")
Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 7
Immunologic checkpoints in cancer therapy: focus on the programmed death-1 (PD-1) receptor pathway
Received 10 August 2014
Accepted for publication 15 September 2014
Published 15 November 2014 Volume 2014:7 Pages 357—365
DOI https://doi.org/10.2147/PGPM.S53163
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Parisa Momtaz,1,2 Michael A Postow1,2
1Department of Medicine, Memorial Sloan Kettering Cancer Center, New York, NY, USA; 2Weill Cornell Medical College, New York, NY, USA
Abstract: T-lymphocytes have the potential to recognize cancer antigens as foreign and therefore eliminate them. However, immune checkpoints such as cytotoxic T-lymphocyte-associated antigen (CTLA)-4 and programmed cell death (PD)-1 receptor and its ligands (PD-L1, PD-L2) suppress the activity of T-lymphocytes. Advances in the understanding of immunology and its role in cancer have led to the development of immune checkpoint inhibitors that block CTLA-4 and PD-1 and result in durable responses in patients with a wide range of cancers. PD-1 and PD-L1 inhibitors are currently in many stages of clinical investigation, and the anti-PD-1 antibody, pembrolizumab, was recently approved by the US Food and Drug Administration. Many questions remain to be answered, such as the optimal administration schedule, biomarkers that associate with benefit, and potential for use of PD-1 agents in combination approaches. Nonetheless, immunotherapy with PD-1 blocking antibodies is now becoming an integral part in the management of cancer.
Keyword: immune checkpoints, immunotherapy, programmed cell death protein-1, cytotoxic T-lymphocyte antigen 4
Immunology and cancer
Activation of the immune system is recognized as an important treatment strategy against cancer.1 Cancer is characterized by genetic mutations and alterations in cellular regulatory processes that can lead to the expression of various tumor-related antigens. These antigens can be presented to cytotoxic T-lymphocytes by way of antigen-presenting cells (APCs). One role of T-lymphocytes is to differentiate between “self” and “non-self” antigens and aid in the removal of “non-self” antigens without over-activating the immune system and causing damage to “self” antigens.2 Therefore, T-lymphocytes have the potential to recognize cancer-related antigens as “non-self” and eradicate these cancer cells.3 The immune response against cancer occurs in three stages, known as the three ‘E’s: elimination, equilibrium, and escape.4,5 The goal is elimination of cancer cells by the immune system; however, when complete elimination cannot be attained, persistent activation of the immune system can maintain a state of equilibrium. Escape is the state of cancer growth, when a tumor cell has been successful in evading immune destruction.
A series of steps must be carried out in order for an effective immune response against cancer to occur. First, the tumor-related antigen must be picked up and processed by the APC. APCs include macrophages, B-lymphocytes, and dendritic cells. Of these, dendritic cells are the most effective, as their dendrites increase their ability to capture antigens for presentation.6 APCs travel to the lymph node to present the processed antigen bound to major histocompatibility complex (MHC) molecules to the T-lymphocytes. To activate and prime the T-cell for its effector phase (ie, to respond against the cancer-related antigens), two signals need to occur. The first signal is the binding of the T-cell receptor (TCR) to the MHC-bound antigen. Simultaneously, a second signal involving the interaction between co-stimulatory molecules, such as B7 on activated APCs and cluster of differentiation (CD)-28 expressed on T-lymphocytes, occurs, and the combination of these two signals allow for the proliferation and activation of T-lymphocytes.
