")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 11 » Issue 1
Impact of tobacco smoking on cytokine signaling via interleukin-17A in the peripheral airways
Authors Levänen B, Glader P, Dahlén B, Billing B, Qvarfordt I, Palmberg L, Larsson K, Lindén A
Received 1 March 2016
Accepted for publication 8 May 2016
Published 6 September 2016 Volume 2016:11(1) Pages 2109—2116
DOI https://doi.org/10.2147/COPD.S99900
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Richard Russell
Bettina Levänen,1 Pernilla Glader,2 Barbro Dahlén,3,4 Bo Billing,4 Ingemar Qvarfordt,2 Lena Palmberg,1 Kjell Larsson,1 Anders Lindén1,2,4
1Unit for Lung and Airway Research, Institute of Environmental Medicine, Karolinska Institutet, Stockholm, 2Department of Internal Medicine & Clinical Nutrition, Institute of Medicine, Sahlgrenska Academy at the University of Gothenburg, Gothenburg, 3Centre for Allergy Research, Institute of Environmental Medicine, Karolinska Institutet, Stockholm, 4Lung Allergy Clinic, Karolinska University Hospital, Stockholm, Sweden
Abstract: There is excessive accumulation of neutrophils in the airways in chronic obstructive pulmonary disease (COPD) but the underlying mechanisms remain poorly understood. It is known that extracellular cytokine signaling via interleukin (IL)-17A contributes to neutrophil accumulation in the airways but nothing is known about the impact of tobacco smoking on extracellular signaling via IL-17A. Here, we characterized the impact of tobacco smoking on extracellular cytokine signaling via IL-17A in the peripheral airways in long-term smokers with and without COPD and in occasional smokers before and after short-term exposure to tobacco smoke. We quantified concentrations of IL-17A protein in cell-free bronchoalveolar lavage (BAL) fluid samples (Immuno-quantitative PCR) and cytotoxic T-cells (immunoreactivity for CD8+ and CD3+) in bronchial biopsies. Matrix metalloproteinase-8 and human beta defensin 2 proteins were also quantified (enzyme-linked immunosorbent assay) in the BAL samples. The concentrations of IL-17A in BAL fluid were higher in long-term smokers without COPD compared with nonsmoking healthy controls, whereas those with COPD did not differ significantly from either of the other groups. Short-term exposure to tobacco smoke did not induce sustained alterations in these concentrations in occasional smokers. Long-term smokers displayed higher concentrations of IL-17A than did occasional smokers. Moreover, these concentrations correlated with CD8+ and CD3+ cells in biopsies among long-term smokers with COPD. In healthy nonsmokers, BAL concentrations of matrix metalloproteinase-8 and IL-17A correlated, whereas this was not the case in the pooled group of long-term smokers with and without COPD. In contrast, BAL concentrations of human beta defensin 2 and IL-17A correlated in all study groups. This study implies that long-term but not short-term exposure to tobacco smoke increases extracellular cytokine signaling via IL-17A in the peripheral airways. In the smokers with COPD, this signaling may involve cytotoxic T-cells. Long-term exposure to tobacco smoke leads to a disturbed association of extracellular IL-17A signaling and matrix metalloproteinase-8, of potential importance for the coordination of antibacterial activity.
Keywords: BAL, COPD, IL-17, tobacco, long-term smoking, occasional smoking
Introduction
An excessive local accumulation of neutrophils and their potentially tissue damaging effector molecules is a common finding in the peripheral airways of smokers with chronic obstructive pulmonary disease (COPD).1 While displaying this accumulation of innate immune cells and their effector molecules, patients with COPD at the same time suffer from increased susceptibility to bacterial infections.2,3 Tentatively, more knowledge about the mechanistic rationale for this immunological phenomenon may prove important for understanding the pathophysiology of COPD.
