")
Back to Archived Journals » Oncolytic Virotherapy » Volume 8
Impact of Induced Syncytia Formation on the Oncolytic Potential of Myxoma Virus
Authors Burton C, Bartee MY, Bartee E
Received 22 June 2019
Accepted for publication 18 November 2019
Published 9 December 2019 Volume 2019:8 Pages 57—69
DOI https://doi.org/10.2147/OV.S220420
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Tommy Alain
Chase Burton, Mee Y Bartee, Eric Bartee
Department of Microbiology and Immunology, Medical University of South Carolina, Charleston, SC, USA
Correspondence: Eric Bartee
Department of Microbiology and Immunology, Medical University of South Carolina, 173 Ashley Ave, Charleston, SC, USA
Email [email protected]
Introduction: Cancer has become one of the most critical health issues of modern times. To overcome the ineffectiveness of current treatment options, research is being done to explore new therapeutic modalities. One such novel treatment is oncolytic virotherapy (OV) which uses tumor tropic viruses to specifically target and kill malignant cells. While OV has shown significant promise in recent clinical trials, the therapeutic use of viruses poses a number of unique challenges. In particular, obtaining effective viral spread throughout the tumor microenvironment remains problematic. Previous work has suggested this can be overcome by forcing oncolytic viruses to induce syncytia formation.
Methods: In the current work, we generated a series of recombinant myxoma viruses expressing exogenous fusion proteins from other viral genomes and examined their therapeutic potential in vitro and in vivo.
Results: Similar to previous studies, we observed that the expression of these fusion proteins during myxoma infection induced the formation of multinucleated syncytia which increased viral spread and lytic potential compared to non-fusogenic controls. Contrary to expectations, however, the treatment of established tumors with these viruses resulted in decreased therapeutic efficacy which corresponded with reduced viral persistence.
Discussion: These findings indicate that enhanced viral spread caused by syncytia formation can actually reduce the efficacy of OV and supports a number of previous works suggesting that the in vitro properties of viruses frequently fail to predict their in vivo efficacy.
Keywords: myxoma virus, syncytia, fusogenic, oncolytic virotherapy, lung cancer
Introduction
Cancer is one of the world’s largest health concerns, accounting for roughly 10 million deaths globally in 2018.1 In addition to this tremendous loss of life, cancer also results in a significant economic burden which is estimated to exceed one trillion dollars per year.2 While numerous treatments for cancer exist, current standards of care, such as chemotherapy and radiation, are often ineffective and can also cause severe toxicities in treated patients.3–6 For these reasons, research has begun looking at novel forms of therapy to combat this disease.
One novel treatment which has shown promise in recent years is oncolytic virotherapy (OV).7–9 This therapy works through the injection of oncotropic viruses into a tumor mass. These viral particles then selectively infect malignant cells resulting in both their acute destruction and the subsequent release of tumor-associated antigens.10 While the recent FDA approval of Imlygic® proves that this approach is clinically viable,11,12 there are still several challenges which limit the overall efficacy of OV.13 For example, limited dissemination of oncolytic viruses within a tumor mass can: restrict the efficacy of direct oncolytic killing, shorten the duration of viral persistence, prevent bystander killing, and reduce the release of tumor antigens from cells.14–16 Studies exploring methods to increase viral spread, therefore, represent one major path to improving OV’s overall therapeutic efficacy.
One proposed means to improve oncolytic viral spread is the use of fusogenic oncolytic constructs. These viruses possess typical oncolytic properties, but also encode some form of exogenous viral fusion (F) protein.17 This F protein is expressed on the surface of infected cells resulting in the fusion of these cells with their neighbors. This causes the formation of large multi-nucleated cell bodies known as syncytia.18 It has been shown that the formation of these syncytia during OV increases both viral spread and bystander killing and that these increases correlate with improved overall efficacies in a number of tumor models.19
We and others have shown that a rabbit-specific poxvirus known as myxoma virus (MYXV) represents an attractive oncolytic agent.20–22 In comparison to other oncolytic agents, MYXV has an excellent safety profile as well as strong immune-stimulatory properties. Its potential efficacy, however, is restricted by the virus’s poor cell to cell spread properties, which, in the case of MYXV, is a result of this virus not producing an appreciable amount of infectious extracellular virions during infection. In the present study, we, therefore, sought to examine the possibility of improving MYXV’s overall oncolytic potential by generating a series of novel constructs which incorporated the F proteins of other, naturally fusogenic viruses in hopes of increasing the viruses direct oncolytic killing potential.
Materials and Methods
Cell Lines and Reagents
BSC40 cells (Cat# CRL-2761) were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). Murine embryonic fibroblasts (MEFs) were a kind gift from Dr. Carl Atkinson. LLC-A9F1 (A9F1) cells were a kind gift from Dr. Mark Rubenstein. All cells were cultured in DMEM supplemented with 10% Fetal Bovine Serum and 1x Penicillin-Streptomycin-L-Glutamine (Corning, Oneonta, NY, USA). Cell viability was measured using the CellTiter-96 Non-Radioactive Cell Proliferation (MTT) assay (Promega, Madison, WI, USA) according to the manufacturer’s recommendations. Antibodies used for this study were purchased from Santa Cruz Biotech (Santa Cruz, CA) and include: actin (clone I19) and c-Myc (clone 9E10).
Generation, Preparation, and Use of Virus
Fusogenic viral constructs were generated as previously described.21 In short, the pBluescript-M135/GFP/M136 plasmid (a kind gift from Dr. Grant McFadden23) was digested at the Nde1 and Pst1 restriction sites. Synthetically generated dsDNA encoding both the transgenes of interest (Table 1) and a synthetic early/late viral promoter (Genewiz, South Plainsfield, NJ) were then ligated into the digested plasmid using DNA ligase. To generate a recombinant virus, the resulting plasmids were transfected into BSC40 cells which were subsequently infected with MYXV (strain Lausanne) at a multiplicity of infection (MOI) of 10. After 48 hrs, cells were harvested and the recombinant virus was purified using repeated isolation of GFP+ foci until clonality was established. A non-fusogenic MYXV expressing GFP (vGFP) isolated using identical methodologies has been previously described.23 Once purified, all viruses were amplified in BSC40 cells and purified through sucrose gradient centrifugation as previously described.24 For all experiments, cells were infected at room temperature for 1 hr at the indicated MOI. After 1 hr, the viral inoculum was removed and replaced with fresh media. Single-step growth curves, foci forming assays, and initial infection assays were each performed as previously described.25–27
 |
Table 1 Origin of F Proteins Used in the Current Study |
Single Step Growth Curve
The cell line of interest was plated onto a 12-well plate. The cells were then infected at the MOI=10. After 1 hr, the inoculum was removed, the cells were washed with sterile 1% PBS and fresh media was added to the well. The cells were harvested at 3, 6, 12, 24, 48 and 72 hrs after the infection. To harvest cells, the media was removed, and cells were pipetted off the well. Cells were then spun at 2500 RPM for 5 mins by centrifugation and the supernatant was discarded. Samples were stored at −80°C. Samples were then titered by repeated cycles of sonication followed by freezing with liquid nitrogen. The cell pellets were then resuspended and 2 μL of the sample was diluted into 2 mL of media. The solution was further diluted from 10−3 to 10−8 in media. From each dilution, 1 mL was added to BSC40 cells plated into a 12-well plate in duplicate. After 48 hrs, the GFP+ foci were counted in order to determine the viral titer of each sample.
