")
Back to Journals » Patient Related Outcome Measures » Volume 9
Impact of novel antifibrotic therapy on patient outcomes in idiopathic pulmonary fibrosis: patient selection and perspectives
Received 5 April 2018
Accepted for publication 29 June 2018
Published 21 September 2018 Volume 2018:9 Pages 321—328
DOI https://doi.org/10.2147/PROM.S144425
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lynne Nemeth
Bridget A Graney, Joyce S Lee
Division of Pulmonary Sciences and Critical Care Medicine, University of Colorado Anschutz Medical Campus, Aurora, CO, USA
Abstract: Patients with idiopathic pulmonary fibrosis, an incurable, progressive fibrotic interstitial lung disease, suffer an impaired quality of life due to symptoms, resultant functional limitations, and the constraints of supplemental oxygen. Two antifibrotic medications, nintedanib and pirfenidone, are approved for the treatment of idiopathic pulmonary fibrosis. Both medications slow the rate of decline of lung function, but their effect on patient-reported outcomes is not yet fully understood. Nintedanib may slow the decline in health-related quality of life for treated patients. Pirfenidone may slow the progression of dyspnea and improve cough. Patients and providers should participate in shared decision-making when starting antifibrotic therapy, taking into consideration the benefits of treatment in addition to drug-related side effects and dosing schedules. Although antifibrotic therapy may have an impact on health-related quality of life, providers should also focus on comprehensive care of the patient to improve health-related outcomes. This includes a multidisciplinary evaluation, diagnosis and treatment of comorbid medical conditions, and referral to and participation in a pulmonary rehabilitation program.
Keywords: interstitial lung disease, idiopathic pulmonary fibrosis, patient-reported outcome measures, quality of life, nintedanib, pirfenidone
Introduction
Idiopathic pulmonary fibrosis (IPF) is a progressive, incurable fibrotic interstitial lung disease (ILD). IPF is a disease of advancing age, with most patients presenting during the sixth and seventh decades of life. Although exactly what causes IPF is unknown, repetitive alveolar epithelial injury leading to fibrosis in a genetically susceptible individual is likely.1 Risk factors for the disease include cigarette smoking, other environmental inhalational exposures, and chronic microaspiration secondary to gastroesophageal reflux disease.1 The diagnosis of IPF is made based on a combination of suggestive clinical history and a usual interstitial pneumonia pattern on high-resolution computed tomography (HRCT) scan. Some cases may require surgical lung biopsy to confirm the diagnosis.2 The clinical course for patients with IPF is heterogeneous, with some experiencing periods of relative stability and others having a more rapid decline in lung function. The overall median survival for patients with IPF is 3–5 years.3
Clinically, as fibrosis advances and further alters the normal pulmonary physiology, affected patients experience a high burden of symptoms. Progressive activity-limiting dyspnea is the hallmark symptom of IPF and leads to significantly impaired physical functioning. Patients also commonly experience nonproductive cough and fatigue. Due to symptoms and the resultant impact on physical, social, and emotional well-being, patients with IPF suffer from decreased health-related quality of life (HRQL).4,5
HRQL refers to a patient’s satisfaction with aspects of life that are impacted by health or disease.6 Measuring HRQL has been described as the “quantification” of the impact of health or disease on a person’s life.7 It can be measured using validated questionnaires that are either generic or disease specific. In IPF research, questionnaires commonly used to measure HRQL include the following: the 36-Item Short Form Questionnaire, a generic HRQL questionnaire; St. George’s Respiratory Questionnaire (SGRQ), an obstructive lung disease-specific HRQL tool that has been widely used in studies of IPF patients;8–11 Living with IPF or A Tool to Assess Quality of Life in IPF, both IPF-specific HRQL questionnaires;12–14 and the King’s Brief Interstitial Lung Disease health status questionnaire, an ILD-specific questionnaire with validity for use in IPF.15,16
Choosing the appropriate tool is dependent on the population and question of interest for a given study. Regardless of the questionnaire used, measuring HRQL is critically important to understanding the impact of disease on patients’ everyday lives. It is also a quantifiable and reproducible measurement that can be used in clinical trials to assess the impact of emerging therapeutics on outcomes that are, arguably, most meaningful to patients.
In October 2014, the US Food and Drug Administration approved two antifibrotic medications – nintedanib and pirfenidone – for the treatment of IPF. In multiple randomized, controlled clinical trials, both drugs slowed the rate of decline in lung function in treated patients.17–20 Over time, this may lead to improved survival. The impact of these medications on patient-reported outcomes (PROs), including symptoms and HRQL, is not yet fully known. Here we review these medications, summarize the literature with respect to PROs and novel antifibrotics, and discuss additional strategies to improve HRQL for patients with IPF.
Nintedanib
Nintedanib is a tyrosine kinase inhibitor that prevents proliferation, migration, and transformation of fibroblasts by targeting upstream receptors important for the development of fibrosis. Specifically, nintedanib competitively blocks the binding sites of platelet-derived growth factor receptor, vascular endothelial growth factor receptor, and fibroblast growth factor receptor.21 Additionally, it may have pleiotropic effects on profibrotic cytokines.
