")
Back to Archived Journals » Comparative Effectiveness Research » Volume 5
Implementation of comparative effectiveness research in personalized medicine applications in oncology: current and future perspectives
Authors IJzerman M , Manca A, Keizer J, Ramsey S
Received 11 July 2015
Accepted for publication 20 August 2015
Published 26 November 2015 Volume 2015:5 Pages 65—72
DOI https://doi.org/10.2147/CER.S92212
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Matthew Maciejewski
Maarten J IJzerman,1,3 Andrea Manca,2,3 Julia Keizer,1 Scott D Ramsey4
1Department of Health Technology and Services Research, University of Twente, Enschede, the Netherlands; 2Centre for Health Economics, University of York, York, UK; 3Department of Population Health, Luxembourg Institute of Health, Strassen, Luxembourg, 4Fred Hutchinson Cancer Research Center, Seattle, WA, USA
Abstract: Personalized medicine (PM) or precision medicine has been defined as an innovative approach that takes into account individual differences in people's genes, environments, and lifestyles in prevention and treatment of disease. In PM, genomic information may contribute to the molecular understanding of disease, to optimize preventive health care strategies, and to fit the best drug therapies to the patient's individual characteristics. Evidence development in the era of genomic medicine is extremely challenging due to a number of factors. These include the rapid technological innovation in molecular diagnostics and targeted drug discoveries, and hence the large number of mutations and multiple ways these may influence treatment decisions. Although the evidence base for PM is evolving rapidly, the main question to be explored in this article is whether existing evidence is also fit for comparative effectiveness research (CER). As a starting point, this paper therefore reflects on the evidence required for CER and the evidence gaps preventing decisions on market access and coverage. The paper then discusses challenges and potential barriers for applying a CER paradigm to PM, identifies common methodologies for designing clinical trials in PM, discusses various approaches for analyzing clinical trials to infer from population to individual level, and presents an example of a clinical trial in PM (The RxPONDER TRIAL) demonstrating good practice. The paper concludes with a future perspective, including modeling approaches for evidence synthesis.
Keywords: personalized medicine, precision medicine, comparative effectiveness research, health technology assessment, oncology, genomics
Introduction
The US Institute of Medicine defines comparative effectiveness research (CER) as the generation and synthesis of evidence that compares the benefits and harms of alternative methods for preventing, diagnosing, treating, and/or monitoring clinical conditions, or for improving the delivery of care. The purpose of CER is to assist consumers, clinicians, purchasers, and policy makers to make informed decisions that will improve health care at both the individual and population levels.1
While CER has emerged in the USA, the European Union has less strictly called for CER as a new paradigm, partly because of the rigorous health technology assessment (HTA) processes that exist in many European Union countries. A central tenet of HTA is the use of pragmatic clinical trials to assess at least two interventions used in clinical practice. European Guidelines in HTA suggest that two or more existing strategies should be compared under real-life circumstances in order to produce meaningful and scientific legitimate (or evidence-based) decision support.
Several authors have discussed the challenges of applying CER to the field of personalized or precision medicine.2–6 Personalized medicine (PM), recently also introduced as “precision medicine” refers to the change from a “one-size-fits-all” approach, to getting the “right treatment for the right individual”. The genomic information may concern the patient’s inherited DNA (germline) or changes to DNA that have occurred over time that are not heritable, including those changes that result in malignancies (somatic DNA and changes). This is particularly true in oncology, where medicines are being developed to target specific mutations found in patients’ tumors. One of the most important developments in PM is genome sequencing, leading to dramatically lower costs and increased speeds of characterizing tumor DNA. This revolution is – despite many clinical and technical hurdles – facilitating this change.7
The evidence base for PM is evolving rapidly, but the question for this article is whether the evidence being generated fits the requirements in CER. The focus in this paper is oncology, arguably the area with the most activity in genomically-targeted treatment. We posit that for oncology the evidence base generally does not fit an ideal for CER. Yet, if CER could demonstrate improved effectiveness of PM at the individual level or in subpopulations in the future, it may stimulate the development of products that are narrowly targeted to patients with specific genetic or other characteristics.8 In this way, PM can contribute to better customized care, with better health outcomes for the individual at a lower cost for society because less people will receive ineffective drug treatment.