Foci Forming Assay
The indicated cell lines were plated into 6-well plates in duplicate. Cells were then infected at MOIs ranging from 0.01 to 0.0001. After 1 hr, the inoculating media was removed and cells were washed with sterile 1% PBS three times before fresh media was added. At 24 hrs interval images of singular GFP+ foci were taken by fluorescent microscopy. Foci area was then measured using ImageJ software. To measure foci image, individual foci were traced in ImageJ and measured using the area measurement command. These values were then averaged for each time point and graphed accordingly.
MTT Assay
Cells were plated to confluency in a 96-well plate as triplicates and infected with the indicated MOI. At 24 hrs intervals after infection, cells were treated with 10 μL of reaction solution containing the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyetraolium bromide) compound. The reaction was then stored in the dark at 37°C until the MTT compound was fully converted to formazan, indicated by the presence of purple precipitate. Once the conversion was completed, 100 μL of solubilization/stop solution was added to lyse the cells and release the formazan product. The plate was then kept from light and absorbance was measured at a wavelength of 595 nm using an Epoch 2 microplate reader. Viability was determined by averaging the readings of each triplicate as a percentage of mock.
Immune Fluorescence
BSC40 cells were plated on glass coverslips and subsequently infected with the indicated virus for 16 hrs. Cells were then fixed with 2% paraformaldehyde, permeabilized with 0.2% Triton X-100, blocked with 3% bovine serum albumin and 0.5% fish gelatin, and stained with anti-c-Myc tag antibody for 2 hrs. After staining, coverslips were mounted on slides and covered with Vectashield H-1200 plus DAPI (4,6 - diamidino-2-phenylindole; Vector Laboratories). Images have been contrast-enhanced and artificially colored to increase visualization of Myc signal.
In vivo Tumor Studies
C57/Bl6 (Charles River Laboratories, Raleigh, NC, USA) or NOD/Scid (The Jackson Laboratory, Bar Harbor, ME, USA) mice, 6 to 8 weeks of age, were injected subcutaneously (SQ) with 4×105 A9F1 cells in 50 μL of sterile phosphate-buffered saline (PBS). Viral treatment was initiated once the tumors reached 25 mm2. Treatment consisted of intratumoral (IT) injections of 1×107 FFU of virus/injection. A total of three injections were given, administered every other day over 5 days (days 0, 2, and 4). For survival studies, the tumor area was monitored with calipers twice weekly until the tumor reached 15 mm in any direction at which point the mouse was euthanized. For analysis of intratumoral viral titer, tumors were excised 24 hrs after the third treatment, transferred to a 40-µm mesh filter, and mechanically separated into a single cell suspension in sterile PBS. Cells within the suspension were mechanically lysed using repeated freeze-thaw and the amount of infectious virus present determined using standard viral titer assays. All experiments were conducted in accordance with the Medical University of South Carolina Institutional Animal Care and Use Committee.
Statistical Analysis
Statistical analysis focused on binary comparisons between individual groups using unpaired Student’s t-test. All results were confirmed using at least two independent experiments. Where possible, data from both experiments were combined into a single data set. Significance was applied to p<0.05 and more significant values were noted.
Results
Generation and in vitro Properties of Fusogenic MYXV Constructs
To test whether the addition of novel fusogenic transgenes would increase the efficacy of MYXV we first generated a series of four new viral constructs encoding the F proteins of other, naturally fusogenic viruses including: newcastle disease virus (NDV), respiratory syncytial virus (RSV), nipah virus (NV), and bovine parainfluenza virus (BPV). Sequences encoding the F protein from each virus were commercially synthesized. For the RSV, NV, and BPV F proteins, the DNA sequence corresponding to the native viral protein was used. For the NDV F protein, a previously identified hyperfusogenic version which induces fusion independent of the NDV accessory hemagglutinin protein was chosen (Table 1). A synthetic early/late promotor was included 5ʹ of the initial start codon of each open reading frame to drive expression during MYXV infection and a C-terminal c-Myc tag was added to each construct to assist in in vitro analysis (Figure 1A). Each open reading frame was then cloned into the existing pBS-135/GFP/136 poxviral recombination plasmid, which inserts both the gene of interest and GFP into the intergenic region between the m135r and m136r viral open reading frames through homologous recombination. Following recombination, each virus was purified to clonality through multiple rounds of selection for GFP+ viral foci.
To test whether each viral construct expressed the encoded exogenous F protein, BSC40 cells were either mock-infected, infected with control virus (vGFP), or infected each new recombinant construct at a multiplicity of infection (MOI) of 1. 24 hrs after infection, cells were harvested and analyzed for transgene expression by immunoblotting lysates for the presence of the c-Myc tag as well as expression of GFP to confirm viral infection. The immunoblot analysis revealed the presence of specific c-Myc-reactive bands in the samples infected with vNDV, vNV, and vBPV which corresponded to the predicted molecular weights of the encoded transgenes. No obvious c-Myc-reactive bands were seen in mock-infected samples or samples infected with either vGFP or vRSV (Figure 1B). A reproducible decrease in the abundance of GFP was also seen in vRSV infected samples which were not observed following infection with the other fusion constructs. Due to the lack of a c-Myc-reactive band in the vRSV-infected samples, we further assayed the expression of the exogenous F proteins using immune fluorescence. BSC40 cells were infected with the indicated viruses for 16 hrs, fixed in paraformaldehyde, and subsequently stained for c-Myc-reactive proteins. This analysis revealed clear c-Myc-reactive signal in cells infected with all four recombinant viruses which were not present in either mock-infected or vGFP-infected cells (Figure 1C). Finally, to determine whether our recombinant viruses induced syncytia formation, BSC40 cells were infected at a low MOI and the resulting foci imaged after 24 hrs. These images indicated an obvious phenotypic change in foci caused by infection with vRSV and vNDV infection while infection with vBPV and vNV resulted in foci which were only slightly different from those caused by vGFP (Figure 1D). Taken together, these data suggest that all four of our recombinant viruses express the encoded fusion transgene and that expression of these transgenes alters MYXV foci formation.