Interleukin (IL)-17A is a cytokine that can be secreted by cytotoxic and T helper lymphocytes and this cytokine is involved in the recruitment of neutrophils to sites of infections in mammals.4 Here, IL-17A stimulates structural cells, such as fibroblasts and epithelial cells, to produce chemokines and growth factors, thereby promoting the local recruitment and accumulation of neutrophils.5,6 Even though it has previously been shown that intracellular IL-17A protein is expressed in peripheral airway tissue and that this tissue expression is higher in smokers with COPD than in nonsmokers, virtually nothing is known with respect to the impact of tobacco smoking on extracellular cytokine signaling via IL-17A in the peripheral airways, the functionally most critical aspect of cytokine production in this compartment.7
We hypothesized that extracellular cytokine signaling via IL-17A in the peripheral airways is detrimentally altered by tobacco smoking and that this also alters critical effector molecules of innate effector cells. To address this hypothesis, we characterized the impact of tobacco smoking on extracellular cytokine signaling via IL-17A in the peripheral airways of well-characterized, current, long-term smokers, with and without COPD, and in occasional smokers, before and after short-term exposure to tobacco smoke. Because of the recent claim that cytotoxic T lymphocytes constitute a source of IL-17A in tobacco smokers, we examined the association of extracellular cytokine signaling via IL-17A with immunoreactivity to CD3+ and CD8+ cells in bronchial biopsies.8 In the peripheral airways, we also quantified matrix metalloproteinase (MMP)-8 and human beta defensin 2 (HBD2), two innate effector molecules, downstream of the coordinating cytokine IL-17A, both being produced by neutrophils.9,10
Materials and methods
Ethics
This study was performed on human subjects after informed oral and written consent in accordance with the Declaration of Helsinki. This study and the clinical study protocol were approved after review by the regional ethical review committee in Stockholm (Diary no 2005/733-31/1-4l) and Gothenburg (Diary no S 313-00; T186-02), respectively.
Cohorts
The study included two cohorts of human subjects as described below.
Long-term exposure
The first cohort was recruited at Karolinska University Hospital in Stockholm and included long-term, current tobacco smokers (referred to as long-term smokers from here and on) with or without COPD and age-matched nonsmoking control subjects (Table 1). The COPD diagnosis was based on Global initiative for chronic Obstructive Lung Disease criteria (post-bronchodilator values of forced expiratory volume in 1 second [FEV1]/forced vital capacity [FVC] <0.7).11 Subjects with COPD had moderate-to-severe COPD (FEV1 >40% of predicted value at two visits) according to Global initiative for chronic Obstructive Lung Disease criteria. All subjects had a transcutaneous arterial oxygen saturation >90%. Local reference values were used.12,13 Current smokers with a FEV1/forced vital capacity >0.7 ratio and FEV1 >80% of predicted values were included in the group of smokers without COPD. Subjects with clinical or laboratory signs of infection within at least 4 weeks prior to bronchoscopy were rescheduled. The control group consisted of nonsmokers with no history of asthma or other lung diseases and all had a normal lung function.
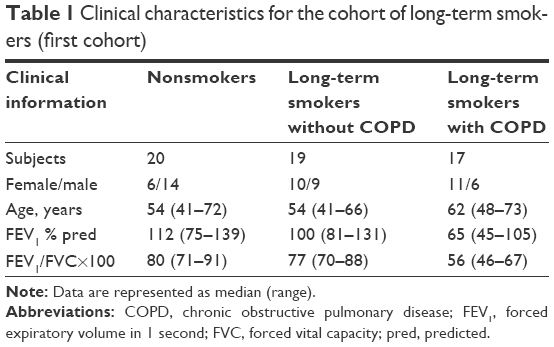 | Table 1 Clinical characteristics for the cohort of long-term smokers (first cohort) |
At the first visit, a bronchodilation was induced by the inhalation of ipratropiumbromid (0.5 mg) and salbutamol (2.5 mg) via an Aiolos Plug-in® inhalator device (Aiolos, Karlstad, Sweden). At the second visit, a bronchodilation test was conducted using only salbutamol (0.1 mg) via a MDI and Volumatic® spacer device (Allen & Hanburys, London, UK). In this cohort, bronchoscopy was performed in accordance with standard procedures as previously described.14 Bronchoalveolar lavage (BAL) was collected by instilling five aliquots of 50 mL sterile saline and gently reaspirating after each aliquot.15 After centrifugation of the BAL sample, the cell pellet was separated from the fluid and cell concentration and viability of the cells were determined. The fluid supernatants of these centrifuged samples (referred to as cell-free BAL fluid) were analyzed for concentrations of extracellular, soluble IL-17A protein, as previously described in detail.16,17 Briefly, before analysis, the fluid was concentrated 20× using Amicon Ultra filters 3K Da (Merck Millipore, Carrigtohill, Co. Cork, Ireland). The concentration of IL-17A protein was then quantified using a high sensitive (limit of detection 1.2–2.5 pg/mL), specific and customized immuno-qPCR (TATAA Biocenter®, Gothenburg, Sweden). For immunohistochemistry of CD3- and CD8-positive cells, bronchial tissue biopsies were obtained from the upper lobe bronchus. The biopsies were fixed in acetone and processed into glycolmethacrylate resin as previously described.18 Thin (2 μm) sections were cut and immunostained in duplicate using monoclonal antibodies (anti-CD3 [M7254] and anti-CD8 [M7103]) (Dako™ Cytomation, Dako Denmark A/S, Glostrup, Denmark.) and the streptavidin–biotin–peroxidase detection system (Dako™ Cytomation).
