising candidates as potential novel antitumor drugs that target the metabolism of isoprenoids and cholesterol of gastric tumors.
Keywords: cholesterol, isoprenoids, gastric cancer, metastasis, simvastatin, terbinafine, avasimibe
Introduction
Mevalonate pathway is one of the most important and conserved biosynthetic processes in animal cells and one of the most highly regulated, probably due to energetic costs and the toxicity of some of the intermediate metabolites and cholesterol itself.1 Membrane lipids are essential compounds that regulate the biophysical and biochemical properties of cell membranes,2–4 including regulation of fluidity and microdomains dynamics.5,6 Cholesterol and other lipids derived from the mevalonate pathway, such as isoprenoids that anchor proteins to the membranes, have also been implicated in the survival and metastatic behavior of several types of cancer.7 Even when cholesterol metabolism has been extensively studied in steroid hormone-dependent tumors such as breast and prostate cancer,8,9 due to the high importance of mevalonate-derived lipids in cell homeostasis, their role is not limited to these tumors.10
Gastric cancer is the third cause of death by cancer in the World.11 Eastern Asia is the region with the highest incidence and mortality, but Eastern and Central Europe and Latin America also present high mortality rates.12
In terms of primary gastric cancer treatment, the main procedure consists of tumor resection (partial or total gastrectomy), but the incidence of local recurrence and peritoneal and distant metastasis is very high, making systemic chemotherapy (mostly palliative) also part of the treatment.13
The liver is one of the main organs of dissemination for gastric tumors, and in their way of reaching other organs, gastric primary tumor cells go through intra- and extravasation, migration and colonization of other tissue environments. Therefore, due to all these new characteristics that metastatic tumors display, they cannot be clinically treated in the same way that primary tumors are,14 since, some of these differences could make them resistant to primary tumor therapies. Also, since metastasis is what, in most of the cases, results in the lethality of the patients, it has been recognized that this is precisely the process that deserves better characterization, to finally develop successful strategies of treatment that specifically recognize the particularities of these tissues.
In the case of gastric cancer, the information regarding cholesterol and other metabolites derived from the mevalonate pathway is very limited. However, an interesting study performed by Chushi and collaborators15 has shown that the first rate-limiting enzyme of the pathway, the hydroxy methyl-glutaryl CoA reductase (HMGCR) is overexpressed in gastric cancer, suggesting that it could be a putative oncogene, as suggested for other tumors.10 They also showed that increasing the expression of HMGCR in gastric cancer cells accelerates their growth and migration abilities. These and other findings related to the participation of lipids such as cholesterol, in the regulation of cell growth and death of tumor cells, add to the widely recognized concept that metabolic reprogramming is one of the hallmarks of cancer.3,16–20
Interestingly, the second-rate limiting enzyme of this pathway, squalene epoxidase, a downstream enzyme that catalyzes the first oxygenation step in sterol biosynthesis,21 has recently been shown to be overexpressed in some tumors.22 This enzyme has also been postulated as a metabolic oncogene due to its effect increasing in vitro cell growth and survival (by apoptosis inhibition) and by accelerating tumor growth in mouse models.22
Some inhibitors of the mevalonate pathway have been clinically tested for gastric cancer23 and the potential anti-proliferative and pro-apoptotic effects of cholesterol-lowering drugs such as statins, is a highly investigated topic.3,7,16,24,25 Other drugs that interfere with the mevalonate pathway such as bisphosphonate zoledronic acid, and farnesyl and geranylgeranyl transferase inhibitors, which affect protein isoprenylation, have also been clinically tested.26
The goal of the present study was to study the potential differences in the role of mevalonate pathway lipids in the homeostasis of two intestinal gastric carcinoma cell lines with characteristics associated with different stages of differentiation, one from a primary tumor and the other from advanced liver metastasis. The obtained results could have implications in future therapeutic decisions based on cholesterol metabolism of gastric tumors.
Materials and Methods
Cell Culture
AGS (CRL-1739) and NCI-N87 (CRL-5822) cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). The AGS cells correspond to a gastric adenocarcinoma located in the stomach whereas the NCI-N87 cells correspond to a gastric carcinoma derived from a metastasis to the liver. Both cell lines were maintained in RPMI-1640 media (R8758; Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS) (F2442; Sigma-Aldrich) and 100 U/mL penicillin-streptomycin (15,140,122; GibcoTM), at 37°C in a humid atmosphere containing 5% CO2.
Growth Curve Analysis
Initially, 1.5×104 AGS and NCI-N87 cells were plated in 24-wells plates, and every two days, the solution with cells was diluted 1:2 in Trypan blue. The cells were counted using a Neubauer camera. For each cell line, the duplication time (DT) between days 2 and 4 was determined according to the following formula: DT = T ln2/ln(Xf/Xi), where T corresponds to the incubation time, Xf to the final cell number and Xi to the initial number of cells.
Inhibition of HMG CoA Reductase
Simvastatin (S6196 Sigma-Aldrich) was the drug applied to inhibit the enzyme. To activate the drug, the protocol described by Dong and collaborators27 was followed. Briefly, 8 mg of simvastatin were dissolved in 0.2 mL of pure ethanol and 0.3 mL of 0.1 N NaOH were added later. Ethanol was evaporated by exposure to nitrogen gas (99.99%). Subsequently, the solution was heated for 2 h at 50°C in a dry bath and then neutralized with HCl to a pH close to 7.2. The solution was brought to a final volume of 4 mL with deionized water (final concentration of 5 mM) and aliquots were stored at −80°C until use.
Suspensions of 8,000 AGS and 16,000 NCI-N87 cells were incubated with different concentrations of the drug (3–100 µM) for 24, 48 and 72 h in media supplemented with 10% FBS.
Mevalonolactone (2.5 µM) (M4667 Sigma-Aldrich), and isoprenoids geranylgeranyl pyrophosphate (GGPP, 7.5µM) (G6025 Sigma-Aldrich) and farnesyl pyrophosphate (FPP, 7.5 µM) (F6892 Sigma-Aldrich) were incorporated to evaluate the role of some of the intermediary metabolites of the mevalonate pathway in the effect induced by simvastatin. Cells were incubated simultaneously for 48 h with simvastatin and each one of the metabolites in media supplemented with 10% FBS.
Inhibition of Squalene Epoxidase
Terbinafine (T8826 Sigma-Aldrich) was dissolved in dimethyl sulfoxide (DMSO) to a final concentration of 150 mM. DMSO concentration was less than 0.1% and no toxic effect was observed on the cells. Suspensions of 8,000 AGS and 16,000 NCI-N87 cells were incubated for 48 h with different concentrations of the inhibitor (5–150 µM) dissolved in media supplemented with 10% FBS. Additionally, 48 h-cell viability was determined in the presence of terbinafine in Advanced RPMI media (12633012; Thermo Fisher Scientific, Inc) containing 1% FBS.