Nintedanib was approved for use in IPF as a result of one Phase II clinical trial (To Improve Pulmonary Fibrosis with BIBF 1120, TOMORROW)22 and two Phase III clinical trials (Investigating the Safety and Efficacy of Nintedanib in IPF, INPULSIS 1 and 2).17 Compared with placebo, treatment with nintedanib 150 mg twice daily demonstrated a significant decrease in the rate of decline of lung function as measured by forced vital capacity (FVC).17 In pooled analysis of data from the TOMORROW and INPULSIS 1 and 2 trials, the annual rate of decline of FVC was 110.9 mL/year less in the treatment group when compared with placebo (95% CI: [78.5, 143.3]; P<0.0001). Furthermore, treated patients had greater time to an acute exacerbation and a reduction in both all-cause and respiratory-specific mortality at 52 weeks.23
Despite achieving these clinically meaningful endpoints, the impact of nintedanib on symptoms and HRQL is not as well understood. In the TOMORROW and INPULSIS studies, HRQL was assessed using the SGRQ. Scores range from 0 to 100, with higher scores indicating worse quality of life. In prior studies, the minimal important difference (MID) – the smallest difference in score on a measure that patients experience as a change in clinical status – of the total SGRQ score in IPF is seven points.10
In the TOMORROW and INPULSIS studies, the total SGRQ score increased at 52 weeks (suggesting deterioration in HRQL) for both the treatment and placebo arms (by 2.92 and 4.97 points, respectively). This between-group difference of –2.05 points was statistically significant (95% CI: [-3.59,–0.50]; P=0.0095),23 suggesting less decline in HRQL in the treatment group. However, as neither group changed by the MID, inferences regarding the impact of treatment on HRQL are difficult to assess. This relatively smaller change in SGRQ may indicate a slower rate of decline in HRQL for patients treated with nintedanib, but it did not improve HRQL.
For some practitioners, there is concern that treatment with nintedanib may impair HRQL due to side effects. The most commonly reported side effects are gastrointestinal (GI) including diarrhea, nausea, abdominal pain, and vomiting. Diarrhea affected over 60% of patients treated with nintedanib when compared with approximately 18% in the placebo arm. Diarrhea is typically limited to the first 3 months of therapy and can be managed with dose reduction and antimotility agents. Due to effective alternative management strategies, only 5% of treated patients discontinued the drug as a result of diarrhea.23 Additional side effects included elevated liver enzymes and weight loss secondary to GI side effects. It is recommended to monitor liver enzymes at initiation, monthly for the first 3 months, and every 3 months thereafter. The effect of nintedanib on extrapulmonary HRQL is unknown.
Pirfenidone
Pirfenidone has been shown to have antiinflammatory and antifibrotic properties, but the exact mechanism of action in IPF is not completely understood. Pirfenidone suppresses the activity of multiple proinflammatory cytokines and TNF-α, and similar to nintedanib, inhibits TGF-β with downstream reduction of fibroblast proliferation.24
Pirfenidone was approved for use in IPF after results from the CAPACITY (Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis, I and II)20 and ASCEND (A Phase III Trial of Pirfenidone in Patients with Pulmonary Fibrosis)18 trials demonstrated a significantly slower decline in FVC when compared with placebo at 1 year. Pooled analysis of the CAPACITY and ASCEND trials demonstrated a mean between-group difference in rate of FVC decline of 148 mL/year less in the treatment group (P<0.001).25 Furthermore, treatment with pirfenidone when compared with placebo reduced the incidence of all-cause mortality at 1 year by 48%, improved progression-free survival,25 and lowered the risk of respiratory-related hospitalizations.26 Importantly, given the variable clinical course of IPF, patients who remained on pirfenidone therapy, even after evidence of disease progression, had a slower subsequent rate of lung function decline when compared with placebo.27
The impact of pirfenidone on HRQL was not directly measured in either the CAPACITY or ASCEND trials. However, dyspnea scores were collected using the University of California San Diego Shortness of Breath Questionnaire (UCSD-SOBQ).28 The UCSD-SOBQ is a 24-item questionnaire used to assess self-perceived levels of dyspnea while performing various physical activities and how much dyspnea or fear of dyspnea limits patients in their everyday lives. Higher scores indicate greater levels of dyspnea.
In pooled analysis, both the treatment and placebo arms had increased UCSD-SOBQ scores at 1 year when compared with baseline. However, fewer patients in the pirfenidone group (24.0%) when compared with placebo (31.4%)25 had an increase in greater than 20 points at 1 year (MID for UCSD-SOBQ=8 points in IPF). Although the UCSD-SOBQ is not a quality of life-specific measure, it does quantify dyspnea, which is the primary driver of impaired quality of life in IPF.29 Therefore, if treatment slows the rate of decline in dyspnea as evidenced by a smaller increase in UCSD-SOBQ score, this may translate into a meaningful benefit for HRQL. Importantly, however, the treatment group did not have an improvement in their dyspnea scores – dyspnea still worsened over the study period for both groups.
In addition to a possible impact on dyspnea scores, pirfenidone has recently been associated with improvements in cough severity in a small, multicenter observational study.30 Patients treated with pirfenidone showed improvements in both objective cough counts and patient-reported cough severity at 12 weeks. Although pirfenidone has not been shown to directly improve HRQL, improving symptoms is an alternative mechanism to impact HRQL for patients with IPF.