The concentrations of extracellular, soluble HBD2 (sensitivity 7.8 pg/mL) (Phoenix Pharmaceuticals, Inc, Burlingame, CA, USA) and MMP-8 (sensitivity 78 pg/mL) (R&D Systems, Inc., Minneapolis, MN, USA) proteins were quantified in cell-free BAL fluid using commercially available enzyme-linked immunosorbent assay (ELISA) kits. Sample values in the MMP-8 ELISA that were below the standard curve were arbitrarily set to half of the lowest value of the standard curve divided by 20 to compensate for the concentration procedure of the BAL fluid samples. Sample values in the HBD2 ELISA that were below the standard curve were set to half of the lowest value of the standard curve. Two samples in the HBD2 ELISA (from the group of smokers without COPD) were above the highest value of the standard curve and these samples were set at the highest value of the standard curve (ie, 500 pg/mL).
Short-term exposure
The second cohort was recruited and investigated at Sahlgrenska University Hospital in Gothenburg. This cohort included healthy occasional tobacco smokers (referred to as occasional smokers from here on) and healthy nonsmoking controls (Table 2). Here, all subjects underwent two bronchoscopies, including BAL; the first at day 1 (termed BAL1) and a second at day 14 (termed BAL2). On days 12 and 13, all occasional smokers smoked ≥10 filter cigarettes of a commercial brand (Marlboro™, tar 10 mg, nicotine 0.8 mg) that were purchased (not given as a gift). Occasional smokers all smoked at least once a month, but not more than four times a month or more than ten cigarettes per week. Moreover, all occasional smokers had refrained from smoking 4 weeks prior to the first bronchoscopy. The nicotine intake of the occasional smokers was confirmed by measuring cotinine levels in the urine at day 14.19 A detailed description of this study protocol was included in the original publication by Glader et al.19 A subgroup of the clinical data from the referred study has been used for the current study as well.
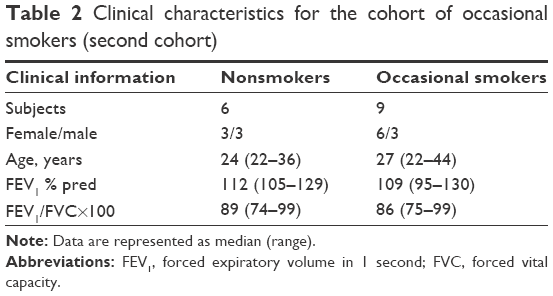 | Table 2 Clinical characteristics for the cohort of occasional smokers (second cohort) |
Statistics
Statistical analyses for more than two groups were performed using Kruskal–Wallis test with Dunn’s multiple comparison test as post hoc test. A nonparametric Wilcoxon signed rank test was used for matched pairs and a nonparametric Mann–Whitney t-test, comparing two groups. A P-value <0.05 was considered statistically significant. Median and range (min–max) were used for all descriptive statistics in the text, whereas median and individual values were used in the figures. The correlations between two observations were tested using the Spearman rank correlation test. Due to the novelty of the key parameters (in particular, the immuno-qPCR for IL-17A), we were unable to perform exact calculations of statistical power.
Results
Group-related differences for IL-17A protein
Long-term smokers
The IL-17A protein was detectable in 55 out of 56 of the cell-free BAL fluid samples of all study groups in the first cohort and these concentrations of IL-17A were clearly higher in long-term smokers without COPD than in nonsmoking healthy control subjects, whereas long-term smokers with COPD displayed corresponding concentrations at a level between the two other groups of cohort 1 (Figure 1). As a result, the pooled group of long-term smokers with and without COPD (15.4 [1.7–95.3] pg/mL, n=36) displayed higher concentrations of IL-17A in cell-free BAL fluid than the nonsmoking healthy control subjects (9.1 [1.6–58.3] pg/mL, n=20) of cohort 1 (P=0.014). Moreover, we did not find any clear, reproducible difference for treatment with (23.6 [5–77.1] pg/mL, n=9) and without (10.2 [1.7–95.3] pg/mL, n=8) corticosteroids (P=0.3655). All the results from these long-term smokers with COPD were therefore pooled and regarded as “ever smokers” in the study. There was no correlation between BAL sample return volumes and the concentrations of IL-17A in cell-free BAL fluid in the pooled group of long-term smokers with and without COPD (data not shown).