Inhibition of Cholesterol Esterification
For this purpose, the drug avasimibe (PZ0190 Sigma-Aldrich) (CI-1011), an inhibitor of Acyl-CoA: Cholesterol O-Acyltransferase (ACAT-1), was used. The same number of cells utilized previously was incubated for 48 h with the inhibitor at different concentrations (1.25–80 µM) in RPMI media containing 10% FBS. A combination of Avasimibe and Terbinafine was also tested at a low dose (80% viability) of Avasimibe.
Cell Viability Assay
To assess the effect of each of the enzymatic inhibitors in cell viability, MTT assay was performed. After the treatments, media with the inhibitors (and additional metabolites) was removed and MTT solution (0.5 mg/mL final concentration) dissolved in the RPMI media was applied for 2 h at 37°C. Then, the media was removed, and the formazan crystals that precipitated were dissolved in 96% ethanol. The absorbance was measured at 570 nm. As a second approach to determining mainly the effect on cell proliferation, Sulforhodamine B assay as described by Vichai and Kirtkara,28 was performed.
After the cells were incubated with simvastatin or terbinafine, they were fixed in 10% trichloroacetic acid (TCA) for 1 h at 4°C. Cells were stained with 0.04% Sulforhodamine B solution for 30 min at room temperature. Then, the wells were rinsed with 1% acetic acid. Protein-bound dye was dissolved in 10 mM Tris base solution and the absorbance was measured at 530 nm. The absorbance values of cells incubated with only media were referenced as 100% of viability. To determine the percentage of cell growth relative to a non-treated control, a plate without treatment was also fixed before the addition of the inhibitors.
Free Cholesterol Staining and Quantification
AGS and NCI-N87 cells were plated in black 96-well plates. Then, cells were treated with the inhibitors: simvastatin or terbinafine for 48 h. Cells were fixed with 4% paraformaldehyde for 10 min at room temperature and to neutralize it, cells were incubated in 1.5 mg/mL glycine solution for 10 min at room temperature. Cells were incubated in a 0.05 mg/mL solution of filipin (F9765 Sigma-Aldrich) in phosphate-buffered saline (PBS) supplemented with 10% FBS for 2 h protected from light at room temperature. For a positive control, the cells were incubated with 5 mM methyl-β-cyclodextrin (MβCD) (C4555 Sigma-Aldrich) for 1 h. Images were acquired by fluorescence microscopy with a 20x objective under the same conditions of LED exposure, integration time and gain in BioTek Cytation 3 Imaging Multi-Mode Reader. Gen5 Image 3.09 software was used to quantify the fluorescence intensity. All images from each cell line were included in the experiment and an automatic setting was applied for preprocessing for DAPI and Brightfield channels. The Cellular analysis tool was used to select only the areas containing cells in the brightfield images. For this, the threshold value for AGS and NCI-N87 was 750 and 4000, respectively. The object size selection was set between 15 and 500 µm for both cell lines. The software estimated The Object Sum Intensity [Tsf (DAPI 377,447)] and the Object Sum Area [Tsf (Bright Field)] and the relative Object Sum Intensity/Object Sum Area was calculated for all images (at least 2 images from 5 independent wells). The mean value and standard deviation were determined, and statistical analyses were performed.
Intracellular Lipid Droplets Staining and Fluorescence Quantification
AGS cells were plated in black 96-well plates. Then, cells were treated with the inhibitors: terbinafine or avasimibe for 48 h. Cells were fixed with 4% paraformaldehyde and incubated in a 0.5 µg/mL solution of Nile red (19123 Sigma-Aldrich) in PBS for 10 minutes at 37°C. Images were acquired by fluorescence microscopy in the RFP filter (531, 593 nm) with a 4 and 20X objective under the same conditions of LED exposure, integration time and gain, in a BioTek Cytation 3 Imaging Multi-Mode Reader. Gen5 Image 3.09 software was used to quantify the fluorescence intensity. Fluorescence quantification was performed as described in the previous section. For each treatment, at least 4–6 pictures (4X) were analyzed with a minimum of 300 events counted per image. The threshold value was 4000 and the object size selection was set between 5 and 100 µm. Two independent experiments were performed. For each experiment, the mean value and standard deviation were determined, and statistical analyses were performed.
RNA Extraction and cDNA Synthesis
AGS cells (350,000) and NCI-N87 cells (700,000) were plated in 6-well plates and allowed to adhere overnight. Then, they were treated with simvastatin or terbinafine for 48 h in cell culture media supplemented with 10% FBS. Cell lysates were stored in RNAlaterTM Stabilization Solution (AM7020; InvitrogenTM) at −80°C, until use. For the RNA isolation, the NucleoSpin RNA II kit (740955; Macherey Nagel) was used, as recommended by the manufacturer. For cDNA synthesis, the RevertAid First Strand cDNA Synthesis kit (K1622; Thermo ScientificTM) was used.
RT-PCR
PCR reactions were carried out in a total volume of 25 µL, using 22.5 µL of PCR SuperMix (10,572; InvitrogenTM), 0.5 µL of Nuclease Free Water and 0.5 µL of each primer (GAPDH 10µM; HMGCR 50 µM and LDLR 50 µM) on a 2720 Applied Biosystems Thermal Cycler with the following conditions: 94°C for 3 min, followed by 35 cycles of denaturing at 94°C for 15 s, annealing at 53.4°C for 30 s and extension at 72°C for 35 s. PCR products were analyzed in 1.8% agarose gels and DNA was stained with SYBR® Safe DNA gel stain (Invitrogen; S33102). The acquisition of gel images was performed using ImageQuantTM LAS 500 (GE Healthcare Life Sciences). Band intensity quantification was carried out by scanning densitometry using ImageJ v1.49 software (National Institutes of Health). The ratio between the sample RNA to be determined and GAPDH was calculated to normalize for initial variations in sample concentration.
The sequences of the primers and the expected product sizes are summarized in Table 1.
