")
Back to Journals » International Journal of Nanomedicine » Volume 14
Impact Of Penetratin Stereochemistry On The Oral Bioavailability Of Insulin-Loaded Solid Lipid Nanoparticles
Authors Alsulays BB , Anwer MK, Soliman GA, Alshehri SM , Khafagy ES
Received 28 July 2019
Accepted for publication 25 October 2019
Published 25 November 2019 Volume 2019:14 Pages 9127—9138
DOI https://doi.org/10.2147/IJN.S225086
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Bader B Alsulays,1 Md Khalid Anwer,1 Gamal A Soliman,2,3 Sultan M Alshehri,4 El-Sayed Khafagy1,5
1Department of Pharmaceutics, College of Pharmacy, Prince Sattam Bin Abdulaziz University, Al-Kharj 11942, Saudi Arabia; 2Department of Pharmacology, College of Veterinary Medicine, Cairo University, Cairo 12211, Egypt; 3Department of Pharmacology, College of Pharmacy, Prince Sattam Bin Abdulaziz University, AlKharj 11942, Saudi Arabia; 4Department of Pharmaceutics, College of Pharmacy, King Saud University, Riyadh, Saudi Arabia; 5Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, Suez Canal University, Ismailia 415-22, Egypt
Correspondence: El-Sayed Khafagy
Department of Pharmaceutics, College of Pharmacy, Prince Sattam Bin Abdulaziz University, PO Box 173, AlKharj 11942, Saudi Arabia
Tel +966 533564286
Fax +966 115886001
Email [email protected]
Purpose: This study evaluated the stereoisomeric effect of L- and D-penetratin—cell-penetrating peptides (CPPs)—incorporated insulin-loaded solid lipid nanoparticles (INS-SLNs) on the bioavailability (BA) of oral insulin (INS).
Methods: Insulin-loaded solid nanoparticles, L-penetratin-INS-SLNs (LP-INS-SLNs), and D-penetratin-INS-SLNs (DP-INS-SLNs) were formulated by double emulsification. The developed SLNs were evaluated for particle size, zeta potential (ZP), and drug encapsulation and subjected to differential scanning calorimetry (DSC), Fourier transform infrared spectroscopy (FTIR), and evaluated for stability against enzymatic degradation in rat intestinal fluid. Finally, the SLNs were administered to rats to evaluate the BA of INS-SLNs that contained L- and D-penetratin.
Results: The mean particle size, PDI, and ZP values of INS-SLNs, LP-INS-SLNs, and DP-INS-SLNs ranged from 618.5 to 973.0 nm, 0.227 to 0.734, and −17.0 to −23.7 mV, respectively. The encapsulation efficiency (%EE) and drug loading (%DL) of INS-SLNs, LP-INS-SLNs, and DP-INS-SLNs ranged from 59.03% to 67.42% and from 1.62% to 1.82%, respectively. Differential scanning calorimetry and FTIR analyses indicated that INS was successfully encapsulated in SLNs. Enzymatic degradation of DP-INS-SLNs was slower in intestinal fluid, and the half-life (t1/2) was significantly prolonged, compared to all other SLNs. The pharmacological availability (PA) and BA of orally administered LP-INS-SLNs, which were the most effective SLNs, were 13.1% and 15.7% relative to s.c. administration, respectively.
Conclusion: Penetratin stereochemistry significantly impacted oral BA of INS-SLNs, which are promising carriers for oral INS administration.
Keywords: cell-penetrating peptides, penetratin, stereochemistry, solid lipid nanoparticles, enzymatic degradation, oral insulin bioavailability
Introduction
Despite significant improvements to large-scale production of insulin (INS), the universal drug clinically used to treat diabetes, development of effective oral INS delivery systems remains a major challenge.1,2 Daily subcutaneous (s.c.) injections are the standard of treatment for insulin-dependent patients with diabetes. Oral INS delivery would be convenient and improve patient compliance. Furthermore, oral administration could result in reduced risk of hyperinsulinemia, weight gain, and hypoglycemia.3 However, oral absorption of INS is inefficient, resulting in low bioavailability (BA), primarily due to degradation by proteolytic enzymes in the gastrointestinal (GI) tract and poor permeability across the intestinal mucosal membrane.4 To exert therapeutic effects, oral INS must be efficiently absorbed from the lumen of the intestine into the general circulation.5 Therefore, development of strategies to enhance absorption of INS through the intestinal epithelial membrane is essential for production of suitable oral dosage forms of INS.
Cell-penetrating peptides (CPPs) have been shown to be potential carriers for systemic delivery of INS across the intestinal mucosal membrane, resulting in dramatically enhanced oral BA of INS.6,7 A previous study showed that penetratin, a CPP derived from Drosophila Antennapedia homeoprotein, is the most effective CPP for delivery of oral INS.8 The hypoglycemic response demonstrated in this study occurred due to intermolecular interactions between INS and L- and/or D-penetratin. Further characterization of these interactions is required for this solid dosage form to progress to preclinical and clinical studies.
Nanoparticles (NPs) have been shown to increase the stability and oral BA of peptides and proteins.9 The physicochemical properties of NPs (e.g., size, surface charge, chemical composition) dictate whether NPs cross the intestinal barrier.10,11 Dual delivery technologies that combine the beneficial properties of CPPs and NPs may improve oral INS delivery.12,13 Recently, several approaches to oral delivery of INS using CPPs/NPs have been developed, and a number of recent studies have demonstrated significant improvements in pharmacological availability (PA) following oral administration of INS using delivery systems such as nanoparticles,14–18 nanocapsules,19 nanoemulsions,20 and liposomes,21 with PA values ranging from 11% to 17%. However, the hypoglycemic impacts observed in these studies were achieved using a relatively complicated chemical conjugation-based formulation with enteric-coated capsules.22 We developed a simple, solution-based formulation based on spontaneous intermolecular interactions between INS and CPPs. This formulation resulted in improved oral delivery of INS, as evidenced by improved BA and PA. The simplicity of preparation and administration of this formulation has significant potential for use in preclinical and clinical studies.
Solid lipid nanoparticles (SLNs) are a proposed alternative to other drug delivery technologies such as microemulsions, microspheres, liposomes, and polymeric nanoparticles. SLNs can incorporate lipophilic and hydrophilic drugs, exhibit improved physical stability, and are simple to manufacturing on a large scale.23–25 Furthermore, SLNs are a versatile system for oral delivery of peptides and proteins.26–29 Several studies of INS-loaded SLNs (INS-SLNs) formulated with CPPs have been published, but none of these studies reported the BA of orally administered INS.30,31
We hypothesized that modified SLNs loaded with INS/L- or D-penetratin complexes would result in better oral delivery of INS. The impacts of the stereoisomeric differences between L-and D-penetratin on BA following oral delivery of INS have not been previously studied. Therefore, we designed INS-SLNs that contained either L- or D-penetratin using a double emulsification technique. The properties of INS-SLNs containing L- or D-penetratin were studied. We also evaluated stability of INS in INS-SLNs that contained L- or D-penetratin against enzymatic degradation in rat intestinal fluid. Finally, the pharmacokinetic parameters of INS-SLNs with or without L- and D-penetratin were evaluated in rats following oral administration.