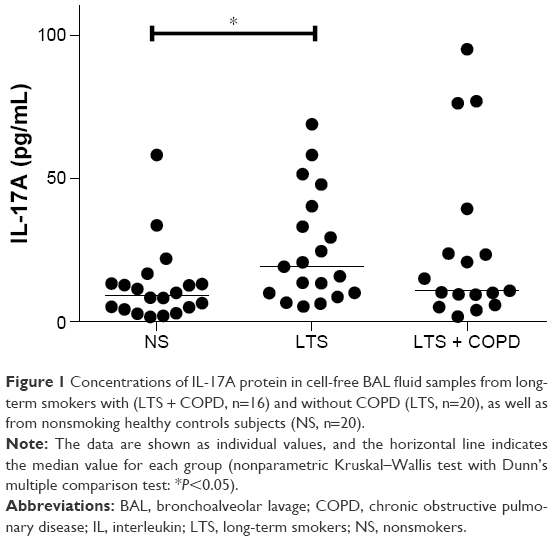 | Figure 1 Concentrations of IL-17A protein in cell-free BAL fluid samples from long-term smokers with (LTS + COPD, n=16) and without COPD (LTS, n=20), as well as from nonsmoking healthy controls subjects (NS, n=20). |
Occasional smokers
We detected no difference for the concentrations of IL-17A protein in cell-free BAL fluid from occasional smokers before and after short-term exposure to tobacco smoke (Figure 2A). However, in these occasional smokers of cohort 2, the concentrations of IL-17A measured prior to short-term exposure were higher than the corresponding concentrations in the nonsmoking controls (Figure 2B). Although these concentrations of IL-17A in occasional smokers prior to short-term exposure were higher than in never-smokers, these concentrations were significantly lower than those in long-term smokers (Figure 3). We performed two bronchoscopies in the nonsmoking controls as well, but here without any cigarette exposure, and found that this procedure per se did not cause any sustained alteration of the concentrations of IL-17A in cell-free BAL fluid (data not shown).
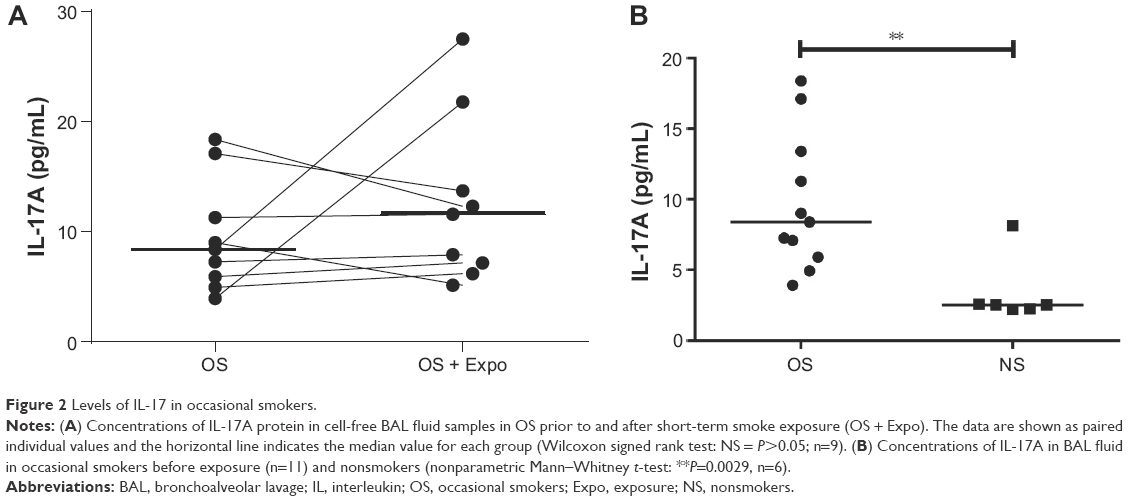 | Figure 2 Levels of IL-17 in occasional smokers. |
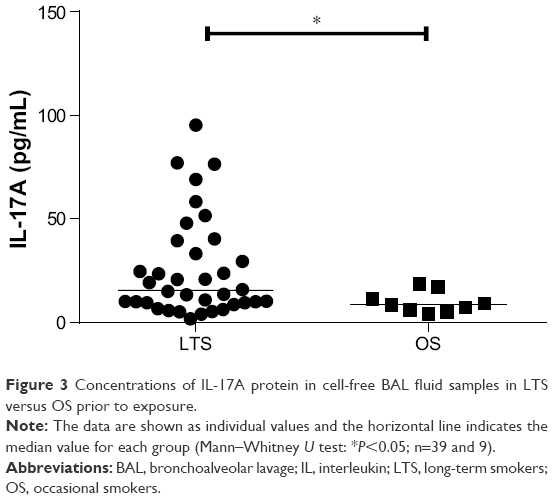 | Figure 3 Concentrations of IL-17A protein in cell-free BAL fluid samples in LTS versus OS prior to exposure. |
Cytotoxic T lymphocytes in long-term smokers
CD8-positive cells
The immunohistochemistry signal for CD8-positive cells (per mm2 submucosa) in bronchial biopsies and the concentrations of IL-17A in cell-free BAL fluid samples displayed a strong positive correlation in the group of long-term smokers with COPD only (Figure 4A). Immunohistochemistry images displayed a clear distinction between positive and negative staining (Figure 4B).