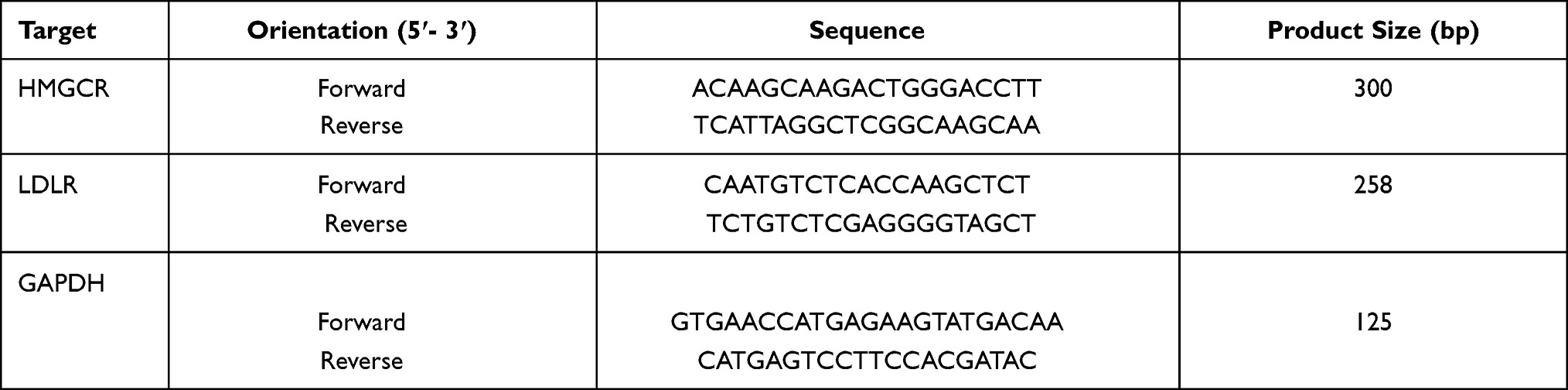 |
Table 1 Primers Used in RT-PCR Reactions for AGS and NCI-N87 Cells |
Statistical Analysis
For all the viability assays, dose–response curves were done, and the half-maximal inhibitory concentration (IC50) was calculated using the Slide Write Plus 6.10 software. All quantitative experiments were repeated three independent times (in triplicate each) and data are expressed as the mean ± standard error. To evaluate the significance of the results, an independent t-test or one-way ANOVA followed by Bonferroni´s post hoc test was applied, using GraphPad Prism 5 and IBM SPSS Statistics 22 software. The level of significance was set at p < 0.05.
Results
Characteristics of Gastric Carcinoma Cell Lines
Cell lines used in this study belong to the intestinal subtype of gastric carcinoma, being AGS from a primary tumor and NCI-N87 from a secondary tumor that metastasized to the liver. AGS cells are moderately differentiated and conserve a rapid rate of division. Metastatic NCI-N87 cells are highly differentiated and divide more slowly (Figure 1A and B). Both cell lines have the same basal cholesterol content (Figure 1C).
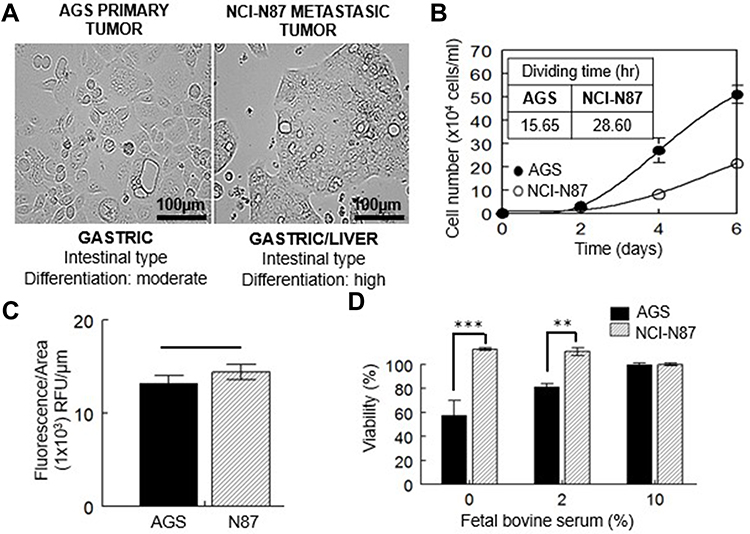 |
Figure 1 Characterization of gastric cancer cell lines. (A) Light image of AGS and NCI-N87 cells; both cell lines are classified in the same histological subtype of tumor according to Lauren’s criteria. (B) Growth curve and proliferation rates of both cell lines. (C) Free cholesterol content measured by filipin staining. (D) Viability at 48 h of AGS and NCI-N87 cells after levels of FBS were reduced. Values are expressed as the mean ± standard error of three independent experiments performed in triplicate. **p<0.01, ***p<0.001. |
When these cells were deprived of FBS for 48 h (0 or 2% of serum present in culture media), only AGS cells showed a decrease in cell viability (Figure 1D), which could be related to the different rate of division. When serum was reduced to 2%, AGS cell viability was close to 80%, in comparison to the cells grown under standard conditions (10% FBS). The total lack of serum reduced AGS viability to almost half (~57%). On the contrary, there was no effect on the viability of NCI-N87 cells grown in serum-free media or at 2% FBS (Figure 1D).
Inhibition of HMGCR by Treatment with Simvastatin and Incorporation of Mevalonolactone, FPP and GGPP
The role of isoprenoids and cholesterol in the proliferation and viability of these two cell lines was determined by testing the effect of some inhibitors on its endogenous synthesis (Figure 2A and B). The experiments were done in the presence of full serum concentration (10% FBS) to ensure cells contained normal levels of cholesterol and isoprenoids.
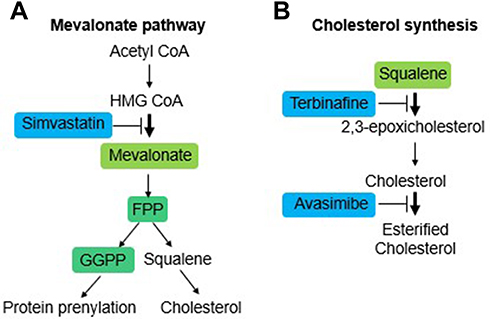 |
Figure 2 Schematic representation of the mevalonate pathway and cholesterol synthesis/esterification. (A) Upstream inhibition of the mevalonate pathway with the HMGCR inhibitor drug simvastatin. (B) Specific inhibition of cholesterol synthesis with the drug terbinafine and cholesterol esterification with the drug avasimibe. |
Figure 3 shows the effect of simvastatin treatment on AGS and NCI-N87 cells. The statin was able to induce a very rapid decrease in cell growth/viability in AGS cells at 24, 48 and 72 h, whereas NCI-N87 cells were significantly more resistant to the drug (Table 2; Figure 3A and B).