To validate whether the altered foci phenotypes observed in our previous experiment constituted the formation of true syncytia we further infected BSC40 cells with each viral construct at an MOI of 0.001 and subsequently stained the cells for either nuclei (Hoescht stain) or actin filaments (phalloidin) after 24 hrs (Figure 2). Cells were then imaged using an Olympus FV10i laser scanning confocal microscope. Interestingly, the results indicated that foci derived from all four constructs were structurally distinct. Foci from vNDV infections displayed a concentration of actin filaments around a central core made up of two nuclei. These actin filaments then expanded out into a large, single body containing numerous other nuclei. In contrast, foci from vRSV infections displayed a single ring of actin which largely excluded other actin filaments from the GFP+ foci region. Similar to vNDV foci, however, multiple distinct nuclei were readily observed within this actin ring. The vNV construct exhibits a less pronounced phenotype characterized by multiple large GFP+ cells each containing several distinct nuclei. In contrast to foci formed by vNDV and vRSV, however, these larger cells did not appear to fuse together and instead remained groups of smaller syncytia. Foci caused by infection with vBPV were slightly phenotypically distinct from those caused by vGFP; however, they did not display either actin clustering/exclusion or appear to contain multiple nuclei and therefore appear unlikely to represent true syncytia.
Fusogenic MYXV’s Display Increased Spread in vitro
Having validated that at least three of our MYXV constructs induced syncytia formation during infection, we next sought to test whether this induction would alter viral replication or spread in vitro. To determine whether syncytia formation or transgene expression had a negative impact on the generation of infectious viral progeny, BSC40 cells were infected with each viral construct at an MOI of 10. Samples were harvested at 3, 6, 12, 24, 48, and 72 hrs after infection and the amount of infectious progeny was assessed by titering each sample on BSC40 cells. Consistent with all viruses displaying normal intracellular replication, we found no significant differences in the number of infectious viruses produced by any construct at any time point (Figure 3A). Similar results were observed following an identical experiment conducted in the murine lung cancer cell line A9F1 (Figure 3A) suggesting that the inclusion of exogenous fusion proteins did not drastically inhibit MYXV’s intracellular replication.
We next asked whether inducing syncytia formation would alter the direct lytic potential of MYXV in infected cells. To test this, BSC40 cells were infected with each viral construct at an MOI of 10. The lytic potential of each construct was then measured by assaying cellular viability 48 hrs after infection using MTT assay (Figure 3B). While infection with all viral constructs, including vGFP, decreased cellular viability compared to mock, infection with either the vNDV or vRSV constructs resulted in a further decrease in cellular viability compared to vGFP (p < 0.001 for both viruses). A significant decrease from vGFP was also seen in the vNV construct; however, the magnitude of this decrease was less robust than that seen with vNDV or vRSV. Again, similar results were seen in the A9F1 model (Figure 3B) although interestingly, vNV did not obviously display improved lytic potential in A9F1 cells.
Finally, since the goal of inducing syncytia formation during OV is to improve viral dissemination throughout the tumor, we next asked whether forcing cell:cell fusion would increase MYXV spread. BSC40 cells were infected at MOIs ranging from 0.01 to 0.0001 and images of individual GFP+ foci were taken at 24 hrs intervals. Viral spread was then assayed by analyzing the overall area of distinct GFP+ foci using ImageJ software. We observed that individual foci resulting from infection with either vNDV or vRSV grew significantly faster than foci caused by infection with vGFP (Figure 3C and D, p < 0.01 at 72 hrs). Interestingly, despite its apparent syncytial phenotype, foci derived from vBPV grew at the same rate as those from vGFP as did foci from the non-syncytial forming vNV (Figure 3C and D). Similar results were observed following an identical experiment conducted in A9F1 cells (Figure 3C and D). Taken together, these results suggest that fusogenic MYXV’s display improved oncolytic properties in vitro.
Syncytia Forming MYXV’s Maintain Their Interferon Mediated Cancer Tropism
It has been previously shown that fusogenic oncolytic viruses can induce syncytia formation while still retaining their cancer-specific replication properties.19 To determine whether this was also true for fusogenic MYXV’s, we asked whether our viral constructs would infect and/or cause syncytia formation in non-malignant cells. To test this, MEFs were infected with each fusogenic construct in the presence of IFN-β added either 1 hr prior to viral infection or 1 hr after viral infection. This protocol allows us to examine the effects of IFN-β on both the prevention of initial infection and the spread of established infection. GFP+ foci were then imaged at 24 hrs intervals and analyzed using ImageJ (Figure 4). Consistent with previous reports,32 pretreatment with IFN-β completely eliminated MYXV infection in MEF cells. In contrast, treatment with IFN-β post-infection resulted in the establishment of infection in individual cells but completely abrogated the spread of infection into neighboring cells (Figure 4A). In the absence of IFN-β, infection of MEF’s proceeded similarly to what was observed in BSC40 cells (Figure 4A and B). Critically, while the speed of viral spread was similar for all constructs (Figure 4B), the altered foci phenotypes observed in our previous experiments were also observed in non-IFN-β treated MEF’s (Figure 4A). These data suggest that syncytia formation can occur in non-malignant cells; however, this formation is prevented by the presence of a functional IFN response which would prevent any significant toxicity concerns in vivo.