Materials And Methods
Materials
Recombinant human insulin (INS, 24 IU/mg) was purchased from Sigma-Aldrich Chemical Company (St. Louis, MO, USA). L- and D-penetratin (purity: ≥95% for each peptide) were purchased from Bachem AG (Bubendorf, Switzerland). Glyceryl trimyristate (Dynasan™ 114) was purchased from Sasol Germany GmbH (Witten, Germany). Soya lecithin was purchased from AppliChem (Darmstadt, Germany). Polyvinyl alcohol (PVA, M.W. 22000) was obtained from BDH Laboratories (Poole, England). All chemicals and reagents were of analytical grade.
Animals
Male Wistar rats weighing 180–220 g were used in this study. The animals were housed in rooms with controlled temperature (23±1°C) and relative humidity (55±5%) and were allowed free access to water and food during acclimatization. Animals were fasted for 24 hrs before the experiments but could drink water ad libitum. The animal study protocol was approved by the Ethical Committee, Pharmacy College, Prince Sattam Bin Abdulaziz University, Al-Kharj, KSA (approval number: PHARM-21-11-2017).
Preparation Of Insulin (INS)-Loaded SLNs
Insulin was incorporated into SLNs with L-penetratin (LP-INS-SLNs) or D-penetratin (DP-INS-SLNs) by double emulsification (Table 1).32,33 Briefly, 1 mM INS (5.8 mg) and 2.5 mM L-penetratin or D-penetratin (5.6 mg) were dissolved in 200 µL of 0.1 N HCl, then 600 µL of pH 7.4 phosphate-buffered saline (PBS) containing 0.001% methylcellulose, which prevents adsorption of CPP to the tube surface, was added. The solution was neutralized with 200 µL of 0.1 N NaOH. The prepared INS solution was emulsified in an organic phase containing glyceryl trimyristate and soya lecithin in dichloromethane using a probe sonicator (Ultrasonic processor, gx-130, Bandelin, Germany) for 180 s at 40% voltage efficiency with pulse on/off for 10 s in an ice bath. The formed emulsion (w/o) was then emulsified in 20 mL of aqueous PVA solution (0.5%, w/v) added dropwise. The mixture was stirred at 3420 rcf for 45 mins to evaporate the organic solvent. SLNs were removed from the bulk media by centrifugation at 22,800 rcf for 15 mins at 4°C (2015 Centurion scientific, Chichester, UK). The SLNs were then washed three times with cold distilled water and freeze-dried using a lyophilizer (Martin Christ Alpha-1-4LD freeze-drier, Osterode, Germany).
 |
Table 1 Compositions Of Insulin (INS)-Loaded Solid Lipid Nanoparticles (SLNs) |
Measurement Of Particle Size, Zeta Potential (ZP), And Polydispersity Index (PDI)
The mean particle size, ZP, and PDI of the INS-SLNs, LP-INS-SLNs, and DP-INS-SLNs were measured using a Malvern particle size analyzer (Zetasizer Nano ZS; Holtsville, NY, USA) maintained at 25±1°C. The light scattering angle was set at 90ºC. The SLNs were dispersed in distilled at a 1:200, which resulted in an aqueous dispersion. The SLN dispersions were sonicated for 5 mins, then transferred to a disposable transparent plastic cuvette for particle size and PDI measurements. The same procedure was followed for measurement of ZP, except the SLN dispersions were transferred to a glass electrode sample holder.
Measurement Of Drug Encapsulation
Encapsulation efficiency (EE) and drug loading (DL) were measured by centrifuging a freshly prepared SLN suspension at 21,380 rcf for 5 mins at 4°C. The unencapsulated INS in the supernatant was measured using HPLC (Waters 600 System, Waters Corporation, Milford, MA, USA). The mobile phase was 55:45 (v/v) pH 3.2 water with phosphoric acid:acetonitrile. The injection volume was 10 μL, the flow rate was 1.0 mL/min, the detector was set to 214 nm, and the column was a 4.6 × 150 mm, 5 μm Symmetry® RP-C18 (Waters Corporation, Milford, MA, USA). The column was maintained at room temperature.33
Encapsulation efficiency was calculated using the following Eq. (1):
(1)
Drug loading was calculated using the following Eq. (2):
(2)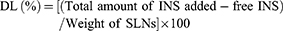
Differential Scanning Calorimetry (DSC)
DSC was performed using a thermal analyzer (Shimadzu DSC-60, Kyoto, Japan). Samples sealed in an aluminum pan (test) and an empty pan (reference) were heated at a rate of 10°C min−1 from 25°C to 250°C in an inert nitrogen gas atmosphere.
Fourier-Transform Infrared (FTIR) Spectroscopy
FTIR spectra of INS, glyceryl trimyristate, soya lecithin, and INS-loaded SLNs (INS-SLNs, LP-INS-SLNs, and DP-INS-SLNs) were recorded from 400 to 4000 cm−1 using the KBr pellet technique (4100 Jasco FTIR spectrophotometer, Tokyo, Japan). The spectra were interpreted by evaluating the different vibrational and functional peaks.
In Vitro Release Studies
The release profiles of INS-SLNs, LP-INS-SLNs, and DP-INS-SLNs were studied in simulated gastric fluid (pH 1.2) and in phosphate buffer (pH 7.4). In vitro release studies were performed using a biological shaker (LBS-030S-Lab Tech, Korea). SLN samples equivalent to 5 mg of INS were dispersed in 5 mL of each medium separately and incubated at 37±0.5°C while shaking at 150 rcf. Aliquots (0.5 mL) were withdrawn at 0.5, 1, 2, 4, and 6 hrs. The samples were centrifuged for 10 mins at 4320 rcf, and supernatants were diluted and analyzed by HPLC.32,33
Stability Of SLNs In Intestinal Fluid
Intestinal fluid was obtained from male Wistar rats by implanting a sonde needle into the upper part of the small intestine. The intestine was then cannulated at the lower portion (~20 cm) to collect intestinal fluid. The lumen of the small intestine was washed with 20 mL of PBS. The collected solution was treated with two volumes of methylene chloride to eliminate any lipids that might interfere with HPLC analysis of INS.34 The lipid extraction procedure was repeated 5 times. Total peptidase concentration in the intestinal fluid was determined using the BCA protein assay kit (Thermo Fisher Scientific Inc., Waltham, MT, U.S.A.). L- and D-penetratin (0.25 mM final concentration) were mixed with INS (10 IU/mL final concentration) and incubated in intestinal fluid at 37°C. Fifty microliters was collected at 5, 15, 30, and 60 mins, and added to 50 µL of ice-cold mobile phase solution to stop enzymatic reactions. The samples were then analyzed for INS content by HPLC.