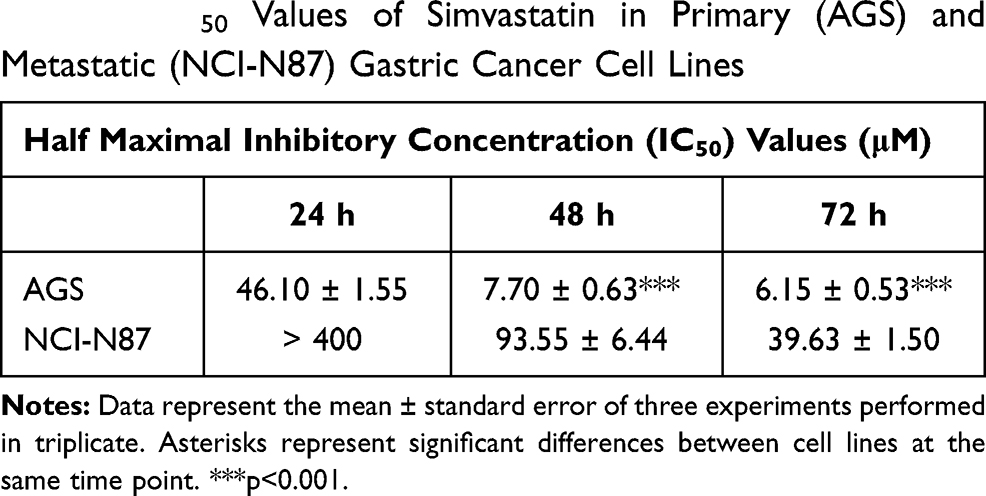 |
Table 2 IC50 Values of Simvastatin in Primary (AGS) and Metastatic (NCI-N87) Gastric Cancer Cell Lines |
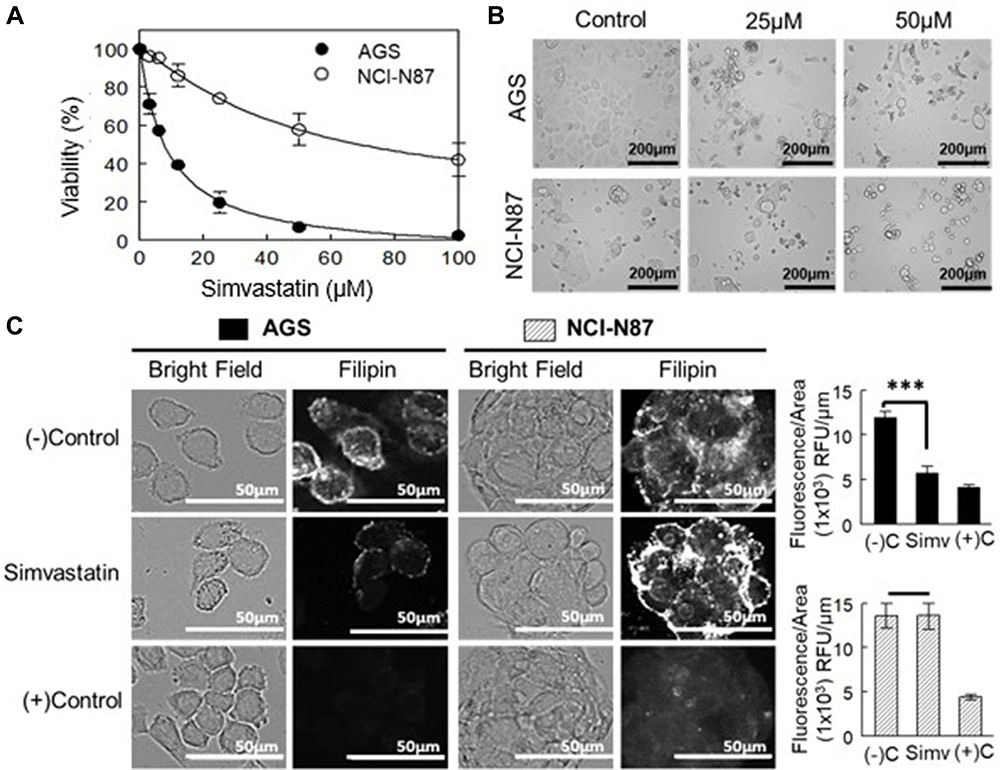 |
Figure 3 Effect of HMGCR inhibition in the proliferation and viability of AGS and NCI-N87 cells. (A) Dose–response curve of cells after 48 h of treatment with simvastatin. Values are expressed as the mean ± standard error of three independent experiments performed in triplicate. (B) Morphology of cell lines after treatment with simvastatin for 48 h. (C) Left panel: cholesterol content determination by filipin staining of cells treated with simvastatin at IC50 values for 48 h. As a positive control, cells were treated with 5 mM MβCD. Right panel: filipin fluorescence quantification adjusted by area. Values are expressed as mean ± standard error of at least ten independent images. ***p<0.001. |
An additional experiment using sulforhodamine B, instead of MTT, was performed to determine whether the decrease in cell viability was due to growth inhibition or cytotoxicity. This difference is calculated based on the results of an untreated plate, so normal proliferation can be followed during the time of the experiment. The effect of simvastatin was mainly due to the inhibition of cell growth (Supplementary Figure 1A and B).
To determine the cholesterol content in cells treated with simvastatin, AGS, and NCI-N87 cells were stained with fluorescent filipin. A simvastatin-induced reduction in fluorescence intensity was observed only in AGS cells (Figure 3C). In NCI-N87 cells, there was no effect on cholesterol levels after the treatment with simvastatin. As a positive control, both cell lines were treated with MβCD (5 mM) for 1 h, to remove all membrane cholesterol and did the filipin staining.
To determine the role of cholesterol or other mevalonate-derived molecules (isoprenoids) in cell proliferation, the experiments were performed with simvastatin at the IC50 for 48 h, in the presence of mevalonolactone, FPP, or GGPP. The recovery of cell growth was measured comparing the IC50 values after the simvastatin treatment alone. A complete recovery was observed in both cell lines when mevalonolactone was added to the media (Figure 4A).
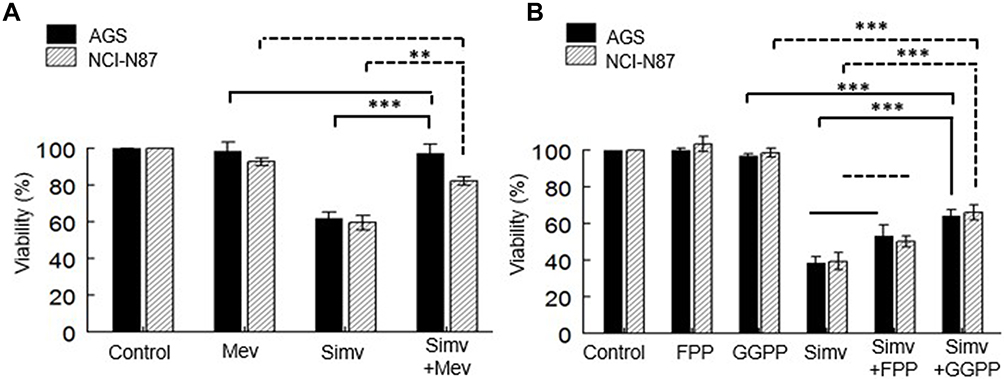 |
Figure 4 Replacement of intermediary metabolites of the mevalonate pathway. (A) Cell lines were co-incubated with simvastatin and mevalonolactone for 48 h. (B) Cell lines were co-incubated with simvastatin and FPP or GGPP for 48 h. Values are expressed as mean ± standard error of three independent experiments performed in triplicate. **p<0.01, ***p<0.001. |
The replenishment of the cells with FPP or GGPP in a co-treatment with simvastatin resulted in GGPP restoring the viability in both cells. The incorporation of FPP did not affect the decrease in cell proliferation (Figure 4B).
