")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 16
Impact of Visceral Obesity on Structural and Functional Alterations of Gut Microbiota in Polycystic Ovary Syndrome (PCOS): A Pilot Study Using Metagenomic Analysis
Authors Bai X , Ma J, Wu X, Qiu L, Huang R, Zhang H, Huang H, Chen X
Received 2 September 2022
Accepted for publication 26 November 2022
Published 11 January 2023 Volume 2023:16 Pages 1—14
DOI https://doi.org/10.2147/DMSO.S388067
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Juei-Tang Cheng
Xuefeng Bai,1,* Jiangxin Ma,1,* Xiaohong Wu,1 Lingling Qiu,2 Rongfu Huang,3 Haibin Zhang,1 Huibin Huang,1 Xiaoyu Chen1
1Department of Endocrinology, Second Affiliated Hospital of Fujian Medical University, Quanzhou City, Fujian Province, People’s Republic of China; 2Department of Reproductive Medicine, Second Affiliated Hospital of Fujian Medical University, Quanzhou City, Fujian Province, People’s Republic of China; 3Department of Clinical Laboratory, Second Affiliated Hospital of Fujian Medical University, Quanzhou City, Fujian Province, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Huibin Huang; Xiaoyu Chen, Department of Endocrinology, the Second Affiliated Hospital of Fujian Medical University, No. 950 Donghai Street, Fengze District, Quanzhou City, Fujian Province, 362000, People’s Republic of China, Tel +86-13313872001 ; +86-13600739755, Email [email protected]; [email protected]
Objective: We aimed to identify structural and functional alterations of gut microbiota associated with visceral obesity in adult women with polycystic ovary syndrome (PCOS).
Methods: Twenty-seven adults with PCOS underwent stool and fasting blood collection, oral glucose tolerance testing, and visceral fat area (VFA) measurement via dual-bioimpedance technique. Metagenomic analysis was used to analyze gut microbiota.
Results: PCOS patients were divided into three groups: visceral obesity group (PCOS-VO, n=9, age 28.33± 5.68 years, BMI 37.06± 4.27 kg/m2, VFA 128.67± 22.45 cm2), non-visceral obesity group (PCOS-NVO, n=10, age 25.40± 4.53, BMI 30.74± 3.95, VFA 52.00± 24.04), normal BMI group (PCOS-NB, n=8, age 27.88± 2.53, BMI 21.56± 2.20, VFA 27.00± 21.18), with no statistical difference in age (P> 0.05) and significantly statistical differences in BMI and VFA (P< 0.05). The groups showed a significant difference in microbial β-diversity between PCOS-VO and PCOS-NVO (P=0.002) and no difference between PCOS-NVO and PCOS-NB (P=0.177). Bacteroidetes was the phylum with the highest relative abundance among all patients, followed by Firmicutes. Those with visceral obesity had a higher abundance of Prevotella, Megamonas, and Dialister genera, positively correlated with metabolic markers (r> 0.4, P< 0.05), and lower abundance of Phascolarctobacterium and Neisseria genera, negatively correlated with metabolic markers (r<-0.4, P< 0.05). Functional annotation analysis showed significant differences in relative abundance of ribosome pathway, fatty acid biosynthesis pathway, and sphingolipid signaling pathway between groups, affecting lipid homeostasis and visceral fat accumulation.
Conclusion: Alteration in β-diversity of gut microbiota exists in PCOS with visceral obesity versus those without visceral obesity and relates to functional differences in ribosomes, fatty acid biosynthesis, and sphingolipid signaling pathways.
Keywords: polycystic ovary syndrome, visceral obesity, gut microbiota, visceral fat area, metagenomic analysis
Introduction
Polycystic ovary syndrome (PCOS) is a highly prevalent disease in reproductive-aged women, characterized by menstrual disorder, abnormally high androgen, polycystic ovary morphology, and infertility.1 Although genetic, gestational environmental and lifestyle factors are thought to be involved in the development of PCOS, how do these factors pathologically trigger biochemical and metabolic disorders remains widely unknown. Limited therapeutic options have focused mainly on symptoms, resulting in unsatisfactory treatment outcomes and long-term metabolic complications. Therefore, there is an urgent need to develop new therapeutic strategies.
Recent studies have reported alterations of gut microbiota in PCOS patients2–5 and provided convincing evidence for a causal correlation between dysbiosis of gut microbiota and onset of ovarian and metabolic dysfunction.2,4 Dysbiosis of gut microbiota can increase secretion of ovarian androgen, which then interfere normal follicular development by triggering a chronic inflammatory response and insulin resistance.6 Further evidence has suggested that gut microbiota and their metabolites can cause imbalances in energy intake, activate inflammatory pathways, stimulate secretion of brain gut peptides, and proliferate pancreatic β-cells, leading to abnormal or excessive fat accumulation, insulin resistance, and compensatory hyperinsulinemia.2,7 Fecal microbiota transplantation (FMT) from the healthy, addition of probiotics, alteration of bile acid metabolism, and/or increasing IL-22 level have been demonstrated to be valuable in treatment for PCOS and may help improve symptoms of PCOS, ameliorate metabolic effect of obesity, and reconstitute normal gut microbiota.2,8,9
Obesity has a marked impact on the metabolic complications of PCOS,10 and its occurrence is 2.8 times higher than that in women without PCOS.11 Obesity affects the relationship between PCOS and gut microbiota. However, inconsistent results exist in microbial diversity and enriching microbiota between obesity and non-obesity,5,6,12 which may be attributed to the key factor BMI for grouping, in addition to geography, ethnicity, and dietary habits. Excess accumulation of intra-abdominal adipose tissue, termed visceral obesity, is part of a phenotype that is significantly correlated with increased inflammatory cytokines and cardiometabolic risk.13 According to perspective of precision medicine, there should be essentially different applications of gut microbiota in the treatments for PCOS complicated with visceral obesity and non-visceral obesity, rather than solely based on BMI.
In this study, visceral fat area (VFA) and subcutaneous fat area (SFA) of patients with PCOS were measured by dual-bioimpedance method easily performed in clinical practice. Visceral obesity is defined by BMI combined with VFA. We obtained fecal metagenomic sequencing from clinical practice in PCOS patients with visceral obesity, non-visceral obesity, and normal BMI, to explore microbial factors associated with susceptibility to visceral obesity. By comparing differences in enriching Kyoto Encyclopedia of Functional Genes and Genomes (KEGG) pathways and KEGG Orthology (KO) between PCOS patients with visceral obesity and controls, we described possible pathological mechanisms for the development of PCOS patients with visceral obesity. The design idea of this study has not been reported, and the findings may provide more guidance for improving treatment and prevention of PCOS patients with visceral obesity.
Methods
Participants
From April 2021 to January 2022, we enrolled participants with untreated PCOS at the Department of Endocrinology and Metabolism’s outpatient clinic at the Second Affiliated Hospital of Fujian Medical University. We chose patients aged from 20 to 35 who were diagnosed with PCOS according to the Rotterdam 2003 criteria. The occurrences of diabetes mellitus, congenital adrenal hyperplasia, Cushing’s syndrome, hyperprolactinemia, thyroid dysfunction, other diseases causing hyperandrogenemia, and severe liver and kidney dysfunction were in exclusion criteria. Other exclusion factors included the use of hormonal drugs, antibiotics, insulin enhancers, probiotics, laxatives within the previous 3 months, pregnant or breastfeeding women, being in weight loss or weight management programs, smokers, or alcoholics.