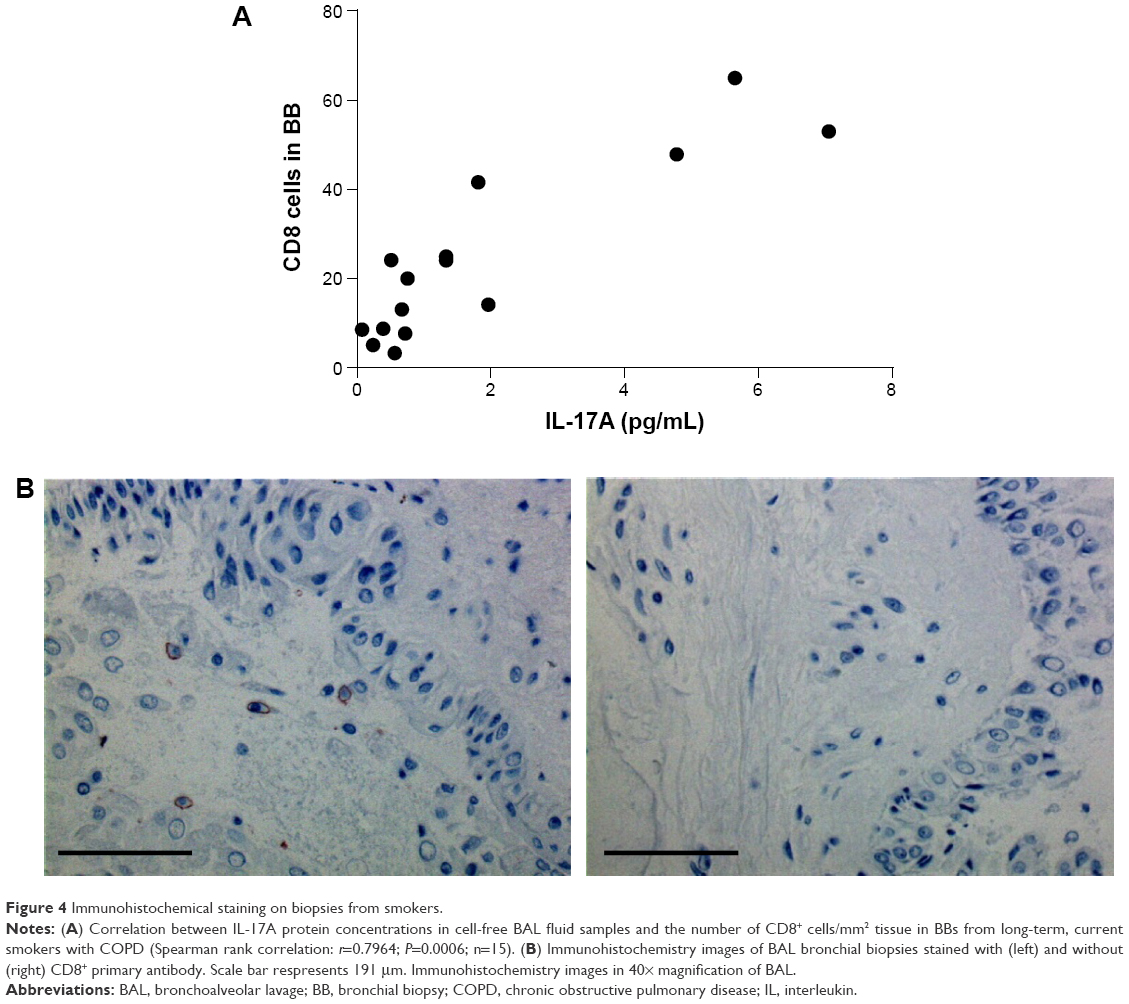 | Figure 4 Immunohistochemical staining on biopsies from smokers. |
CD3-positive cells
In analogy to CD8-positive cells, the corresponding immunohistochemistry signal for CD3-positive cells and IL-17A BAL concentrations displayed a positive correlation in the group of long-term smokers with COPD (r=0.508, P<0.05, data not shown). Thus, no corresponding correlation was detected in the group of long-term smokers without COPD or in nonsmoking healthy subjects.