Inhibition of Squalene Epoxidase by Treatment with Terbinafine
A drug that affects the downstream enzyme in the mevalonate pathway, the squalene epoxidase (Figure 2B), was used to determine whether the effect in cell viability was directly associated with the presence of the cholesterol molecule (and not to isoprenoids). Resistance to the drug was observed in both cell lines (Figure 5A, Table 3). In AGS cells (cultured in advanced media to avoid cell death due to the lack of other sera-containing factors), viability significantly decreased when the FBS was reduced to 1%, an effect that was not observed on NCI-N87 cells (Table 3).
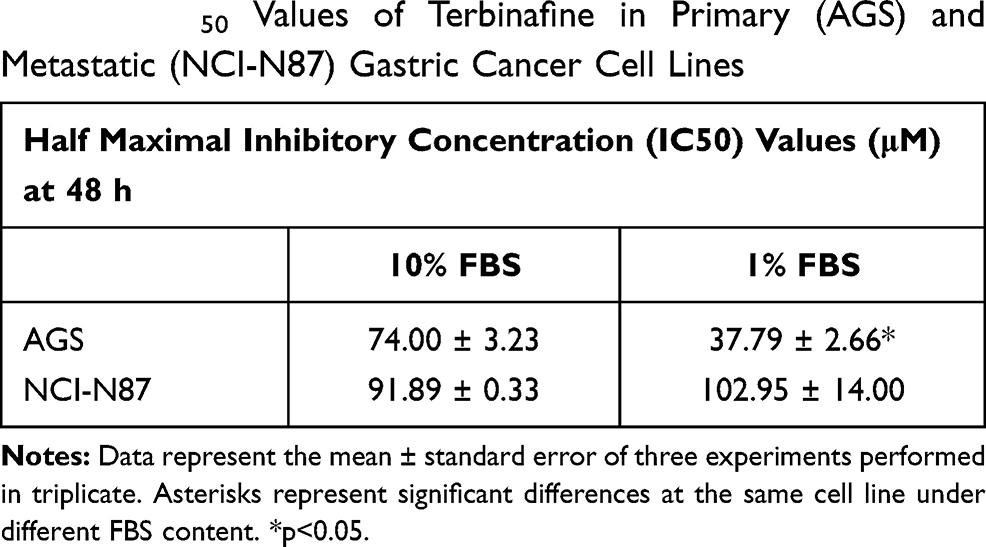 |
Table 3 IC50 Values of Terbinafine in Primary (AGS) and Metastatic (NCI-N87) Gastric Cancer Cell Lines |
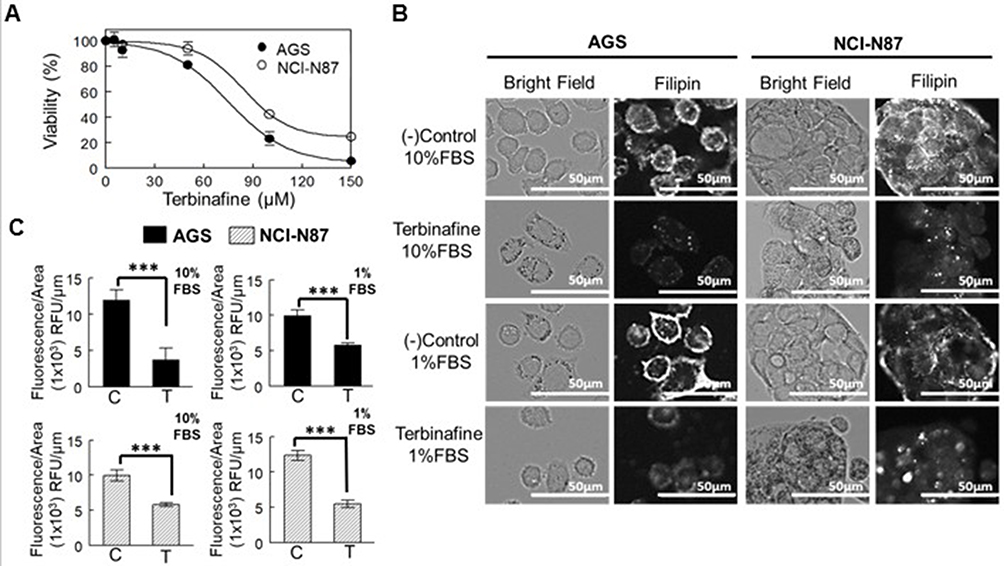 |
Figure 5 Inhibitory effect of squalene epoxidase in AGS and NCI-N87 cell viability. (A) Dose–response curve of cells after treatment with terbinafine for 48 h in media supplemented with 10% FBS. Values are expressed as mean ± standard error of three independent experiments performed in triplicate. (B) Determination of cholesterol after staining AGS and NCI-N87 cells with filipin. Cells were treated with terbinafine at IC50 value for 48 h in media supplemented with 1% (advanced media) or 10% FBS. (C) Filipin fluorescence quantification was adjusted by area. Values are expressed as mean ± standard error of at least ten independent images. ***p<0.001. |
When cell cholesterol was quantified by filipin staining at the terbinafine IC50 concentration (at 48 h), the levels of this lipid decreased in both cell lines, independently of the concentration of FBS in the media (Figure 5B and C).
Potential Regulatory Feedbacks After Inhibition of the Mevalonate Pathway
Determination of mRNA levels of HMGCR by RT-PCR showed a similar behavior when cells were treated with simvastatin or terbinafine, evidenced by an increase in gene expression in both cell lines, indicating that normal feedback in the mevalonate pathway due to a decrease in cholesterol levels was triggered (Figure 6).