Fusogenic MYXV’s Show Decreased Treatment Efficacy in vivo
Having verified both the efficacy and specificity of our fusogenic MYXV’s in vitro, we finally sought to determine whether the induced syncytia formation caused by these constructs would impact clinical efficacy in an in vivo animal model. To test this, we chose the LLC-A9F1 model which we have previously shown is partially susceptible to MYXV-based oncolytics. C57/B6 mice were subcutaneously injected with 4×105 A9F1 cells. Once the tumors reached 25 mm2, mice were randomly sorted into six cohorts and treated with intratumoral injection of either saline or 1x107 FFU of each viral construct. A total of three injections were given, administered every other day over 5 days. Overall tumor burden was measured using calipers and animals were euthanized when their tumor reached 15 mm in any direction. Consistent with previous publications, A9F1 tumors in mock-treated mice grew rapidly once established and all mice in this cohort reached criteria for euthanasia by day 31. Treatment with vGFP significantly delayed tumor growth; however, it was unable to eradicate established tumors in these experiments. In stark contrast to our hypothesis, however, we observed an inverse relationship between a virus's fusogenicity in vitro and its therapeutic potential in vivo. Instead of reducing tumor burden and prolonging survival compared to the control virus, treatment of tumors with fusogenic MYXV’s was associated with reduced overall efficacy characterized by: increased rates of tumor growth (Figure 5A), increased overall tumor burden at day 31 (Figure 5B), and reduced overall survival (Figure 5C). To test whether this might be due to reduced direct oncolytic killing, we subsequently performed a second experiment in which we harvested tumors 24 hrs after the final treatment and analyzed the amount of infectious virus present using foci forming assays. The results demonstrated that the amount of infectious virus present was significantly higher in vGFP treated tumors than in tumors treated with any of the fusogenic constructs. Additionally, the amount of virus recovered from tumors treated with each fusogenic construct was inversely correlated to that viruses' apparent fusogenicity in vitro (Figure 5D). To test whether this reduction in viral titer was due to enhanced immune clearance, we assayed the efficacy and persistence of our fusogenic viruses in an immune-deficient setting. NOD/Scid mice were subcutaneously injected with 4×105 A9F1 cells. After tumors reached 25 mm2, the mice were sorted into six cohorts and treated with intratumoral injection of either saline or 1×107 FFU of each viral construct. Tumor growth and viral titers were then analyzed as above. Consistent with MYXV achieving therapeutic efficacy primarily through induction of adaptive anti-tumor immunity, in NOD/Scid mice we did not observe a significant decrease in tumor volume following treatment with any viral construct (Figure 6A–C). Consistent with our results in C57/Bl6 mice, however, tumors treated with fusogenic viral constructs displayed both a trend towards reduced efficacy and significantly lower viral titers compared to vGFP treated tumors (Figure 5D). Again, the decrease in viral titers was more pronounced in the highly fusogenic constructs (vNDV and vRSV) than in the less fusogenic viruses (vNV or vBPV).
Discussion
While recent studies have shown OV to be an efficacious form of cancer therapy, several hurdles must still be overcome for this treatment to be fully adapted into the clinic. One of these hurdles is the inefficient spread of the viral agents within the tumor microenvironment. To combat this issue, several groups have previously explored the use of fusogenic oncolytic viruses which can induce large syncytia during infection. These fusogenic viruses have been shown to display: increased rates of viral spread, syncytial-mediated infection of normally resistant cell populations, and improved direct lytic potential. Together these traits have allowed fusogenic viruses to obtain improved therapeutic efficacies in multiple preclinical models. Interestingly, all of these works have tested the efficacy of a single fusogenic construct compared to a non-fusogenic control. This has left a major unanswered question concerning fusogenic oncolytic constructs in terms of how the choice of fusogenic protein impacts outcomes. In this regard, our results indicate that not all fusogenic F proteins are equivalent, either in terms of their ability to induce syncytia formation or the phenotype of the syncytia which are induced. In our case, the inclusion of the F proteins from either NDV or RSV resulted in a profound syncytial phenotype characterized by the generation of large, multinucleated bodies. Interestingly, despite having the strongest syncytial phenotype, no c-Myc tagged transgene corresponding to RSV-F could be detected in vRSV infected samples by Western blot (Figure 1B). This result was reproduced in fourth independent experiments suggesting it was not a technical artifact. Since the RSV-F protein could be detected in immune fluorescence, this lack is not caused by a failure of the virus to express the exogenous F protein. Instead, we hypothesize that the RSV-F transgene becomes highly insoluble during infection and cannot be accurately analyzed using Western blot. Alternatively, the syncytia produced by this virus could be fragile resulting in loss of transgene expressing cells during processing. Regardless, we are confident that the clear formation of syncytia by this construct indicates a successful expression of the RSV-F transgene. In contrast, the inclusion of the F protein from BPV resulted in the formation of cell:cell fusions which appeared to be made up of multiple smaller syncytia. These phenotypes were consistent across infection of multiple cell lines suggesting they were driven by the properties of the encoded F protein and not the context of its expression. These data suggest that the inclusion of a fusogenic protein can improve oncolytic treatment in some instances; however, the choice of F protein could play a critical role in determining this outcome.
Similar to the previous works, the initial results with our fusogenic MYXV constructs suggest that they display promising oncolytic traits in vitro. The two constructs which displayed the most pronounced syncytial phenotypes (vNDV and vRSV) also displayed significant increases in both viral spread and direct lytic killing, two traditional hallmarks used to predict oncolytic potential. Interestingly, while these two constructs were clearly superior to the others in BSC40 and A9F1 cells, all four fusogenic constructs displayed similarly enhanced lytic potential in GL261 glioblastoma cells (Figure S1) suggesting that the efficacy of fusogenic viruses might be, at least in part, cell type specific. One potential reason for this could again have to do with the differences between the F proteins of various viruses. Each F protein typically has a unique set of binding partners which are required for its fusion activity. It is, therefore, possible that the binding partners required for NV-F and BPV-F are not present in either BSC40 or A9F1 cells. These data again suggest that the concept of fusogenic viruses having inherently improved oncolytic properties might be an oversimplification and that therapeutic efficacy might be significantly impacted by the specific choice of F protein.
Critically, while multiple previous studies have demonstrated the potential to improve the efficacy of various oncolytic viruses by forcing them to induce syncytia formation, our results suggest that doing this in the context of oncolytic MYXV actually decreases efficacy. This appears to be correlated with a significant decrease in either the replication or persistence of fusogenic-MYXV’s in vivo (Figure 5). Critically, this loss of viral titer was seen at similar levels in both immune-competent and immune-deficient settings suggesting that it is likely not the result of immune-mediated viral clearance. Instead, our finding that fusogenic MYXV’s displays significantly enhanced the lytic potential in vitro suggests that the reduced viral titers seen following fusogenic therapies might be due to syncytia formation resulting in rapid cell lysis. This might be beneficial immediately following therapy; however, if this cell death occurs too rapidly, it would prevent viral replication and spread. Interestingly, a similar phenomenon was observed following MYXV treatment of multiple myeloma cells.27,33 In these cells, viral infection induces an apoptotic response so rapid it eliminates infected malignant cells prior to the completion of the viral replication cycle. Interestingly, in the case of myeloma, viral replication appeared to be dispensable for achieving oncolytically induced immunotherapy. However, it is possible that more persistent infection might be required to induce immunotherapy in solid tumors such as the A9F1 tumor used in our study.