With this immunologic framework in mind, historically, several immunotherapeutic agents have been used in cancer therapy, including cancer vaccines and cytokines such as interferon-alpha and interleukin (IL)-2. Though there have been some successes in cancer vaccine development such as the FDA approval of sipuleucel-T,7 generally the efficacy of therapeutic cancer vaccines is felt to be modest.8 Interferon-alpha and IL-2 have also shown modest benefit. Interferon-alpha is the only FDA-approved agent for the adjuvant treatment of melanoma. This approval was based on initial data that showed an improvement in relapse-free survival (RFS) and overall survival (OS); however, subsequent studies confirmed an improvement in RFS but not necessarily in OS.9,10 IL-2 is approved for treatment of metastatic melanoma. Response rates to IL-2 range from 16% to 23%, and 5%–10% of patients achieve profound, durable benefits. Nevertheless, both interferon-alpha and IL-2 are associated with toxicity, and better therapies that help more patients are needed.11,12
It is possible that prior immunotherapeutic approaches such as vaccines and cytokines had only limited success due to the high level of immunosuppressive networks involved in patients with cancer that overpowered these prior attempts at enhancing anti-tumor immunity. A greater understanding of immune activation, particularly T-lymphocyte activation, has now identified multiple co-stimulatory and co-inhibitory pathways that regulate this process.6 One important mechanism relevant to immunotherapy are the co-inhibitory receptors such as cytotoxic T-lymphocyte-associated antigen (CTLA)-4 and programmed cell death 1 (PD-1) receptor.6 These co-inhibitory molecules serve to dampen the immune response to maintain immunologic homeostasis. During antigen presentation, T-lymphocytes are also affected by the microenvironment, thus the immune response generated is a result of several factors, including stimulation and inhibition in the context of the microenvironment.3,6
Cancers have many ways of evading and thus escaping an otherwise effective immune response. Tumor-related antigens may not be properly presented, antigens may be recognized as “self” and induce anergy, T-lymphocytes may not be appropriately activated, or T-lymphocytes may be excessively inhibited.3
Cytotoxic T-lymphocyte antigen 4 as the prototypical immunologic checkpoint
In order to maintain immune homeostasis and avoid complications from immune over-activation, several mechanisms of negative regulation are put in place. One such mechanism involves immune checkpoints, which include the receptors CTLA-4 and PD-1, receptors that are expressed on the T-lymphocyte surface. Tumor cells are capable of resisting the immune system by expressing ligands such as PD-L1 or PD-L2, which interact with the PD-1 receptor to suppress immunity.3,13
CTLA-4 (also known as CD152) and PD-1 (also known as CD279) were the first two immune checkpoints to be evaluated extensively in the setting of clinical cancer immunotherapy. They differ in the manner and level at which they negatively regulate the immune system (Figure 1).13
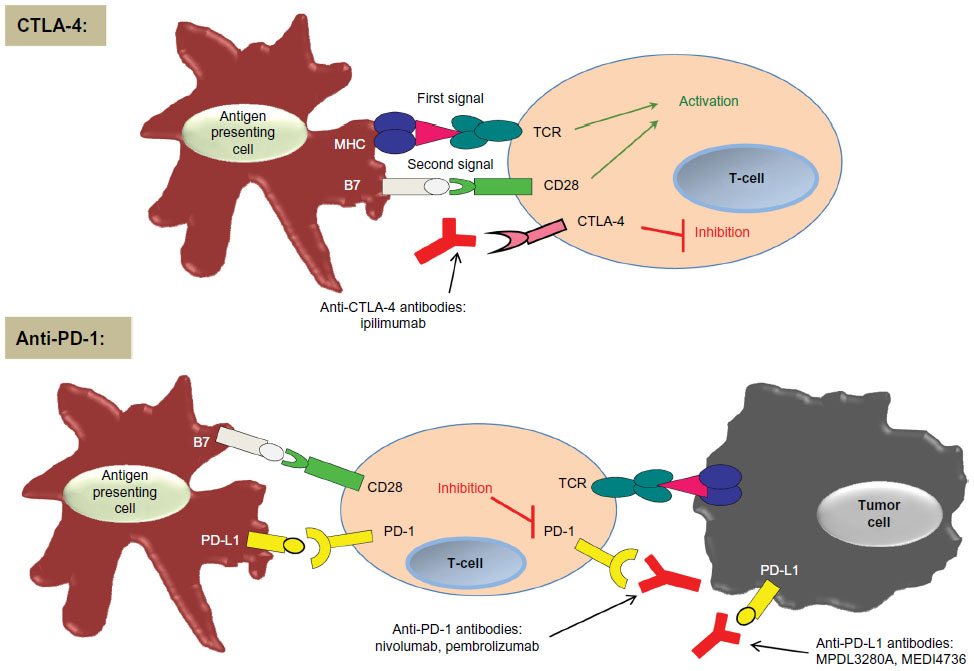 | Figure 1 Simplified concept of CTLA-4 and PD-1 immune checkpoints. |
CTLA-4 was the first immune checkpoint receptor to be targeted by a therapeutic agent. It is expressed only on T-lymphocytes, and it negatively regulates T-lymphocyte activation by competing with the co-stimulatory molecule CD28 in binding the ligands B7.1 (also known as CD80) and B7.2 (also known as CD86). CTLA-4 has a greater affinity for these ligands and is also capable of independently sending inhibitory signals to the T-lymphocyte.14–16 Pre-clinical models of CTLA-4 blockade showed an anti-tumor immune response.17,18