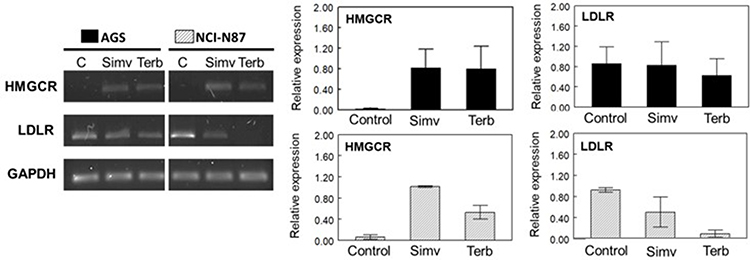 |
Figure 6 Semiquantitative RT-PCR of HMGCR and LDLR in AGS and NCI-N87 cells. Left panel: effect of simvastatin and terbinafine at IC50 values after 48 h on HMGCR and LDLR mRNA expression. GAPDH expression was used as an internal control of expression. Right panel: relative expression of HMGCR and LDLR determined against GAPDH. Values are expressed as mean ± standard error of two independent experiments. |
To test whether HMGCR could be affected by squalene accumulation due to terbinafine inhibition of its target enzyme,29 a control experiment was carried out in the presence of mevalonolactone. No compensatory effect in cell viability was observed, indicating the absence of an indirect effect of terbinafine on HMGCR activity (data not shown).
Since the effect of terbinafine was performed in media containing different concentrations of LDL-cholesterol (1% and 10% FBS-supplemented media), mRNA levels of LDLR were measured and, surprisingly, a high reduction in gene expression was observed on NCI-N87 cells. The same effect was not detected on AGS cells, suggesting some differences in the compensatory mechanism of cholesterol uptake between both cell lines (Figure 6).
Inhibition of Cholesterol Esterification by Treatment with ACAT-1 Inhibitor Avasimibe
An inhibitor of cholesterol esterification was used to determine whether free (non-esterified) cholesterol accumulation affected cell viability in primary and metastatic gastric cancer cell lines (Figure 2B). At 48 h, the IC50 value for the AGS cell line was 35.8 ± 7.4 µM, and for the NCI-N87 cell line was 60.7±4.5, showing a significant difference (p < 0.001) (Figure 7A).
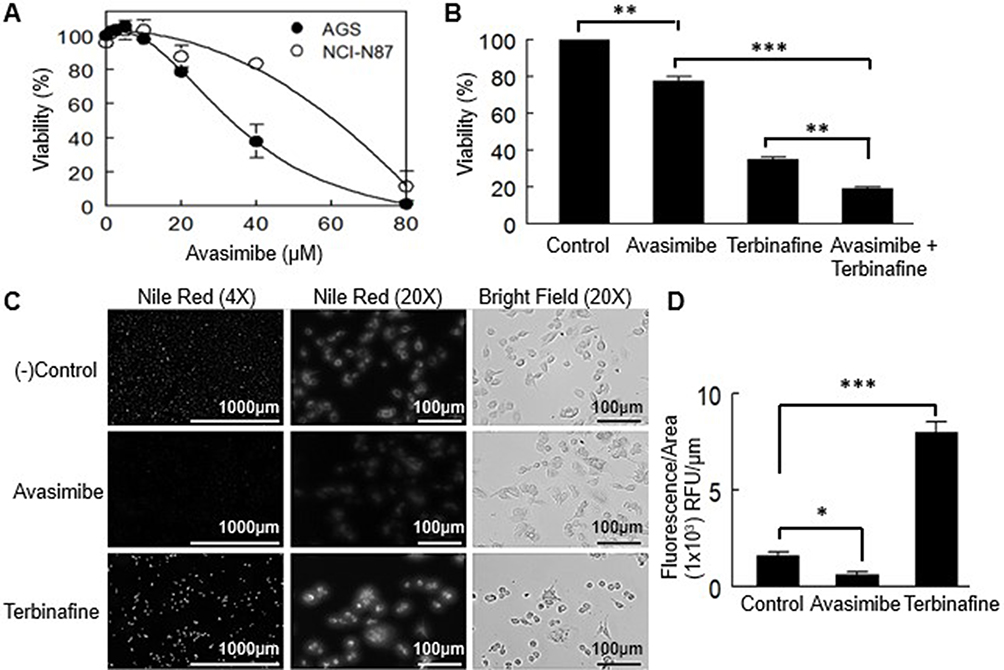 |
Figure 7 Effect of the inhibition of cholesterol esterification in the viability of AGS and NCI-N87 cells. (A) Incubation of the cells with avasimibe was done for 48 h. Values are expressed as mean ± standard error of three experiments performed in triplicate. (B) Co-incubation of avasimibe at a low dose (80% viability) and terbinafine. Values are expressed as mean ± standard error of three independent experiments each performed in triplicate. **p<0.01, ***p<0.001. (C) Nile red staining of intracellular lipid droplets in AGS cells. (D) Fluorescence quantification adjusted by area. Values are expressed as mean ± standard error of at least 4 independent images. *p<0.05, ***p<0.001. |
To determine whether the loss of cell viability induced by terbinafine in AGS cells was due to squalene-induced toxicity, a combination of both treatments (terbinafine at its IC50 and 15 µM avasimibe) was applied, and potentiation of the effect was observed (Figure 7B). That was expected if the toxicity was due to squalene accumulation, because of the reported protective effect carried out on the cells, by lipid droplets.29
To measure squalene accumulation, we stained AGS cells with Nile red, a fluorescent probe that stains lipid droplets, the reservoirs of esterified cholesterol and, as mentioned, the squalene buffering structures. As expected, terbinafine increased Nile red staining whereas avasimibe caused a decrease in lipid droplets observed as a decline in the fluorescence staining (Figure 7C and D).
Discussion
Several alterations in cancer cell metabolism, with important consequences in growth and survival, have been extensively described, some of them as the result of the dysregulation of key enzymes of specific cellular pathways.4,7 This special pattern of characteristics displayed by tumors has been included as one of the hallmarks of cancer, which is described as metabolic reprogramming and specifically in the case of this study, lipid metabolism reprogramming.16–20
The mevalonate pathway is a very conserved and essential process, which not only culminates with the synthesis of cholesterol but also produces several other very important molecules required to cell proliferation, survival, migration and invasion.2,3 The effects of a dysregulated mevalonate pathway and other aspects of cholesterol cell homeostasis in human cancers, especially in the case of metastatic tumors, remain virtually unexplored.
In the mevalonate pathway, the two enzymes that have been postulated as rate-limiting ones are HMGCR (inhibited by statins), which, from a regulatory point of view, represents the most important enzyme of the process,2 and squalene epoxidase (also known as squalene monooxygenase), an enzyme that recently has gained some attention as the second-rate limiting enzyme of the process.22 Both enzymes are regulated by cell cholesterol content, but with some variations in their control mechanisms.21 Squalene epoxidase participates in the cholesterol synthesis-committed segment of the pathway, so in the presence of terbinafine, a clinically used inhibitor of this enzyme, sterol production could be directly separated from isoprenoid synthesis. Interestingly, although this enzyme is far from being as well characterized as HMGCR, it has been demonstrated that very promising chemopreventive natural compounds, such as resveratrol and green tea polyphenols, for instance, do inhibit the monooxygenase and decrease serum cholesterol,21 which makes it a very interesting target for cancer prevention.