This paper starts with a discussion about the specifics of CER in PM by reviewing the literature and identifying known evidence gaps. Second, several issues for developing clinical trials for PM are discussed while specifically looking into some specific design challenges and statistical methods to infer from the population to the individual level. As an example, a trial was designed for PM application and was introduced as a best practice. Finally, a future perspective is offered identifying some recent methodological progress generating better information on the comparative effectiveness of genomically-targeted therapies relative to other accepted cancer care.
From personalized and precision medicine to personalized health care
While the terms PM and precision medicine are adopted by large clinical and research communities, its definition is still vague and subject to controversy. This is mainly (or partly) due to the fact that PM can be observed from different points of view. The following two definitions illustrate the issue. Schleidgen et al (re)define PM as: “to improve stratification and timing of health care by utilizing biological information and biomarkers on the level of molecular disease pathways, genetics, proteomics as well as metabolomics”.9 The Obama administration defines PM as “[…] an innovative approach to disease prevention and treatment that takes into account individual differences in people’s genes, environments, and lifestyles” (https://www.whitehouse.gov/the-press-office/2015/01/30/fact-sheet-president-obama-s-precision-medicine-initiative). These different points of view have led to some discussion whether the term “precision” medicine indeed is different from PM.10
What is even more interesting, though, is that a large group of people prefer the term “personalized health care” to make explicit that there is a general trend of tailoring health care to the specific needs of patients. In personalized health care, managing patient heterogeneity in terms of their biology, values and preferences is important. Stratification of patients based on molecular profile alone would be sub optimal, given differences in clinical characteristics, patients’ preferences and values which affect the effectiveness of treatment.11 The personalization of health care should therefore be seen as an integrated whole, tailoring health care to individual patients.
Personalized health care and CER
To inform regulatory agencies about approval of new pharmaceuticals, three relevant developments have emerged over the past decade, including evidence-based medicine, CER, and HTA. In their insightful overview, Luce et al contrast CER, evidence-based medicine, and HTA.12 Although there are many similarities, a main difference is that HTA traditionally takes the costs into the equation and more specifically informs reimbursement decisions.12 However, both CER and HTA aim to inform decision makers about whether an intervention will work in practice (Figure 1).
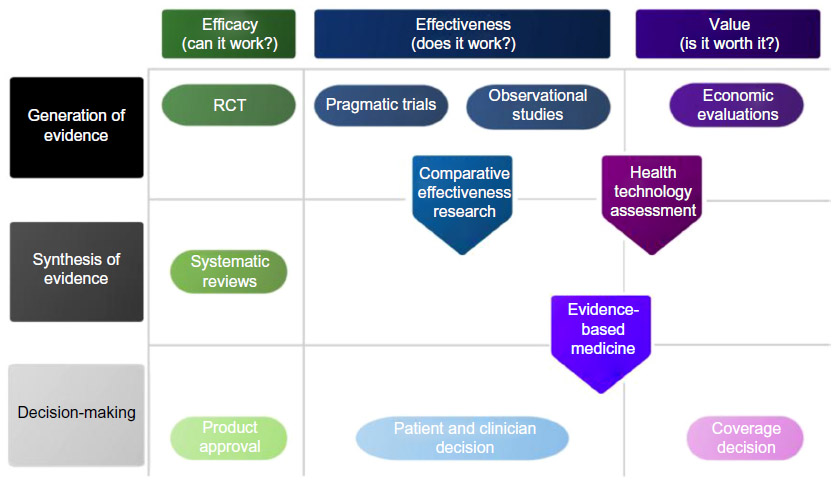 | Figure 1 Comparison of comparative effectiveness research, evidence-based medicine, and health technology assessment. |
The introduction of CER has led to an intense discussion amongst academics and policy makers, since CER and HTA both analyze the effectiveness of medical treatment, with the intent of informing clinical decisions and policies affecting health care.5 The common methodological framework in CER is the randomized controlled trial (RCT), although several other clinical research strategies may be used.13,14 While comparison of two or more drugs at the same time is essential for CER, the US Food and Drug Administration (FDA) enabling law (FDC Act, as amended in 1962) does not require assessment of comparative effectiveness for decision support. Furthermore, the US legislative history made it very clear there was no requirement to determine relative effectiveness. A new drug does not have to be better than, or even as good as, existing treatment to be approved by the FDA. This is similar to the European Medicines Agency regarding their market authorization process, which basically evaluates the therapeutic safety and harm of a new drug.
