")
Back to Journals » International Journal of Nanomedicine » Volume 16
Immunotherapy of Tumor RNA-Loaded Lipid Nanoparticles Against Hepatocellular Carcinoma
Authors Zhang Y, Xie F, Yin Y, Zhang Q, Jin H, Wu Y, Pang L, Li J , Gao J
Received 10 November 2020
Accepted for publication 21 December 2020
Published 25 February 2021 Volume 2021:16 Pages 1553—1564
DOI https://doi.org/10.2147/IJN.S291421
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yan Shen
Yake Zhang,1,2,* Fangyuan Xie,3,* You Yin,4,* Qin Zhang,1,* Hong Jin,5 Yan Wu,6 Liying Pang,7 Jun Li,2 Jie Gao1,2
1Institute of Translational Medicine, Shanghai University, Shanghai, 200444, People’s Republic of China; 2Laboratory of Drug Discovery and Design, School of Pharmacy, Liaocheng University, Liaocheng, 252000, People’s Republic of China; 3Department of Pharmacy, Shanghai Eastern Hepatobiliary Surgery Hospital, Shanghai, 200438, People’s Republic of China; 4Department of Neurology, Changzheng Hospital, Second Military Medical University, Shanghai, 200003, People’s Republic of China; 5Department of Laboratory Medicine, Hongqi Hospital of Mudanjiang Medical College, Mudanjiang, 157011, People’s Republic of China; 6Heilongjiang Key Laboratory of Anti-Fibrosis Biotherapy, Mudanjiang Medical College, Mudanjiang, 157011, People’s Republic of China; 7The First Clinical Medical College of Mudanjiang Medical College, Mudanjiang, 157011, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jie Gao
Institute of Translational Medicine, Shanghai University, Shanghai, 200444, People’s Republic of China
Tel/Fax + 86-0577-88816381
Email [email protected]
Jun Li
Laboratory of Drug Discovery and Design, School of Pharmacy, Liaocheng University, Liaocheng, 252000, People’s Republic of China
Tel/Fax + 86-0577-88816381
Email [email protected]
Purpose: Hepatocellular carcinoma (HCC) is one of the leading causes of cancer-related deaths worldwide. Most current therapeutic strategies primarily include localized treatment, lacking effective systemic strategies. Meanwhile, recent studies have suggested that RNA vaccines can effectively activate antigen-presenting cells (APCs) and lymphocytes to produce a strong systemic immune response and inhibit tumor growth. However, tumor vaccines loaded with a single tumor antigen may induce immunosuppression and immune evasion, while identifying tumor-specific antigens can require expensive and laborious procedures. Therefore, the use of whole tumor cell antigens are currently considered to be promising, potentially effective, methods. Previously, we developed a targeted liposome-polycation-DNA (LPD) complex nanoparticle that possess a small size, high RNA encapsulation efficiency, and superior serum stability. These particles were found to successfully deliver RNA to tumor sites. In the current study, we encapsulated total tumor-derived RNA in lipid nanoparticles (LNPs) to target dendritic cells (DCs) to incite expeditious and robust anti-tumor immunity.
Methods: Total tumor-derived RNA was extracted from liver cancer cells (Hepa1-6 cells). LNPs loaded with tumor RNA were then prepared thin-film hydration method. The ability of RNA LNPs to induce DC maturation, cytotoxicity, and anti-tumor activity, was investigated in vitro and in vivo.
Results: The average particle size of LNPs and RNA LNPs was 102.22 ± 4.05 nm and 209.68 ± 6.14 nm, respectively, while the zeta potential was 29.97 ± 0.61 mV and 42.03 ± 0.42 mV, respectively. Both LNPs and RNA LNP vaccines exhibited good distribution and stability. In vitro, RNA LNP vaccines were capable of promoting DC maturation and inducing T lymphocytes to kill Hepa1-6 cells. In vivo, RNA LNP vaccines effectively prevent and inhibit HCC growth.
Conclusion: RNA LNPs may serve as an effective antigen specific vaccine to induce anti-tumor immunity for HCC.
Keywords: RNA lipid nanoparticles, tumor vaccine, dendritic cells, cancer immunotherapy
Introduction
Hepatocellular carcinoma (HCC) is one of the most common malignant cancers worldwide, with frequent recurrence and poor prognosis. Conventional therapy has limited efficacy against HCC. Therefore, an urgent need exists to develop effective therapies.1 The advent of immunotherapy has revolutionized cancer treatment due to its mild side effects and significant therapeutic effects. In fact, it is now considered the fourth most commonly administered tumor treatment method, after surgery, radiotherapy, and chemotherapy.2–4
Tumor vaccines, specifically, have attracted increasing attention in cancer immunotherapy. These vaccines can activate host immunity against tumors by exploiting the different protein expressions of tumor and normal cells to effectively present tumor antigens and activate specific cytotoxic T cells (CTLs) with high specificity and low side effects.5–8 However, numerous factors hinder the application of tumor vaccines, including poor antigenicity, as well as systemic and intratumoral immunosuppression.
To overcome these obstacles, RNA vaccines have been developed to incite expeditious and robust anti-tumor immunity.9 Compared with traditional vaccines, RNA vaccines are capable of bypassing MHC classification restriction, eliciting immunogenicity without the need for adjuvant, and are readily produced and safely stored. In addition, RNA vaccines do not integrate into the genome, exhibiting a high safety profile.10 However, RNA vaccines targeting tumor-associated, or tumor-specific epitopes, can cause profound intratumoral and systemic immunosuppression that may facilitate immune evasion. Moreover, although tumor cells may be heterogeneous, it is difficult to identify specific antigens expressed by all tumor cells.11,12 To address this challenge, dendritic cell (DC)-based tumor vaccines loaded with RNA derived from the whole transcriptome have been designed. In 2018, a randomized controlled Phase III clinical trial reported that the DC Vax-L vaccine (dendritic cell vaccine loaded with autologous tumor cell lysate from glioma patients) significantly improved the survival rate of newly diagnosed glioblastoma patients.13 Mu et al also reported that among 19 patients with androgen-resistant prostate cancer treated with DC vaccines loaded with total tumor-derived mRNA, 8 patients progressed, 11 patients were considered to have stable disease, and 13 patients showed a decrease in serum prostate-specific antigen levels.14 Therefore, DC-based tumor vaccines loaded with whole transcriptome RNA may offer potential clinical significance.