Clear differences between these cell lines in response to the treatment with simvastatin were observed since AGS were sensitive whereas NCI-N87 cells were more resistant. Interestingly, at 48 hours of treatment, cholesterol levels were significantly lowered only in AGS cells, and since LDL-cholesterol was highly present in the media, both cells should be able to easily internalize this lipid. However, highly proliferating cancer cells, such as AGS, could have increased requirements of cholesterol (and isoprenoids), the reason why statins have been promoted as chemopreventive agents and a treatment option for several tumors.7 Inhibition of cell proliferation was the main effect observed with this treatment and our results in the presence of mevalonolactone and isoprenoids suggest that protein geranylgeranylation was relevant (in both cell lines), for this process. However, another metabolite is likely to be considered important because the compensation with GGPP was not total, as with mevalonolactone.
Simvastatin is affecting both cell lines since HMGCR transcript concentration increased as expected, due to the normal feedback triggered by low cholesterol, but perhaps in the case of AGS, due to a higher requirement, the normal levels of this lipid could not be reached as easily.
After targeting the lower part of the mevalonate pathway with terbinafine, both cell lines were closer in terms of resistance, since this treatment was less effective than the one obtained with simvastatin, and filipin staining shows that membrane cholesterol decreased in both cell lines. In AGS cells, the resistance was significantly lower at 1% FBS, making the cells more sensitive to terbinafine. However, the differential effect induced by FBS did not correlate with the decrease in membrane cholesterol, which could indicate that the effect in cell viability induced by terbinafine could be due to other factors.
Mahoney and collaborators29 have recently shown that, in neuroendocrine cancer cells, an inhibitor of squalene monooxygenase decreases cell viability, but that the effect was not due to cholesterol decrease but to squalene accumulation. They also postulated a protective effect of lipid droplets (formed by esterified cholesterol and phospholipids) against the toxicity caused by squalene. Following this line of thought, an inhibitor of ACAT-1, the main enzyme responsible for cholesterol esterification and the formation of lipid droplets, was tested, and toxicity was observed on both cell lines. As was predicted if squalene accumulation was responsible for the toxicity, terbinafine potentiated the effect of avasimibe. To confirm our hypothesis about the direct role of squalene in the cytotoxicity observed, we quantified lipid droplets and evidenced an increase in the presence of these structures after terbinafine treatment, which clearly suggests squalene accumulation inside the cells.
In the case of metastatic cells such as NCI-N87, they could have a different capacity to deal with free cholesterol (non-esterified), an alteration that resulted in less sensitivity to avasimibe than in AGS. NCI-N87 cells, on the other hand, did not change their sensitivity to terbinafine at low FBS, probably because they are more resistant to the accumulation of squalene than AGS cells.
Contrary to expected, NCI-N87 cells displayed a decrease in the expression of LDLR due to drug treatments. Interestingly, lower levels of LDLR have been observed in advanced-stage prostate cancer cells, suggesting that, to provide cholesterol, some advanced and metastatic cancer cells could rely more on this lipid synthesis than on its uptake.30 It is possible that the regulatory mechanisms triggered in gastric metastatic cells to increase cell cholesterol could be different than the ones present in primary gastric cancer cells, favoring, in the metastasis, the synthesis of new cholesterol over the uptake. This interesting possibility needs to be clarified in the future through more sensitive approaches such as RT-qPCR, to better understand the effect of the drug treatments on gene expression of key proteins involved in cholesterol homeostasis, such as the LDLR, for instance, and others.
These results show that isoprenoids and another metabolite, probably cholesterol, are both important for maintaining cell growth and viability in AGS cells, but NCI-N87 are both resistant to simvastatin and terbinafine, which indicated that these lipids are less relevant for the survival of these metastatic cells. Another alternative could be that metastatic cells display distinct compensatory mechanisms to adjust their lipid homeostasis, something that has been suggested at least, for prostate cancer cells.
Some studies have previously evidenced that not all cancer cells are sensitive to statins and it has been suggested that sensitivity to these drugs is associated with epithelial–mesenchymal transition (EMT), postulating that statins are more effective on more aggressive (invasive-metastatic) cancer cells.31 Also, Kuzu and collaborators32 showed that statins were more effective in mesenchymal-like cancer cells. Even when AGS cells were originated from a primary tumor, according to their phenotype, they have EMT features, such as the absence of normal E-cadherin expression. NCI-N87 cells, on the contrary, which are derived from a tumor that had already metastasized (to the liver), have a more epithelial-like phenotype.22 NCI-N87 cells are also highly differentiated and show a lower proliferation rate than AGS cells. Interestingly, in recent studies, Raghu et al33 have suggested that the expression of E-cadherin could be associated with resistance to atorvastatin-induced growth suppression of several cancer cells and Warita et al34 observed that E-cadherin exogenous expression transformed statin-sensitive cells into partially resistant ones.
Another cell feature that could be important in the resistance of NCI-N87 to these treatments is the presence of HER2. However, the inhibition of this pathway alone was not enough to induce a significant decrease in cell growth and viability, but there is a possibility that it could overcome, at least partially, the resistance to chemotherapy. Cholesterol is the main component of lipid rafts and it is very well established that modulating membrane cholesterol content strongly alters the movement, interactions and activities of lipid raft proteins, such as death receptors, growth factor receptors and oncogenes, such as EGFR and HER2, for instance, important for several types of cancer, including gastric tumors.
In terms of clinical treatment, gastric cancer patients get a very late diagnosis and a poor prognosis, so it is imperative to find more effective treatment options, especially for advanced and metastatic gastric cancer. For aggressive and highly invasive primary gastric tumors and secondary recurrences, statins could be an interesting treatment option, maybe in combination with other drugs, since, according to our results, the IC50 of simvastatin for AGS cells is relatively low and in the predicted anti-tumor in vivo range. That is not the case for the metastatic cell line NCI-N87, which is clearly highly resistant to the drug.
For more differentiated metastatic gastric cancer, on the other hand, other therapeutic strategies must be developed to successfully sensitize these tumors and overcome their resistance to conventional therapies. The cholesterol esterification process or the LDL-cholesterol uptake could be interesting target options that remained to be explored.
Funding
This study was partially supported by Vicerrectoría de Investigación (grant 422‑B7‑098) and Sistema de Estudios de Posgrado, both from the Universidad de Costa Rica.
Disclosure
The authors reported no conflicts of interest for this work.