Several papers have been published discussing the challenges in evidence development for PM. Conti et al identified five major challenges that need to be overcome while translating the promise of genomics into PM, based on a panel discussion with experts.3 Most of these are related to issues as discussed in CER, including: 1) the limited quantity and quality of available evidence of effectiveness, 2) the complexity of information provided by currently available tests (for success, many genes should be linked to many diseases), 3) the rapid commercialization of new genetic tests with limited oversight or regulation of genetic testing, 4) apprehension of genomics by physicians, patients, and payers, and 5) the uncertainty about clinical utility. With regard to the latter, many other experts agree that clinical utility – the impact of the PM test on patient outcomes – is more important15 and should drive CER.16,17
While these five issues are generally acknowledged, many experts have argued that, while evidence development in theory is not different, there are several other barriers that impede the translation of PM to clinical practice.
One such barrier, preventing evidence development of PM, is pointed out by Towse and Garrison. They argued that there is generally a disincentive for manufacturers to generate the evidence for molecular tests, as more detailed information about biomarkers will reduce the target population size for a drug and thus will decrease the returns for industry.18 The complexity of getting the incentives right, for translating PM to the clinic, requires the involvement of stakeholders to examine unexpected implementation problems and to better design CER to inform decision-making, supporting the argument of a multiple stakeholder approach in designing CER.19
Similar with the notion of the importance of clinical utility, another discussion arises, ie, how to determine value. In this respect, the choice for a clinical end point in clinical trials is far from trivial. CER aims to inform clinicians, patients, and policy makers about the value of diagnostic tests, and PM may be quite different to these stakeholders. Phillips et al pointed out that for effective translation to the market, a systematic analysis of utilization, patient preferences, and economic value is required.20 Although this paper is not specifically about the economic value of PM, we pose measuring patient preferences to measure value in addition to clinical outcomes to be important.21 Typically, discrete-choice experiments or multi-criteria decision analysis are used to quantify stakeholder weights for clinical outcomes and process of care.22,23
Consequences for trial design and analysis in PM
One of the biggest challenges to PM is that RCTs are designed to draw conclusions at the population level, where PM specifically is interested in the individual level. The complexity arises because trials cannot be designed for a specific subgroup, as these groups may not be known a priori. And if those subgroups would be identified a priori, treatments are selected based on the presence or absence of molecular markers and thus additional diagnostic tests.
I-Spy was one of the first unique trials dealing with the complex issues of designing RCTs allowing treatment selection based on specific marker expressions.24,25 In general, three challenges related to RCT design in PM are discussed here: 1) including identifying and handling of multiple subgroups, 2) complexity of mutations and tumor biology and 3) statistical approaches to analyze data at the individual level.
The large number of biomarkers creates a large quantity of subgroups
In the first place, it is not possible to design RCTs for subgroups that are unknown at the design stage. RCTs need to be certain about which markers to target.26 Companion diagnostic tests are expected to identify potential responders, but it is hard to do a trial comparing potential responders to nonresponders. Such a trial needs to be designed smart, and requires many patients to avoid low statistical power. As treatment is based on the presence or absence of a particular mutation, it is hard to design controlled trials randomizing patients with all different and unique combinations of mutations. Such trials are only possible in case of either a single or small number of mutations. However, with the availability of whole genome sequencing in clinical patient care, the amount of information that will become available is extremely complex to handle in RCTs.