Pharmacokinetics Of Orally Administered SLNs
The animals were anesthetized with intraperitoneal (i.p.) sodium pentobarbital injection (50 mg/kg; Nembutal®; Abbott Laboratories, IL, USA), then dosed with INS solution, INS-SLNs, LP- INS-SLNs, or DP- INS-SLNs (10 IU/kg) by oral gavage or with INS solution (1 IU/kg) by s.c. injection. All animals were tested for blood-glucose concentration prior to dosing. After oral administration of INS solution or INS-SLNs, approximately 0.3 mL of blood was withdrawn from the jugular vein before and 0.25, 0.5, 1, 2, 3, 4, 6, 8, and 12 hrs after dosing. Fifteen microliters of blood was removed and analyzed for glucose levels using a glucometer (Accu-Check Active; Roche Diagnostics GmbH, Mannheim, Germany). The remainder of the blood was kept at room temperature, then centrifuged (14,250 rcf for 10 mins at 4°C) to isolate serum. One hundred microliters of serum was used to determine INS concentration using a human INS ELISA kit (Sigma-Aldrich Co., St. Louis, MO, USA). Absorbance at 450 nm was measured using a microplate reader (BioTek Instruments Inc., Winooski, VT, USA).
Data Analysis
The total area under the concentration curve (AUC) from 0 to 12 hrs was evaluated using the sum of consecutive trapezoids between each data point. The peak serum concentration (Cmax) and the time taken to reach the peak serum concentration (Tmax) were directly determined using the serum concentration–time curve for INS.
The biological activity of INS was represented as the percentage of the glucose concentration before dosing compared to the blood-glucose concentration in the control group (INS solution). The hypoglycemic effect was estimated using the trapezoidal method for the area above the curve (AAC) from 0 to 12 hrs ([AAC]oral and [AAC]s.c. for oral and s.c. INS, respectively). The pharmacological availability (PA (%)) of INS was calculated using Eq. (3).
(3)
The AUC of the INS concentration–time profile ([AUC]oral and [AUC]s.c.) from 0 to 12 hrs was represented by the sum of successive trapezoids between each data point. The BA (%) of INS was calculated relative to the s.c. injections using Eq. (4).
(4)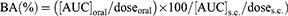
Statistical Analysis
All data were normally distributed and were expressed as the mean ± SEM. For group comparisons, one-way ANOVA with duplication was used. Significant differences in mean values were determined using Student’s unpaired t-test. P≤0.05 was considered statistically significant.
Results And Discussion
Preparation And Characterization Of SLNs
SLNs were prepared by W/O/W double emulsion, which is commonly used for loading hydrophobic and hydrophilic macromolecules.35,36 The primary emulsion (W/O) was first prepared, then emulsified in a continuous aqueous phase to form the double emulsion (W/O/W) containing INS and penetratin in the internal aqueous phase, lipid matrix (glyceryl trimyristate) in the oil phase, and PVA as the emulsifier in the external aqueous phase. To prevent loss of INS pharmacological activity, INS–penetratin complexes were formed via noncovalent interactions. This approach relies on electrostatic interactions between penetratin and INS under physiological conditions, and hydrophobic residues can contribute to hydrophobic interaction.37 The quantities of INS and penetratin incorporated into SLNs were determined by absorption in INS in vivo, and the absorption-enhancing efficiency of penetratin. The optimal ratio of INS to penetratin was approximately 1:2.5.37 A previous study showed that the combination of glyceryl trimyristate and soybean lecithin resulted in increased crystallinity, which may prevent premature dissolution of INS.33
Particle size, PDI, ZP, EE, and DL data for the SLNs are summarized in Table 2. The INS-loaded SLNs (INS-SLNs, LP-INS-SLNs, and DP-INS-SLNs) ranged in size from 618.5±8.4 (INS-SLN) to 973.0±15.3 nm (DP-INS-SLN), which was within the nanoparticulate range of ≤1000 nm. The size of the SLNs increased with addition of L-penetratin and D-penetratin. The PDI values of the SLNs ranged from 0.227 to 0.734. Values less than 1.0 indicated that the particles were suitable for analysis using differential light scanning analysis. Larger PDI values can occur due to aggregation/agglomeration resulting from electrostatic interactions.38
 |
Table 2 Particle Characterization Of Insulin (INS)-Loaded Solid Lipid Nanoparticles (SLNs) |
The ZP values were −17.0±1.53, −23.7±2.13, and −20.9±1.63 mV for INS-SLNs, LP-INS-SLNs, and DP-INS-SLNs, respectively (Table 2). These results demonstrated that the SLNs produced in this study were a stable system under dynamic conditions.38 Negative ZP values may have been due to slightly negatively charged lipid matrix and soybean phospholipid on the particle surfaces at neutral pH. Penetratin contributes a positive charge to the SLNs. Negative ZP values of LP-INS-SLNs and DP-INS-SLNs similar to those of INS-SLNs indicated that the positive charge of penetratin was neutralized by interactions with INS within the SLNs.
Measurement Of Drug Encapsulation
The results of INS EE and DL are summarized in Table 2. The EE and DL of INS in the SLNs (INS-SLNs, LP-INS-SLNs, and DP-INS-SLNs) ranged from 59.03±4.21 to 67.42±3.12% and 1.62±0.09 to 1.82±0.12%, respectively. Inclusion of L-penetratin resulted in increased entrapment of INS, likely due to electrostatic interactions between densely positively charged penetratin amino acids (arginine and lysine) and negatively charged INS, which resulted in penetration of INS into the lipid matrix. Both LP-INS-SLNs and DP-INS-SLNs exhibited fine particle sizes, absolute value above negative of ZP, and excellent INS EE and DL. These results confirmed that stable and highly dispersible nanoparticulate dispersions were formulated by double emulsion in our study.
DSC Thermal Analysis
Figure 1 shows the DSC spectra of pure INS and lipids (glyceryl trimyristate, soya lecithin), freeze-dried INS-SLNs, LP-INS-SLNs, and DP-INS-SLNs across the range of 25–250°C. The thermogram of pure INS showed a broad endothermic peak at 95.39°C that represented denaturation of the drug, and an exothermic peak at 212.46°C that corresponded to decomposition.39,40 The DSC spectrum of glyceryl trimyristate had a distinct peak at 78°C, and the DSC spectrum of soya lecithin had a merged peak across the temperature range of 190-210°C.32 The endothermic peaks of pure INS were absent in the freeze-dried INS-SLNs, LP-INS-SLNs, and DP-INS-SLNs, which confirmed that INS was completely encapsulated. However, the peak associated with glyceryl trimyristate was observed in all SLNs due to its solid crystalline state.