However, one disadvantage of RNA is its inherent instability. Indeed, naked RNA administered to patients in vivo will be degraded and inactivated before localizing to antigen-presenting cells (APCs), making it difficult to stimulate a specific tumor immune response. Meanwhile, nanocarriers, a new type of nano-drug delivery system, have been developed as delivery vehicles for RNA, which can efficiently deliver tumor antigen RNA to DCs and induce immune responses.15–18 Compared with common tumor vaccines, nanocarriers that deliver tumor antigen RNA have many advantages: 1) Nanocarriers can effectively prevent RNA degradation and increase stability. 2) RNA can efficiently encode multiple tumor antigens. 3) Nanocarriers can simultaneously encapsulate immune adjuvants and enhance the immunogenicity of the vaccine. 4) Compared with free drugs, nanocarriers are more readily recognized and engulfed by APCs. 5) Nanocarriers can increase the cross-presentation of antigens, thereby increasing the CTL response. 6) Ligand modification improves the targeting of APCs.19–21
Due to the large amount of preclinical toxicity data and promising safety data, nanoliposomes have been developed as delivery vehicles for RNA in human trials. Kranz et al encapsulated RNA encoding tumor antigens into lipoplexes, which target DCs and activate tumor-specific T cell immune effects.22 After successfully verifying the anti-tumor effect of the lipoplexes in mice, a clinical trial was carried out in three patients with advanced skin cancer, and a strong tumor immune response was detected. Currently, many RNA-liposomal cancer vaccines are in human clinical trials and have shown encouraging early results.23 Therefore, RNA-nanoliposomes are expected to achieve rapid and effective anti-tumor immunity.
Previously, we developed targeted LPD (liposome-polycation-DNA complexes) for siRNA delivery, which are small in size, with high siRNA encapsulation efficiency, superior serum stability, and the ability to successfully deliver siRNA to tumor sites.24–26 Similarly, Leaf Huang’s research group also used LPD nanoparticles to target delivery of the large nucleic acid molecule, PD-L1 trap plasmid, to locally, and transiently, produce PD-L1 trap fusion protein in tumor tissues.27 Therefore, in this study, we extracted the total RNA of liver cancer cells and loaded them on the lipid nanoparticles (LNPs) to construct a DC-targeted RNA LNP cancer vaccine. These vaccines are expected to be effectively translate, process, and present RNA on MHC molecules in DCs, inducing high-efficiency anti-liver cancer cell-specific immune responses. Hence the results of this research can provide a new tumor vaccine with high-efficiency and low toxicity for immunotherapy of liver cancer, which has important theoretical significance and potential clinical applications for tumor immunotherapy of HCCs and other tumors.
Materials and Methods
Materials, Cell Culture, and Mice
DOTAP and CHOL were purchased from Avanti Co., Ltd. (AL, USA; purity ≥ 98%). Dimethyl sulfoxide (DMSO) and lipopolysaccharide (LPS) were provided by Sigma-Aldrich (MO, USA). Dulbecco’s modified Eagle’s medium (DMEM) with high glucose, DMEM-F12, penicillin-streptomycin, fetal bovine serum (FBS), MEM NEAA, HEPES, sodium pyruvate, trypsin, and phosphate-buffered saline (PBS) were provided by Thermo Fisher Scientific (Waltham, MA, USA). The APC-CD40, APC-CD80, APC-CD86, FITC-CD11c, PE-MHCII, and FITC-CD8 antibodies were purchased from BD Bioscience Co., Ltd. (USA). GM-CSF and IL-4 were obtained from Pepro Tech (USA). CD8 immunomagnetic bead kits were purchased from Intervention (USA). The Cell Counting Kit-8 from Gibco (USA) and Cyto Tox 96 Non-Radioactive Cytotoxicity Assay Kit was purchased from Promega (Japan).
Hepa1-6 cells were purchased from the Institute of Chinese Academy of Sciences and cultured in a humidified atmosphere of 5% CO2 at 37°C. The cells were cultured in DMEM supplemented with 10% FBS and 100 μg/mL penicillin-streptomycin. C57BL/6 mice were purchased from Jinan Pengyue Laboratories. All animal studies were approved by the Ethics Committee of Liaocheng University. All animal procedures were performed in accordance with the guidelines of the Committee on Animal of Liaocheng University.
RNA Isolation
RNA was isolated as previously described.15 Briefly, total tumor-derived RNA from Hepa1-6 cells was isolated using TRIzol reagent following the manufacturer’s instructions. RNA was spectrophotometrically quantified at 260 and 280 nm using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, USA).
Lipid Nanoparticle Preparation
The film hydration method was used to prepare cationic lipid nanoparticles (LNPs) with a final concentration of 2.5 mg/mL. First, 5 mg of DOTAP was dissolved in 10 mL of chloroform solution in an eggplant-shaped flask. The solution was rotated and evaporated in a vacuum at 45 °C for 30 min to generate a uniform lipid film. Next, 2 mL of PBS was added to resuspend the lipid film in a water bath at 50 °C for 2 h. The mixture was extruded through 200 nm, 100 nm, and 50 nm polycarbonate membranes to obtain LNPs.
Lipid Nanoparticle Stability
LNPs were stored at 4 °C for two weeks, and the particle size was measured each day. The storage stability was determined by changing the particle size.
RNA Lipid Nanoparticle Preparation
A certain volume of RNA and LNPs were mixed and incubated at room temperature for 15–20 min to obtain the RNA-DOTAP complex (the mass ratio of RNA to LNPs was 1:15). In vitro, 200 μM RNA was added to every 1 × 105 cells in PBS buffer. In vivo, 25 μg RNA was administered to each mouse; 200 μL of RNA LNPs was injected into C57Bl/6 mice through the tail vein. In both cases, the mixture was stored at room temperature (15−20 min) to facilitate complex formation.
RNA Lipid Nanoparticle Gel Retardation Assay
A certain volume of DL2000 marker, RNA, LNPs, RNA LNPs with 5% TritonX-100, and RNA LNPs with DEPC water were made to a final volume of 20 μL with 1 × loading buffer, and quickly added to 1% agarose gel. Gel electrophoresis was carried out at 120 V for 20 min, and the gel was imaged.
Dendritic Cells and T Cells
Bone marrow was harvested from naive C57BL/6 mice, mature DCs were then generated by RNA LNPs and 1 μg/mL LPS and activated ex vivo based on previously published methods.28 DCs were cultured in RPMI1640 medium supplemented with 100 μg/mL GM-CSF and 100 μg/mL IL-4. T cells were acquired from the spleens of C57BL/6 mice. CD8+ T cells were isolated using a CD8 immunomagnetic bead kit, cultured in RPMI1640 medium buffer with (100X) MEM NEAA, (1000X) HEPES, and (100 mM) sodium pyruvate and expanded and activated ex vivo based on previously published methods.28
Detecting the Cytotoxicity of RNA Lipid Nanoparticles on Dendritic Cells
Cytotoxic effects of RNA LNPs against DCs were measured using CCK-8 assays. Briefly, immature DCs on the fifth day were seeded at a density of 8 × 104 cells/well in 96-well plates and incubated for 24 h or 48 h. The medium was then replaced with fresh medium containing 100 μL of RNA LNPs at varying concentrations (0.2 μM, 2 μM, 20 μM, 200 μM) and incubated for 24 h or 48 h. Next, 20 μL of CCK-8 solution was added to each well to evaluate cytotoxicity. After incubation for approximately 4 h, the absorbance of each well was read at 450 nm using a microplate reader (Thermo Scientific, USA).