Similar to nintedanib, there are known side effects to treatment with pirfenidone that may impact HRQL. Pirfenidone is associated with GI side effects including nausea, appetite suppression, abdominal pain, diarrhea, and vomiting. Taking the medication with meals or a dose reduction strategy helps to ameliorate some of the GI side effects that patients experience. Skin rash is another commonly reported side effect of pirfenidone. Patients should avoid or minimize sun exposure and use sun-protection including sunblock and protective clothing. Liver function elevations may also be seen with pirfenidone, and enzyme monitoring at initiation, monthly for the first 6 months, and every 3 months thereafter is recommended. Pirfenidone interacts with multiple hepatic enzyme systems; so, monitoring for drug–drug interactions, particularly as patients with IPF are often elderly with multiple comorbid conditions requiring medications, is of utmost importance. The impact of these side effects on HRQL in patients with IPF is unknown.
Interventions to improve health-related quality of life in IPF
With the approval and increasing use of novel antifibrotic agents that slow disease progression and may allow patients to live longer, determining how to help IPF patients live better is of utmost importance given their overall poor HRQL. Symptoms, including cough, fatigue, and dyspnea in particular, are the most influential drivers of impaired HRQL in IPF (Figure 1).31 The resultant physical functional limitations due to deconditioning, as patients move less, and eventual social isolation also contribute to impaired quality of life.32 Furthermore, the constraints of supplemental oxygen – tubing, heavy and difficult to move tanks, oxygen needs that cannot be met by a portable concentrator – exacerbate these challenges.33
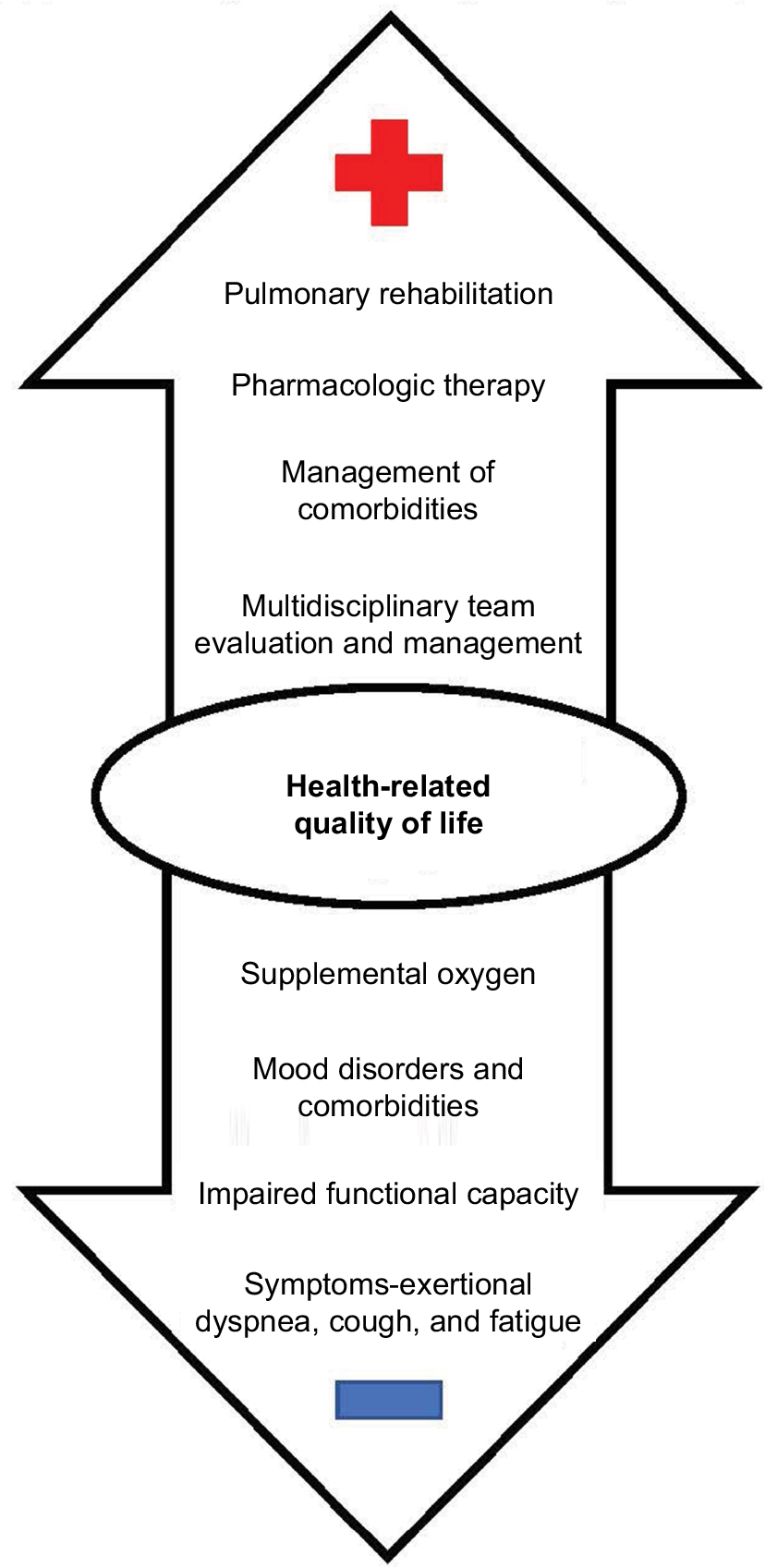 | Figure 1 Positive and negative influences on HRQL in IPF. Notes: Symptoms, and dyspnea in particular, are the most influential drivers of impaired HRQL in IPF. Other drivers of impaired HRQL include the resultant physical functional limitations due to deconditioning, mood disorders and comorbidities, and the constraints of supplemental oxygen. Pulmonary rehabilitation is associated with improved HRQL in IPF. Pharmacologic therapies, management of comorbid conditions, and a multidisciplinary approach to diagnosis and management may also improve HRQL for some patients. Abbreviations: HRQL, health-related quality of life; IPF, idiopathic pulmonary fibrosis. |
Currently, pulmonary rehabilitation (PR) is the only intervention shown to improve HRQL for patients with IPF. By definition,34 PR is a multidisciplinary program for patients with chronic respiratory disease that, when integrated into a broader treatment strategy, improves functional status, reduces symptoms, increases participation in social and physical activities, and decreases health care costs. These goals are accomplished by combining exercise training, education, counseling, and behavior modification techniques. The delivery of PR is not standardized and can be inpatient, outpatient, or home based, with each program having certain benefits.34