In contrast to CTLA-4, which regulates T-lymphocytes at the level of initial activation, PD-1 regulates immunity at multiple phases of the immune response, including exerting its effect on effector T-lymphocyte activity in the peripheral tissues.
Programmed cell death receptors as another critical immunologic checkpoint
In addition to its activity in cancer immunotherapy, PD-1 has been shown to play a role in allergy, autoimmunity, infectious disease, and transplantation immunity.2 PD-1 is highly expressed on tumor-infiltrating lymphocytes (TILs) in the effector phase and serves to inhibit T-lymphocyte activity during chronic antigen exposure when it is engaged by its ligands. Tumor cells monopolize on this immune-resistance mechanism. In peripheral tissues, tumor cells and other cells in the tumor microenvironment express PD-1 ligands, which are believed to protect the tumor cells from immune destruction (Figure 1).11,19
PD-L1 (also known as B7-H1 or CD274) and PD-L2 (also known as B7-DC or CD273) are the two ligands for PD-1. When one of the PD-1 ligands engages the PD-1 receptor, the ligand/receptor interaction dampens the T-lymphocyte response in several ways. It inhibits the kinases involved in T-lymphocyte activation via phosphatase activity and other signaling pathways.20 Although it predominately regulates T-lymphocyte effector activity distally in the tissue and tumors, data from PD-1-deficient T-lymphocytes suggest that PD-1 likely plays a role at multiple steps in the immune response.21 In addition, PD-1 is expressed on other activated cells, including B-lymphocytes and natural killer (NK) cells.13 Lastly, chronic antigen exposure can lead to persistent PD-1 expression. Enhanced PD-1 expression via chronic antigen exposure can therefore both change the duration of T-lymphocyte/APC interaction and can lead to T-lymphocyte anergy or exhaustion.13,22
As noted above, PD-1 is expressed on TILs from many types of cancers. PD-1 ligands are also expressed on different types of tumors. PD-L1 is most commonly expressed on solid tumors, including melanoma, ovarian, lung, and renal carcinomas.23 PD-L2 has been reported to be upregulated in different types of lymphoma.24 Pre-clinical data have shown that deliberate expression of PD-L1 on mouse tumor cells inhibits anti-tumor T-lymphocyte response.13,23 Yet analysis by immunohistochemistry (IHC) or flow cytometry has shown that PD-L1 level expression on tumor cells is variable and reasons for its heterogeneity are multifactorial.25
Mechanisms that regulate tumor cell PD-L1 expression include the ‘innate immune resistance’ and the ‘adaptive immune resistance’ mechanisms. The innate immune-resistance mechanism describes tumor cell PD-L1 expression that may be related to oncogenic signaling pathways inherent in the tumor cell. This mechanism does not depend on inflammatory signals in the microenvironment. One example of innate immune resistance includes constitutive anaplastic lymphoma kinase (ALK) signaling in lung cancer that has been reported to drive PD-L1 expression through signal transducer and activator of transcription (STAT)-3 signaling.26,27,28
The adaptive immune-resistance mechanism describes tumor cell PD-L1 expression that is induced in response to immune activity within the tumor microenvironment, thereby leading to a non-uniform expression of PD-L1.29 With adaptive immune resistance, the tumor cells take advantage of the PD-1/PD-L1interaction that under normal circumstances protects cells from immune-mediated harm. Inflammatory signals (such as interferons) produced by an active antitumor immune response in the tumor microenvironment can lead to increased expression of PD-L1, which in turn protects the tumor cell by then inhibiting that very same antitumor response.13,25