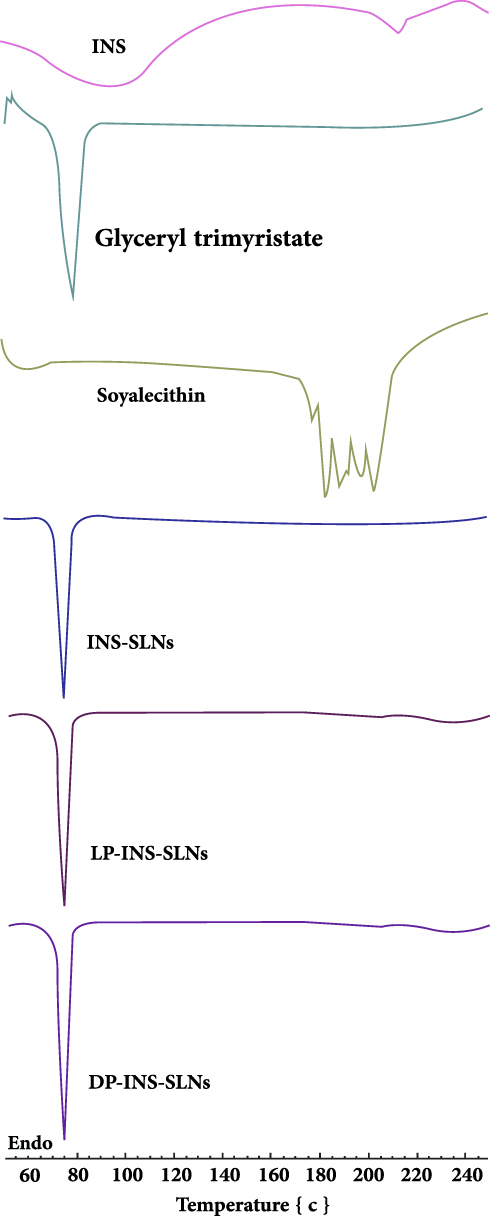 |
Figure 1 Comparative DSC spectra of insulin (INS)-loaded solid lipid nanoparticles (SLNs). |
FTIR Spectral Analysis
FTIR spectra were collected for pure INS, glyceryl trimyristate, soya lecithin, and SLNs that contained L-penetratin and D-penetratin to evaluate drug–lipid interactions (Figure 2). The main peaks assigned to pure INS were associated with symmetrical and asymmetrical N-H bending of amino acids at 1533 cm−1 and 1661 cm−1, respectively.39 A slight shift, and decreased intensity, of these peaks was observed in the spectra of INS-SLNs, LP-INS-SLNs, and DP-INS-SLNs (Figure 2). These results indicated that INS was successfully encapsulated inside the lipid matrix. Peaks near 2910, 2806, and 1763 cm−1 were observed in all the SLNs due to presence of glyceryl trimyristate.32
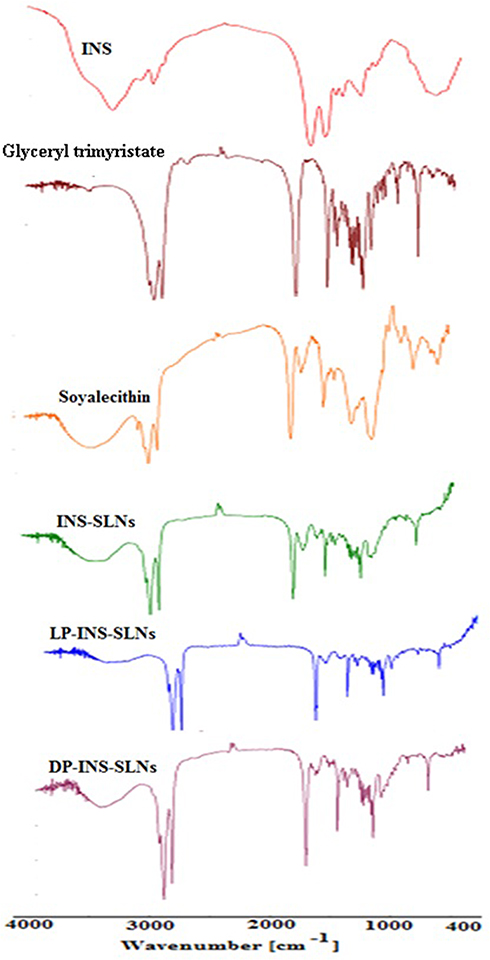 |
Figure 2 Comparative FTIR spectra of insulin (INS)-loaded solid lipid nanoparticles (SLNs). |
In Vitro Release Studies
The in vitro release profiles of INS-SLNs, LP-INS-SLNs, and DP-INS-SLNs in simulated gastric fluid (pH 1.2) and simulated intestinal fluid (pH 7.4) are presented in Figures 3 and 4. Insulin release from INS-SLNs, LP-INS-SLNs, and DP-INS-SLNs at pH 1.2 was 77%, 91%, and 82%, respectively. Insulin release from INS-SLNs, LP-INS-SLNs, and DP-INS-SLNs at pH 7.4 was 62%, 76%, and 69%, respectively.33 Dissolution of INS occurred more readily from LP-INS-SLNs and DP-INS-SLN than from INS-SLNs, which may have been due to the presence of penetratin.
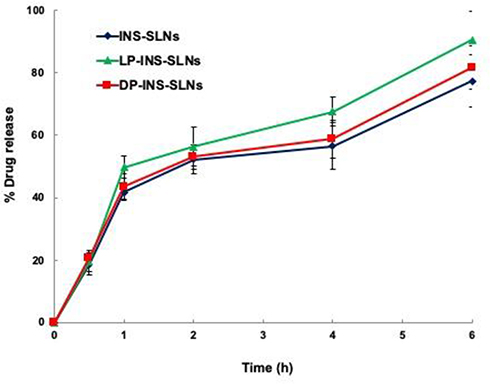 |
Figure 3 Release profile of INS from different SLNs in simulated gastric fluid (pH 1.2). Each data point represents the mean±SEM (n=3). |
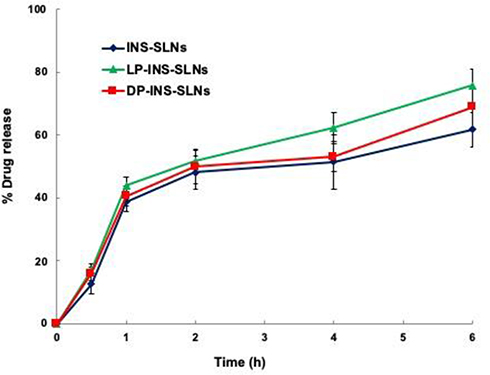 |
Figure 4 Release profile of INS from different SLNs in simulated intestinal fluid (pH 7.4). Each data point represents the mean±SEM (n=3). |
The performance of our formulation leveraged electrostatic and/or hydrophobic interactions between penetratin and INS under physiologic conditions.37 Release of penetratin from the INS/penetratin complex occurred in intestinal fluid due to proteolytic enzymes. As such, penetratin release is difficult to measure in vitro due to the lack of proteolytic enzymes.