In two randomized controlled clinical trials of outpatient-based PR for IPF patients, there was an improvement in overall HRQL scores during the intervention period.35,36 Similar improvements in HRQL have been reported by several observations trials37–39 and in a subgroup analysis of PR in ILD.40 Unfortunately, these improvements in HRQL are not sustained following completion of PR programs.
The only pharmacologic therapy shown to impact HRQL in IPF is sildenafil, a phosphodiesterase-5 inhibitor that may preferentially improve blood flow to better ventilated areas of the lung. In one study of patients with advanced IPF, those treated with sildenafil maintained their baseline level of dyspnea and HRQL as measured by the UCSD-SOBQ and SGRQ, respectively. Comparatively, patients in the placebo group had statistically significant declines in dyspnea and HRQL during the study period.41 Despite these possible benefits, current guidelines recommend against the use of sildenafil due to a lack of benefit on mortality and acute exacerbations in the face of potential adverse events and cost related to therapy.42
Management strategies for patients with IPF
Despite the paucity of interventions that improve HRQL for patients with IPF, comprehensive specialty care is important for accurate diagnosis and appropriate therapeutic management. Making the initial diagnosis of IPF is often challenging. Patients are usually elderly and present with progressive dyspnea. This symptom is often attributed to a comorbid condition such as coronary artery disease or congestive heart failure, delaying targeted evaluation for pulmonary causes. Even after confirming the presence of an ILD with pulmonary function tests or HRCT, multidisciplinary discussion (MDD) with the treating pulmonologist, radiologist, and pathologist is recommended to improve accuracy of the diagnosis.2 For cases that are recommended for biopsy after initial discussion, MDD should be repeated to incorporate pathology results and refine the diagnosis. Patients with a suspected or confirmed diagnosis of ILD should be referred to a center with expertise in the management of these conditions. Delayed evaluation at a specialty center is associated with higher risk of death in IPF, specifically, regardless of disease severity.43
After the diagnosis of IPF has been confirmed, practitioners and patients should discuss treatment options and participate in shared decision-making regarding the use of antifibrotic medications. Patients with mild-to-moderate disease (based on FVC), who do not have liver disease, should consider treatment with antifibrotics. The decision of which antifibrotic to choose, if both are available, is primarily driven by three factors: contraindications if present, side-effect profile, and dosing (Table 1). Regardless of the choice of antifibrotic, patients must be counseled that these medications may slow the progression of the disease but are not likely to improve their symptoms of breathlessness or improve their HRQL.