Study Protocol
Participants received case information registration, physical measurements, and laboratory evaluations to establish eligibility. We recorded case information including age at menarche, menstrual cycle, duration of menstruation, infertility history, medication history, dietary habit, physical activity, and sleep habit. Physical measurements included height, weight, waist and hip circumference, assessment of body hair and acne referring to modified Ferriman-Gallwey (mFG) score and Global Acne Grading System (GAGS), acanthosis nigricans, and also included VFA and SFA, which were accurately calculated by dual-bioimpedance technique using the device DUALSCAN HDS-2000 (OMRON HEALTHCARE Co., Ltd). mFG score ≥4 is defined as hirsutism in Chinese.14 VFA ≥100 cm2 is defined as visceral obesity according to the recommended VFA cutoff from the Japanese Society for the Study of Obesity.15 Using BMI 25 kg/m2 (the Asian-Pacific standard) and VFA 100 cm2 as cut points, PCOS patients were divided into three groups in this study: visceral obesity group (PCOS-VO, BMI ≥25 kg/m2, VFA ≥100 cm2), non-visceral obesity group (PCOS-NVO, BMI ≥25 kg/m2, VFA <100 cm2), normal BMI group (PCOS-NB, BMI <25 kg/m2, VFA <100 cm2).
Laboratory evaluations included stool and fasting blood collection, oral glucose tolerance testing (OGTT), and color ultrasound scan of ovaria-utero-liver. Fasting intravenous blood were tested for lipids, liver and kidney functions, sex hormones, 25-hydroxy vitamin D, and thyroid function. Sex hormones, such as total testosterone (TT), free testosterone (FT), luteinizing hormone (LH), follicle-stimulating hormone (FSH), 17-alpha hydroxyprogesterone (17-α OHP), and sex hormone-binding globulin (SHBG) were measured by chemiluminescence method, while dehydroepiandrosterone (DHEA), dihydrotestosterone (DHT), and anti-Mullerian hormone (AMH) were measured by enzyme-linked immunosorbent assay. Androstenedione was measured by high-performance liquid chromatography-tandem mass spectrometry. 25-Hydroxy vitamin D was measured by chemiluminescence method. It should be noted that testing of sex hormones was performed on days 3–5 of menstrual cycle (not limited to amenorrhea).
The calculation formulas used in the study were as follows,
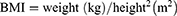
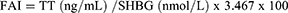
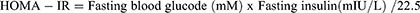
where FAI represents Free androgen index, HOME-IR represents Homeostatic Model Assessment for Insulin Resistance.
Stool Collection and Metagenomic Sequencing
Fecal samples were collected on non-menstrual morning. We placed 300 mg of each sample into Longsee fecal sampling tube containing preservative fixatives provided by Wekemo Tech Group Co., Ltd. (Shenzhen, China) and froze them at −80°C. Microbial genomic DNA was extracted from the stools using cetyltrimethylammonium bromide (CTAB) method and then were sequenced on the Illumina Novaseq 6000 platform. Quality control and preprocessing of metagenomic data were implemented to obtain clean readings.16
Microbial Taxonomy and Functional Annotation
Kraken2 and the self-build microbial database by searching in NT nucleic acid database and RefSeq whole-genome database of NCBI were used to identify microbiota from the clean readings in all samples. Bracken was used to predict the actual relative abundance of microbial taxa at seven levels of classification (kingdom, phylum, class, order, family, genus, and specie). The relative abundance refers to the sequence number percent (SN%) for each microbial taxon at different taxonomic levels, which indicates the ratio of the sequence number to the total annotated data at that level (total value of 1 for each taxonomic level in each sample). Meanwhile, the clean readings were matched to protein sequences from UniRef90 Database using HUMAnN2 software. Based on the corresponding relationships between UniRef90 ID and functional databases, such as KEGG pathway database and KO database, the annotation information and relative abundance data were obtained.
Statistical Analysis
The clinical data were processed using SPSS statistical software (Version 19.0). One-way ANOVA or Kruskal–Wallis test was used to compare quantitative variables. The quantitative metagenomics analysis was implemented via online Wekemo Bioincloud (https://www.bioincloud.tech) provided by Wekemo Tech Group Co., Ltd. We used Principal Coordinate analysis (PCoA) based on Bray-Curtis distance and permutation multivariate analysis of variance (PERMANOVA) to evaluate microbial β-diversity between groups. Linear discriminant analysis (LDA) effect size (LEfSe) and Dunn test analysis were further applied to identify significantly enriching taxa, KEGG pathways, and KOs. We also calculated Spearman correlation coefficients between clinical parameters and enriching taxa at the genus level (top 200 in abundance) to determine significant correlations. Thirty enriching genera with the most significant correlations were displayed by correlation heat map. All statistical analysis and graphics were conducted using R software (V.4.1.0) and QIIME (version 2). All the tests were two-sided and p <0.05 was considered statistically significant.
Results
Clinical Characteristics of PCOS Patients
A total of 27 PCOS patients aged 20 to 35 were enrolled in this study. To explore the relationship between visceral obesity and gut microbiota, patients were divided into three groups: PCOS-VO group (n=9, age 28.33±5.68 years, BMI 37.06±4.27 kg/m2, VFA 128.67±22.45 cm2), PCOS-NVO group (n=10, age 25.40±4.53 years, BMI 30.74±3.95 kg/m2, VFA 52.00±24.04 cm2), PCOS-NB group (n=8, age 27.88±2.53 years, BMI 21.56±2.20 kg/m2, VFA 27.00±21.18 cm2), with no statistical difference in age (P>0.05) and significantly statistical differences in BMI and VFA (P<0.05) (Table 1). Free testosterone and FAI were higher in PCOS-VO group compared with PCOS-NVO and PCOS-NB groups, but only FAI was statistically significant between PCOS-VO and PCOS-NB groups (P=0.032). Comparing metabolic parameters such as blood glucose (glucose 0H, glucose 2H), lipids (total cholesterol, triglyceride, low-density lipoprotein, apolipoprotein B), and uric acid among the three groups, there were no statistical differences (P<0.05). However, insulin 0H and HOMA-IR were significantly increased in PCOS-VO group and PCOS-NVO group compared to PCOS-NB group, respectively (all P<0.05). Deficiency of 25-hydroxy vitamin D was more severe in PCOS-VO group (12.51±2.62 ng/mL) and PCOS-NVO group (14.80±5.03 ng/mL) compared to PCOS-NB group (21.61±7.57 ng/mL), both P<0.05.