SLN Stability In Intestinal Fluid
Figure 5 shows the effects of penetratin (0.25 mM) on stability of INS-SLNs (10 IU/mL) in rat intestinal fluid. After 1 hr of incubation in intestinal fluid, the percentages of remaining INS, INS-SLNs, LP-INS-SLNs, and DP-INS SLNs were 41.9%, 51.6%, 51.8%, and 94.9%, respectively. Elimination of INS followed apparent first-order kinetics. Insulin in solution was rapidly degraded in intestinal fluid. Incorporation of INS into SLNs, particularly DP-INS-SLN, considerably reduced INS degradation. Table 3 summarizes the elimination rate constant (kel) and half-life (t1/2) of INS solution and INS-SLNs with or without penetratin in intestinal fluid. The kel was calculated using the slope of the curve for each SLN formulation (Figure 3). The formula for half-life was t1/2= 0.693/kel. The kel (2.7×10−2 min−1) and t1/2 (25.2 min) for INS-SLNs were not significantly different from those of INS in solution (kel = 3.4×10−2 min−1, t1/2=20.1 min). In contrast, the kel of DP-INS-SLNs was significantly lower (0.5×10−2 min−1) and the t1/2 (136.7 min) was significantly longer than that of INS in solution and INS-SLNs. Furthermore, the t1/2 of DP-INS-SLNs was significantly longer than that of LP-INS-SLNs (28.1 min).
 |
Table 3 Elimination Constant And Half-Life Of Insulin (INS) Solution And INS-Loaded Solid Lipid Nanoparticles (INS-SLNs) In Rat Intestinal Fluid |
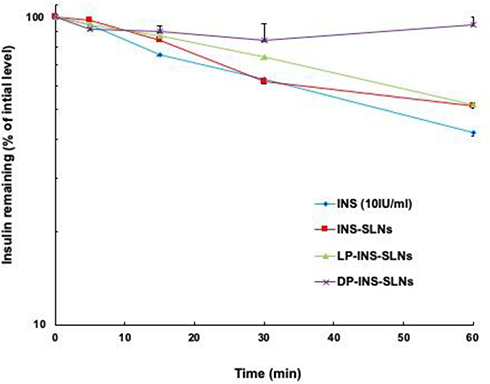 |
Figure 5 Degradation profile of INS solution (10 IU/mL) and INS-SLNs over time in rat intestinal fluid. Each data point represents the mean±SEM (n=3). |
These results suggested that the SLNs formulated in our study improved the stability of encapsulated INS. Rapid degradation of INS in intestinal fluid in our study was consistent with other reports.7,8 In contrast, INS in SLNs was protected from enzymatic degradation. Moreover, inclusion of penetratin significantly protected the SLNs in intestinal fluid.17 D-penetratin may protect INS from degradation by forming a stable physical complex with INS, resulting in increased resistance to proteolytic enzymes.7 In a previous study, an INS–CPP complex may have maintained intermolecular interactions in the small intestine. In contrast to D-penetratin, the INS–L-penetratin complex underwent degradation by intestinal enzymes due to low resistance of L-penetratin to enzymatic degradation.7 In this study, DP-INS-SLNs showed the greatest resistance to enzymatic degradation in rat intestinal fluid among all of the formulations evaluated. These results showed that inclusion of a CPP, particularly D-penetratin, in INS-containing SLNs protected against enzymatic degradation in the GI tract.
Assessment Of The Oral Bioavailability Of INS-Loaded SLNs
The oral pharmacological effects and pharmacokinetic parameters of different SLNs were determined in rats. Figure 6A shows the blood-glucose level-time profiles following oral administration of different INS (10 IU/kg) formulations. No obvious hypoglycemic response was detected after oral administration of INS in solution, which indicated that no INS was absorbed by the intestinal lumen. Oral administration of INS-SLNs, LP-INS-SLNs, and DP-INS-SLNs resulted in significant hypoglycemic responses, which suggested that INS encapsulated in SLNs was protected against diverse pH conditions and against enzymatic degradation in the GI tract. The hypoglycemic effect peaked at 3 hrs (70.9% of the initial glucose levels) after oral administration of INS-SLNs equivalent to 10 IU/kg of INS. Surprisingly, LP-INS-SLNs induced the greatest hypoglycemic effect among the formulations evaluated. The maximum hypoglycemic effects in response to oral LP-INS-SLNs and DP-INS-SLNs occurred at 3 hrs and 2 hrs (47.2% and 79.9% of the initial level), respectively. Figure 6B shows the serum profile following oral administration of INS solution, INS-SLNs, LP-INS-SLNs, and DP-INS-SLNs (10 IU/kg). No apparent absorption was observed following oral administration of INS solution. When INS-SLNs were orally administered by gavage, INS absorption was greater than that following oral administration of INS solution. In contrast, DP-INS-SLNs administration resulted in slightly greater INS absorption than INS-SLNs. However, LP-INS-SLNs administration resulted in the greatest absorption of INS.
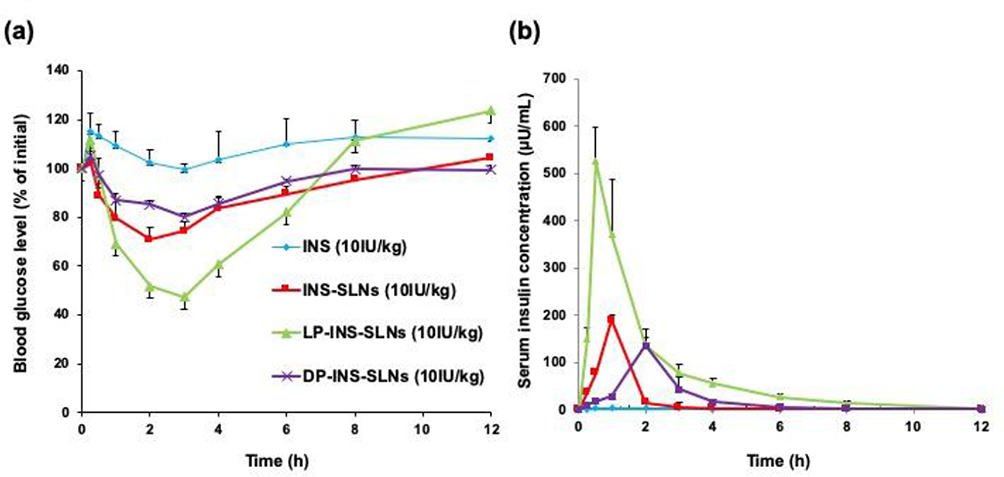 |
Figure 6 Blood glucose (A) and serum insulin (B) concentration vs time profiles following oral administration of INS solution (10 U/kg) and INS-SLNs. Each data point represents the mean±SEM (n=5). |
Table 4 shows the pharmacodynamic and pharmacokinetic parameters derived from INS concentration–time profiles following oral administration of INS solution and various SLN formulations. Comparison of the area above the hypoglycemic curve versus time profile (AAC) obtained following oral INS formulations with that following s.c. injection of INS solution (1 IU/kg) was used to determine the relative pharmacological availability (PA%) of the oral INS formulations. The PA% obtained following oral administration of INS solution, INS-SLNs, LP-INS-SLNs, and DP-INS-SLNs was 0.3%, 7.5%, 13.1%, and 4.6%, respectively. The PA% following oral administration of LP-INS-SLNs was significantly higher (p < 0.01) than that of all SLNs or INS solution. Therefore, LP-INS-SLNs significantly increased the Cmax, AUC, and BA compared to s.c. injection of INS. The AUC values were increased from 12.4±1.8, 207.6±4.1, 109.6±32.5 μU h/mL for INS solution, INS-SLNs, and DP-INS-SLNs, respectively, to 295.9±31.0 μU h/mL for LP-INS-SLNs. The oral bioavailability of LP-INS-SLNs was 26.1, 1.4, and 2.7 times higher than that of INS solution, INS-SLNs, and DP-INS-SLNs, respectively.