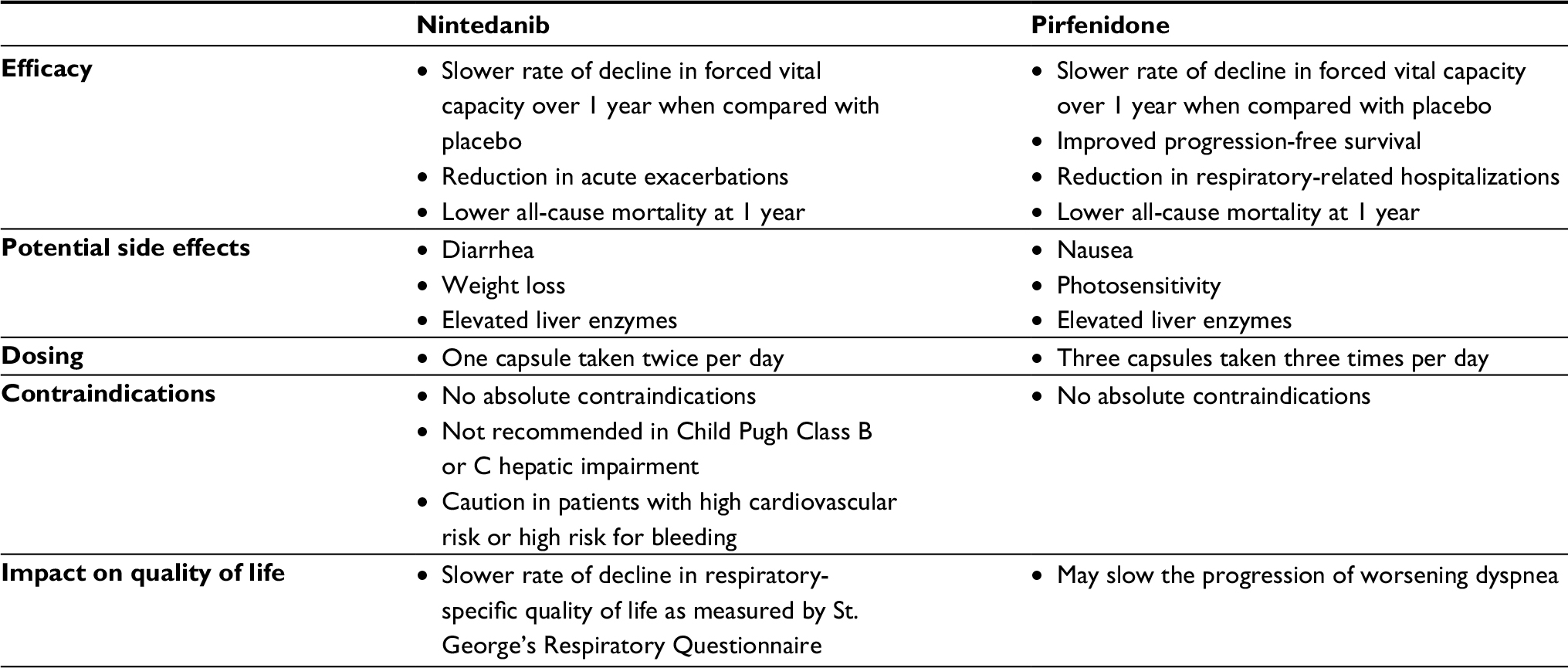 | Table 1 Comparison of antifibrotic agents approved for the treatment of IPF Abbreviation: IPF, idiopathic pulmonary fibrosis. |
Nintedanib should not be used in patients with moderate-to-severe hepatic impairment. Current use of anticoagulation is a relative contraindication to nintedanib, and patients should be cautioned that their risk of bleeding may be increased with concurrent use. There was also a small but increased risk of cardiovascular events reported in patients taking nintedanib compared with placebo. There are no absolute contraindications to treatment with pirfenidone. Pirfenidone is metabolized primarily by the CYP1A2 substrate. Monitoring for drug–drug interactions is very important, and the dose should be reduced if patients are taking CYP1A2 inhibitors (eg, ciprofloxacin).
Both medications, despite known possible side effects, are generally well tolerated. Many side effects, if they do occur, can be managed with supportive care or dose reduction strategies and rarely require drug discontinuation. Both drugs require monitoring for liver enzyme abnormalities after initiation. Patients should be informed of possible side effects, but also counseled that these side effects can be managed and often diminish with time.
With respect to dosing, pirfenidone is titrated up to a final dose of three capsules three times per day when compared with nintedanib, which is one capsule twice per day. Patients’ ability to adhere to the dosing schedule and number of tablets may be a consideration in drug choice.
Irrespective of the decision to initiate therapy with antifibrotic medications, optimal management of IPF requires a comprehensive approach, including attention to management of symptoms and treatment of comorbid medical conditions, which are highly prevalent44 in this population.
The most common symptoms that affect patients with IPF are exertional dyspnea, cough, and fatigue. Each of these symptoms is associated with impaired HRQL.29,31,45,46 Unfortunately, symptom management in IPF is challenging with a paucity of effective, durable interventions currently available. PR has a positive impact on lessening symptoms of dyspnea and fatigue,37,47 but the benefits typically are not sustainable following the completion of the program. Supplemental oxygen may improve dyspnea for some patients with IPF, but it is unknown who will benefit and who will not.48,49 The burdens of supplemental oxygen may worsen these symptoms for some patients33,50,51 and is independently associated with impaired HRQL.5,12,52
In addition to the small observational study demonstrating a positive impact on cough severity in patients treated with pirfenidone, thalidomide has also been shown to lessen symptoms of cough in IPF.53 Further, a novel formulation of inhaled sodium cromoglicate (PA101) showed promising findings on reducing chronic cough in patients with IPF in a Phase II trial.54 Additional research is needed to develop effective, durable therapies to better manage the symptoms patients with IPF often experience.