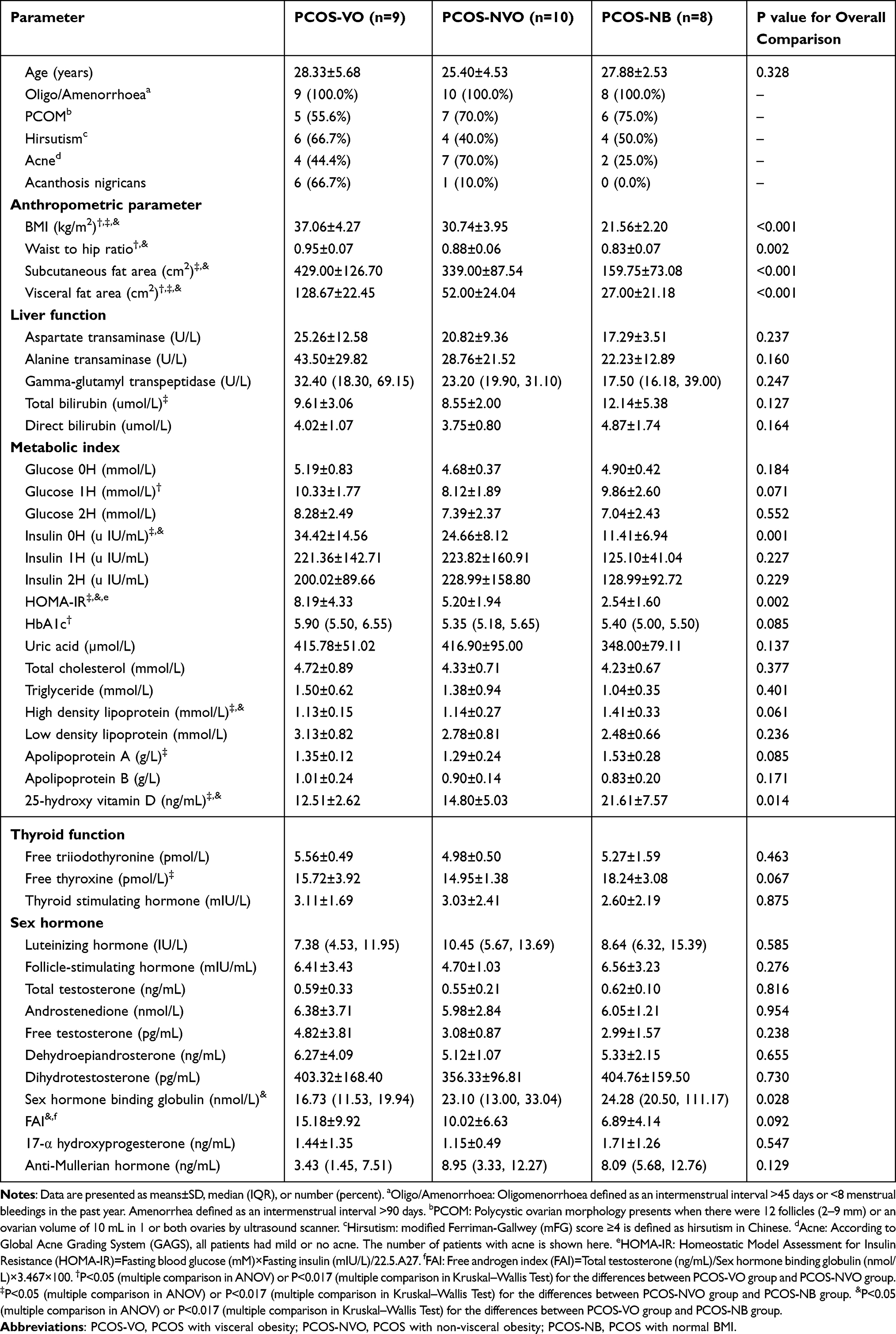 |
Table 1 Clinical Characteristics in Patients with Untreated PCOS |
In addition, we obtained information on each participant’s daily dietary, physical activity, and sleep habit through a questionnaire, which did not differ between groups. These investigations excluded the effect of daily habits on gut microbiota between groups.
Abundance Summary and Diversity Analysis of Gut Microbiota
Each microbial taxon was identified for its relative abundance at seven taxonomic levels (kingdom, phylum, class, order, family, genus, species). A total of 2767 species, 521 genera, and 38 phyla were counted in this study. This study showed significant microbial β-diversity among PCOS-VO, PCOS-NVO, and PCOS-NB groups (F=2.13, P=0.003, Figure 1A) by PERMANOVA method. This difference was observed between PCOS-VO group and PCOS-NVO group (F=3.209, P=0.002), and between PCOS-VO group and PCOS-NB group (F=1.854, P=0.039). However, there was no significant difference in microbial β-diversity between PCOS-NVO group and PCOS-NB group (F=1.299, P=0.177). At the species level, there were 1089 common species in three groups, and 364/495/278 unique species in PCOS-VO, PCOS-NVO, and PCOS-NB groups, respectively (Figure 1B).
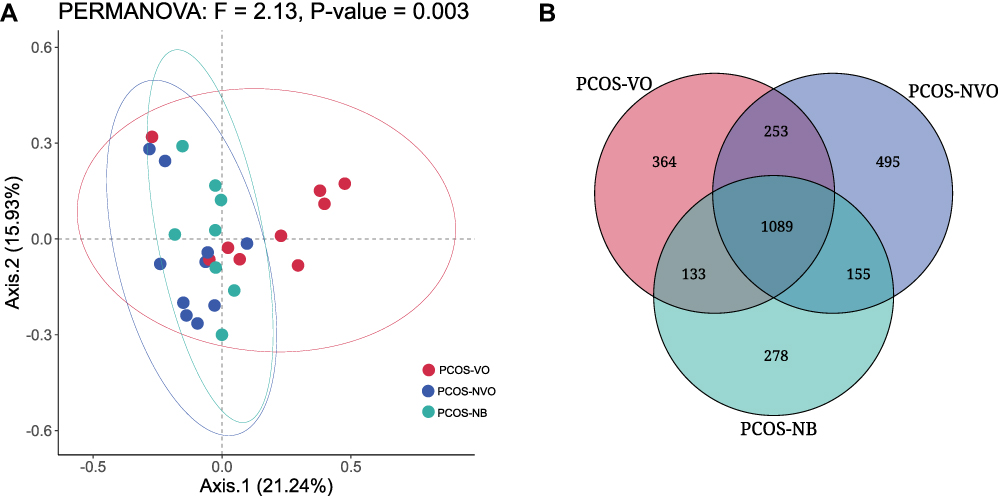 |
Figure 1 Comparison of microbial community characteristics between three groups with PCOS from fecal metagenomic sequencing. (A) β-diversity was evaluated by Principal Coordinate analysis (PCoA) based on Bray-Curtis distance and permutation multivariate analysis of variance (PERMANOVA) between groups. (B) Venn diagram of unique or common species between groups. Abbreviations: PCOS, polycystic ovary syndrome; PCOS-VO, PCOS with visceral obesity; PCOS-NVO, PCOS with non-visceral obesity; PCOS-NB, PCOS with normal BMI. |
Abundance Comparison of Gut Microbiota
Figure 2 shows relative abundance of gut microbiota in patients with PCOS. Bacteria and Heunggongvirae were the two main kingdoms in all samples (Figure 2A). At the phylum level, Bacteroidetes (PCOS-VO, sequence number percent 58.20%; PCOS-NVO, 60.25%; PCOS-NB, 53.28%), Firmicutes (PCOS-VO, 28.85%; PCOS-NVO, 26.64%; PCOS-NB, 31.11%), Proteobacteria (PCOS-VO, 1.98%; PCOS-NVO, 3.96%; PCOS-NB, 5.99%), and Uroviricota (PCOS-VO, 5.33%; PCOS-NVO, 4.42%; PCOS-NB, 4.64%) were abundant taxa (Figure 2B), with the first three phyla coming from Bacteria kingdom and the last one coming from Heunggongvirae kingdom.