Immune checkpoint inhibitors
A greater understanding of immune checkpoints led to the development of several immune checkpoint inhibitors. Ipilimumab (trade name Yervoy®), an anti-CTLA-4 monoclonal antibody, was the first agent to demonstrate a survival benefit in patients with metastatic cutaneous melanoma and gained FDA approval in 2011.
Clinical trials with ipilimumab showed a modest response rate of 10%–15% at the expense of ipilimumab immune-related adverse events (most commonly colitis and dermatitis). However two Phase III trials showed that ipilimumab improves OS. The median OS rate for patients treated with ipilimumab was prolonged compared with patients receiving a gp100 cancer vaccine (median OS 10.9 versus 6.4 months), and patients treated with ipilimumab and dacarbazine chemotherapy had improved OS compared with those treated with dacarbazine alone (median OS 11.2 versus 9.1 months).30,31
Patients who achieved a response were often found to have a durable response, lasting 1.5–2 years, and 18% of patients treated with ipilimumab survived beyond 2 years, some patients even many years longer.30–32 In addition, the kinetics of response to immunotherapy differs as compared with targeted therapy or chemotherapy. Many different radiographic patterns may be seen after ipilimumab. On occasion, responses to ipilimumab can be delayed, sometimes not occurring until 6 months following initiation of therapy. Other times, the tumor burden appears to have increased on initial imaging but then ultimately may subsequently regress. These differences in response kinetics led to the consideration and subsequent development of the immune-related response criteria as a method for monitoring response to immunotherapy.33
The majority of ipilimumab-related adverse events are immune related, and the most common observed side effects include diarrhea, colitis, and dermatitis. Less common severe immune-related adverse events include hypophysitis, thyroiditis, and hepatitis. In the Phase III trial of ipilimumab with or without gp100 peptide vaccine compared with gp100 peptide vaccine alone, immune-related adverse events were noted in 60% of patients treated with ipilimumab, and grade 3 and grade 4 immune-related adverse events occurred in 10%–15% of these patients.30
The therapeutic benefit achieved with CTLA-4 blockade led to the effort in identifying other potential immune checkpoint inhibitors – inhibitors that would be more specific, equally efficacious, and have less immune toxicity.3 Given that PD-1 and PD-L1 are often thought to be more distal immune modulators, PD-1 and PD-L1 inhibitors were identified as potentially fulfilling those needs. Although agents against PD-1 or PD-L1 (Table 1) are not yet FDA approved, they are currently in clinical trials, with promising results including high objective durable response rates (ORR) and a favorable side effect profile.34
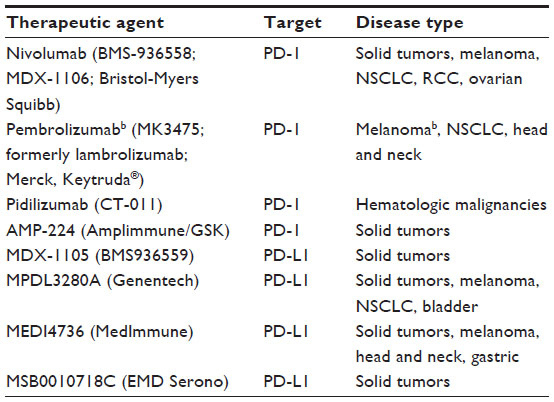 | Table 1 PD-1 and PD-L1 therapeutic agents in clinical developmenta |
Clinical potential of anti-PD-1 and anti-PD-L1 antibodies
Although PD-1- and PD-L1-directed therapy is currently undergoing investigation in several types of malignancy, including both solid tumors and hematologic malignancies, PD-1 and PD-L1 therapy has been most studied in patients with metastatic melanoma. Antibodies targeting PD-1 in clinical development include nivolumab (Opdivo), pembrolizumab (also known as MK-3475, formerly lambrolizumab), and pidilizumab (CT-011). Of these, nivolumab and pembrolizumab have been most extensively studied in patients with solid tumors.