Innate effector molecules in long-term smokers
The MMP-8 protein concentration was detectable in 52 out of 56 of the cell-free BAL fluid samples of all study groups in the first cohort. However, the MMP-8 BAL concentrations displayed no statistically significant differences between the study groups in the first cohort (data not shown). There was a correlation between MMP-8 and IL-17A concentrations in the BAL fluid when all three study groups were included (Figure 5A). Moreover, we observed a strong correlation when we analyzed the group of nonsmokers from the first cohort separately (Figure 5B) whereas no such correlation was evident in the pooled group of long-term smokers with and without COPD (data not shown).
 | Figure 5 MMP-8 protein levels measured in BAL fluid. |
The HBD2 protein concentration was detectable in 55 out of 56 of the cell-free BAL fluid samples of all study groups in the first cohort; they did, however, not display any statistically significant differences between the study groups in the first cohort (data not shown). However, here we found a statistically significant correlation between HBD2 and IL-17A concentrations in all study groups (Figure 6A–C).
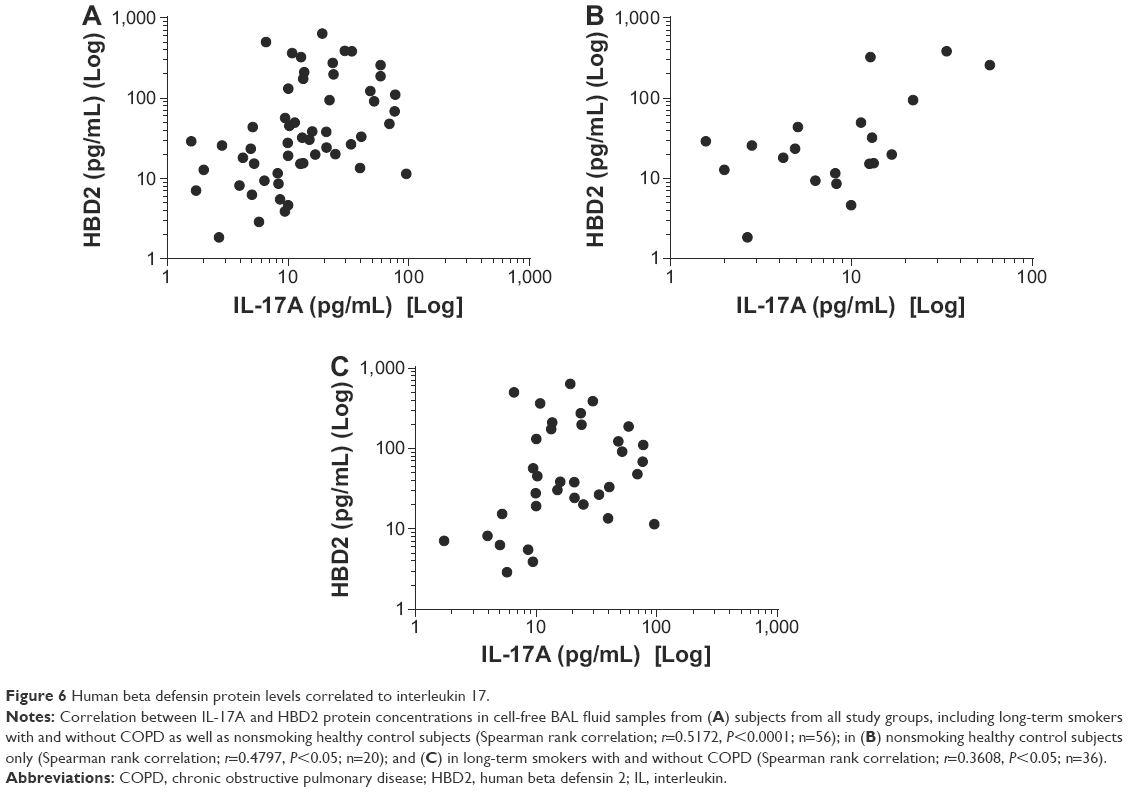 | Figure 6 Human beta defensin protein levels correlated to interleukin 17. |
Discussion
In this first study on cytokine signaling via IL-17A in the peripheral airways of long- and short-term smokers, the results showed that the concentration of extracellular IL-17A protein in cell-free BAL fluid is increased in long-term smokers without COPD, whereas those with COPD only tended to display an increase, the lack of significance possibly due to the limited material available. Moreover, there was no increase in the referred extracellular IL-17A protein after short-term exposure in occasional smokers and this is, to the best of our knowledge, completely novel information on how much smoking is required for the smoking-related alteration in IL-17A signaling.20–24 We also obtained confirmatory evidence that cytokine signaling via IL-17A may relate to cytotoxic lymphocytes, at least in long-term smokers with COPD.8 We think that our strict matching of tobacco load and sex among the long-term smokers with and without COPD, as well as our use of a highly sensitive and specific Immuno quantitative-PCR method made our key findings possible.19
A novel smoking-related mechanism of interest was indicated by the observation that the relationship between extracellular IL-17A signaling and an innate effector molecule in the peripheral airways is altered in long-term smokers. The case was that the correlation between the concentrations of IL-17A and the neutrophil protease MMP-8, a protease that is known to be affected by smoking, was evident in cell-free BAL fluid from nonsmokers but not in the cell-free BAL fluid from the pooled group of long-term smokers with and without COPD.25,26 It can be speculated that this loss of correlation reflects a disturbance in the IL-17A-mediated control of anti-bacterial activity originating from neutrophils, a possibility that clearly warrants further investigation in mechanistic studies. This is supported by the finding by Roos et al23 that neutrophil accumulation in the lungs caused by tobacco smoking is dependent on IL-17A and IL-1R1. For the innate effector molecule HBD2, our results argue that the relationship with extracellular IL-17A signaling was intact in all study groups.