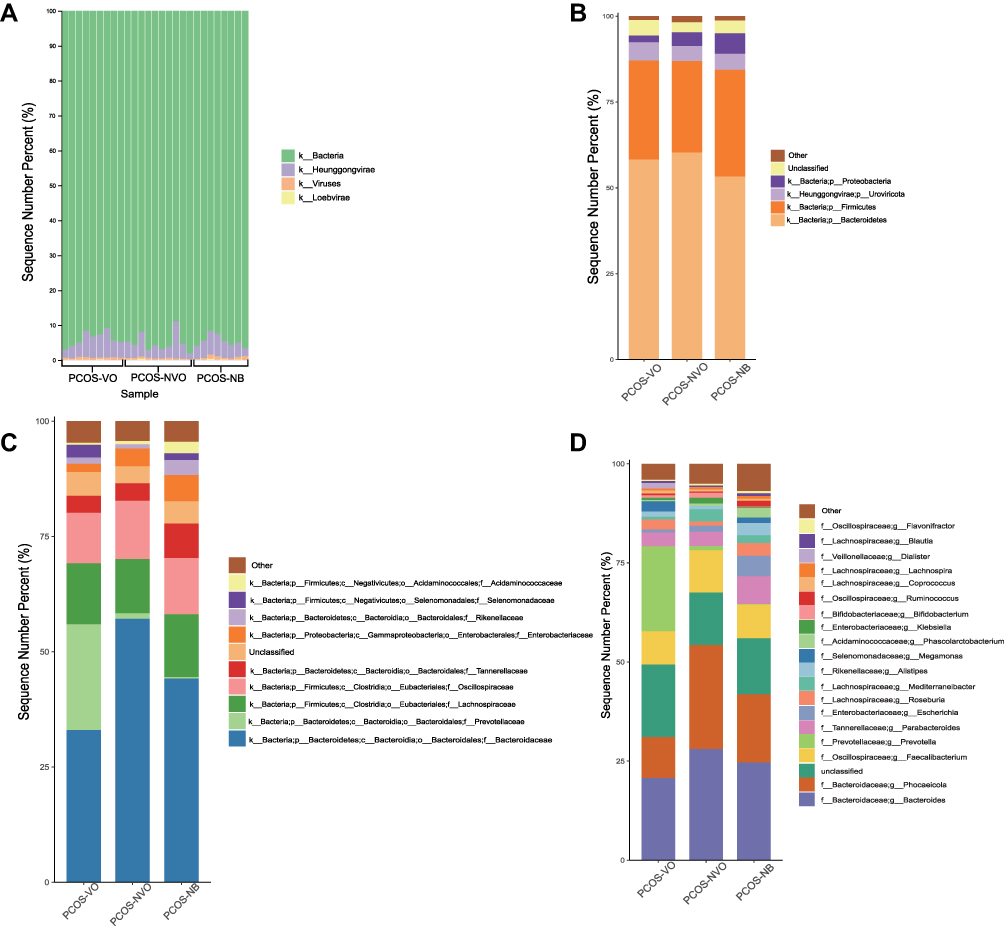 |
Figure 2 Relative abundance of gut microbiota in 27 patients with PCOS. (A) Microbial histogram analysis at the kingdom level for each sample. Microbial histogram analysis at the phylum level (B, top 5), the family level (C, top 10), and the genus level (D, top 20) for each group. The abbreviations for kingdom, phylum, class, order, family, genus, and species are k, p, c, o, f, g and s, respectively. Abbreviations: PCOS, polycystic ovary syndrome; PCOS-VO, PCOS with visceral obesity; PCOS-NVO, PCOS with non-visceral obesity; PCOS-NB, PCOS with normal BMI. |
When it comes to Bacteroidetes phylum, different families were observed to be significantly abundant in different groups, with Prevotellaceae for PCOS-VO group (LDA=5.0, P<0.05), Bacteroidaceae for PCOS-NVO group (LDA=5.07, P<0.05), Odoribacteraceae for PCOS-NB group (LDA=3.64, P<0.05) (Figures 2C and 3A). There were three dominant genera, including Bacteroides, Phocaeicola, and Prevotella (Figure 2D). Abundant genera were further compared by Dunn test (Table 2). Compared to PCOS-NVO group, Prevotella genus was more abundant in PCOS-VO group, while Phocaeicola genus was less abundant (both P<0.05).
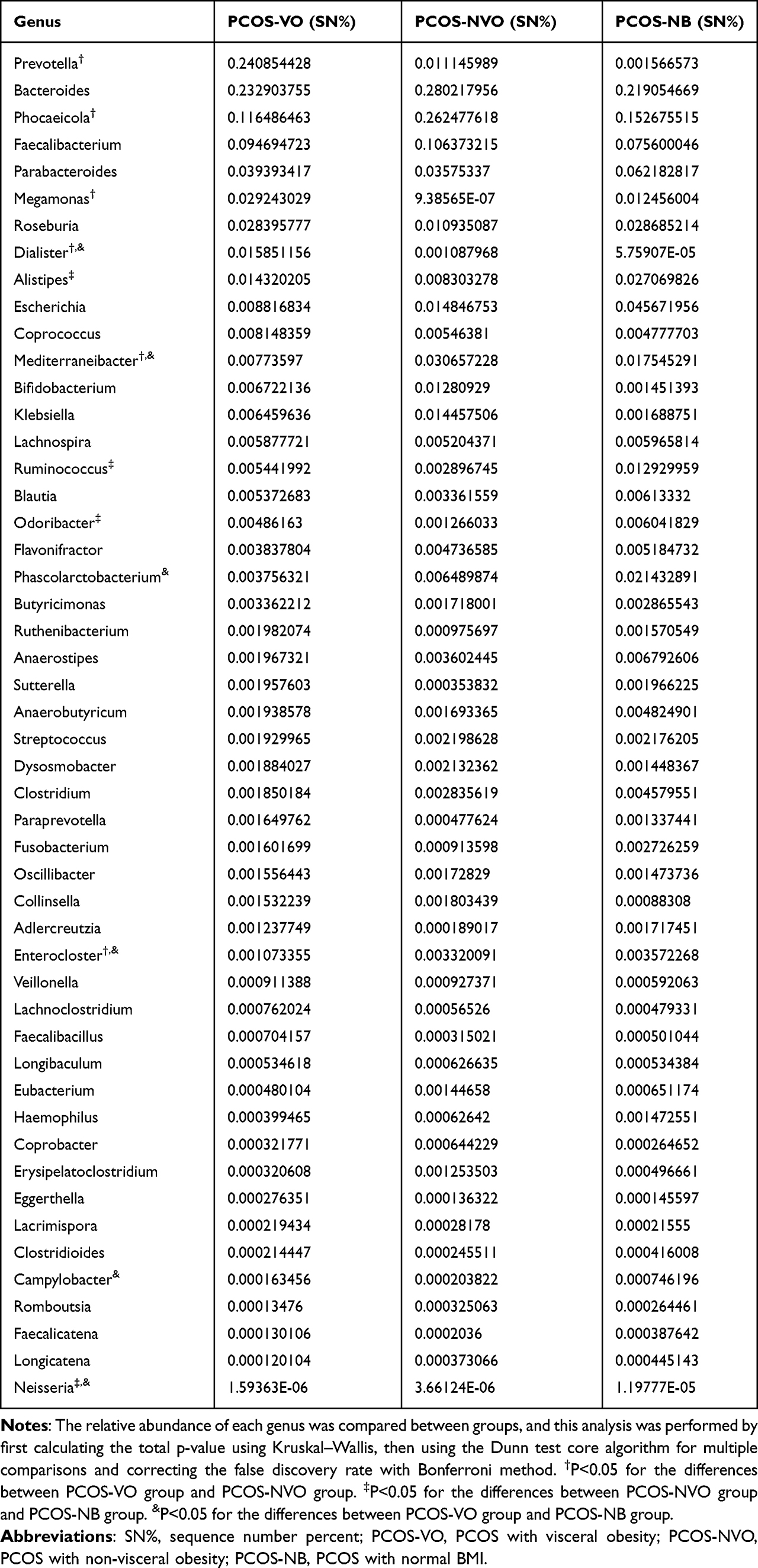 |
Table 2 Comparison of Relative Abundance of Microbial Genera Among PCOS Patients |
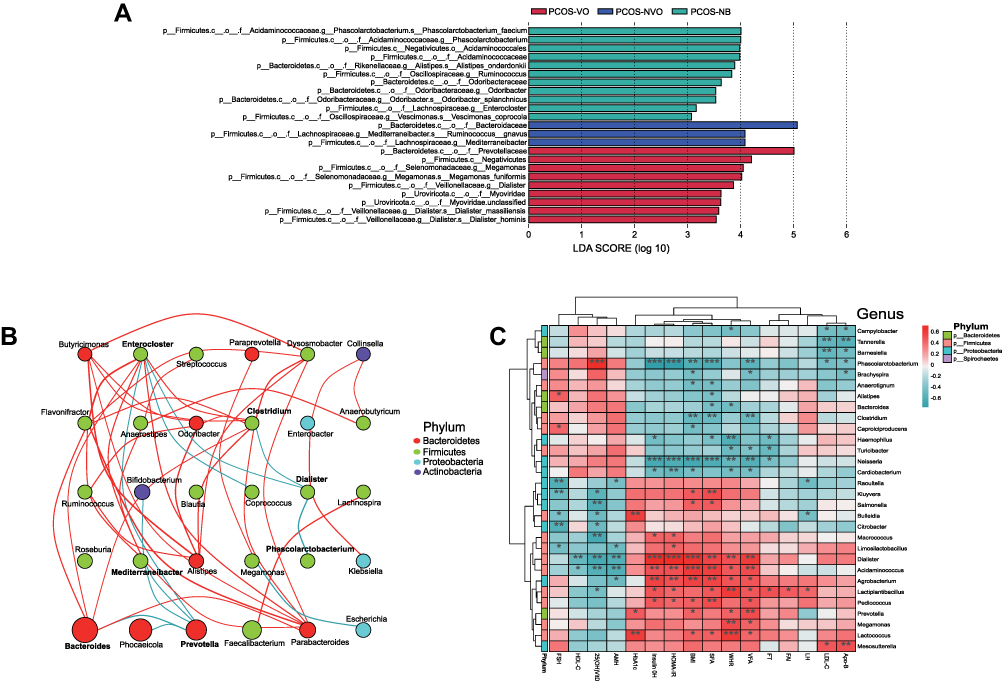 |
Figure 3 Enriching microbial taxa in PCOS patients of each group and their correlations with clinical parameters. (A) LEfSe identified significant abundant taxa in each group (LDA score (log 10)>3, p<0.05). The abbreviations for kingdom, phylum, class, order, family, genus, and species are k, p, c, o, f, g and s, respectively. (B) Network analysis of correlations among enriching microbial taxa at the genus level. The size of ellipse denotes the relative abundance of the genus. The red and green lines denote positive and negative correlations, respectively. Only top 30 genera in abundance are counted and mapped. Only r>0.4 or r<-0.4, and P<0.05 is displayed a line. Microbial taxa from different phyla are shown by different colors on the right. (C) Heatmap of correlations between clinical parameters and microbial taxa at the genus level. Microbial taxa with top 30 abundance detected in samples are listed on the left, and clinical parameters are shown on the bottom. Microbial taxa from different phyla are shown by different colors on the right. R values are shown in different colors, and only r>0.4 