Taken together this study shows that while the use of F proteins provides increased efficacy to non-fusogenic viruses in vitro, it brings into question whether this efficacy can be transferred to every oncolytic virus in vivo.
 |
Figure 1 Construction of fusogenic MYXV constructs. (A) Genomic structure of generated fusogenic viruses. (B) Expression of c-Myc-tagged F proteins during infection measured by immunoblot. Expression of GFP is included to track overall infection and expression of actin is included as a loading control. Data are representative of three independent experiments. (C) Expression of c-Myc-tagged F proteins during infection measured by immune fluorescence for c-Myc (red). Infected cells can be identified by GFP expression. Note that images have been contrast-enhanced to increase visualization of c-Myc. (D) Images of GFP+ foci resulting from infection of BSC40 cells with the indicated viral construct. |
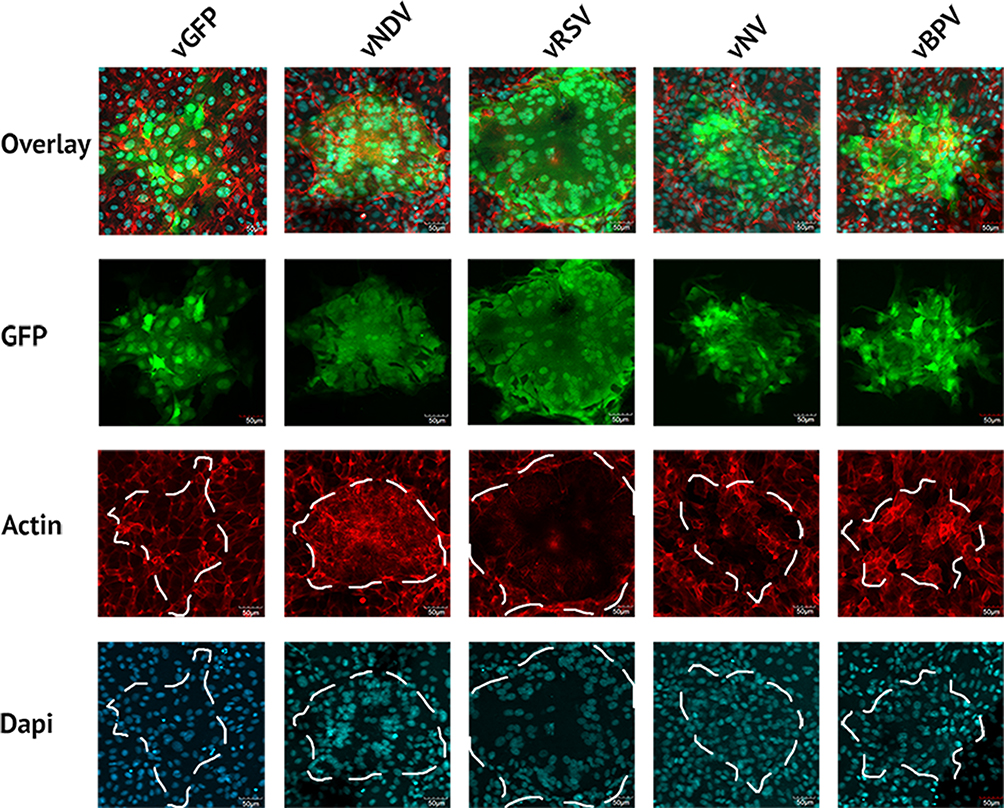 |
Figure 2 Fusogenic MYXV’s form multinucleated syncytia. BSC40 cells were infected with the indicated viral construct. After 48 hrs, cells were stained with phalloidin and Hoechst stain to identify the cytoskeleton and nucleus, respectively, and fluorescent images of infected regions were then taken. |
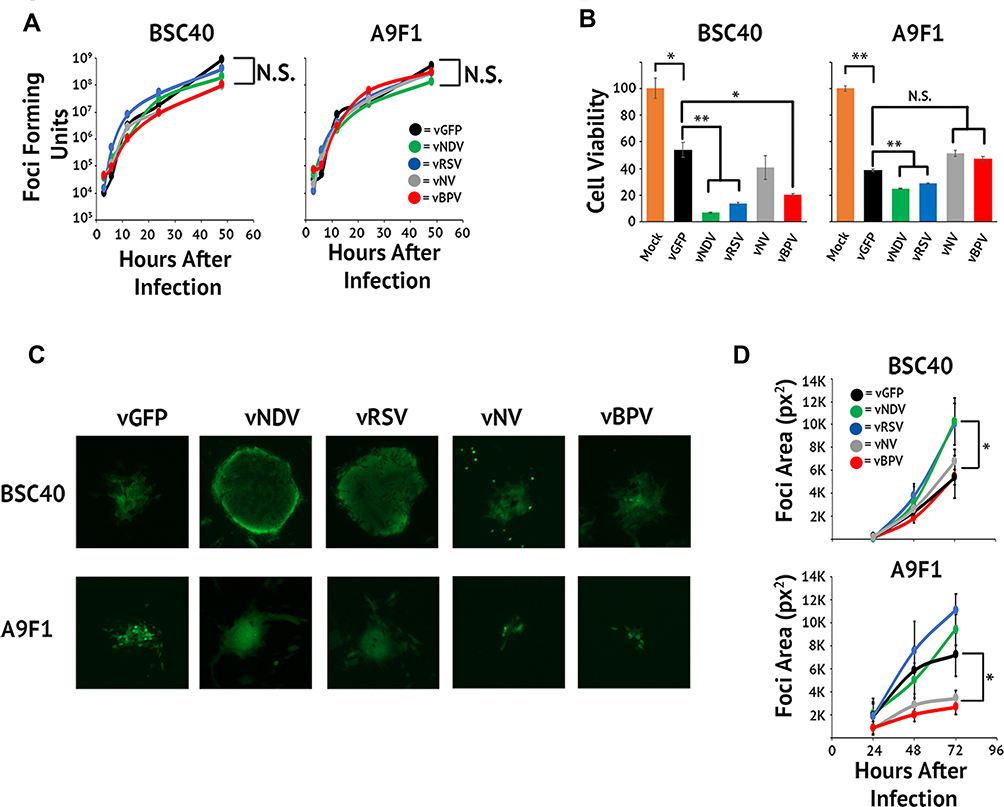 |
Figure 3 In vitro characterization of fusogenic constructs. (A) BSC40 and A9F1 cells were infected with the indicated virus at an MOI of 10. Cells were harvested 3, 6, 12, 24, 48, and 72 hrs post-infection and the amount of infectious virus present analyzed using standard foci forming