Genetic mutations are less tumor specific, and expressions are dynamic and can differ between early cancer and metastatic disease
The information provided by sequencing, combined with the growing literature cataloging how these disruptions affect the biological function of the cancer cells across tumor types, is hinting at a future where cancer therapy will be defined not only by the site of the cancer (eg, breast or lung) but also by matching the patient’s aberrant genomic morphology to one or more drugs that have been shown to be effective in targeting those disruptions in those tumors. A roadmap has been published outlining the process by which oncology will move from the current model of patients being managed through well-defined and referenced therapy “pathways” to highly variable individualized combinations of treatments that are based on a genomic signature specific to the patient’s tumor.
Part of this is the growing evidence base that genomic expressions are dynamic over time and differ between early stage tumors and metastatic disease.27,28 Many biologicals are being developed targeting the same molecular mechanism, yet the expressions may change over time making the drug less or more effective.29 For clinical trial design, such dynamic behavior is hard to control for and potentially may lead to misclassification affecting the power of the study. In addition to these considerations, there is a need for further research on the identification and interpretation of response markers for clinical decision-making. For instance, circulating tumor cells or cell-free DNA may be considered as markers of treatment response, to guide switching therapies.30 Currently, clinical trials are being prepared investigating the use of such markers in addition to clinical examination and imaging biomarkers (studies on switching from first-line [docetaxel] to second-line [cabazitaxel] chemotherapy in metastatic castration-resistant prostate cancer in the USA [clinicaltrials.gov: NCT01718353] and the CTC-Stop trial in the UK are led by Prof Johann de Bono at Institute for Cancer Research, London).
Statistical approaches to infer from the population to the individual
One specific problem in the analysis of RCT data for PM has to do with moving away from population mean treatment effect to focus on subgroup – or even individual-level treatment effect. Several statistical approaches can be identified in this respect; however, the statistical analysis of RCT data to derive PM recommendations requires careful considerations. Before starting with the analysis, researchers need to consider the RCT design used that generated the data at hand; a general principle of good practice and applied research31 valid both for population-average and individualized treatment decisions alike. Conditional on the study design, researchers will then need to define the analytical paradigm within which they intend to operate, identifying the individualized treatment rule (ie, a model that gives the optimal treatment for each patient [this is often based on Rubin’s potential outcomes framework]);32 the (type of) predictor and prognostic variables to be used in the model; evaluating the performance of the models (in terms of goodness-of-fit and internal validation); choosing a method of inference based on the type of outcome variable and the data dimension; fitting the model to a training data set (to identify key covariates and estimate model parameters) and applying the treatment rules to future patients (to assess the prognostic and predictive ability of the model and select the optimal treatment).33 In this sense, the trial design will be influential in determining the analytical derivation of individualized treatment rules. The above analytical sequence is appropriate when the individualized treatment rule is to take place at one time point only. A number of more general (possibly realistic) cases encompassing sequential treatment decisions can be imagined to take place in oncology, whereby, following treatment failure, patients and their clinicians may choose to switch drug therapy.34 The analysis of these types of trials require not only careful design35 but more sophisticated dynamic treatment regimen analytical models,36 which take into account information from the patient’s past history and any other relevant factors (eg, treatment preferences). Finally, more recently authors have argued that the analysis of RCT data for PM is an evolutionary process in which system biology-based in-silico models are developed.37 These models integrate RCT data and multiple complementary information from other sources (eg, biobanks) and constantly update with more information as this is generated.
The RxPONDER trial: a multi-stakeholder perspective
From the previous sections it emerges that clinical trial design in PM is challenging in multiple ways. The RxPONDER38 trial describes good practice while referring to the issues discussed in the section “Personalized health care and CER”, by: 1) overcoming the complexity of information provided by currently available tests (clear linkage of specific genes to therapy); 2) apprehension of genomics by physicians, patients, and payers by involving multiple stakeholders; and 3) emphasizing the clinical utility with regard to treatment consequences.