or r<-0.4, and P<0.05 are marked. *Means 0.01≤P<0.05, ** means 0.001≤P<0.01, *** means P<0.001. Abbreviations: LEfSe, linear discriminant analysis effect size; LDA, linear discriminant analysis; FSH, follicle-stimulating hormone; HDL-C, high-density lipoprotein cholesterol; AMH, anti-Mullerian hormone; HOMA-IR, Homeostatic Model Assessment for Insulin Resistance; SFA, subcutaneous fat area; WHR, waist-to-hip ratio; VFA, visceral fat area; FT, free testosterone; FAI, free androgen index; LH, luteinizing hormone; LDL-C, low-density lipoprotein cholesterol; Apo-B, apolipoprotein B. |
Firmicutes phylum included several enriching families, such as Lachnospiraceae, Oscillospiraceae, Selenomonadales, Acidaminococcales (Figure 2C). Figures 2D and 3A show that there were different genera with high relative abundance in different groups, such as Megamonas and Dialister in PCOS-VO group (LDA=4.05 and 3.54, P<0.05), Mediterraneibacter in PCOS-NVO group (LDA=4.08, P<0.05), Ruminococcus, Phascolarctobacterium, and Enterocloster in PCOS-NB group (all LDA>3.0, P<0.05).
Proteobacteria phylum was the third most abundant Bacteria kingdom. Enterobacteriaceae family and Escherichia genus were more abundant in PCOS-NB group compared with the other two groups (Figure 2C and D), but without significance (Dunn test, P>0.05, Table 2). Neisseria genus was more abundant in PCOS-NB group compared to PCOS-VO and PCOS-NVO groups (Dunn test, P<0.05, Table 2). Network analysis of correlation among enriching microbial taxa at the genus level can be found in Figure 3B.
Association Between Microbial Taxa and Clinical Parameters
Heatmap shows that multiple metabolic markers were significantly correlated with relative abundance of microbial taxa at the genus level (only r>0.4 or r<-0.4, and P<0.05 are marked, Figure 3C). For example, the relative abundance of Prevotella, Megamonas, and Dialister genera, which were enriched in PCOS-VO group, were positively correlated with certain parameters of BMI, WHR, VFA, SFA, HbA1c, insulin 0H, and HOMA-IR, respectively. In contrast, Phascolarctobacterium and Neisseria genera, which were enriched in PCOS-NB group, showed a significantly negative association with insulin 0H, HOMA-IR, SFA, VFA, and BMI. In addition, the relative abundance of Phascolarctobacterium genus was positively correlated with 25-hydroxy vitamin D, while Dialister, Lactiplantibacillus, Macrococcus, Salmonella, and Kluyvera genera were negatively correlated with 25-hydroxy vitamin D.