References
1. Lim SC, Parajuli KR, Duong HQ, Choi JE, Han SI. Cholesterol induces autophagic and apoptotic death in gastric carcinoma cells. Int J Oncol. 2014;44:805–811. doi:10.3892/ijo.2014.2246
2. Bathaie SZ, Ashrafi M, Azizian M, Tamanoi F. Mevalonate pathway and human cancers. Curr Mol Pharmacol. 2017;10:77–85. doi:10.2174/1874467209666160112123205
3. Clendening JW, Penn LZ. Targeting tumor cell metabolism with statins. Oncogene. 2012;31:4967–4978. doi:10.1038/onc.2012.6
4. Sharpe LJ, Brown AJ. Controlling cholesterol synthesis beyond 3-Hydroxy-3-methylglutaryl-CoA reductase (HMGCR). J Biol Chem. 2013;288:18707–18715. doi:10.1074/jbc.R113.479808
5. Hryniewicz-Jankowska A, Augoff K, Biernatowska A, Podkalicka J, Sikorski AF. Membrane rafts as a novel target in cancer therapy. Biochim Biophys Acta Rev Cancer. 2014;1845:155–165. doi:10.1016/j.bbcan.2014.01.006
6. Murai T. Cholesterol lowering: role in cancer prevention and treatment. Biol Chem. 2014;396:1–11. doi:10.1515/hsz-2014-0194
7. Iannelli F, Lombardi R, Milone MR, et al. Targeting mevalonate pathway in cancer treatment: repurposing of statins. Recent Pat Anticancer Drug Discov. 2018;13:184–200. doi:10.2174/1574892812666171129141211
8. Danilo C, Frank PG. Cholesterol and breast cancer development. Curr Opin Pharmacol. 2012;12:677–682. doi:10.1016/j.coph.2012.07.009
9. Pelton K, Freeman MR, Solomon KR. Cholesterol and prostate cancer. Curr Opin Pharmacol. 2012;12:751–759. doi:10.1016/j.coph.2012.07.006
10. Clendening JW, Pandyra A, Boustros PC, El Ghamrasni S, Khosravi F. Dysregulation of the mevalonate pathway promotes transformation. Proc Natl Acad Sci USA. 2010;107:15051–15056. doi:10.1073/pnas.0910258107
11. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
12. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2014;136:E359–E386. doi:10.1002/ijc.29210
13. Dittman Y, Settmacher U. Individualized treatment of gastric cancer: impact of molecular biology and pathohistological features. World J Gastrointest Oncol. 2015;7:292–302. doi:10.4251/wjgo.v7.i11.292
14. Riihimäki M, Thomsen H, Hemminki A, Sundquist K, Hemminki K. Comparison of survival of patients with metastases from known versus unknown primaries: survival in metastatic cancer. BMC Cancer. 2013;13:36. doi:10.1186/1471-2407-13-36
15. Chushi L, Wei W, Kangkang X, Yongzeng F, Ning X, Xiaolei C. HMGCR is up-regulated in gastric cancer and promotes the growth and migration of the cancer cells. Gene. 2016;587:42–47. doi:10.1016/j.gene.2016.04.029
16. Cheng C, Geng F, Cheng X, Guo D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun. 2018;38:27. doi:10.1186/s40880-018-0301-4
17. Göbel A, Rauner M, Hofbauer LC, Rachner TD. Cholesterol and beyond-the role of the mevalonate pathway in cancer biology. Biochim Biophys Acta Rev Cancer. 2020;1873:188351.
18. Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science. 2020;368. doi:10.1126/science.aaw5473
19. Matsushita Y, Nakagawa H, Koike K. Lipid metabolism in oncology: why it matters, how to research and to treat. Cancers. 2021;13:474. doi:10.3390/cancers13030474
20. Ohshima K, Morii E. Metabolic reprogramming of cancer cells during tumor progression and metastasis. Metabolites. 2021;11:28. doi:10.3390/metabo11010028
21. Gill S, Stevenson J, Kristiana I, Brown AJ. Cholesterol-dependent degradation of squalene monooxygenase, a control point in cholesterol synthesis beyond HMG-CoA reductase. Cell Metab. 2011;13:260–273. doi:10.1016/j.cmet.2011.01.015
22. Liu D, Wong CC, Fu L, et al. Squalene epoxidase drives NAFLD-induced hepatocellular carcinoma and is a pharmaceutical target. Sci Transl Med. 2018;10(437):eaap9840. doi:10.1126/scitranslmed.aap9840
23. Goulitquer S, Croyal M, Lalande J, et al. Consequences of blunting the mevalonate pathway in cancer identified by a pluri-omics approach. Cell Death Dis. 2018;9:745. doi:10.1038/s41419-018-0761-0
24. Ahmadi Y, Ghorbanihaghjo A, Argani H. The balance between induction and inhibition of mevalonate pathway regulates cancer suppression by statins: a review of molecular mechanisms. Chem Biol Interact. 2017;273:e285.
25. Ortiz N, Díaz C. Mevalonate pathway as a novel target for the treatment of metastatic gastric cancer. Oncol Lett. 2020;20:320. doi:10.3892/ol.2020.12183
26. Mazieres J, Pradines A, Favre G. Perspectives on farnesyl transferase inhibitors in cancer therapy. Cancer Lett. 2004;206:159–167. doi:10.1016/j.canlet.2003.08.033
27. Dong W, Vuletic S, Albers JJ. Differential effects of simvastatin and pravastatin on expression of Alzheimer's disease-related genes in human astrocytes and neuronal cells. J Lipid Res. 2009;50:2095–2102. doi:10.1194/jlr.M900236-JLR200
28. Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1:1112–1116. doi:10.1038/nprot.2006.179
29. Mahoney CE, Pirman D, Chubukov V, et al. A chemical biology screen identifies a vulnerability of neuroendocrine cancer cells to SQLE inhibition. Nat Commun. 2019. doi:10.1038/s41467-018-07959-4
30. Huang B, Song B, Xu C. Cholesterol metabolism in cancer: mechanisms and therapeutic opportunities. Nat Metab. 2020;2:132–141. doi:10.1038/s42255-020-0174-0
31. Yu R, Longo J, van Leeuwen JE, et al. Statin-induced cancer cell death can be mechanistically uncoupled from prenylation of RAS family proteins. Cancer Res. 2018;78:1347–1357. doi:10.1158/0008-5472.CAN-17-1231
32. Kuzu OF, Noory MA, Robertson GP. The role of cholesterol in cancer. Cancer Res. 2016;76:2063–2070. doi:10.1158/0008-5472.CAN-15-2613
33. Raghu VK, Beckwitt CH, Warita K, Wells A, Benos PV, Oitvai ZN. Biomarker identification for statin sensitivity of cancer cell lines. Biochem Biophys Res Commun. 2018;495:659–665. doi:10.1016/j.bbrc.2017.11.065
34. Warita K, Warita T, Beckwitt CH, et al. Statin-induced mevalonate pathway inhibition attenuates the growth of mesenchymal-like cancer cells that lack functional E-cadherin mediated cell cohesion. Sci Rep. 2014;4:7593. doi:10.1038/srep07593
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.