Obstructive sleep apnea (OSA), emphysema, pulmonary hypertension (PH), and mood disorders are particularly prevalent comorbidities and may impact HRQL. OSA is also highly prevalent in this population,5,12,52,55 and may worsen other comorbidities (eg, PH), if untreated. Poor sleep as a result of OSA may also worsen symptoms of depression and anxiety. Overall, effective treatment of OSA in patients with IPF has been shown to improve fatigue, symptoms of depression, and overall HRQL.56
Emphysema and PH may exacerbate symptoms of IPF, particularly dyspnea and impaired functional capacity. Furthermore, emphysema or PH in combination with IPF may make resting or exertional hypoxemia more likely and more severe, often requiring supplemental oxygen therapy. This combination is particularly deleterious on HRQL given the additive impact of supplemental oxygen therapy.50
Mood disorders are also common in patients with IPF, with as many as 49% of patients having depression and 31% having anxiety.57 The dyspnea that IPF patients experience is strongly associated with and drives symptoms of depression and anxiety. Furthermore, mood symptoms may heighten the perception of dyspnea, leading to a reinforcing negative cycle and worsening HRQL. Pharmacologic treatment of depression and anxiety has not been studied in IPF, necessitating an individualized approach for each patient. PR and disease-specific support groups may provide patients with a sense of validation and shared experience and may improve symptoms.47 These modalities, in addition to cognitive-behavioral therapy and palliative care, may be useful adjuncts for the treatment of mood disorders.
Screening IPF patients for these prevalent comorbid conditions is crucial to optimizing medical management, decreasing symptom burden, and potentially improving outcomes, including HRQL.
In addition to decisions regarding antifibrotic medications and treatment of symptoms and comorbidities, IPF patients should be monitored for disease progression with regular pulmonary function tests. All patients with IPF should be evaluated for the need for supplemental oxygen therapy with an oxygen titration study. If supplemental oxygen is necessary, patients and caregivers should be educated about realistic expectations for oxygen use including a variable impact on symptoms and the likely overall negative impact on quality of life for both.48,51,58,59 Patients should also be referred to PR programs for exercise training to improve functional capacity,37,40,60 to gain information about their disease, and to improve HRQL as previously discussed. Appropriate patients should be referred for lung transplant evaluation at a dedicated center. Furthermore, depending on the stage and pace of disease, patient functional status and goals, palliative care involvement may be appropriate for symptom management and end-of-life planning.
Conclusion
Despite recent advances in the treatment of IPF with antifibrotic agents that slow the decline in lung function, it remains an insidiously progressive and incurable disease. Patients suffer with significantly impaired quality of life due to the impact of disease on their physical, functional, emotional, and psychosocial well-being. Though antifibrotics have been shown to slow the rate of disease progression, they have not been shown to improve patients’ quality of life. Until there are curative treatment options available, future research should focus on the impact of symptom-targeted therapies, combination therapy with existing antifibrotics, and multidisciplinary treatments including PR and palliative care on HRQL.
A patient-centered, comprehensive approach to care including attention to symptoms, comorbidities, and advanced care planning remains the cornerstone of care of patients with IPF. As both disease- and symptom-specific therapeutics continue to advance, it is increasingly important to assess not only the impact on disease progression and mortality, but the impact on patients’ lives on a day-to-day basis. With the ultimate goal of improving both quality and quantity of life for patients with IPF, it is imperative to understand the impact of emerging interventions on patient-centered outcomes.
Acknowledgment
BAG thanks the generous contribution of the Reuben M Cherniak Fellowship Award from the National Jewish Health for providing ongoing support for her research.
Author contributions
All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
JSL is supported by National Institutes of Health/National Heart, Lung, and Blood Institute K23-HL138131. The authors report no conflicts of interest in this work.
References
Ahluwalia N, Shea BS, Tager AM. New therapeutic targets in idiopathic pulmonary fibrosis. Aiming to rein in runaway wound-healing responses. Am J Respir Crit Care Med. 2014;190(8):867–878. | ||
Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824. | ||