assays. (B) BSC40 and A9F1 cells were plated in triplicate into 96-well plates and infected as described. Cellular viability was measured via MTT assay at 24 hrs intervals post-infection. (C) BSC40 and A9F1 cells were infected at an MOI of 0.001 and images of GFP+ foci taken at 24, 48, and 72 hrs post-infection. (D) Size of individual foci were quantitated using ImageJ software. Data presented are representative of three individual experiments. Significance was determined using Student’s t-test (*p < 0.05, **p<0.005). |
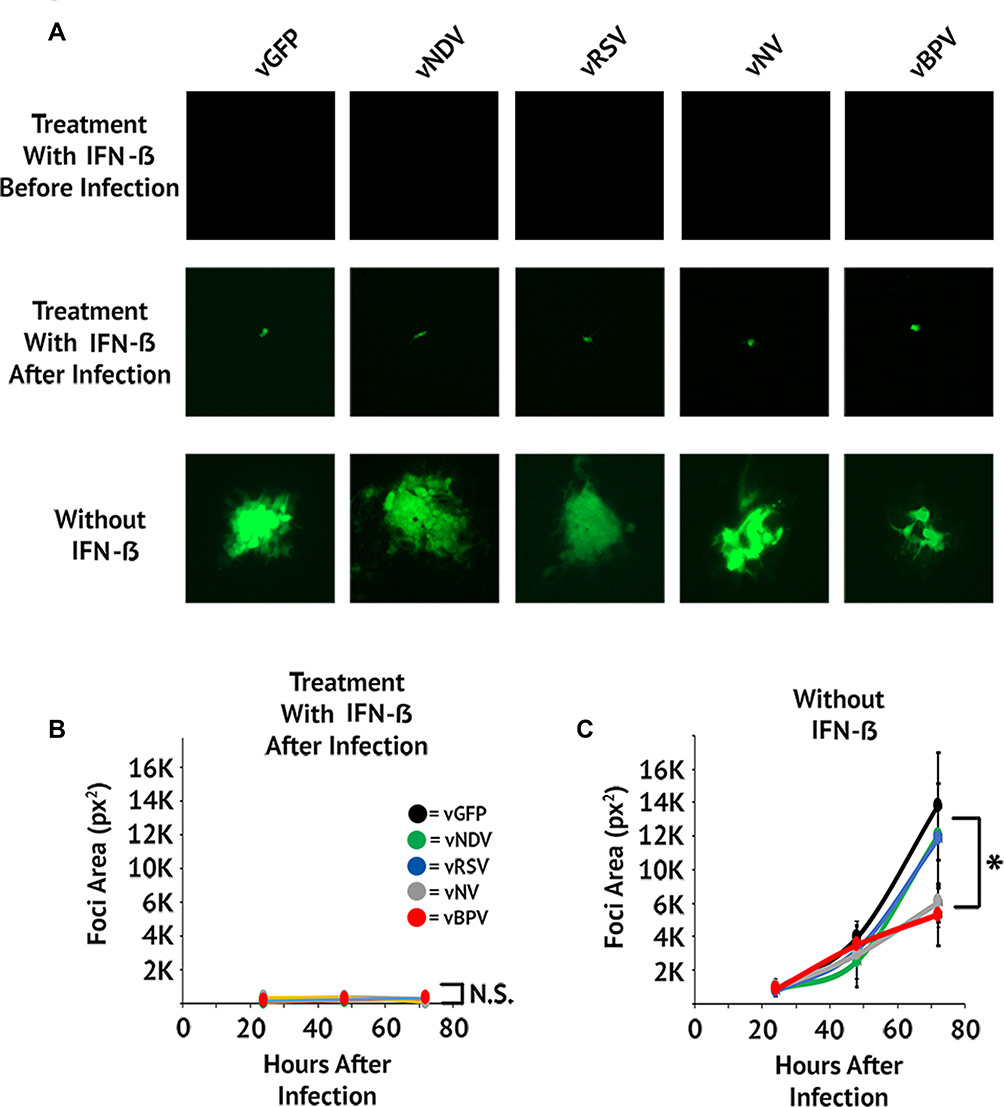 |
Figure 4 Fusogenic MYXV’s retain their IFN mediated cancer tropism. (A) MEF cells were infected at an MOI of 0.001. Cells were treated with IFN-β either 1-hr pre-infection or 1-hr post-infection. Images of GFP+ foci were then taken at 24, 48, and 72 hrs post-infection. (B and C) Size of individual foci were quantitated using ImageJ software. Data presented are representative of two individual experiments. Significance was determined using Student’s t-test (*p < 0.05). |
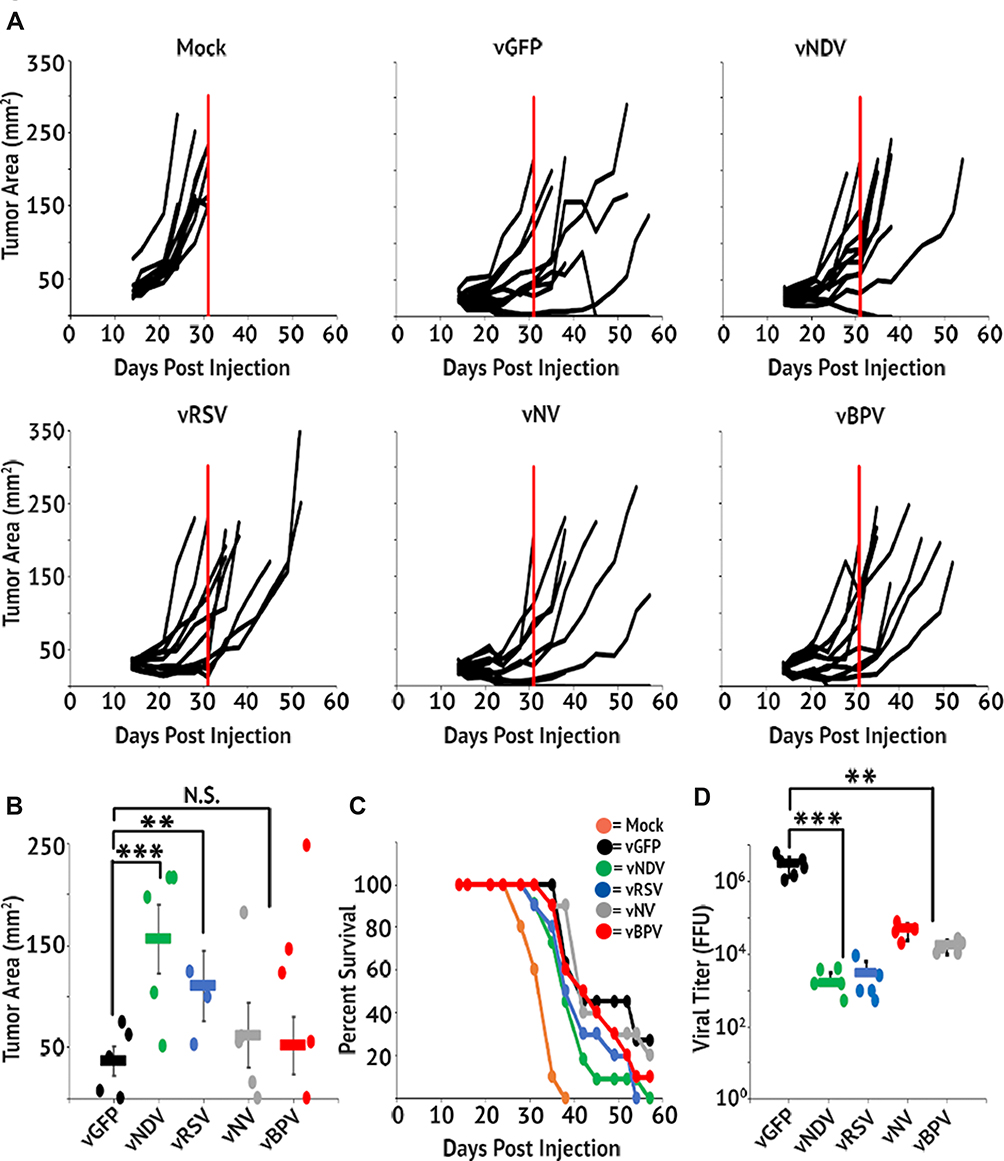 |
Figure 5 Fusogenic MYXV’s display reduced therapeutic potential in vivo. C57/Bl6 mice bearing A9F1 tumors were treated with three IT injections of the indicated viral constructs (n=10 or 11 per group). (A) Area of tumors from individual mice over time. Vertical red line marks the euthanasia of the last mock-treated animal and is included for reference. (B) Average tumor burden on day 31 (the day the last mock-treated animal was euthanized). (C) Survival of mice treated with fusogenic constructs. (D) Amount of infectious virus present in tumors 24 hrs after the last viral treatment. Data shown represent the summation of two individual experiments. Significance was determined using Student’s t-test (**p < 0.01, ***p < 0.001). |