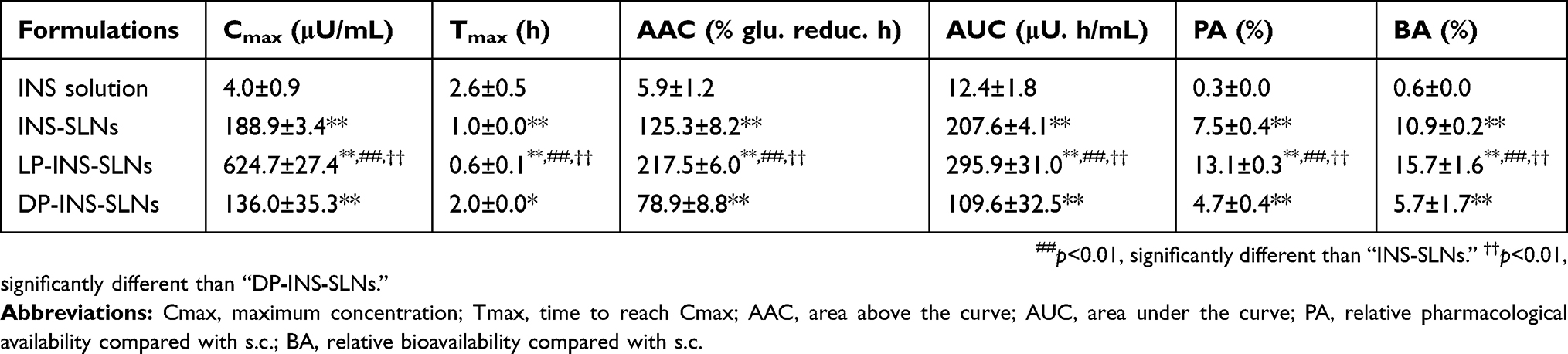 |
Table 4 Pharmacokinetic And Pharmacodynamic Parameters In Rats Following Oral Administration Of Insulin (INS) Solution And INS-Loaded Solid Lipid Nanoparticles (SLNs) |
Many studies have shown that CPPs could cross cell membranes in a receptor-independent manner.41,42 The uptake mechanism of CPPs has not been characterized, but mechanisms such as standard endocytosis, inverted micelles, caveolae, clathrin, macropinocytosis, and interactions with cell surface heparin sulfate proteoglycans have been suggested.43,44 Despite the absence of a clear understanding of the uptake mechanisms, CPPs such as penetratin have been shown to enhance delivery of macromolecular drugs into cells or tissues through covalent linkage with these drugs.45,46 Although these covalent binding approaches enhance delivery, covalent modification often results in alteration in bioactivity of the delivered drug.47 To overcome this obstacle, we developed novel SLNs loaded with the CPPs L- and D-penetratin. We encapsulated INS in these SLNs without covalent linkage and observed enhanced oral INS delivery. This formulation strategy, which resulted in encapsulation of INS and penetratin, protected INS against enzymatic degradation in the GI tract, resulting in absorption of the INS-penetratin complex in the intestine.
L- and D-penetratin enhanced the intestinal absorption of INS to different extents. Proteolytic stability of CPPs is a critical parameter for consideration during formulation development, susceptibility to proteolytic cleavage will reduce delivery of the drug cargo by exposing it to GI luminal fluids.6–8 The differences in susceptibility to enzymatic degradation between the L- and D-forms influenced the ability of penetratin to improve intestinal INS absorption. In the intestinal lumen, L-penetratin is less metabolically stable than D-penetratin.48,49 It is possible that formation of complexes through electrostatic and/or hydrophobic interactions may have resulted in protection against enzymatic degradation. In addition, INS must be dissociated from complexes in the lumen to permeation through the epithelial basal membrane.50 The dissociation rate of INS from complexes is a critical factor for determining INS absorption. A balance of interactions within the complex, and readiness of dissociation may result in optimal absorption.51
Our results were consistent with a previous study that showed that INS/L-penetratin resulted in better oral INS absorption than INS/D-penetratin.6 To further evaluate this phenomenon, aggregates of INS/penetratin complexes were evaluated in the presence of intestinal enzyme fluid. In the presence of intestinal fluid, L-penetratin aggregates gradually disappeared and the solution became nearly clear after 60 mins. In comparison, aggregates of the D-penetratin complex remained throughout the study period. Another study showed that oral administration of enveloped nanocomplexes (ENCPs) containing INS and the hydrophobically modified CPP, octaarginine (r8), whereas of ENCPs by a protecting polymer was poly (glutamic acid)-poly (ethylene glycol) (PGA-PEG) showed insignificant hypoglycemic effect compared with the INS.52 In this study, only 2% of the INS was transported across the Caco-2 cell monolayers, which resulted in a moderate response to INS following oral administration to rats. The author assumed that the ENCPs were internalized in the intestinal epithelium where they formed a depot reservoir from which INS might be slowly released. We are currently evaluating this phenomenon in other studies.
A previous study showed that D-penetratin exhibited higher pharmacological availability than L-penetratin in an in vivo study of INS absorption.8 This result was inconsistent with the results of our study. Despite the reduced stability of peptides containing L-amino acids, L-penetratin enhanced absorption of INS to a greater extent than D-penetratin. In previous studies, L-penetratin did not protect INS against degradation.6,7,49 However, INS associated with D-penetratin was comparatively stable, and intestinal fluid may promote dissociation of INS from D-penetratin in the small intestine. In contrast, inclusion of INS and penetratin in a lipid matrix in SLN formulations may have maintained the intermolecular interaction between INS and penetratin in the GI tract in our study. The INS–L-penetratin complex was degraded to a greater extent than the INS–D-penetratin complex by intestinal enzymes due to lower resistance of the L-form of the peptide to enzymatic degradation.
The internalization efficiency of L-penetratin is maintained even when its structure is partly altered, which suggests that partially degraded L-penetratin may maintain the ability to transport drugs across membranes.53 In our study, L-penetratin enhanced intestinal INS absorption to a greater extent than D-penetratin, which was consistent with previous studies that evaluated intestinal INS-CPP absorption.6,49 Differences in the penetratin concentrations used in both studies may have influenced the rate of INS release from the INS–CPP complex in the GI tract. Moreover, the INS-SLNs did not protect against enzymatic degradation as well as LP-INS-SLNs (Figure 3 and Table 3). In addition, enhanced absorption of LP-INS-SLNs may have depended on interactions between L-penetratin and the SLNs, the penetratin content in the SLNs, and penetratin distribution characters within the SLNs. Therefore, further studies to characterize the mechanisms by which penetratin INS-SLNs enhance INS release are needed. The LP-INS-SLNs developed in our study represent a promising drug delivery platform to improve oral BA of INS.