Flow Cytometric Analysis
The maturity of DCs was analyzed by flow cytometry (BD Canto10C, USA). Briefly, RNA-DCs and RNA LNP-DCs were harvested from immature DCs (iDCs) within 48 h of incubation with RNA and RNA LNPs and washed and centrifuged twice with PBS. Next, 1 μg of fluorescent antibodies (CD40, CD86, MHC II, CD80, CD11c) was added to samples for surface expression and incubated in the dark for 30 min at room temperature. Finally, 500 μL PBS was added to resuspend the cells for flow cytometric analysis.
T Cell Proliferation Analysis
T cell proliferation was determined using the CCK-8 assay following the manufacturer’s instructions. Briefly, RNA-DCs, RNA LNP-DCs, and mature DCs (mDCs) were cultured with CD8+ T lymphocytes at a ratio of 10:1 for 7 days. Subsequently, 10 μL of CCK-8 solution was added to each well to evaluate cytotoxicity. After incubation for 4 h, the absorbance was measured at 450 nm using a microplate reader (Thermo Scientific, USA).
T Cell Cytotoxicity Analysis
T cell cytotoxicity was evaluated using the Cyto Tox 96 Non-Radioactive Cytotoxicity Assay Kit according to the manufacturer’s instructions. Briefly, Hepa1-6 cells at a density of 1 × 104 cells/well were seeded in 48-well plates. T cells were then divided into three groups: i) T cells stimulated by mDC alone (DC-CTLs); ii) T cells stimulated by RNA-DC-CTLs; iii) T cells stimulated by RNA LNP-DC-CTLs. The T cells were then cultured with Hepa1-6 cells at various ratios (100:1, 40:1, 10:1) for 4 h. Finally, the absorbance was measured at 490 nm using a microplate reader (Thermo Scientific, USA). The following formula was used to calculate cell viability: ([A – B]/[C – B]) × 100%. Where A, B, and C are defined as the absorbance of experimental groups, total natural release groups, and largest release groups, respectively.
Tumor Implantation
Tumor cells were harvested with trypsin and suspended in serum-containing media before being washed in PBS. For subcutaneous tumors, cell pellets were resuspended in the appropriate PBS concentration, 4 × 106 cells per mouse. Tumors were injected subcutaneously into the lateral back of C57BL/6 mice.
In vivo Anti-Tumor Assays
The preventive and therapeutic effects of RNA LNPs in vivo were evaluated in mice bearing subcutaneous tumors derived from liver cancer cells. To assess the preventive effects of vaccines, mice were first injected with tumor vaccines, followed by tumor cells. The C57BL/6 mice (male, 4-week-old, approximately 13 g) were randomly assigned to two groups (n = 6 mice per group). Briefly, mice were injected intravenously with 200 μL of PBS or RNA LNPs on days 0, 3, and 6, and inoculated subcutaneously in the right back with 4 × 106 Hepa1-6 cells on day 10. Finally, the tumors were harvested on day 31. In addition, to determine the therapeutic effects of the vaccines, mice were first injected with tumor cells, followed by tumor vaccines. The C57BL/6 mice (male, 4-week-old, approximately 13 g) were randomly assigned to two groups (n = 6 mice per group). Briefly, mice were inoculated subcutaneously in the right back with 4 × 106 Hepa1-6 cells on day 0 and injected intravenously with 200 μL of PBS or RNA LNPs on days 3, 6, and 10. Finally, the tumors were harvested on day 21. Tumor volumes were evaluated using calipers to assess the maximum longitudinal (length) and transverse (width) diameters and calculated by the modified ellipsoidal formula [0.5 (length × width2)]. The body weight of the mice was monitored every 3 days.
Statistical Analysis
All data were expressed as the mean value of standard deviations, and statistical analysis was performed using SPSS 25.0 software. Student’s unpaired t-tests and one-way analysis of variance were used to detect statistical differences between groups. A p < 0.05 was considered statistically significant.
Results
Characterization of Lipid Nanoparticles
To characterize the formulation, we performed dynamic light scattering on LNPs and RNA LNPs. The average particle size of the LNPs was 102.22 ± 4.05 nm and the zeta potential was 29.97± 0.61 mV (Figure 1A and C). Meanwhile, when total tumor-derived RNA was encapsulated, the size increased to 209.68 ± 6.14 nm, and the zeta potential was 42.03 ± 0.42 mV (Figure 1B and D). The stability of the LNPs was also investigated, however, their size was unchanged over two weeks and was maintained at approximately 100 nm (Figure 1E).
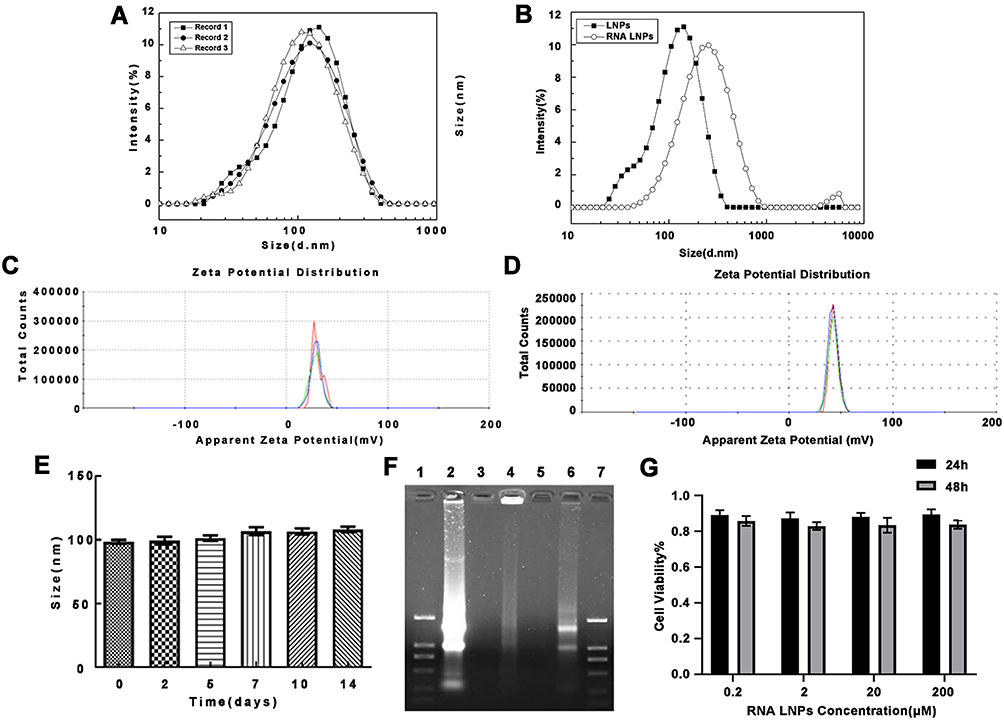 |
Figure 1 Characterization of lipid nanoparticles and RNA lipid nanoparticles. (A) Size distribution of LNPs and (B) RNA LNPs. (C) Zeta potential of LNPs and (D) RNA LNPs. (E) Stability of LNPs. (F) Gel retardation assay demonstrating the complexation efficiency of RNA by LNPs. Lane 1: DL2000 Marker, Lane 2: free RNA control, Lane 3: LNPs, Lane 4: RNA LNPs, Lane 5: blank, Lane 6: RNA LNPs with 5% Triton X-100, Lane 7: DL200 Marker. DL2000 Marker bands from top to bottom were 2000, 1000, 750, 500, 250, 100 bp. (G) Cytotoxicity of RNA LNPs against DCs at 24 and 48 h. Data are presented as means ± standard deviations (n = 3). |
Next, the gel retardation assay was used to evaluate the binding ability of LNPs to RNA. Results show that RNA was trapped within the gel well, however, became released after RNA LNPs were destroyed by 5% TritonX-100, resulting in the generation of a bright band in the gel image (Figure 1F). Hence, LNPs exhibited a strong binding capacity for RNA.