The first antibody to target PD-L1 in clinical trials was MDX-1105. Antibodies currently in clinical development that target PD-L1 include MPDL3280A (Genentech), MEDI4736 (MedImmune), and MSB0010718C (EMD Serono). Lastly, novel approaches such as AMP-224 (Amplimmune), a PD-1 decoy, are also under investigation.
Nivolumab
The first-in-human Phase I study of PD-1 monotherapy with nivolumab (Opdivo, formerly BMS936558; MDX1106) was conducted in patients with refractory solid tumors.35 One durable complete response (CR) was seen in a patient with colorectal cancer. Two partial responses (PRs) were also seen (one in a patient with melanoma and the other in a patient with renal cell carcinoma).
A subsequent Phase I trial investigated the safety and activity of nivolumab in 296 patients with pre-treated advanced melanoma (n=104), non-small cell lung cancer (NSCLC) (n=122), castration-resistant prostate cancer (n=17), renal cell cancer (n=34), or colorectal cancer (n=19). Data from this study showed that objective responses were seen in advanced melanoma (ORR 28%), renal cell cancer (ORR 27%), and NSCLC (ORR 18%).36 Durable objective responses were noted in 20/31 (65%) of patients lasting ≥1 year. For patients with advanced melanoma, durable responses of ≥1 year were noted in 13/18 (72%) who received nivolumab for more than 1 year and stable disease that lasted ≥24 weeks was noted in 6/94 patients. A follow-up analysis of this trial in patients with melanoma was notable for an ORR of 31%, a median duration of response of 2 years, and a median OS of 16.8 months for all dose cohorts (with 1- and 2-year survival rates of 62% and 43%, respectively) and 20.3 months at the 3 mg/kg dose.37
Common treatment-related adverse events included fatigue, diarrhea, pruritus, rash, nausea, and decreased appetite. Grade 3 or 4 treatment-related adverse events were seen in 14% of patients. Treatment-related serious adverse events were noted in 11% of patients and included pneumonitis (3%, and grade 3 or 4 in 1%), colitis, hepatitis, thyroiditis, and hypophysitis.36
Another Phase I study looked at safety and efficacy of nivolumab with a vaccine in ipilimumab-refractory or ipilimumab-naïve patients with melanoma.38 The ORR was 25% for both groups of patients. The median duration of response was not reached at the median 8.1-month follow-up, and some responses lasted up to 140 weeks. Lastly, in this study, 12 of the 18 ipilimumab-naïve patients who progressed on nivolumab were subsequently treated with ipilimumab. Two of the 12 patients experienced a PR, and two patients had a mixed response, suggesting that patients who progress on PD-1 may still respond to CTLA-4 blockade. This is consistent with findings in another study where patients who progressed on prior ipilimumab were found to still respond to nivolumab.38,39
Pembrolizumab
A Phase I trial investigated the safety and tumor response with pembrolizumab (MK-3475, formerly lambrolizumab, Keytruda®) in patients with both pre-treated and treatment-naïve advanced melanoma.40 A total of 135 patients were treated, and the ORR confirmed across all dose cohorts irrespective of prior treatment was 38%, with the highest confirmed ORR of 52% in the 10 mg/kg cohort. As with nivolumab, responses were durable (11 months at median follow-up), and 81% of patients who had a response were still receiving treatment at the time of data analysis.