Conclusion
In summary, we developed a novel SLN for efficient oral absorption of INS by incorporating INS with penetratin, a CPP. A double emulsification technique was used to prepare LP-INS-SLNs and DP-INS-SLNs. These nanoparticles were characterized by uniform size, negative ZP values of ZP, and high INS EE. The chirality of penetratin significantly influenced the absorption-enhancing efficacy and stabilization of INS in rat intestinal fluid of rat. Among the INS-SLNs examined in this study, DP-INS-SLNs was the most stable against enzymatic degradation in intestinal fluid. However, LP-INS-SLNs most effectively enhanced INS absorption, resulting in improved hypoglycemic response in rats following oral administration. In conclusion, we found that inclusion of L- and D-penetratin into INS-SLNs may be a promising formulation strategy for oral administration of INS.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Gedawy A, Martinez J, Al-Salami H, Dass CR. Oral insulin delivery: existing barriers and current counter-strategies. J Pharm Pharmacol. 2018;70(2):197–213. doi:10.1111/jphp.12852
2. Wong CY, Martinez J, Dass CR. Oral delivery of insulin for treatment of diabetes: status quo, challenges and opportunities. J Pharm Pharmacol. 2016;68(9):1093–1108. doi:10.1111/jphp.12607
3. Khan Ghilzai NM. New developments in insulin delivery. Drug Dev Ind Pharm. 2003;29(3):253–265. doi:10.1081/DDC-120018199
4. Smart AL, Gaisford S, Basit AW. Oral peptide and protein delivery: intestinal obstacles and commercial prospects. Expert Opin Drug Deliv. 2014;11(8):1323–1335. doi:10.1517/17425247.2014.917077
5. Lundquist P, Artursson P. Oral absorption of peptides and nanoparticles across the human intestine: opportunities, limitations and studies in human tissues. Adv Drug Deliv Rev. 2016;106(Pt B):256–276. doi:10.1016/j.addr.2016.07.007
6. Kamei N, Morishita M, Eda Y, Ida N, Nishio R, Takayama K. Usefulness of cell-penetrating peptides to improve intestinal insulin absorption. J Control Release. 2008;132(1):21–25. doi:10.1016/j.jconrel.2008.08.001
7. Khafagy E-S, Iwamae R, Kamei N, Takeda-Morishita M. Region-dependent role of cell-penetrating peptides in insulin absorption across the rat small intestinal membrane. AAPS J. 2015;17(6):1427–1437. doi:10.1208/s12248-015-9804-y
8. Nielsen EJ, Yoshida S, Kamei N, et al. In vivo proof of concept of oral insulin delivery based on a co-administration strategy with the cell-penetrating peptide penetratin. J Control Release. 2014;189:19–24. doi:10.1016/j.jconrel.2014.06.022
9. Gamboa JM, Leong KW. In vitro and in vivo models for the study of oral delivery of nanoparticles. Adv Drug Deliv Rev. 2013;65(6):800–810. doi:10.1016/j.addr.2013.01.003
10. Fan W, Xia D, Zhu Q, Hu L, Gan Y. Intracellular transport of nanocarriers across the intestinal epithelium. Drug Discov Today. 2016;21(5):856–863. doi:10.1016/j.drudis.2016.04.007
11. Beloqui A, des Rieux A, Préat V. Mechanisms of transport of polymeric and lipidic nanoparticles across the intestinal barrier. Adv Drug Deliv Rev. 2016;106(PtB):242–255. doi:10.1016/j.addr.2016.04.014
12. Silva S, Almeida AJ, Vale N. Combination of cell-penetrating peptides with nanoparticles for therapeutic application: a review. Biomolecules. 2019;9(1):
13. Farkhani SM, Valizadeh A, Karami H, Mohammadi S, Sohrabi N, Badrzadeh F. Cell penetrating peptides: efficient vectors for delivery of nanoparticles, nanocarriers, therapeutic and diagnostic molecules. Peptides. 2014;57:78–94. doi:10.1016/j.peptides.2014.04.015
14. Guo F, Zhang M, Gao Y, et al. Modified nanoparticles with cell-penetrating peptide and amphipathic chitosan derivative for enhanced oral colon absorption of insulin: preparation and evaluation. Drug Deliv. 2016;23:2003–2014. doi:10.3109/10717544.2015.1048489
15. Guo F, Ouyang T, Peng T, et al. Enhanced oral absorption of insulin using colon-specific nanoparticles co-modified with amphiphilic chitosan derivatives and cell-penetrating peptides. Biomater Sci. 2019;7:1493–1506. doi:10.1039/c8bm01485j
16. Liu X, Liu C, Zhang W, Xie C, Wei G, Lu W. Oligoarginine-modified biodegradable nanoparticles improve the intestinal absorption of insulin. Int J Pharm. 2013;448:159–167. doi:10.1016/j.ijpharm
17. Zhu S, Chen S, Gao Y, et al. Enhanced oral bioavailability of insulin using PLGA nanoparticles co-modified with cell-penetrating peptides and engrailed secretion peptide (Sec). Drug Deliv. 2016;23(6):1980–1991. doi:10.3109/10717544.2015.1043472
18. Sheng J, He H, Han L, et al. Enhancing insulin oral absorption by using mucoadhesive nanoparticles loaded with LMWP-linked insulin conjugates. J Control Release. 2016;233:181–190. doi:10.1016/j.jconrel.2016.05.015
19. Yang L, Li M, Sun Y, Zhang L. A cell-penetrating peptide conjugated carboxymethyl-β-cyclodextrin to improve intestinal absorption of insulin. Int J Biol Macromol. 2018;111:685–695. doi:10.1016/j.ijbiomac.2018.01.077
20. Barbari GR, Dorkoosh FA, Amini M, et al. A novel nanoemulsion-based method to produce ultrasmall, water-dispersible nanoparticles from chitosan, surface modified with cell-penetrating peptide for oral delivery of proteins and peptides. Int J Nanomedicine. 2017;12:3471–3483. doi:10.2147/IJN.S116063
21. Manosroi A, Tangjai T, Sutthiwanjampa C, et al. Hypoglycemic activity and stability enhancement of human insulin-tat mixture loaded in elastic anionic niosomes. Drug Deliv. 2016;23:3157–3167. doi:10.3109/10717544.2016.1157840
22. Barbari GR, Dorkoosh F, Amini M, et al. Synthesis and characterization of a novel peptide-grafted Cs and evaluation of its nanoparticles for the oral delivery of insulin, in vitro, and in vivo study. Int J Nanomedicine. 2018;13:5127–5138. doi:10.2147/IJN.S161240
23. Müller RH, Mäder K, Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery - a review of the state of the art. Eur J Pharm Biopharm. 2000;50:161–177. doi:10.1016/S0939-6411(00)00087-4
24. Mehnert W, Mäder K. Solid lipid nanoparticles: production, characterization and applications. Adv Drug Deliv Rev. 2001;47:165–196. doi:10.1016/S0169-409X(01)00105-3
25. Souto EB, Müller RH. Lipid nanoparticles: effect on bioavailability and pharmacokinetic changes. Handb Exp Pharmacol. 2010;197:115–141. doi:10.1007/978-3-642-00477-3_4