To evaluate the safety of RNA LNPs, the CCK-8 assay was used to determine the effects of various concentrations of RNA LNPs on the viability of DCs at 24 and 48 h. Results demonstrate that RNA NPs had no obvious cytotoxicity at the indicated concentrations, demonstrating that RNA LNPs were safe at concentrations below 200 μM (Figure 1G).
RNA Lipid Nanoparticles Promote Dendritic Cell Maturation in vitro
The expression of CD40, CD80, CD86, CD11c, and MHCII molecules on DCs was determined by flow cytometry. Both RNA-DCs and RNA LNP-DCs were found to have significantly increased expression of surface molecules compared to iDCs, which was similar to that of mDCs (Figure 2). This demonstrated that RNA LNPs could efficiently promote DC maturation in vitro.
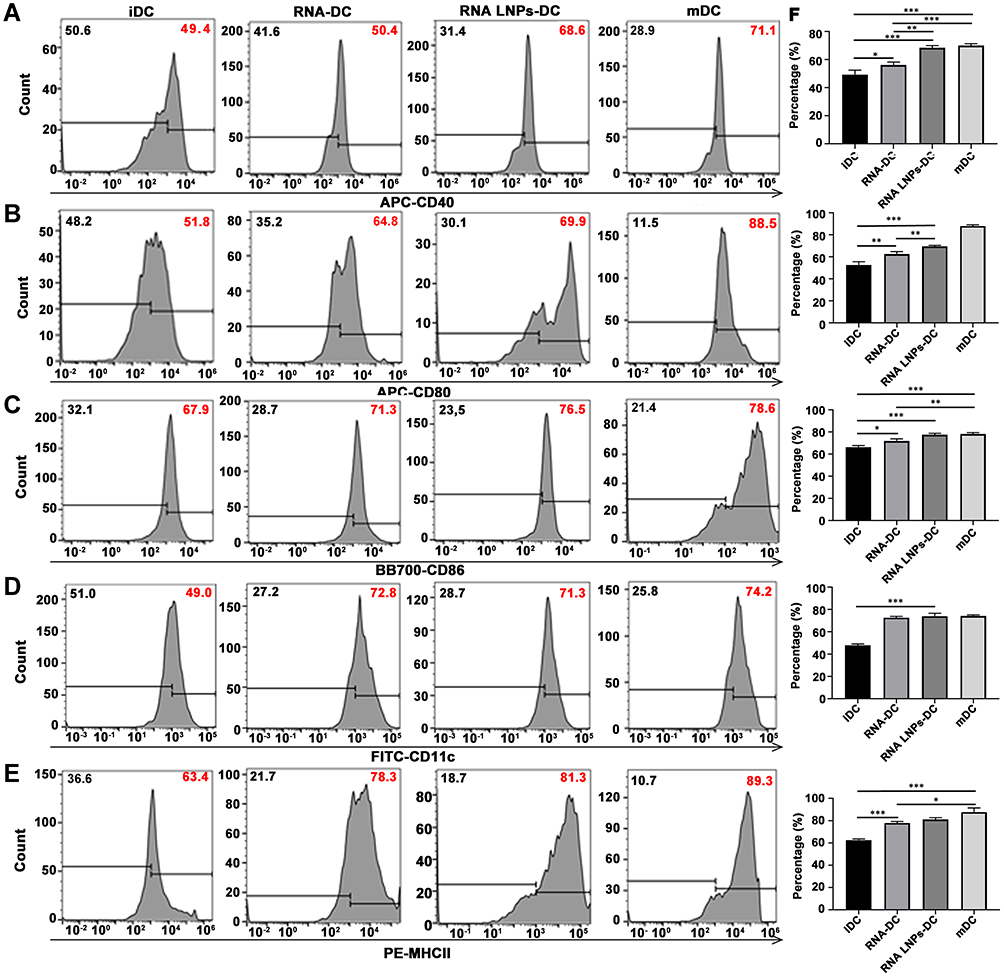 |
Figure 2 RNA lipid nanoparticles promote dendritic cell maturation in vitro. Surface protein expression of (A) CD40, (B) CD80, (C) CD86, (D) CD11c, and (E) MHCII, as measured by flow cytometry. (F) Percentage of positive cells. Data are presented as means ± standard deviations (n = 3). *p < 0.05; **p < 0.01; ***p < 0.001. Abbreviations: iDC, immature dendritic cell; mDC, mature dendritic cell. |
RNA Lipid Nanoparticles Induce Dendritic Cell and T Cell Activation in vitro
Since CD8+ T lymphocytes have a strong ability to specifically inhibit tumor growth, obtaining higher purity CD8+ T cells can facilitate more efficient killing of tumor cells. First, we isolated CD8+ T cells by magnetic-activated cell sorting (MACS), the purity of which was determined to be 81.4%, which was significantly improved compared to the 19.3% before isolation (Figure 3A and B). Next, RNA-DCs, RNA LNP-DCs, and mDCs were cultured with CD8+ T lymphocytes at a ratio of 10: 1 for 7 days. T cell proliferation stimulated by DCs was measured using the CCK-8 assay. Results show that RNA-DCs, RNA LNP-DCs, mDCs, and iDCs stimulated CD8+ T lymphocytes to expand by 51.8%, 51.3%, 50.2%, and 50.1%, respectively (Figure 3C). Meanwhile, the cytotoxicity of RNA LNP-DC-CTLs, RNA-DC-CTLs, and DC-CTLs targeting Hepa1-6 cells was evaluated by Cyto Tox 96 Non-Radioactive Cytotoxicity Assay. The results showed that RNA LNP-DC-CTLs were the most efficient against Hepa1-6 cells, followed by RNA-DC-CTLs and DC-CTLs (Figure 3D). This suggests that RNA LNP vaccines could induce specific CTLs against Hepa1-6 cells.