In this study, 79% of patients reported drug-related adverse events of any kind (primarily low grade), and 13% of patients reported grade 3 or 4 drug-related adverse events. Grade 3 or 4 adverse events included hypothyroidism, diarrhea, abdominal pain, fatigue, decreased appetite, transaminitis, renal failure, rash, and pruritus. In regards to immune-related adverse events, pneumonitis was noted in 4% (all below grade 3), grade 3 or 4 transaminitis in 1%, grade 3 nephritis in 1%, grade 3 hyperthyroidism in 1%, grade 2 adrenal insufficiency in 1%, and hypothyroidism in 1%. Diarrhea was reported in 20% of the patients; however, only one case was grade 3. Pembrolizumab has also been extensively evaluated in patients with NSCLC, but no published results are yet available. Pembrolizumab was recently FDA approved for patients with advanced or unresectable melanoma on 4 September 2014.
Pidilizumab (CT-011)
A Phase I trial evaluated the safety and pharmacokinetics of pidilizumab (CT-011) in 17 patients with advanced acute myeloid leukemia, chronic lymphocytic leukemia, non-Hodgkin’s lymphoma, Hodgkin’s lymphoma, and multiple myeloma who had progressed despite chemotherapy or stem cell transplantation.41 This study showed that pidilizumab was safe, well tolerated, and clinical benefit was demonstrated in 33% of patients.
Subsequently, pidilizumab has been investigated in two Phase II trials in patients with hematologic malignancies. The first was an international Phase II trial that evaluated pidilizumab following autologous hematopoietic stem-cell transplantation (AHSCT) for diffuse large B-cell lymphoma.42 A total of 66 patients were treated. In 35 patients who had measurable disease after AHSCT and received pidilizumab, the ORR was 51% (34% CR and 17% PR). The PFS was 0.72 at 16 months following the first treatment. Neutropenia (19%) and thrombocytopenia (8%) were the most frequent grade 3 or 4 toxicity reported; 4% of patients experienced a related serious adverse event.42
The second Phase II study evaluated the safety and activity of pidilizumab in combination with rituximab in patients with relapsed follicular lymphoma.43 32 patients were enrolled, and the median follow-up was 15.4 months. The study’s primary endpoint was objective response, for which 29 patients were evaluable. An objective response was noted in 66% (19/26) of patients, with 52% CR and 14% PR. The combination of pidilizumab and rituximab was well tolerated, with no immune-related adverse events or grade 3 or 4 treatment-related events.
MDX-1105
A multicenter Phase I study of the anti-PD-L1 agent, MDX-1105, was investigated in patients with advanced solid tumors: melanoma (n=55), NSCLC (n=75), colorectal cancer (n=18), renal cell carcinoma (n=17), ovarian cancer (n=17), pancreatic cancer (n=14), gastric cancer (n=7), and breast cancer (n=4).19 Amongst these disease types, an objective response was noted in 17% of melanoma, 12% of renal cell carcinoma, 10% of NSCLC, and 5% of ovarian cancer patients. In patients with at least 1 year follow-up, 50% (8/16) had a durable response lasting ≥1 year.19 The most common treatment-related adverse events were diarrhea, arthralgia, fatigue, rash, nausea, pruritus, and headache, a majority of which were low grade. Grade 3 or 4 treatment-related toxicities were noted in 9% of patients. A total of 39% of patients experienced a possible immune-related adverse event, which included rash, hypothyroidism, hepatitis, endophthalmitis, diabetes mellitus, myasthenia gravis, and one case of sarcoidosis. Though clinical development of MDX-1105 is no longer continuing as monotherapy, this clinical experience was the first published evidence on the potential benefits of PD-L1 blockade.