26. Muchow M, Maincent P, Müller RH. Lipid nanoparticles with a solid matrix (SLN, NLC, LDC) for oral drug delivery. Drug Dev Ind Pharm. 2008;34:1394–1405. doi:10.1080/03639040802130061
27. Niu Z, Conejos-Sánchez I, Griffin BT, O’Driscoll CM, Alonso MJ. Lipid-based nanocarriers for oral peptide delivery. Adv Drug Deliv Rev. 2016;106:337–354. doi:10.1016/j.addr.2016.04.001
28. McClements DJ. Encapsulation, protection, and delivery of bioactive proteins and peptides using nanoparticle and microparticle systems: a review. Adv Colloid Interface Sci. 2018;253:1–22. doi:10.1016/j.cis.2018.02.002
29. du Plessis LH, Marais EB, Mohammed F, Kotzé AF. Applications of lipid based formulation technologies in the delivery of biotechnology-based therapeutics. Curr Pharm Biotechnol. 2014;15(7):659–672. doi:10.2174/1389201015666140804163143
30. Zhang Z, Lv H, Zhou J. Novel solid lipid nanoparticles as carriers for oral administration of insulin. Pharmazie. 2009;64:574–578. doi:10.1691/ph.2009.9051
31. Zhang ZH, Zhang YL, Zhou JP, Lv HX. Solid lipid nanoparticles modified with stearic acid-octaarginine for oral administration of insulin. Int J Nanomedicine. 2012;7:3333–3339. doi:10.2147/IJN.S31711
32. Yassin AE, Anwer MK, Mowafy HA, El-Bagory IM, Bayomi MA, Alsarra IA. Optimization of 5-flurouracil solid-lipid nanoparticles: a preliminary study to treat colon cancer. Int J Med Sci. 2010;7(6):398–408. doi:10.7150/ijms.7.398
33. Ansari MJ, Anwer MK, Jamil S, et al. Enhanced oral bioavailability of insulin-loaded solid lipid nanoparticles: pharmacokinetic bioavailability of insulin-loaded solid lipid nanoparticles in diabetic rats. Drug Deliv. 2016;23(6):1972–1979. doi:10.3109/10717544.2015.1039666
34. Asada H, Douen T, Mizokoshi Y, et al. Stability of acyl derivatives of insulin in the small intestine: relative importance of insulin association characteristics in aqueous solution. Pharm Res. 1994;11:1115–1120. doi:10.1023/A:1018928613837
35. Chen C, Fan T, Jin Y, et al. Orally delivered salmon calcitonin-loaded solid lipid nanoparticles prepared by micelle-double emulsion method via the combined use of different solid lipids. Nanomedicine (Lond). 2013;8(7):1085–1100. doi:10.2217/nnm.12.141
36. Aditya NP, Aditya S, Yang H, Kim HW, Park SO, Ko S. Co-delivery of hydrophobic curcumin and hydrophilic catechin by a water-in-oil-in-water double emulsion. Food Chem. 2015;173:7–13. doi:10.1016/j.foodchem.2014.09.131
37. Khafagy E-S, Morishita M, Takayama K. The role of intermolecular interactions with penetratin and its analogue on the enhancement of absorption of nasal therapeutic peptides. Int J Pharm. 2010;388(1–2):209–212. doi:10.1016/j.ijpharm.2009.12.060
38. Budhian A, Siegel SJ, Winey KI. Haloperidol-loaded PLGA nanoparticles: systematic study of particle size and drug content. Int J Pharm. 2007;336(2):367–375. doi:10.1016/j.ijpharm.2006.11.061
39. Kassem M, Ali A, El-Assal M, El-badrawy A. Formulation, characterization and in vivo application of oral insulin nanotechnology using different biodegradable polymers: advanced drug delivery system. Int J Pharm Sci Res. 2018;9(9):3664–3677. doi:10.13040/IJPSR.0975-8232.9(9).3664-77
40. Azevedo JR, Sizilio RH, Brito MB, et al. Physical and chemical characterization insulin loaded chitosan-TPP nanoparticles. J Therm Anal Calorim. 2011;106(3):685–689. doi:10.1007/s10973-011-1429-5
41. Torchilin VP. Tat peptide-mediated intracellular delivery of pharmaceutical nanocarriers. Adv Drug Deliv Rev. 2008;60(4–5):548–558. doi:10.1016/j.addr.2007.10.008
42. Gupta B, Torchilin VP. Transactivating transcriptional activator-mediated drug delivery. Expert Opin Drug Deliv. 2006;3(2):177–190. doi:10.1517/17425247.3.2.177
43. Zorko M, Langel U. Cell-penetrating peptides: mechanism and kinetics of cargo delivery. Adv Drug Deliv Rev. 2005;57(4):529–545. doi:10.1016/j.addr.2004.10.010
44. Chung SK, Maiti KK, Lee WS. Recent advances in cell-penetrating, non-peptide molecular carriers. Int J Pharm. 2008;354(1–2):16–22. doi:10.1016/j.ijpharm.2007.08.016
45. Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. The design, synthesis and evaluation of molecules that enable or enhance cellular uptake: peptoid molecular transporters. Proc Natl Acad Sci USA. 2000;97(24):13003–13008. doi:10.1073/pnas.97.24.13003
46. Kosuge M, Takeuchi T, Nakase I, Jones AT, Futaki S. Cellular internalization and distribution of arginine-rich peptides as a function of extracellular peptide concentration, serum, and plasma membrane associated proteoglycans. Bioconjug Chem. 2008;19(3):656–664. doi:10.1021/bc700289w
47. Meade BR, Dowdy SF. Exogenous siRNA delivery using peptide transduction domains/cell penetrating peptides. Adv Drug Deliv Rev. 2007;59(2–3):134–140. doi:10.1016/j.addr.2007.03.004
48. Pappenheimer JR, Dahl CE, Karnovsky ML, Maggio JE. Intestinal absorption and excretion of octapeptides composed of D amino acids. Proc Natl Acad Sci U S A. 1994;91(5):1942–1945. doi:10.1073/pnas.91.5.1942
49. Khafagy E-S, Morishita M, Kamei N, Eda Y, Ikeno Y, Takayama K. Efficiency of cell-penetrating peptides on the nasal and intestinal absorption of therapeutic peptides and proteins. Int J Pharm. 2009;381(1):49–55. doi:10.1016/j.ijpharm.2009.07.022
50. Kamei N, Aoyama Y, Khafagy E-S, Henmi M, Takeda-Morishita M. Effect of different intestinal conditions on the intermolecular interaction between insulin and cell-penetrating peptide penetratin and on its contribution to stimulation of permeation through intestinal epithelium. Eur J Pharm Biopharm. 2015;94:42–51. doi:10.1016/j.ejpb.2015.04.030
51. Khafagy E-S, Morishita M, Isowa K, Imai J, Takayama K. Effect of cell-penetrating peptides on the nasal absorption of insulin. J Control Release. 2009;133(2):103–108. doi:10.1016/j.jconrel.2008.09.076
52. Niu Z, Samaridou E, Jaumain E, et al. PEG-PGA enveloped octaarginine-peptide nanocomplexes: an oral peptide delivery strategy. J Control Release. 2018;276:125–139. doi:10.1016/j.jconrel.2018.03.004
53. Khafagy E-S, Morishita M, Ida N, Nishio R, Isowa K, Takayama K. Structural requirements of penetratin absorption enhancement efficiency for insulin delivery. J Control Release. 2010;143(3):302–310. doi:10.1016/j.jconrel.2010.01.019
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.