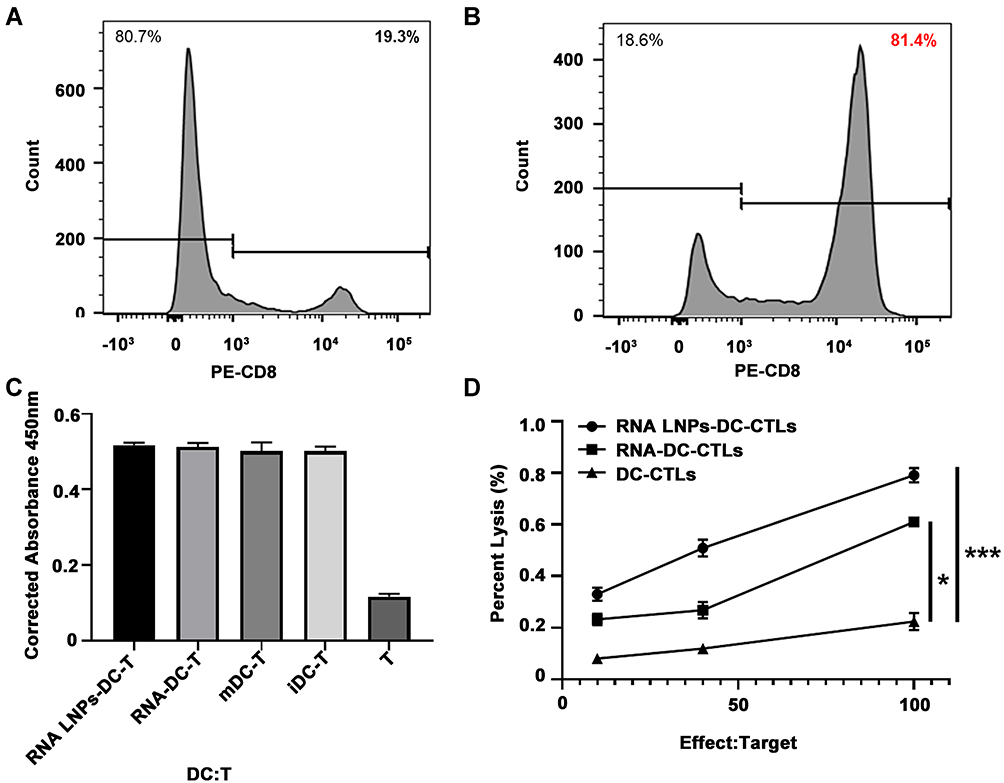 |
Figure 3 RNA lipid nanoparticles induce dendritic cell and T cell activation in vitro. Positive CD8 T lymphocyte cells were isolated and cultured. Purity of positive CD8 T lymphocytes before (A) and after (B) isolation, as determined by flow cytometry. (C) Proliferation assay for T cells activated by RNA LNPs-DC, RNA-DC, mDC, or iDC at a ratio of 10:1 for seven days. (D) Cytotoxicity assay for effector T cells activated by RNA LNPs-DC, RNA-DCs, or DCs against Hepa1-6 target cells at different effect to target ratios. Data are presented as means ± standard deviations (n = 3). *p < 0.05; ***p < 0.001. |
Preventive and Therapeutic Effects of Tumor Vaccines on Hepatocellular Carcinoma
To determine the preventive effects of RNA LNPs on HCC, C57BL/6 mice were immunized intravenously with RNA LNPs three times at 3 day intervals, followed by subcutaneous challenge with 4 × 106 Hepa1-6 cells; tumors were harvested on day 31 (Figure 4A). Compared with the PBS treatment group, significantly delayed tumor growth was observed in RNA LNP-treated mice, and no significant difference was noted in the body weight of the mice (Figure 4B–D).
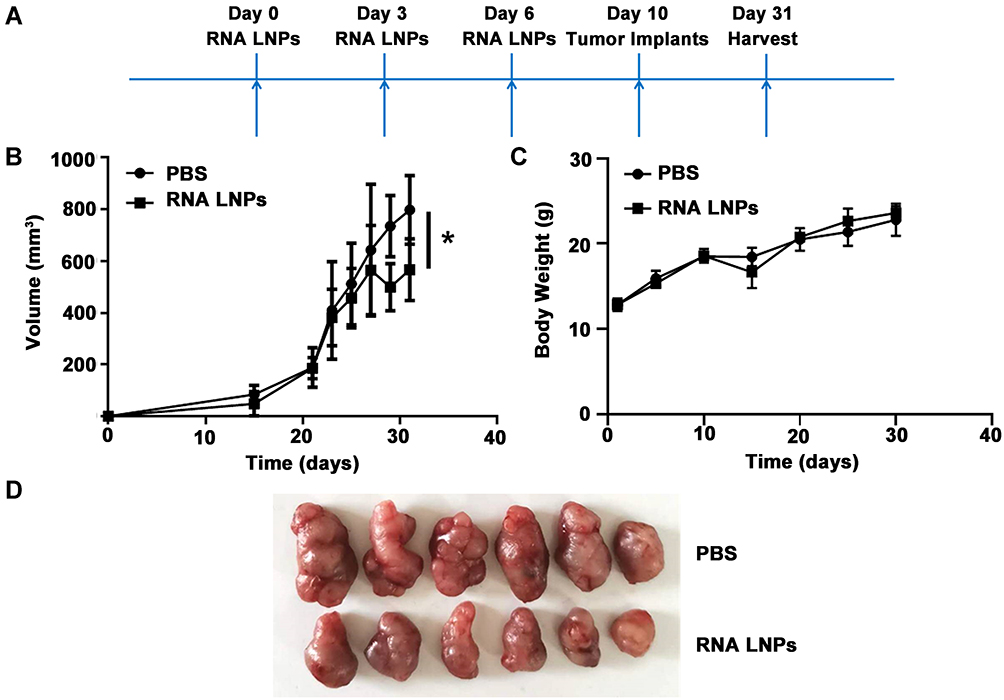 |
Figure 4 Preventive effects of tumor vaccines on hepatocellular carcinoma. (A) A total of 200 μL RNA LNPs were injected on days 0, 3, 6 via the tail vein. Then 4 × 106 Hepa1-6 cells were implanted in the flanks of C57BL/6 mice on day 10. Finally, tumor cells were harvested on day 31. (B) Tumor growth curves for mice treated with PBS and RNA LNPs. (C) Body weight measurements for study duration. (D) Images of excised tumors at the end point. Data are presented as means ± standard deviations (n = 6). *p < 0.05. |
Next, the anti-tumor activity of RNA LNPs was investigated in a Hepa1-6 tumor-bearing mouse model. C57BL/6 mice were implanted subcutaneously with Hepa1-6 tumors, followed by tail vein injection of RNA LNPs three times at 3 day intervals. Tumors were harvested from surviving animals on day 21 (Figure 5A). The results were similar to the preventive effect with RNA LNPs exhibiting a stronger inhibitory effect on tumor growth than PBS (Figure 5B–D). These results demonstrate that RNA LNPs can effectively prevent and inhibit the growth of Hepa1-6 tumors in vivo.