MPDL3280A (Genentech), MEDI4736 (MedImmune), MSB0010718C (EMD Serono), and AMP-224 (Amplimmune/GlaxoSmithKline)
Other anti-PD-L1 agents currently in Phase I clinical trials include MPDL3280A, MEDI4736, and MSB0010718C.
In a Phase I MPDL3280A trial, an expansion cohort of 38 patients with metastatic melanoma has thus far shown an ORR of 29%, with a 24-week PFS of 43%.44 This agent has also been studied in patients with NSCLC and renal cancer, and the Phase I expansion cohort of 52 NSCLC patients and 55 patients with renal cancer showed an ORR of 22% and 13%, respectively.45,46
MEDI4736 is also being investigated in patients with NSCLC, gastric, and head and neck cancers, in whom responses have been seen. MSB0010718C is also currently in early phase clinical trials in solid tumors.47–49
AMP-224 is a recombinant B7-DC-Fc fusion protein that modulates the PD-1 axis by depleting PD-1 high expressing effector T-cells. The hypothesis is that this decoy removes the exhausted T-cells, allowing the functional effector T-cells to restore immune function. The potential benefit of this novel mechanism of action is still under investigation.50
Combination therapies
To improve the number of patients who benefit from immunotherapy with PD-1/PD-L1 agents, combinations with other immunotherapy and standard anticancer treatments have been pursued. The combination of nivolumab and ipilimumab was investigated in a Phase I trial.39 The concurrent administration of both agents showed a manageable side effect profile and resulted in an ORR of ~40% across all dose level cohorts, with a duration of response ranging from 6 to 72 weeks at the time of publication.39 Immune-related toxicities were numerically greater with the combination therapy: grade 3 or 4 adverse events were noted in 53% of patients, but many of these grade 3/4 toxicities were asymptomatic laboratory toxicities of uncertain clinical relevance, such as elevated lipase values without associated symptoms of pancreatitis. Although the combination of nivolumab and ipilimumab has been the most extensively reported PD-1/PD-L1 combination study thus far, ongoing trials are combining PD-1/PD-L1 agents with small-molecule targeted therapy, chemotherapy, and vaccines in a variety of tumor types and disease settings.
Re-treatment with anti-CTLA-4 or PD-1 blockade
Although responses to both CTLA-4 and PD-1 blockade can be durable, some patients ultimately develop progressive disease. Though the reasons for ultimate progression in these patients are unclear, one theory suggests that ‘immunoediting’ can occur, wherein persistent immune response can change the phenotype of the tumor to make it less immunogenic.51 Re-treatment or a re-challenge with immunotherapy in this setting can in theory potentiate the immune response to recognize the more ‘edited’ or progressive tumor.51
Clinical data support re-treating select patients with immunologic checkpoint-blocking antibodies, such as those that target CTLA-4 and PD-1. Some patients who initially had an objective response or stable disease to ipilimumab but ultimately progressed were re-treated with ipilimumab as part of the Phase III trial of ipilimumab plus gp100 vaccine versus ipilimumab alone versus gp100 vaccine alone.51 Of the 31 re-treatment-eligible patients in the two ipilimumab-containing arms of the trial, the best ORR to re-treatment was 3/23 (13%) in the ipilimumab plus gp100 vaccine and 3/8 (37.5%) in the ipilimumab-alone groups.
Successful re-treatment has also been reported with anti-PD-1 therapy. One publication reported a patient with metastatic melanoma who was treated on a Phase I nivolumab trial35 who initially experienced a PR that lasted for months after discontinuing treatment but eventually had progressive disease. This patient received re-treatment with nivolumab that resulted in regaining a PR that lasted 16 additional months at the time of publication.52 The experience with both CTLA-4 and PD-1 re-treatment indicates that re-treatment with antibodies that target CTLA-4 and PD-1 is a reasonable option for patients who had some initial benefit with treatment and no significant toxicities.