Olson AL, Swigris JJ, Lezotte DC, Norris JM, Wilson CG, Brown KK. Mortality from pulmonary fibrosis increased in the United States from 1992 to 2003. Am J Respir Crit Care Med. 2007;176(3):277–284. | ||
Yount SE, Beaumont JL, Chen SY, et al. Health-related quality of life in patients with idiopathic pulmonary fibrosis. Lung. 2016;194(2):227–234. | ||
Tomioka H, Imanaka K, Hashimoto K, Iwasaki H. Health-related quality of life in patients with idiopathic pulmonary fibrosis--cross-sectional and longitudinal study. Intern Med. 2007;46(18):1533–1542. | ||
van Manen MJ, Geelhoed JJ, Tak NC, Wijsenbeek MS. Optimizing quality of life in patients with idiopathic pulmonary fibrosis. Ther Adv Respir Dis. 2017;11(3):157–169. | ||
Olson AL, Brown KK, Swigris JJ. Understanding and optimizing health-related quality of life and physical functional capacity in idiopathic pulmonary fibrosis. Patient Relat Outcome Meas. 2016;7:29–35. | ||
Swigris JJ, Esser D, Wilson H, et al. Psychometric properties of the St George’s Respiratory Questionnaire in patients with idiopathic pulmonary fibrosis. Eur Respir J. 2017;49(1):1601788. | ||
Swigris JJ, Brown KK, Behr J, et al. The SF-36 and SGRQ: validity and first look at minimum important differences in IPF. Respir Med. 2010;104(2):296–304. | ||
Swigris JJ, Esser D, Conoscenti CS, Brown KK. The psychometric properties of the St George’s Respiratory Questionnaire (SGRQ) in patients with idiopathic pulmonary fibrosis: a literature review. Health Qual Life Outcomes. 2014;12:124. | ||
Kreuter M, Swigris J, Pittrow D, et al. Health related quality of life in patients with idiopathic pulmonary fibrosis in clinical practice: insights-IPF registry. Respir Res. 2017;18(1):139. | ||
Swigris JJ, Wilson SR, Green KE, Sprunger DB, Brown KK, Wamboldt FS. Development of the ATAQ-IPF: a tool to assess quality of life in IPF. Health Qual Life Outcomes. 2010;8:77. | ||
Yorke J, Spencer LG, Duck A, et al. Cross-Atlantic modification and validation of the A Tool to Assess Quality of Life in Idiopathic Pulmonary Fibrosis (ATAQ-IPF-cA). BMJ Open Respir Res. 2014;1(1):e000024. | ||
Graney B, Johnson N, Evans CJ, et al. Living with idiopathic pulmonary fibrosis (L-IPF): developing a patient-reported symptom and impact questionnaire to assess health-related quality of life in IPF. Am J Respir Crit Care Med. 2017;195:A5353. | ||
Patel AS, Siegert RJ, Keir GJ, et al. The minimal important difference of the King’s Brief Interstitial Lung Disease Questionnaire (K-BILD) and forced vital capacity in interstitial lung disease. Respir Med. 2013;107(9):1438–1443. | ||
Patel AS, Siegert RJ, Brignall K, et al. The development and validation of the King’s Brief Interstitial Lung Disease (K-BILD) health status questionnaire. Thorax. 2012;67(9):804–810. | ||
Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071–2082. | ||
King TE, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2083–2092. | ||
Taniguchi H, Ebina M, Kondoh Y, et al. Pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J. 2010;35(4):821–829. | ||
Noble PW, Albera C, Bradford WZ, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. 2011;377(9779):1760–1769. | ||
George G, Vaid U, Summer R. Therapeutic advances in idiopathic pulmonary fibrosis. Clin Pharmacol Ther. 2016;99(1):30–32. | ||
Richeldi L, Costabel U, Selman M, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. 2011;365(12):1079–1087. | ||
Richeldi L, Cottin V, du Bois RM, et al. Nintedanib in patients with idiopathic pulmonary fibrosis: combined evidence from the TOMORROW and INPULSIS(®) trials. Respir Med. 2016;113:74–79. | ||
Kim ES, Keating GM. Pirfenidone: a review of its use in idiopathic pulmonary fibrosis. Drugs. 2015;75(2):219–230. | ||
Noble PW, Albera C, Bradford WZ, et al. Pirfenidone for idiopathic pulmonary fibrosis: analysis of pooled data from three multinational phase 3 trials. Eur Respir J. 2016;47(1):243–253. | ||
Ley B, Swigris J, Day BM, et al. Pirfenidone reduces respiratory-related hospitalizations in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2017;196(6):756–761. | ||
Nathan SD, Albera C, Bradford WZ, et al. Effect of continued treatment with pirfenidone following clinically meaningful declines in forced vital capacity: analysis of data from three phase 3 trials in patients with idiopathic pulmonary fibrosis. Thorax. 2016;71(5):429–435. | ||
Eakin EG, Resnikoff PM, Prewitt LM, Ries AL, Kaplan RM. Validation of a new dyspnea measure: the UCSD Shortness of Breath Questionnaire. University of California, San Diego. Chest. 1998;113(3):619–624. | ||
Swigris JJ, Gould MK, Wilson SR. Health-related quality of life among patients with idiopathic pulmonary fibrosis. Chest. 2005;127(1):284–294. | ||
van Manen MJG, Birring SS, Vancheri C, et al. Effect of pirfenidone on cough in patients with idiopathic pulmonary fibrosis. Eur Respir J. 2017;50(4):1701157. | ||
Nishiyama O, Taniguchi H, Kondoh Y, et al. Health-related quality of life in patients with idiopathic pulmonary fibrosis. What is the main contributing factor? Respir Med. 2005;99(4):408–414. | ||