The RxPONDER trial is designed to evaluate the role of a gene expression profile test, the 21-gene assay (Oncotype DX®), for women with early stage breast cancer involving the lymph nodes.38 Guidelines suggest that women with lymph node-positive breast cancer should be routinely offered chemotherapy but there is evidence that many patients who receive chemotherapy do not benefit in terms of reducing their risk of cancer recurrence.1 OncotypeDX® is now being marketed as an option for patients with hormone-receptor-positive breast cancer involving zero to three positive nodes, but the test’s role among women with node-positive disease has not been established. Existing evidence includes retrospective evaluations of two randomized clinical trials finding that the 21-gene assay recurrence score (RS) had reasonable predictive value in identifying women who will benefit from chemotherapy with node-positive breast cancer.2,3
While reducing the use of chemotherapy in women who will not benefit from it could potentially spare thousands from chemotherapy-related morbidity and reduce the related expenditures substantially, it is important to have high-quality evidence from a prospective clinical trial due to the risks of not giving chemotherapy to those who would otherwise benefit. Another way to describe this issue is that better evidence is needed to find an RS value that produces an acceptable level “false negative” RS. Existing retrospective data are insufficient to determine whether the RS can be calibrated to provide a balance that would be acceptable to women and providers.4
To evaluate the comparative effectiveness of management using OncotypeDX® versus current practice, South-West Oncology Group (SWOG), a large US-based cancer clinical trials cooperative group, in conjunction with the US National Cancer Institute, designed a prospective clinical trial, SWOG S1007 (clinical trial registry: NCT01272037), given the acronym RxPONDER.38 This multicenter Phase III trial targets women with hormone-receptor-positive and HER2-negative breast cancer involving 1–3 lymph nodes. Women with a 21-gene assay RS of 25 or less are randomized to endocrine therapy alone versus chemotherapy followed by endocrine therapy. Uniquely, the trial design phase involved active participation of an external stakeholder group prior to finalizing the trial design and end points for the purpose of ensuring that the study results would be informative to patients, clinicians, and payers while balancing considerations such as internal validity and trial feasibility. Another important feature addressed the problem that reimbursement for genomic tests in the context of a clinical trial is unclear.
Several stakeholders identified the importance of looking into patient preferences. It is known that some patients ignore the results of their 21-gene RS test, which potentially may reduce its clinical utility and thus cost-effectiveness. Therefore, more specific information about the patients will help practitioners to understand influences on patient decision-making in breast cancer care.
Working with the stakeholder group and many health insurers, a joint funding arrangement involving health insurers and the National Cancer Institute was created for the RxPONDER study. The trial began enrolling patients in September 2013. Table 1 lists the primary aim and study hypothesis, while comparative effectiveness questions for S1007 can be found in the original publication.38
 | Table 1 Primary and secondary objectives and hypotheses for the RxPONDER study |
Future perspectives
While several good practices for designing clinical trials in PM can be identified and presented, the ongoing development and application of its precepts is dynamic. Over time, current evidence for development paradigms are likely to be insufficient, because they are largely based on a standardized comparison between two or more treatments in a well-identified population. These principles are challenged in PM. In this perspective, we pose that three methodological developments are required and may contribute to advancement of PM, which include 1) modeling approaches, 2) use of “big data”, and 3) resource constraints in health service delivery.
First, the target population may not be well defined, and the treatment strategies can be dynamic, sequential, and very specific for the individual. In such cases where standard RCTs cannot be carried out easily, simulation modeling approaches may be useful.39 However, standard modeling such as being employed in CER and HTA is not appropriate in PM for three distinct reasons. Standard cohort modeling methods, such as Markov state-transition models, may not be sufficient because such models have difficulties in accommodating time-dependent behavior (eg, the patient’s reaction and behaviour as a result of specific clinical events, test results, etc), often an important feature of PM. As cancer increasingly is considered a chronic disease, it is required to model entire care pathways using patient-level data. In addition, molecular diagnostics for evaluating mutations in tumor biology are increasingly being employed along the clinical care pathway as a response marker to guide switches from first-line to second-line chemotherapy and further. These dynamics require more understanding of the tumor biology, mechanism to capture tumor tissue and analytic capacity to identify treatment response, how patients’ preferences change over time, and to integrate these in CER.