Functional Characteristics of Gut Microbiota Between Groups
We annotated the fecal gene catalogs using KEGG pathway and KO databases. The most abundant pathway was metabolism pathway at KEGG level-1. A total of 57 level-2 pathways were identified in this study, and the top three were amino acid metabolism, carbohydrate metabolism, metabolism of cofactors and vitamins, all of which belong to metabolism pathways (Supplementary Table 1). In PCOS-VO group, it showed significant enrichments in map03010 (ribosome), map00780 (biotin metabolism), and map00061 (fatty acid biosynthesis) at KEGG level-3 (LAD=3.59, 3.26, 3.12, all P<0.05, Figure 4A). We also identified several KOs that differed significantly in relative abundance between groups with the results shown in Figure 4B. Most of these KOs were involved in the map03010 (ribosome), such as ribosomal protein S9 (RPS9), RPS4, RPS19, RPS11, RPS13, RPL1, RPL2, RPL5, RPL14, RPL22. In addition, LEfSe method also suggested that map04071 (sphingolipid signaling pathway) was enriched in PCOS-NB group (LAD=2.18, P<0.05, Figure 4A).
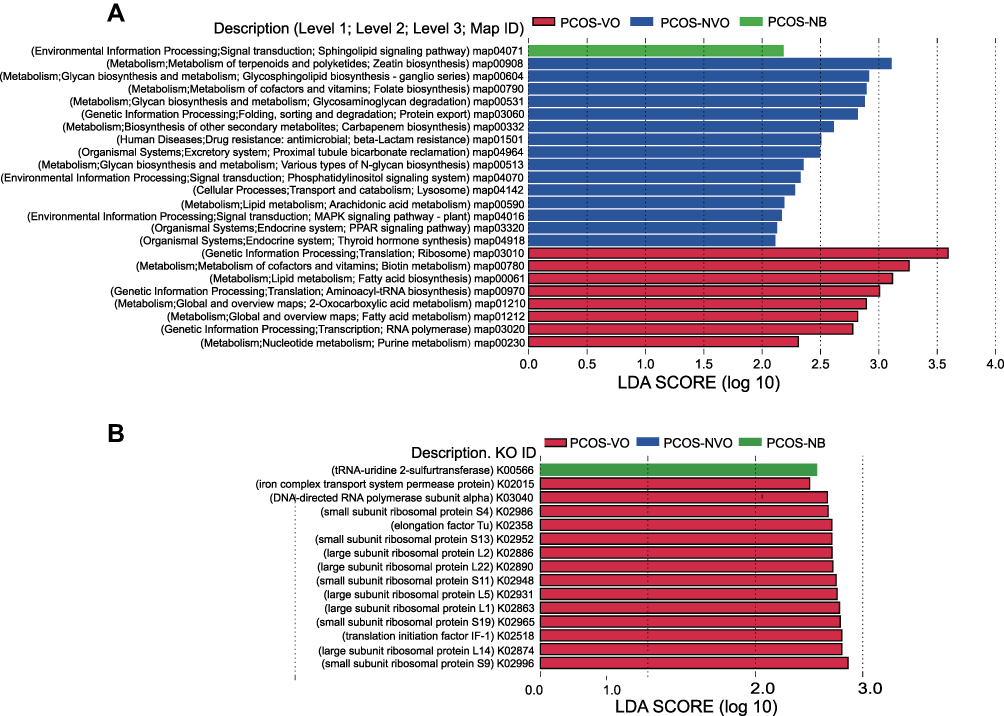 |
Figure 4 Functional annotation of gut microbiota of 27 PCOS patients from metagenomic sequencing data. Linear discriminant analysis (LDA) effect size (LEFSe) was used to identify KEGG pathways (A) and KOs (B) with significant differences in relative abundance between groups. LDA>2 for KEGG pathways or LDA>2.5 for KOs, and p<0.05 are listed. Abbreviations: KEGG, Kyoto Encyclopedia of Functional Genes and Genomes; KO, KEGG Orthology. |
Discussion
We first studied the gut microbiota of PCOS patients with visceral obesity using fecal metagenomic sequencing. Microbial β-diversity is an index to measure the similarity of microbial composition among individuals. Previous studies showed significant differences in microbial β-diversity between PCOS patients and healthy controls,5,6 while some studies suggested an opposite finding.4,17 There were also opposite findings on whether microbial diversity is homogenized among PCOS population. Some studies showed no significant difference in β-diversity based on Bray-Curtis distance between PCOS-HB (BMI ≥24 kg/m2) and PCOS-LB (BMI <24 kg/m2),5,17 but Insenser et al indicated microbial β-diversity of obese PCOS (BMI ≥30 kg/m2) was significantly lower than that of non-obese PCOS (BMI <30 kg/m2).12 In regard to literature, the effect of obesity on microbial β-diversity yielded conflicting results, which may be attributed to a key factor BMI for grouping. Our study shows that microbial β-diversity was significantly different between PCOS-VO and PCOS-NVO groups but not different between PCOS-NVO and PCOS-NB groups. Thus, PCOS phenotype with visceral obesity may essentially affect the change of microbial diversity rather than obesity distinguished only by BMI.
Bacteroidetes is the main phyla in gut microbiota of all PCOS patients in this study. Different families were observed abundantly in different groups, with Prevotellaceae for PCOS-VO, Bacteroidaceae for PCOS-NVO, and Odoribacteraceae for PCOS-NB. Relative abundances of Prevotella genus and Bacteroides genus were negatively correlated (Figure 3B), in other words, the existence of one kind seemed to lead to the rejection of the other kind.18 One clinical study19 reported a higher abundance of Prevotella genus in PCOS with obesity, consistent with ours. Our study also indicated that Prevotella genus was positively correlated with BMI, VFA, WHR. Meanwhile, Bacteroides genus was negatively correlated with WHR and SFA (Figure 3C). Carolina Serena et al conducted a clinical trial noting that a higher relative abundance of succinate-producing Prevotellaceae and a lower relative abundance of succinate-consuming Odoribacteraceae were found in obese individuals. Obesity concomitant with impaired glucose metabolism is associated with increased levels of circulating succinate.20 Therefore, Prevotellaceae family, Prevotella genus, and Odoribacteraceae family may be involved in the development of fat accumulation and severe metabolism in PCOS with visceral obesity.
Firmicutes was another dominant phylum of gut microbiota in this study, which together with Bacteroidetes and Proteobacteria account for more than 90% of total gut microbiota. Dialister, Phascolarctobacterium, and Clostridium genera all belong to Firmicutes phylum. Dialister was more abundant in PCOS-VO group, which was positively correlated with VFA, SFA, BMI, insulin 0H, and HOMA-IR. Phascolarctobacterium and Clostridium both increased negatively with Dialister (Figure 3B), and they were negatively correlated with VFA, SFA, and BMI. These are in line with literature studies on differences in gut microbiota between obese and non-obese participants.21,22 In insulin-sensitive individuals, the abundance of Dialister was lower,21 while Phascolarctobacterium was higher.23 Dialister is a kind of gram-negative cocci, which has been shown to produce short-chain fatty acids (SCFAs) such as acetate and propionate.24 Circulating SCFAs produced in gut can trigger endogenous secretion of hunger-regulating peptides such as glucagon-like peptide 1 (GLP-1) and peptide YY (PYY),25 which may promote energy uptake and lead to obesity. Phascolarctobacterium is a kind of succinate-consuming taxa,20,26 thus the decrease of Phascolarctobacterium abundance may lead to an increase in intestinal succinate metabolites. Previous studies showed correlation between abundance of Clostridium, reducing body fat accumulation and enhancing insulin sensitivity.27,28 Zhao et al confirmed that Clostridium_sensu_stricto_1 might be one of the key pathogens causing PCOS-IR rat model.29 Thus, Dialister, Phascolarctobacterium, and Clostridium genera may play an important role in the mechanism of morbid obesity in PCOS patients.