When we quantified IL-17A protein concentrations in cell-free BAL fluid from occasional smokers prior to and after exposure, we did not obtain any evidence for a sustained impact of short-term smoking on extracellular cytokine signaling via IL-17A protein. Thus, the observed increase in the concentrations of IL-17A in cell-free BAL fluid in long-term smokers is likely to be the result of long-term exposure mainly. In line with this idea, we also obtained evidence that long-term smoking causes more of an increase in extracellular cytokine signaling via IL-17A than occasional smoking. We did this by comparing concentrations of IL-17A in cell-free BAL fluid from occasional smokers prior to exposure to those in long-term smokers with and without COPD. We found that these concentrations of IL-17A were higher in long-term smokers and that occasional smokers had higher concentrations than nonsmoking healthy control subjects.
We cannot rule out that the lack of a difference in the concentration of IL-17A for long-term smokers with and without COPD relates to the use of corticosteroids in some of the smokers with COPD, even though we failed to prove a statistically significant impact of this treatment. Of note, however, the previous study by Doe et al20 forwarded evidence supporting the impact of long-term smoking but also the lack of a substantial difference for the smokers with and without COPD, although this study measured immunoreactivity in tissue and not extracellular protein.20 Another study showed opposing results in terms of an association between COPD progression and elevated IL-17A assessed by immunohistochemical methods on lung tissues.22
In fact, even though it is original, the assessment of extracellular cytokine signaling via IL-17A in the peripheral airways of long-term smokers that we present here is compatible with all previous studies on IL-17 cytokines in other compartments from central and peripheral airways, although the impact of short-term exposure has not previously been addressed at all.3,12–14,20 The results of our current study thereby add to the growing body of evidence that forwards IL-17A as a mediator of interest for understanding inflammation caused by tobacco smoking in several compartments of human lungs.
Conclusion
The current study forward evidence that long-term but not short-term tobacco smoking exerts a substantial impact by increasing extracellular cytokine signaling via IL-17A protein in the peripheral airways, whereas the impact of COPD per se remains uncertain. Moreover, the current study also indicates that this long-term smoking may be associated with a disturbance in the IL-17A-mediated control of antibacterial activity originating from neutrophils. We think that our findings have important implications for the understanding of the pathogenesis of airway inflammation caused by tobacco smoke, including COPD, given that repeated bacterial infections in the airways constitute a hallmark and a negative prognostic factor in these smokers. New studies are required to validate whether the current findings illustrate the mechanistic rationale for the immunological paradox that there are more innate effector cells and, at the same time, more bacterial infections in long-term smokers with COPD.
Acknowledgments
The authors wish to acknowledge the excellent technical assistance by Ann-Sofie Lantz, BSc and Elisabeth Henriksson, BSc, and the skillful recruitment of study subjects by Maria Skedinger, MD and Anita Simhag, MD, all from Karolinska University Hospital. The authors also wish to acknowledge the skillful recruitment and investigation of study subjects by Britt-Marie Eldh from Sahlgrenska University Hospital. Project funding was obtained from the Swedish Research Council, the Swedish Heart-Lung Foundation, the Foundation for Rehabilitation and Medical Research in Gothenburg (FRF-stiftelsen), Stockholm County Council, Apotekare Hedbergs Foundation, King Gustaf V’s and Queen Victoria’s Freemason Research Fund, and Karolinska Institutet.
Disclosure
The authors report no conflicts of interest in this work.