Large observational studies and databases that contain extensive genomic, phenotype, socioeconomic, clinical, and outcomes data for thousands of patients (“big data”) are potentially useful additions to more commonly used health outcomes data collected in trials and other planned clinical research.40 Although planned data collection is reliable and less prone to all sorts of bias, it is also of limited value to evaluate the dynamics in the health system. In particular, observational studies and other big data sources would allow more detailed analysis of health operations, compliance to guidelines, and the dynamic interactions in the system, and how they impact patients in the context of PM. These data sources may be useful in addition to other registries, with an emphasis on the use of claims and administrative data and electronic medical records. Claims databases and electronic medical records provide a rich source of data to analyze trends in health care and health care utilization. Claims and administrative data sources would allow evaluation of access to care, and also monitor the amount of physician–physician interactions as a proxy for quality of care evaluating the effect of care coordination programs.41 Several computational approaches are used and explored to mine big data that may also apply to PM.42 But other statistical approaches that deal with observational data sets are promising, such as propensity scores or the instrumental variable approach.43 In particular, the instrumental variable approach is gaining popularity as a method to translate the average treatment effect into a patient-level treatment effect, accounting for patient-level heterogeneity.44
Finally, CER intends to generalize findings from clinical trials to inform health policy. Resource constraints (eg, lack of sequencing capacity) determine utilization, and should also be captured in models to inform health policy. One future direction that has found its way into outcomes research is dynamic simulation modeling.45,46 Dynamic simulation modeling methods, such as discrete-event simulation and agent-based modeling, may be used to incorporate the complex interactions in to the health care delivery system, the emergent behavior of the system, and the dynamics of diagnostic and treatment interactions, which are very useful in PM. In addition to such dynamic modeling approaches, the complexity of translating PM to the clinic requires an examination and involvement of stakeholders, supporting the argument of a multiple stakeholder approach in designing CER.17,19
Executive summary
Evidence development in the era of genomic medicine is extremely challenging due to a number of factors. These include the rapid technological innovation in molecular diagnostics and targeted drug discoveries, and hence the large number of mutations and multiple ways these may influence treatment decisions. The evidence base for PM is evolving rapidly, but the question for this article is whether the evidence being generated fits the requirements in CER.
Although several best practices can be identified, such as the RxPONDER trial, current evidence for development paradigms may be insufficient, because they are largely based on a standardized comparison of two or more treatments in a well-identified population. These principles are challenged in PM. In this perspective, we pose that three methodological developments are required and may contribute to advancement of PM, which include: 1) modeling approaches, 2) use of big data, and 3) methods to incorporate resource and system constraints in health care delivery models.
Disclosure
The authors report no conflicts of interest.