Proteobacteria phylum, a kind of gram-negative bacteria, was more abundant in PCOS-NB group, especially Gammaproteobacteria class, Enterobacteriaceae family, and Escherichia genus, but without significance. Chen et al suggested that a higher abundance of Proteobacteria phylum was observed in PCOS-HB group compared to PCOS-LB group, with the grouped BMI set at 24 kg/m2, which may be a factor contributing to the opposite result to our study.5 Debédat et al indicated that increase in Proteobacteria, Gammaproteobacteria, and Escherichia coli was correlated with the amount of weight loss or post-bariatric surgery, as well as reduced systemic inflammation and improved glucose homeostasis.30 Therefore, some taxa of Proteobacteria phylum may be involved in improving morbid obesity in PCOS patients. In this study, Spearman correlation analysis showed that Neisseria and Cardiobacterium genera were negatively correlated with BMI, VFA, WHA, Insulin 0H, and HOMA-IR.
In order to comprehensively understand the differences in gut microbiota between visceral obesity group and non-visceral obesity group, we analyzed the function of microbial taxa. The ribosome pathway was significantly enriched in PCOS-VO group. Ghosh et al conducted whole-genome expression profiling of whole blood and showed that ribosome pathway was the top ranked pathway in the obese cohort.31 The ribosome pathway is related to ribosome composition, protein synthesis, and hematopoietic cell survival. While ribosomes have long been considered as complex machines with unchanging composition, in recent years researchers have proposed that ribosomes can have heterogeneous composition, and this heterogeneity can be caused by variations of any component, such as rRNA modifications, rRNA variants, stoichiometric ratios of ribosomal proteins, post-translational modifications, and ribosome-associated proteins.32 Complex ribosomes consist of ribosomal proteins with ribosomal RNAs, and perturbations of these checkpoints in ribosome pathway may lead to excessive activation of ribosomal organisms. Analysis of KOs in this study showed that ribosome pathway included several ribosomal protein genes (PRL14, PRS9, PRL5, et al Figure 4B), which were enriched in PCOS-VO group. Wu et al conducted experiments on Caenorhabditis elegans and demonstrated that nucleolus stress promotes lipid synthesis and accumulation through the ribosomal protein RPL11/RPL5, which upregulates PHA-4 expression located in the nucleus and transcriptionally activates the expression of lipid synthesis genes.33 Therefore, it is reasonable to speculate that ribosome pathway may be involved in fat accumulation in PCOS with visceral obesity and aggravate severe metabolism.
This study also showed a higher relative abundance of fatty acid biosynthesis but a lower relative abundance of sphingolipid signaling pathway in PCOS-VO group, which may be correlated with lipid homeostasis and insulin resistance in PCOS. Excess circulating fatty acids (FAs) are characteristic of obesity and can induce insulin resistance, steatosis, β-cell dysfunction, and apoptosis.34 Ceramide plays a key role in lipotoxicity. The imbalance between ceramide and sphingosine-1 phosphate (S-1P) is involved in lipid homeostasis in human diseases such as Alzheimer’s disease, type 2 diabetes-induced insulin resistance, and cardiovascular diseases.35
Conclusion
This study suggested that β-diversity of gut microbiota in PCOS was significantly different between visceral obesity and non-visceral obesity. A variety of enriching taxa with significant differences between groups, such as Prevotellaceae family, Odoribacteraceae family, Prevotella genus, Phascolarctobacterium genus, Dialister genus, and Clostridium genus, may participate in the development of PCOS patients with visceral obesity through fat accumulation, SCFA metabolism (including succinate metabolism), and energy uptake. These enriching taxa can be used as future candidate biomarkers for PCOS patients with visceral obesity. We also found that the underlying and functional differences between groups were mainly distributed in ribosome, fatty acid biosynthesis, and sphingolipid signaling pathways, affecting lipid homeostasis and visceral fat accumulation. One obvious limitation of this study is that the sample size was small, allowing only preliminary findings. Another limitation is that the groups showed a statistical difference in BMI between PCOS-VO and PCOS-NVO, and the effect of BMI on gut microbiota could not be excluded. However, VFA and BMI, respectively, define obesity from different perspectives. Clinically, VFA and BMI are often positively correlated, so it is difficult to completely distinguish the effect of the two on gut microbiota. Of course, we will industriously further expand clinical samples to recruit PCOS patients with visceral obesity and non-visceral obesity with matching BMI for exploration of the relationship between VFA, BMI, and gut microbiota. More basic mechanical experiments on cells or animals should also be performed to demonstrate how gut microbiota influences pathological processes of visceral obesity, metabolism, and reproduction in PCOS.
Data Sharing Statement
The data that support the findings of this study are available from the corresponding authors.
Ethics Approval and Informed Consent
The study protocol was approved by the Medical Ethics Committee of the Second Affiliated Hospital of Fujian Medical University (Ethics number 321). All participants completed stool collection for fecal metagenomic sequencing and signed informed consent. The study protocol was conducted in accordance with the Declaration of Helsinki.
Acknowledgments
We thank Wekemo Tech Group Co., Ltd. (Shenzhen, China) for high-throughput metagenomic sequencing.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by Startup Fund for scientific research, Fujian Medical University (2018QH1102), Fujian Provincial Health Research Talents Training Project (2019-1-48), Fujian Provincial Health Research Talents Training Project (2019-ZQN-66), and Clinical Key Specialty Construction Project of Fujian Province.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Azziz R. Polycystic ovary syndrome. Obstet Gynecol. 2018;132(2):321–336. doi:10.1097/AOG.0000000000002698
2. Qi X, Yun C, Sun L, et al. Gut microbiota-bile acid-interleukin-22 axis orchestrates polycystic ovary syndrome. Nat Med. 2019;25(8):1225–1233. doi:10.1038/s41591-019-0509-0
3. Jobira B, Frank DN, Pyle L, et al. Obese adolescents with PCOS have altered biodiversity and relative abundance in gastrointestinal microbiota. J Clin Endocrinol Metab. 2020;105(6):e2134–e2144. doi:10.1210/clinem/dgz263
4. Torres PJ, Siakowska M, Banaszewska B, et al. Gut microbial diversity in women with polycystic ovary syndrome correlates with hyperandrogenism. J Clin Endocrinol Metab. 2018;103(4):1502–1511. doi:10.1210/jc.2017-02153
5. Chen F, Chen Z, Chen M, et al. Reduced stress-associated FKBP5 DNA methylation together with gut microbiota dysbiosis is linked with the progression of obese PCOS patients. NPJ Biofilms Microbiomes. 2021;7(1):60. doi:10.1038/s41522-021-00231-6
6. Liu R, Zhang C, Shi Y, et al. Dysbiosis of gut microbiota associated with clinical parameters in polycystic ovary syndrome. Front Microbiol. 2017;8:324. doi:10.3389/fmicb.2017.00324
7. Barber TM, Dimitriadis GK, Andreou A, Franks S. Polycystic ovary syndrome: insight into pathogenesis and a common association with insulin resistance. Clin Med. 2016;16(3):262–266. doi:10.7861/clinmedicine.16-3-262
8. Yang YL, Zhou WW, Wu S, et al. Intestinal flora is a key factor in insulin resistance and contributes to the development of polycystic ovary syndrome. Endocrinology. 2021;162(10):bqab118. doi:10.1210/endocr/bqab118
9. Torres PJ, Ho BS, Arroyo P, et al. Exposure to a healthy gut microbiome protects against reproductive and metabolic dysregulation in a PCOS mouse model. Endocrinology. 2019;160(5):1193–1204. doi:10.1210/en.2019-00050
10. Shi W, Zhao Q, Zhao X, Xing C, He B. Analysis of endocrine and metabolic indexes in non-obese patients with polycystic ovary syndrome and its compare with obese patients. Diabetes Metab Syndr Obes. 2021;14:4275–4281. doi:10.2147/DMSO.S329108
11. Glueck CJ, Goldenberg N. Characteristics of obesity in polycystic ovary syndrome: etiology, treatment, and genetics. Metabolism. 2019;92:108–120. doi:10.1016/j.metabol.2018.11.002
12. Insenser M, Murri M, Del Campo R, et al. Gut microbiota and the polycystic ovary syndrome: influence of sex, sex hormones, and obesity. J Clin Endocrinol Metab. 2018;103(7):2552–2562.