References
Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350:2645–2653. | ||
Bagaitkar J, Demuth DR, Scott DA. Tobacco use increases susceptibility to bacterial infection. Tob Induc Dis. 2008;4:12. | ||
Nuorti JP, Butler JC, Farley MM, et al. Cigarette smoking and invasive pneumococcal disease. Active Bacterial Core Surveillance Team. N Engl J Med. 2000;342:681–689. | ||
Laan M, Palmberg L, Larsson K, Linden A. Free, soluble interleukin-17 protein during severe inflammation in human airways. Eur Respir J. 2002;19:534–537. | ||
Kolls JK, Linden A. Interleukin-17 family members and inflammation. Immunity. 2004;21:467–476. | ||
Linden A, Laan M, Anderson GP. Neutrophils, interleukin-17A and lung disease. Eur Respir J. 2005;25:159–172. | ||
Eustace A, Smyth LJ, Mitchell L, Williamson K, Plumb J, Singh D. Identification of cells expressing IL-17A and IL-17F in the lungs of patients with COPD. Chest. 2011;139:1089–1100. | ||
Chang Y, Nadigel J, Boulais N, et al. CD8 positive T cells express IL-17 in patients with chronic obstructive pulmonary disease. Respir Res. 2011;12:43. | ||
Kao CY, Chen Y, Thai P, et al. IL-17 markedly up-regulates beta-defensin-2 expression in human airway epithelium via JAK and NF-kappaB signaling pathways. J Immunol. 2004;173:3482–3491. | ||
Murphy G, Reynolds JJ, Bretz U, Baggiolini M. Collagenase is a component of the specific granules of human neutrophil leucocytes. Biochem J. 1977;162:195–197. | ||
National Institute of Health NH LaBI: Global strategy for the diagnosis, management and prevention of chronic obstructive pulmonary disease. NHBLBI/WHO Workshop Report, Update 2009. | ||
Hedenstrom H, Malmberg P, Agarwal K. Reference values for lung function tests in females. Regression equations with smoking variables. Bull Eur Physiopathol Respir. 1985;21:551–557. | ||
Quanjer PH, Tammeling GJ, Cotes JE, Pedersen OF, Peslin R, Yernault JC. Lung volumes and forced ventilatory flows. Report working party standardization of lung function tests, European Community for Steel and Coal. Official Statement of the European Respiratory Society. Eur Respir J Suppl. 1993;16:5–40. | ||
Djukanovic R, Wilson JW, Lai CK, Holgate ST, Howarth PH. The safety aspects of fiberoptic bronchoscopy, bronchoalveolar lavage, and endobronchial biopsy in asthma. Am Rev Respir Dis. 1991;143:772–777. | ||
Larsson BM, Palmberg L, Malmberg PO, Larsson K. Effect of exposure to swine dust on levels of IL-8 in airway lavage fluid. Thorax. 1997;52:638–642. | ||
Glader P, Smith ME, Malmhall C, et al. Interleukin-17-producing T-helper cells and related cytokines in human airways exposed to endotoxin. Eur Respir J. 2010;36:1155–1164. | ||
Lind K, Kubista M. Development and evaluation of three real-time immuno-PCR assemblages for quantification of PSA. J Immunol Methods. 2005;304:107–116. | ||
Britten KM, Howarth PH, Roche WR. Immunohistochemistry on resin sections: a comparison of resin embedding techniques for small mucosal biopsies. Biotech Histochem. 1993;68:271–280. | ||
Glader P, Eldh B, Bozinovski S, et al. Impact of acute exposure to tobacco smoke on gelatinases in the bronchoalveolar space. Eur Respir J. 2008;32:644–650. | ||
Doe C, Bafadhel M, Siddiqui S, et al. Expression of the T helper 17-associated cytokines IL-17A and IL-17F in asthma and COPD. Chest. 2010;138:1140–1147. | ||
Di Stefano A, Caramori G, Gnemmi I, et al. T helper type 17-related cytokine expression is increased in the bronchial mucosa of stable chronic obstructive pulmonary disease patients. Clin Exp Immunol. 2009;157:316–324. | ||
Roos AB, Sanden C, Mori M, Bjermer L, Stampfli MR, Erjefalt JS. IL-17A is elevated in end-stage chronic obstructive pulmonary disease and contributes to cigarette smoke-induced lymphoid neogenesis. Am J Respir Crit Care Med. 2015;191:1232–1241. | ||
Roos AB, Sethi S, Nikota J, et al. IL-17A and the promotion of neutrophilia in acute exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2015;192:428–437. | ||
Shan M, Yuan X, Song LZ, et al. Cigarette smoke induction of osteopontin (SPP1) mediates T(H)17 inflammation in human and experimental emphysema. Sci Transl Med. 2012;4:117ra9. | ||
Atkinson JJ, Lutey BA, Suzuki Y, et al. The role of matrix metalloproteinase-9 in cigarette smoke-induced emphysema. Am J Respir Crit Care Med. 2011;183:876–884. | ||
Kang MJ, Oh YM, Lee JC, et al. Lung matrix metalloproteinase-9 correlates with cigarette smoking and obstruction of airflow. J Korean Med Sci. 2003;18:821–827. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.