References
Institute of Medicine (IOM). Initial National Priorities for Comparative Effectiveness Research. Washington, DC: The National Academies Press; 2009. | |
Annemans L, Redekop K, Payne K. Current methodological issues in the economic assessment of personalized medicine. Value Health. Sep 2013;16(6 Suppl):S20–S26. | |
Conti R, Veenstra DL, Armstrong K, Lesko LJ, Grosse SD. Personalized medicine and genomics: challenges and opportunities in assessing effectiveness, cost-effectiveness, and future research priorities. Med Decis Making. 2010;30(3):328–340. | |
Epstein R, Teagarden JR. Comparative effectiveness and personalized medicine: evolving together or apart? Health Aff (Millwood). 2010; 29(10):1783–1787. | |
Garber AM, Tunis SR. Does comparative-effectiveness research threaten personalized medicine? N Engl J Med. 2009;360(19):1925–1927. | |
Ramsey SD, Veenstra D, Tunis SR, Garrison L, Crowley JJ, Baker LH. How Comparative Effectiveness Research Can Help Advance “Personalized Medicine” In Cancer Treatment. Health Aff (Millwood). 2011;30(12):2259–2268. | |
Tuna M, Amos CI. Genomic sequencing in cancer. Cancer Lett. 2013;340(2):161–170. | |
Downing GJ. Key aspects of health system change on the path to personalized medicine. Transl Res. 2009;154(6):272–276. | |
Schleidgen S, Klingler C, Bertram T, Rogowski WH, Marckmann G. What is personalized medicine: sharpening a vague term based on a systematic literature review. BMC Med Ethics. 2013;14:55. | |
Rubin R. Precision medicine: the future or simply politics? JAMA. 2015;313(11):1089–1091. | |
Espinoza MA, Manca A, Claxton K, Sculpher MJ. The value of heterogeneity for cost-effectiveness subgroup analysis: conceptual framework and application. Med Decis Making. 2014;34(8):951–964. | |
Luce BR, Drummond M, Jönsson B, et al. EBM, HTA, and CER: clearing the confusion. Milbank Q. 2010;88(2):256–276. | |
Lyman GH, Hirsch B. Comparative effectiveness research and genomic personalized medicine. Personalized Medicine. 2010;7(3):223–227. | |
Petticrew M, Roberts H. Evidence, hierarchies, and typologies: horses for courses. J Epidemiol Community Health. 2003;57(7):527–529. | |
Fryback DG, Thornbury JR. The efficacy of diagnostic imaging. Med Decis Making. 1991;11(2):88–94. | |
Ginsburg GS, Kuderer NM. Comparative effectiveness research, genomics-enabled personalized medicine, and rapid learning health care: a common bond. J Clin Oncol. 2012;30(34):4233–4242. | |
Khoury MJ, Berg A, Coates R, Evans J, Teutsch SM, Bradley LA. The Evidence Dilemma In Genomic Medicine. Health Aff (Millwood). 2008;27(6):1600–1611. | |
Towse A, Garrison LP Jr. Economic incentives for evidence generation: promoting an efficient path to personalized medicine. Value Health. 2013;16(6 Suppl):S39–S43. | |
Deverka PA, Schully SD, Ishibe N, et al. Stakeholder assessment of the evidence for cancer genomic tests: insights from three case studies. Genet Med. 2012;14(7):656–662. | |
Phillips KA, Liang S-Y, van Bebber S; Canpers Research Group. Challenges to the translation of genomic information into clinical practice and health policy: Utilization, preferences and economic value. Curr Opin Mol Ther. 200810(3):260–266. | |
van Til JA, IJzerman MJ. Why Should Regulators Consider Using Patient Preferences in Benefit-risk Assessment? PharmacoEconomics. 2014;32(1):1–4. | |
Severin F, Schmidtke J, Mühlbacher A, Rogowski WH. Eliciting preferences for priority setting in genetic testing: a pilot study comparing best-worst scaling and discrete-choice experiments. Eur J Hum Genet. 2013;21(11):1202–1208. | |
Groothuis-Oudshoorn CG, Fermont JM, van Til JA, IJzerman MJ. Public stated preferences and predicted uptake for genome- based colorectal cancer screening. BMC Med Inform Decis Mak. 2014;14:18. | |
Printz C. I-SPY 2 may change how clinical trials are conducted. Cancer. 2013;119(11):1925–1927. | |