13. Tchernof A, Després JP. Pathophysiology of human visceral obesity: an update. Physiol Rev. 2013;93(1):359–404. doi:10.1152/physrev.00033.2011
14. Endocrinology and Metabolism Branch of Chinese Medical Doctor Association. Expert consensus on diagnosis and treatment of polycystic ovary syndrome. Chin J Endocrinol Metab. 2018;34(1):7.
15. Examination Committee of Criteria for ‘Obesity Disease’ in Japan; Japan Society for the Study of Obesity. New criteria for ‘obesity disease’ in Japan. Circ J. 2002;66(11):987–992. doi:10.1253/circj.66.987
16. Bharti R, Grimm DG. Current challenges and best-practice protocols for microbiome analysis. Brief Bioinform. 2021;22(1):178–193. doi:10.1093/bib/bbz155
17. Mammadova G, Ozkul C, Yilmaz Isikhan S, Acikgoz A, Yildiz BO. Characterization of gut microbiota in polycystic ovary syndrome: findings from a lean population. Eur J Clin Invest. 2021;51(4):e13417. doi:10.1111/eci.13417
18. Tett A, Pasolli E, Masetti G, Ercolini D, Segata N. Prevotella diversity, niches and interactions with the human host. Nat Rev Microbiol. 2021;19(9):585–599. doi:10.1038/s41579-021-00559-y
19. Jobira B, Frank DN, Silveira LJ, et al. Hepatic steatosis relates to gastrointestinal microbiota changes in obese girls with polycystic ovary syndrome. PLoS One. 2021;16(1):e0245219. doi:10.1371/journal.pone.0245219
20. Serena C, Ceperuelo-Mallafré V, Keiran N, et al. Elevated circulating levels of succinate in human obesity are linked to specific gut microbiota. ISME J. 2018;12(7):1642–1657. doi:10.1038/s41396-018-0068-2
21. Naderpoor N, Mousa A, Gomez-Arango LF, Barrett HL, Dekker Nitert M, de Courten B. Faecal microbiota are related to insulin sensitivity and secretion in overweight or obese adults. J Clin Med. 2019;8(4):452. doi:10.3390/jcm8040452
22. Pinart M, Dötsch A, Schlicht K, et al. Gut microbiome composition in obese and non-obese persons: a systematic review and meta-analysis. Nutrients. 2021;14(1):12. doi:10.3390/nu14010012
23. Moreno-Indias I, Sánchez-Alcoholado L, García-Fuentes E, Cardona F, Queipo-Ortuño MI, Tinahones FJ. Insulin resistance is associated with specific gut microbiota in appendix samples from morbidly obese patients. Am J Transl Res. 2016;8(12):5672–5684.
24. Wade WG. Dialister. Bergey’s manual of systematics of archaea and bacteria 2015. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/9781118960608.gbm00696.
25. Cox TO, Lundgren P, Nath K, Thaiss CA. Metabolic control by the microbiome. Genome Med. 2022;14(1):80. doi:10.1186/s13073-022-01092-0
26. Nagao-Kitamoto H, Leslie JL, Kitamoto S, et al. Interleukin-22-mediated host glycosylation prevents Clostridioides difficile infection by modulating the metabolic activity of the gut microbiota. Nat Med. 2020;26(4):608–617. doi:10.1038/s41591-020-0764-0
27. Fan S, Raychaudhuri S, Page R, Shahinozzaman M, Obanda DN. Metagenomic insights into the effects of urtica dioica vegetable on the gut microbiota of C57BL/6J obese mice, particularly the composition of Clostridia. J Nutr Biochem. 2021;91:108594. doi:10.1016/j.jnutbio.2021.108594
28. Obanda DN, Husseneder C, Raggio AM, et al. Nutrition. Abundance of the species Clostridium butyricum in the gut microbiota contributes to differences in obesity phenotype in outbred Sprague-Dawley CD rats. Nutrition. 2020;78:110893. doi:10.1016/j.nut.2020.110893
29. Zhao H, Chen R, Zheng D, et al. Modified banxia xiexin decoction ameliorates polycystic ovarian syndrome with insulin resistance by regulating intestinal microbiota. Front Cell Infect Microbiol. 2022;12:854796. doi:10.3389/fcimb.2022.854796
30. Debédat J, Clément K, Aron-Wisnewsky J. Gut microbiota dysbiosis in human obesity: impact of bariatric surgery. Curr Obes Rep. 2019;8(3):229–242. doi:10.1007/s13679-019-00351-3
31. Ghosh S, Dent R, Harper ME, Gorman SA, Stuart JS, McPherson R. Gene expression profiling in whole blood identifies distinct biological pathways associated with obesity. BMC Med Genomics. 2010;3:56. doi:10.1186/1755-8794-3-56
32. Elhamamsy AR, Metge BJ, Alsheikh HA, Shevde LA, Samant RS. Ribosome biogenesis: a central player in cancer metastasis and therapeutic resistance. Cancer Res. 2022;82(13):2344–2353. doi:10.1158/0008-5472.CAN-21-4087
33. Wu J, Jiang X, Li Y, et al. PHA-4/FoxA senses nucleolar stress to regulate lipid accumulation in Caenorhabditis elegans. Nat Commun. 2018;9(1):1195. doi:10.1038/s41467-018-03531-2
34. Bellini L, Campana M, Mahfouz R, et al. Targeting sphingolipid metabolism in the treatment of obesity/type 2 diabetes. Expert Opin Ther Targets. 2015;19(8):1037–1050. doi:10.1517/14728222.2015.1028359
35. Parveen F, Bender D, Law SH, Mishra VK, Chen CC, Ke LY. Role of ceramidases in sphingolipid metabolism and human diseases. Cells. 2019;8(12):1573. doi:10.3390/cells8121573
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.