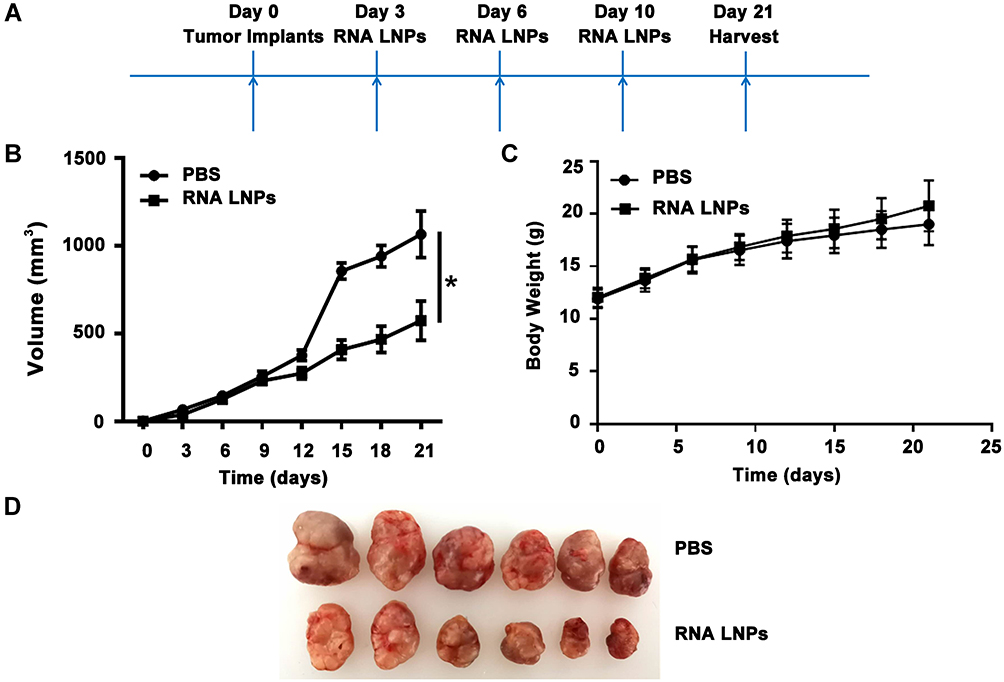 |
Figure 5 Treatment effects of tumor vaccines against hepatocellular carcinoma. (A) 4 × 106 Hepa1-6 cells were implanted in the flanks of C57BL/6 mice on day 0. Then 200 μL of RNA LNPs were injected on days 3, 6, and 10 via the tail vein. Finally, tumor cells were harvested on day 21. (B) Tumor growth curves for mice treated with PBS and RNA LNPs. (C) Body weight measurements for study duration. (D) Images of excised tumors at the end point. Data are presented as means ± standard deviations (n = 6). *p < 0.05. |
Discussion
Liver cancer is a malignant tumor with a poor prognosis and high incidence. To date, its therapeutic strategies have primarily focused on local treatment with no effective systemic treatment. Since the 20th century, methods have been developed that exploit the specificity of the immune system to eliminate tumors. Tumor immunity is based on immunological principles to improve the immunogenicity of tumor cells and the activity of effector cells, stimulate and enhance the body’s anti-tumor immune response, and cooperate with the body’s immune system to inhibit tumor growth.29–32 Over the past few decades, several effective strategies have been developed. Among them, tumor vaccines have been widely used in clinical studies and are a promising approach for cancer immunotherapy. However, many tumor patients are insensitive to these vaccines due to their weak immunogenicity as well as the associated systemic and intratumoral immunosuppression. In this study, total tumor-derived RNA was extracted from liver cancer cells and encapsulated to construct a liver cancer cell RNA LNP vaccine with DC-targeting ability. The RNA LNP vaccine can translate and synthesize tumor antigens in DCs, induce high-efficiency anti-HCC specific immune responses, and inhibit the growth of HCC.
The anti-tumor effect of tumor vaccines depends on their effective presentation of tumor antigens to sensitize and induce specific CTLs. Hence, tumor vaccines based on predetermined tumor antigen peptides or proteins have an inherent risk of not presenting effective antigens, resulting in immunosuppression and potential immune evasion. In addition, the disadvantage of this strategy is that it requires expensive and laborious procedures to identify tumor-specific antigens. To overcome these limitations, tumor vaccines based on antigens derived from whole tumor cells have been developed.33–35 As confirmed by the results of a large number of clinical trials, this type of vaccine has shown broad prospects in cancer treatment. One promising resource for whole tumor antigens is total tumor-derived RNA, which was applied in the current study. Herein, we isolated total RNA from Hepa1-6 cells using TRIzol reagent to promote the potency and durability of anti-tumor immunity.
Nucleic acid vaccines also have the advantages of easy production and operation, stable product efficacy, and good safety. Meanwhile, RNA offers several advantages over DNA. First, RNA must only be delivered to the cytoplasm, not the nucleus, which provides fewer challenges to the development of drug delivery vehicles. Second, RNA does not integrate into the genome and, therefore, is does not carry a risk of mutagenesis. Third, RNA is readily produced in large quantities and with high purity through in vitro transcription. Fourth, RNA is easily degraded in serum, thereby reducing potential safety issues. However, low serum stability also constitutes an obstacle to the development of RNA nanomedicine, thus, a carrier that can adequately protect RNA from premature degradation is required. In addition, the efficacy of RNA must be improved to account for sufficient protein production.
DOTAP is one of the earliest developed liposomes and has been well studied with a promising safety profile.36 We have previously developed DOTAP LPDs for siRNA delivery with superior stability and targeting. Combining this RNA delivery carrier with DCs, the most potent APCs in the body that have been used in many tumor vaccine immunotherapy trials, is considered to be an effective approach to induce potentially therapeutic immune responses. Moreover, compared with free drugs, nanocarriers are more readily recognized and phagocytosed by DCs. Therefore, we encapsulated RNA in LNPs, which not only improved the stability and transfection efficiency of RNA, but also promoted the phagocytosis of DCs and anti-tumor immunity.
DCs can process and present tumor antigens and induce CTLs with specific killing ability, thereby inhibiting tumor growth.37,38 At present, RNA-pulsed DC vaccines have been developed, which have shown promise in clinical trials of tumor patients, however, are limited by their costly and tedious preparation process and short shelf-life. Alternatively, RNA LNPs can be produced within a few days after the patient undergoes a biopsy, thereby shortening the time required to generate a personalized vaccine. Moreover, nanoliposomes (such as DOTAP) have been used in several clinical trials to enhance the delivery of drugs, plasmids, and oligonucleotides, which can be easily made into clinical-grade preparations with favorable safety profiles and can be engineered to upregulate innate and adaptive host immunity. Therefore, RNA LNP vaccines have promising application prospects in the clinical application of tumor immunotherapy.