Barker AD, Sigman CC, Kelloff GJ, Hylton NM, Berry DA, Esserman LJ. I-SPY 2: An Adaptive Breast Cancer Trial Design in the Setting of Neoadjuvant Chemotherapy. Clin Pharmacol Ther. 2009; 86(1):97–100. | |
Schilsky RL. Personalized medicine in oncology: the future is now. Nat Rev Drug Discov. 2010;9(5):363–366. | |
Ligthart ST, Bidard FC, Decraene C, et al. Unbiased quantitative assessment of Her-2 expression of circulating tumor cells in patients with metastatic and non-metastatic breast cancer. Ann Oncol. 2013;24(5):1231–1238. | |
Mao C, Wu XY, Yang ZY, et al. Concordant analysis of KRAS, BRAF, PIK3CA mutations, and PTEN expression between primary colorectal cancer and matched metastases. Sci Rep. 2015;5:8065. | |
Magbanua MJ, Wolf DM, Yau C, et al. Serial expression analysis of breast tumors during neoadjuvant chemotherapy reveals changes in cell cycle and immune pathways associated with recurrence and response. Breast Cancer Res. 2015;17:73. | |
Nalejska E, Maczynska E, Lewandowska MA. Prognostic and predictive biomarkers: tools in personalized oncology. Mol Diagn Ther. 2014; 18(3):273–284. | |
Rubin DB. For objective causal inference, design trumps analysis. Annals of Applied Statistics. 2008;2(3):808–840. | |
Rubin DB. Causal Inference Using Potential Outcomes. Journal of the American Statistical Association. 2005;100(469):322–331. | |
Ma J, Hobbs BP, Stingo FC. Statistical methods for establishing personalized treatment rules in oncology. BioMed Research International. Epub 2015 Feb 9. | |
Murphy SA. Optimal dynamic treatment regimes. J Royal Statistical Soc B. 2003;65(2):331–366. | |
Lavori PW, Dawson R. Dynamic treatment regimes: practical design considerations. Clin Trials. 2004;1(1):9–20. | |
Chakraborty B, Murphy SA. Dynamic Treatment Regimes. Annu Rev Stat Appl. 2014;1:447–464. | |
Corander J, Aittokallio T, Ripatti S, Kaski S. The rocky road to personalized medicine: computational and statistical challenges. Personalized Medicine. 2012;9(2):109–114. | |
Ramsey SD, Barlow WE, Gonzalez-Angulo AM, et al. Integrating comparative effectiveness design elements and endpoints into a phase III, randomized clinical trial (SWOG S1007) evaluating oncotypeDX-guided management for women with breast cancer involving lymph nodes. Contemporary Clinical Trials. 2013;34(1):1–9. | |
Leunis A, Redekop WK, van Montfort KA, Löwenberg B, Uyl-de Groot CA. The development and validation of a decision-analytic model representing the full disease course of acute myeloid leukemia. Pharmacoeconomics. 2013;31(7):605–621. | |
Collins B. Big Data and Health Economics: Strengths, Weaknesses, Opportunities and Threats. Pharmacoeconomics. Epub 2015 Jun 21. | |
Pollack CE, Lemke KW, Roberts E, Weiner JP. Patient sharing and quality of care: measuring outcomes of care coordination using claims data. Med Care. 2015;53(4):317–323. | |
Crown WH. Potential application of machine learning in health outcomes research and some statistical cautions. Value Health. 2015;18(2):137–140. | |
Giordano SH. Comparative effectiveness research in cancer with observational data. Am Soc Clin Oncol Educ Book. 2015;35:e330–e335. | |
Baker SG, Kramer BS, Lindeman KS. Latent class instrumental variables: a clinical and biostatistical perspective. Stat Med. Epub 2015 Aug 4. | |
Marshall DA, Burgos-Liz L, IJzerman MJ, et al. Applying Dynamic Simulation Modeling Methods in Health Care Delivery Research-The SIMULATE Checklist: Report of the ISPOR Simulation Modeling Emerging Good Practices Task Force. Value Health. 2015;18(1):5–16. | |
Marshall DA, Burgos-Liz L, IJzerman MJ, et al. Selecting a Dynamic Simulation Modeling Method for Health Care Delivery Research-Part 2: Report of the ISPOR Dynamic Simulation Modeling Emerging Good Practices Task Force. Value Health. 2015;18(2):147–160. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.