Belkin A, Swigris JJ. Health-related quality of life in idiopathic pulmonary fibrosis: where are we now? Curr Opin Pulm Med. 2013;19(5):474–479. | ||
Graney BA, Wamboldt FS, Baird S, et al. Looking ahead and behind at supplemental oxygen: a qualitative study of patients with pulmonary fibrosis. Heart Lung. 2017;46(5):387–393. | ||
Nici L, Donner C, Wouters E, et al. American Thoracic Society/European Respiratory Society statement on pulmonary rehabilitation. Am J Respir Crit Care Med. 2006;173(12):1390–1413. | ||
Nishiyama O, Kondoh Y, Kimura T, et al. Effects of pulmonary rehabilitation in patients with idiopathic pulmonary fibrosis. Respirology. 2008;13(3):394–399. | ||
Holland AE, Hill CJ, Conron M, Munro P, Mcdonald CF. Short term improvement in exercise capacity and symptoms following exercise training in interstitial lung disease. Thorax. 2008;63(6):549–554. | ||
Swigris JJ, Fairclough DL, Morrison M, et al. Benefits of pulmonary rehabilitation in idiopathic pulmonary fibrosis. Respir Care. 2011;56(6):783–789. | ||
Ozalevli S, Karaali HK, Ilgin D, Ucan ES. Effect of home-based pulmonary rehabilitation in patients with idiopathic pulmonary fibrosis. Multidiscip Respir Med. 2010;5(1):31–37. | ||
Kozu R, Senjyu H, Jenkins SC, Mukae H, Sakamoto N, Kohno S. Differences in response to pulmonary rehabilitation in idiopathic pulmonary fibrosis and chronic obstructive pulmonary disease. Respiration. 2011;81(3):196–205. | ||
Dowman L, Hill CJ, Holland AE. Pulmonary rehabilitation for interstitial lung disease. Cochrane Database Syst Rev. 2014;10:CD006322. | ||
Idiopathic Pulmonary Fibrosis Clinical Research Network, Zisman DA, Schwarz M, et al. A controlled trial of sildenafil in advanced idiopathic pulmonary fibrosis. N Engl J Med. 2010;363(7):620–628. | ||
Raghu G, Rochwerg B, Zhang Y, et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. Am J Respir Crit Care Med. 2015;192(2):e3–e19. | ||
Lamas DJ, Kawut SM, Bagiella E, Philip N, Arcasoy SM, Lederer DJ. Delayed access and survival in idiopathic pulmonary fibrosis: a cohort study. Am J Respir Crit Care Med. 2011;184(7):842–847. | ||
Raghu G, Amatto VC, Behr J, Stowasser S. Comorbidities in idiopathic pulmonary fibrosis patients: a systematic literature review. Eur Respir J. 2015;46(4):1113–1130. | ||
Swigris JJ, Stewart AL, Gould MK, Wilson SR. Patients’ perspectives on how idiopathic pulmonary fibrosis affects the quality of their lives. Health Qual Life Outcomes. 2005;3:61. | ||
van Manen MJ, Birring SS, Vancheri C, et al. Cough in idiopathic pulmonary fibrosis. Eur Respir Rev. 2016;25(141):278–286. | ||
Ryerson CJ, Cayou C, Topp F, et al. Pulmonary rehabilitation improves long-term outcomes in interstitial lung disease: a prospective cohort study. Respir Med. 2014;108(1):203–210. | ||
Cao M, Wamboldt FS, Brown KK, et al. Supplemental oxygen users with pulmonary fibrosis perceive greater dyspnea than oxygen non-users. Multidiscip Respir Med. 2015;10:37. | ||
Nishiyama O, Taniguchi H, Kondoh Y, et al. A simple assessment of dyspnoea as a prognostic indicator in idiopathic pulmonary fibrosis. Eur Respir J. 2010;36(5):1067–1072. | ||
Belkin A, Albright K, Swigris JJ. A qualitative study of informal caregivers’ perspectives on the effects of idiopathic pulmonary fibrosis. BMJ Open Respir Res. 2014;1(1):e000007. | ||
Graney BA, Wamboldt FS, Baird S, et al. Informal caregivers experience of supplemental oxygen in pulmonary fibrosis. Health Qual Life Outcomes. 2017;15(1):133. | ||
Cullen DL, Stiffler D. Long-term oxygen therapy: review from the patients’ perspective. Chron Respir Dis. 2009;6(3):141–147. | ||
Horton MR, Santopietro V, Mathew L, et al. Thalidomide for the treatment of cough in idiopathic pulmonary fibrosis: a randomized trial. Ann Intern Med. 2012;157(6):398–406. | ||
Birring SS, Wijsenbeek MS, Agrawal S, et al. A novel formulation of inhaled sodium cromoglicate (PA101) in idiopathic pulmonary fibrosis and chronic cough: a randomised, double-blind, proof-of-concept, phase 2 trial. Lancet Respir Med. 2017;5(10):806–815. | ||
Lancaster LH, Mason WR, Parnell JA, et al. Obstructive sleep apnea is common in idiopathic pulmonary fibrosis. Chest. 2009;136(3):772–778. | ||
Mermigkis C, Bouloukaki I, Antoniou K, et al. Obstructive sleep apnea should be treated in patients with idiopathic pulmonary fibrosis. Sleep Breath. 2015;19(1):385–391. | ||
King CS, Nathan SD. Idiopathic pulmonary fibrosis: effects and optimal management of comorbidities. Lancet Respir Med. 2017;5(1):72–84. | ||
Bell EC, Cox NS, Goh N, et al. Oxygen therapy for interstitial lung disease: a systematic review. Eur Respir Rev. 2017;26(143):160080. | ||
Visca D, Montgomery A, de Lauretis A, et al. Ambulatory oxygen in interstitial lung disease. Eur Respir J. 2011;38(4):987–990. | ||
Naji NA, Connor MC, Donnelly SC, Mcdonnell TJ. Effectiveness of pulmonary rehabilitation in restrictive lung disease. J Cardiopulm Rehabil. 2006;26(4):237–243. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.