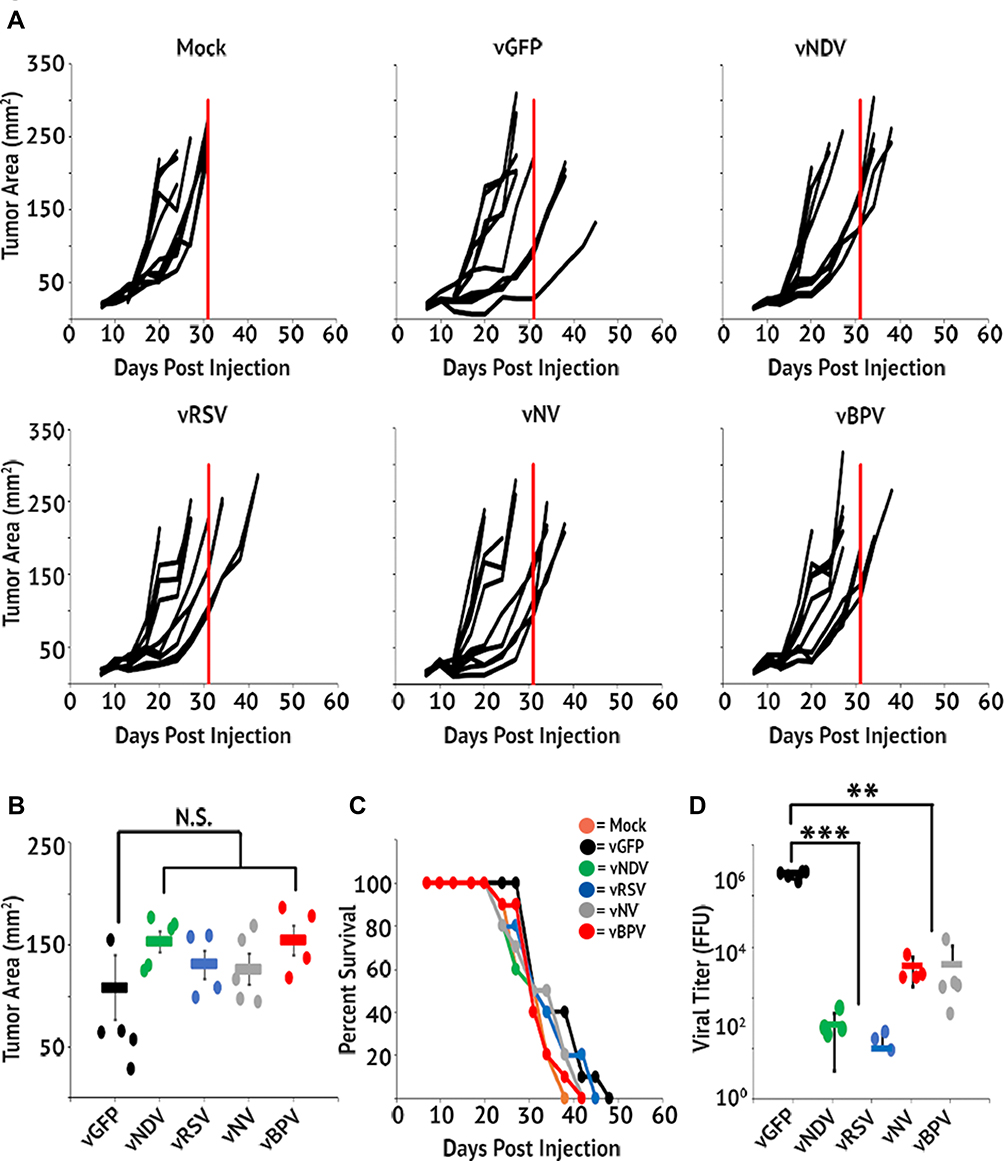 |
Figure 6 Reduced viral persistence is not mediated by immune clearance. Immune deficient NOD/Scid mice bearing A9F1 tumors were treated with three IT injections of the indicated viral constructs (n=10 or 11 per group). (A) Area of tumors from individual mice over time. Vertical red line marks the euthanasia of the last mock-treated animal and is included for reference. (B) Average tumor burden on day 31 (the day the last mock-treated animal was euthanized). (C) Survival of mice treated with fusogenic constructs. (D) Amount of infectious virus present in tumors 24 hrs after the last viral treatment. Data shown represent the summation of two individual experiments. Significance was determined using Student’s t-test (**p < 0.01, ***p < 0.001). |
Ethical Approval
All work was carried out under the supervision and approval of the biosafety and IACUC committees at the Medical University of South Carolina.
Disclosure
All authors declared that they have no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J Clin. 2018. doi:10.3322/caac.21492
2. Orangio GR. The economics of colon cancer. Surg Oncol Clin N Am. 2018;27:327–347. doi:10.1016/j.soc.2017.11.007
3. Baskar R, Yap SP, Chua KL, Itahana K. The diverse and complex roles of radiation on cancer treatment: therapeutic target and genome maintenance. Am J Cancer Res. 2012;2:372–382.
4. Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615–627. doi:10.1146/annurev.med.53.082901.103929
5. Delaney G, Jacob S, Featherstone C, Barton M. The role of radiotherapy in cancer treatment: estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer. 2005;104:1129–1137. doi:10.1002/(ISSN)1097-0142
6. Brockman RW. Mechanisms of resistance to anticancer agents. Adv Cancer Res. 1963;7:129–234.
7. Russell SJ, Peng KW. Viruses as anticancer drugs. Trends Pharmacol Sci. 2007;28:326–333. doi:10.1016/j.tips.2007.05.005
8. Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 2012;30:658–670. doi:10.1038/nbt.2287
9. Roberts MS, Groene WS, Lorence RM, Bamat MK. Naturally occurring viruses for the treatment of cancer. Discov Med. 2006;6:217–222.
10. Vaha-Koskela MJ, Heikkila JE, Hinkkanen AE. Oncolytic viruses in cancer therapy. Cancer Lett. 2007;254:178–216. doi:10.1016/j.canlet.2007.02.002
11. Pol J, Kroemer G, Galluzzi L. First oncolytic virus approved for melanoma immunotherapy. Oncoimmunology. 2016;5:e1115641. doi:10.1080/2162402X.2015.1115641
12. Fountzilas C, Patel S, Mahalingam D. Review: oncolytic virotherapy, updates and future directions. Oncotarget. 2017;8:102617–102639. doi:10.18632/oncotarget.v8i60
13. Stanford MM, Breitbach CJ, Bell JC, McFadden G. Innate immunity, tumor microenvironment and oncolytic virus therapy: friends or foes? Curr Opin Mol Ther. 2008;10:32–37.
14. Freeman SM, Abboud CN, Whartenby KA, et al. The “bystander effect”: tumor regression when a fraction of the tumor mass is genetically modified. Cancer Res. 1993;53:5274–5283.
15. Loya SM, Zhang X. Enhancing the bystander killing effect of an oncolytic HSV by arming it with a secretable apoptosis activator. Gene Ther. 2015;22:237–246. doi:10.1038/gt.2014.113
16. Hyer ML, Sudarshan S, Schwartz DA, Hannun Y, Dong JY, Norris JS. Quantification and characterization of the bystander effect in prostate cancer cells following adenovirus-mediated FasL expression. Cancer Gene Ther. 2003;10:330–339. doi:10.1038/sj.cgt.7700576
17. Bateman A, Bullough F, Murphy S, et al. Fusogenic membrane glycoproteins as a novel class of genes for the local and immune-mediated control of tumor growth. Cancer Res. 2000;60:1492–1497.
18. Higuchi H, Bronk SF, Bateman A, Harrington K, Vile RG, Gores GJ. Viral fusogenic membrane glycoprotein expression causes syncytia formation with bioenergetic cell death: implications for gene therapy. Exp Ther. 2000;60:6396–6402.
19. Krabbe T, Altomonte J. Fusogenic viruses in oncolytic immunotherapy. Cancers (Basel). 2018;10.
20. Stanford MM, Shaban M, Barrett JW, et al. Myxoma virus oncolysis of primary and metastatic B16F10 mouse tumors in vivo. Mol Ther. 2008;16:52–59. doi:10.1038/sj.mt.6300348
21. Bartee MY, Dunlap KM, Bartee E. Tumor-localized secretion of soluble PD1 enhances oncolytic virotherapy. Cancer Res. 2017;77:2952–2963. doi:10.1158/0008-5472.CAN-16-1638
22. Burton C, Das A, McDonald D, et al. Oncolytic myxoma virus synergizes with standard of care for treatment of glioblastoma multiforme. Oncolytic Virother. 2018;7:107–116. doi:10.2147/OV.S179335
23. Johnston JB, Barrett JW, Chang W, et al. Role of the serine-threonine kinase PAK-1 in myxoma virus replication. J Virol. 2003;77:5877–5888. doi:10.1128/JVI.77.10.5877-5888.2003
24. Smallwood SE, Rahman MM, Smith DW, McFadden G. Myxoma virus: propagation, purification, quantification, and storage. Curr Protoc Microbiol. 2010;Chapter 14:Unit 14A 11. doi:10.1002/9780471729259.mc14a01s17
25. Wolfe AM, Dunlap KM, Smith AC, Bartee MY, Bartee E. Myxoma virus M083 is a virulence factor which mediates systemic dissemination. J Virol. 2018;92. doi:10.1128/JVI.02186-17
26. Chan WM, Bartee EC, Moreb JS, Dower K, Connor JH, McFadden G. Myxoma and vaccinia viruses bind differentially to human leukocytes. J Virol. 2013;87:4445–4460. doi:10.1128/JVI.03488-12
27. Bartee E, Chan WM, Moreb JS, Cogle CR, McFadden G. Selective purging of human multiple myeloma cells from autologous stem cell transplantation grafts using oncolytic myxoma virus. Biol Blood Marrow Transplant. 2012;18:1540–1551. doi:10.1016/j.bbmt.2012.04.004
28. Orabi A, Hussein A, Saleh AA, El-Magd MA, Munir M. Evolutionary insights into the fusion protein of Newcastle disease virus isolated from vaccinated chickens in 2016 in Egypt. Arch Virol. 2017;162(10):3069–3079. doi:10.1007/s00705-017-3483-1. Epub 2017 Jul 8
29. Haid S, Grethe C, Bankwitz D, Grunwald T, Pietschmann T. Identification of a Human Respiratory Syncytial Virus Cell Entry Inhibitor by Using a Novel Lentiviral Pseudotype System. J Virol. 2015:30;90(6):3065–3073.
30. Chua KB, Koh CL, Hooi PS, et al Isolation of Nipah virus from Malaysian Island flying-foxes. Microbes Infect. 2002;4:145–151.
31. Neill JD, Ridpath JF, Valayudhan BT. Identification and genome characterization of genotype B and genotype C bovine parainfluenza type 3 viruses isolated in the United States. BMC Vet Res. 2015;11:112.
32. Wang F, Ma Y, Barrett JW, et al. Disruption of Erk-dependent type I interferon induction breaks the myxoma virus species barrier. Nat Immunol. 2004;5:1266–1274. doi:10.1038/ni1132
33. Bartee E, Bartee MY, Bogen B, Yu XZ. Systemic therapy with oncolytic myxoma virus cures established residual multiple myeloma in mice. Mol Ther Oncolytics. 2016;3:16032. doi:10.1038/mto.2016.32
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.