Conclusion
Following administration, tumor RNA LNPs become ingested by DCs, resulting in DC maturation and translation of tumor antigens. Subsequently, DCs migrate to peripheral lymphoid organs, where they activate both tumor antigen specific CD8+ T cells (CTL cells) through the endogenous antigen processing and presentation pathway, and activated CD4+ T cells (Th cells), which can provide a co-stimulatory signal for CTL activation through the cross-presentation pathway. Subsequently, effector T cells home from peripheral lymphoid organs to the tumor site through lymphocyte recirculation and trigger a specific anti-tumor immune response (Figure 6). This indicates the role of tumor RNA LNPs as potent anti-tumor vaccines.
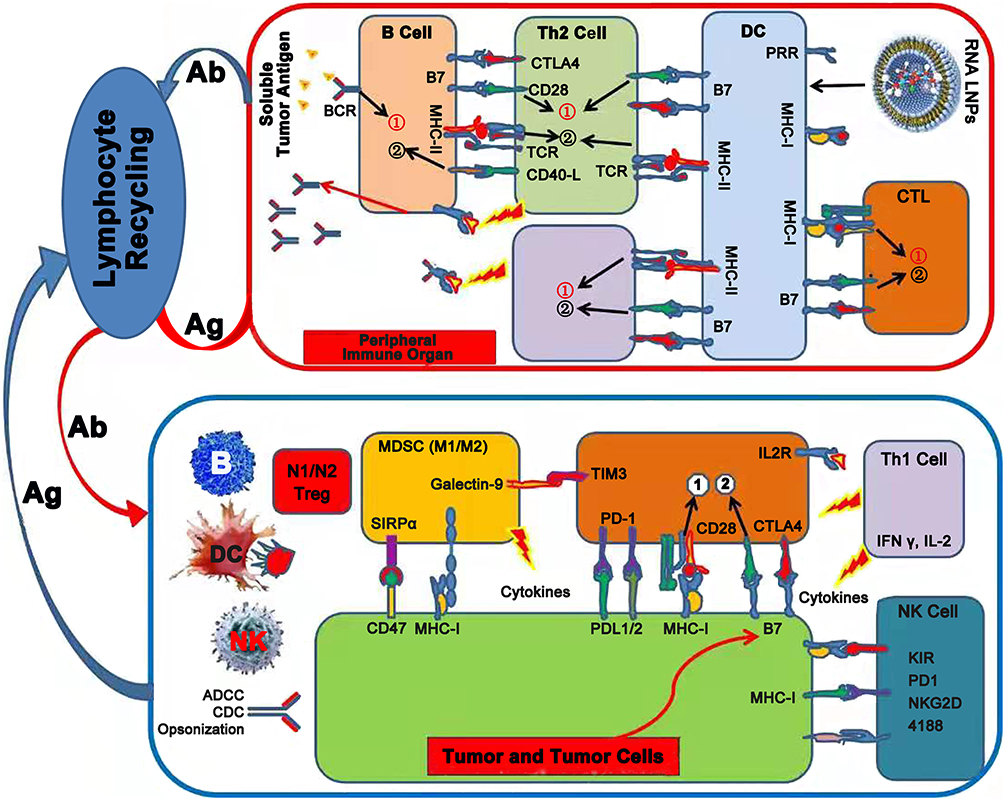 |
Figure 6 Tumor vaccine and tumor immunotherapy. Total tumor RNA was extracted and mixed with an immune adjuvant to formulate tumor vaccine. Tumor antigen was expressed and presented or cross-presented to Th and Tc cells by antigen presenting cells (DCs) in lymph node to generate specific anti-tumor response. Abbreviations: Ab, antibody; Ag, antigen; BCR, B cell receptor; TCR, T-cell receptor; MHC, major histocompatibility complex; PRR, pattern recognition receptors; Th1/2, T helper type 1/2; CTL, cytotoxic T-lymphocyte; CTLA4, cytotoxic T-lymphocyte-associated protein 4; Treg, regulatory T cell; DC, dendritic cell; NK, natural killer cell; MDSC, myeloid-derived suppressor cell; ADCC, antibody-dependent cellular cytotoxicity; CDC, complement-dependent cytotoxicity; SIRPα, signal regulatory protein α; LILRB1, leukocyte immunoglobulin like receptor B1; TIM-3, T-cell immunoglobulin and mucin domain-3; PD1, programmed cell death protein 1; PD-L1, programmed death-ligand 1; IL-2, Interleukin-2; IFNγ, Interferon γ; KIR, killer cell immunoglobulin-like receptor; NKG2D, natural killer group 2 member D. |
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant numbers 82072051, 81771964, 81803450, 81402512); the open project of Shandong Collaborative Innovation Center for Antibody Drugs (grant number CIC-AD1817); Youth Innovative Science and Technology Program of Shandong Colleges and University (grant number 2019KJM012); the special project of clinical research of health industry of Shanghai Municipal Health Commission (grant number 201940178); the scientific research project of Hongkou District Health Committee of Shanghai (grant number 2002-17); and by the research project of Shanghai Fourth People’s Hospital (grant numbers 0443 and 0446).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Chen S, Cao Q, Wen W, Wang H. Targeted therapy for hepatocellular carcinoma: challenges and opportunities. Cancer Lett. 2019;460:1–9. doi:10.1016/j.canlet.2019.114428
2. Busato D, Mossenta M, Baboci L, Di Cintio F, Toffoli G, Dal BM. Novel immunotherapeutic approaches for hepatocellular carcinoma treatment. Expert Rev Clin Pharmacol. 2019;12:453–470. doi:10.1080/17512433.2019.1598859
3. Iñarrairaegui M, Melero I, Sangro B. Immunotherapy of hepatocellular carcinoma: facts and hopes. Clin Cancer Res. 2018;24:1518–1524. doi:10.1158/1078-0432.CCR-17-0289
4. Yang Y. Cancer immunotherapy: harnessing the immune system to battle cancer. J Clin Invest. 2015;125:3335–3337. doi:10.1172/JCI83871
5. Thomas S, Prendergast GC. Cancer vaccines: a brief overview. Methods Mol Biol. 2016;1403:755–761.
6. Aurisicchio L, Pallocca M, Ciliberto G, Palombo F. The perfect personalized cancer therapy: cancer vaccines against neoantigens. J Exp Clin Cancer Res. 2018;37:86. doi:10.1186/s13046-018-0751-1
7. Hu Z, Ott PA, Wu CJ. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat Rev Immunol. 2018;18:168–182. doi:10.1038/nri.2017.131
8. Van der Burg SH, Arens R, Ossendorp F, van Hall T, Melief CJ. Vaccines for established cancer: overcoming the challenges posed by immune evasion. Nat Rev Cancer. 2016;16:219–233.
9. Ulmer JB, Mason PW, Geall A, Mandl CW. RNA-based vaccines. Vaccine. 2012;30(30):4414–4418. doi:10.1016/j.vaccine.2012.04.060
10. McNamara MA, Nair SK, Holl EK. RNA-based vaccines in cancer immunotherapy. J Immunol Res. 2015;2015:794528. doi:10.1155/2015/794528
11. Sayour EJ, Grippin A, De Leon G, et al. Personalized tumor RNA loaded lipid-nanoparticles prime the systemic and intratumoral milieu for response to cancer immunotherapy. Nano Lett. 2018;18:6195–6206. doi:10.1021/acs.nanolett.8b02179
12. Sahin U, Türeci Ö. Personalized vaccines for cancer immunotherapy. Science. 2018;359:1355–1360. doi:10.1126/science.aar7112
13. Stepanenko AA, Chekhonin VP. Recent advances in oncolytic virotherapy and immunotherapy for glioblastoma: a glimmer of hope in the search for an effective therapy? Cancers (Basel). 2018;10:492. doi:10.3390/cancers10120492
14. Mu LJ, Kyte JA, Kvalheim G, et al. Immunotherapy with allotumour mRNA-transfected dendritic cells in androgen-resistant prostate cancer patients. Br J Cancer. 2005;93:749–756. doi:10.1038/sj.bjc.6602761
15. Sayour EJ, De Leon G, Pham C, et al. Systemic activation of antigen-presenting cells via RNA-loaded nanoparticles. Oncoimmunology. 2016;6:e1256527. doi:10.1080/2162402X.2016.1256527
16. Markov V, Mironova NL, Shmendel EV, Maslov MA, Zenkova MA. Systemic delivery of complexes of melanoma RNA with mannosylated liposomes activates highly efficient murine melanoma-specific cytotoxic T cells in vivo. Mol Biol. 2017;51:118–125. doi:10.1134/S0026893317010137
17. Lee TJ, Haque F, Shu D, et al. RNA nanoparticle as a vector for targeted siRNA delivery into glioblastoma mouse model. Oncotarget. 2015;6:14766–14776. doi:10.18632/oncotarget.3632
18. Grabbe S, Haas H, Diken M, Kranz LM, Langguth P, Sahin U. Translating nanoparticulate-personalized cancer vaccines into clinical applications: case study with RNA-lipoplexes for the treatment of melanoma. Nanomedicine (Lond). 2016;11:2723–2734. doi:10.2217/nnm-2016-0275
19. Shao K, Singha S, Clemente-Casares X, Tsai S, Yang Y, Santamaria P. Nanoparticle-based immunotherapy for cancer. ACS Nano. 2015;9:16–30. doi:10.1021/nn5062029
20. Di Gioacchino M, Petrarca C, Gatta A, et al. Nanoparticle-based immunotherapy: state of the art and future perspectives. Expert Rev Clin Immunol. 2020;16:513–525. doi:10.1080/1744666X.2020.1762572
21. Liu J, Zhang R, Xu ZP. Nanoparticle-based nanomedicines to promote cancer immunotherapy: recent advances and future directions. Small. 2019;15:e1900262. doi:10.1002/smll.201900262
22. Kranz LM, Diken M, Haas H, et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature. 2016;534:396–401. doi:10.1038/nature18300
23. Sayour EJ, Mendez-Gomez HR, Mitchell DA. Cancer vaccine immunotherapy with RNA-loaded liposomes. Int J Mol Sci. 2018;19:2890. doi:10.3390/ijms19102890
24. Gao J, Liu W, Xia Y, et al. The promotion of siRNA delivery to breast cancer overexpressing epidermal growth factor receptor through anti-EGFR antibody conjugation by immunoliposomes. Biomaterials. 2011;32:3459–3470. doi:10.1016/j.biomaterials.2011.01.034
25. Gao J, Yu Y, Zhang Y, et al. EGFR-specific PEGylated immunoliposomes for active siRNA delivery in hepatocellular carcinoma. Biomaterials. 2012;33:270–282. doi:10.1016/j.biomaterials.2011.09.035
26. Gao J, Chen H, Yu Y, et al. Inhibition of hepatocellular carcinoma growth using immunoliposomes for co-delivery of adriamycin and ribonucleotide reductase M2 siRNA. Biomaterials. 2013;34:10084–10098. doi:10.1016/j.biomaterials.2013.08.088
27. Song W, Shen L, Wang Y, et al. Synergistic and low adverse effect cancer immunotherapy by immunogenic chemotherapy and locally expressed PD-L1 trap. Nat Commun. 2018;9:2237. doi:10.1038/s41467-018-04605-x
28. Flores C, Pham C, Snyder D, et al. Novel role of hematopoietic stem cells in immunologic rejection of malignant gliomas. Oncoimmunology. 2015;4:e994374. doi:10.4161/2162402X.2014.994374
29. Huang TX, Fu L. The immune landscape of esophageal cancer. Cancer Commun (Lond). 2019;39:79. doi:10.1186/s40880-019-0427-z
30. Liu Y, Wang H, Li D, et al. In situ formation of peptidic nanofibers can fundamentally optimize the quality of immune responses against HIV vaccine. Nanoscale Horiz. 2016;1:135–143. doi:10.1039/C5NH00064E
31. Wang H, Ding Y, Su S, et al. Assembly of hepatitis E vaccine by ‘in situ’ growth of gold clusters as nano-adjuvants: an efficient way to enhance the immune responses of vaccination. Nanoscale Horiz. 2016;1:394–398. doi:10.1039/C6NH00087H
32. Zhang D, Zhang J, Li Q, Song A, Li Z, Luan Y. Cold to hot: rational design of a minimalist multifunctional photo-immunotherapy nanoplatform toward boosting immunotherapy capability. ACS Appl Mater Interfaces. 2019;11:32633–32646. doi:10.1021/acsami.9b09568
33. Chiang CL, Coukos G, Kandalaft LE. Whole tumor antigen vaccines: where are we? Vaccines (Basel). 2015;3:344–372. doi:10.3390/vaccines3020344
34. Yang R, Xu J, Xu L, et al. Cancer cell membrane-coated adjuvant nanoparticles with mannose modification for effective anticancer vaccination. ACS Nano. 2018;12:5121–5129. doi:10.1021/acsnano.7b09041
35. Fan Y, Kuai R, Xu Y, Ochyl LJ, Irvine DJ, Moon JJ. Immunogenic cell death amplified by co-localized adjuvant delivery for cancer immunotherapy. Nano Lett. 2017;17:7387–7393. doi:10.1021/acs.nanolett.7b03218
36. Simberg D, Weisman S, Talmon Y, Barenholz Y. DOTAP (and other cationic lipids): chemistry, biophysics, and transfection. Crit Rev Ther Drug Carrier Syst. 2004;21:257–317. doi:10.1615/CritRevTherDrugCarrierSyst.v21.i4.10
37. Sabado RL, Balan S, Bhardwaj N. Dendritic cell-based immunotherapy. Cell Res. 2017;27:74–95.
38. Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. 2020;20:7–24. doi:10.1038/s41577-019-0210-z
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.