PD-L1 expression: a dynamic biomarker
There has been great interest in finding biomarkers that can help identify which patients would benefit most from anti-PD-1/PD-L1 therapy. PD-L1 expression on tumor cells as detected by immunohistochemical staining has been most extensively studied.
Interest in PD-L1 as a biomarker began after the first correlation between PD-L1 expression positivity and response to nivolumab was published.36 However, in another Phase I study of safety and efficacy of nivolumab with a vaccine in ipilimumab-refractory or ipilimumab-naïve melanoma patients, patients with PD-L1-negative tumors also experienced objective responses. These data indicate that a lack of expression of PD-L1 by the tumor does not preclude response to nivolumab.38 In addition, the Phase I trial of the combination of nivolumab and ipilimumab also evaluated the relationship between PD-L1 expression and response. In this study, PD-L1 positivity did not associate with improved responses to PD-1 therapy (ORR was noted in 6/13 PD-L1-positive and 9/22 PD-L1-negative patients).39
PD-L1 is an inducible biomarker, and it cannot be considered in the same way as other molecular biomarkers in oncology, such as the presence of the estrogen receptor or human epidermal growth factor receptor (HER)-2 positivity in breast cancer or the epidermal growth factor receptor (EGFR) mutation in NSCLC. Varied assays are available to detect PD-L1 with immunohistochemistry using numerous antibodies, some of which stain tumor cells and others that stain additional cells in the tumor microenvironment. The precise cutoffs to determine ‘positivity’ are also not yet known.
In summary, though PD-L1 positivity may indicate an immune active tumor microenvironment that is favorable to PD-1 therapies, this requires further study. Since patients who lack PD-L1 expression may still benefit from anti-PD-1 and anti-PD-L1 therapy, at this time we do not suggest excluding PD-L1-negative patients from clinical trials of these agents.13,36,53
Conclusion and future directions
A greater understanding of immune checkpoint inhibition has led to the discovery of efficacious immunotherapy approaches for patients with a variety of cancers. Therapies targeting CTLA-4, such as ipilimumab, have been the prototypic immune checkpoint inhibitors and have paved the way for perhaps even more efficacious anti-PD-1 and anti-PD-L1 antibodies. Clinical trials are demonstrating that these agents have benefit in many solid as well as hematologic malignancies.
The goal with immunotherapy is immune modulation and restoration of immune function, and the focus of treatment has shifted from targeting the tumor to targeting the patient’s immune system. Further research into optimal biomarkers, combination therapies with novel agents, and/or other immunotherapeutic agents, strategies for sequential therapies, optimal dosing, and duration of treatment is needed.
Disclosure
Michael Postow has the following conflicts of interest to disclose: consultant: Bristol-Myers Squibb and Amgen; and research grant support: Bristol-Myers Squibb. Parisa Momtaz has no conflicts of interest to disclose.
References
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | |
Ribas A. Tumor immunotherapy directed at PD-1. N Engl J Med. 2012;366(26):2517–2519. | |
Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1–10. | |
Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–360. | |
Swann JB, Smyth MJ. Immune surveillance of tumors. J Clin Invest. 2007;117(5):1137–1146. | |
Raval RR, Sharabi AB, Walker AJ, Drake CG, Sharma P. Tumor immunology and cancer immunotherapy: summary of the 2013 SITC primer. J Immunother Cancer. 2014;2:14. | |
Kantoff PW, Higano CS, Shore ND, et al; IMPACT Study Investigators. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–422. | |
Postow M, Callahan MK, Wolchok JD. Beyond cancer vaccines: a reason for future optimism with immunomodulatory therapy. Cancer J. 2011;17(5):372–378. | |
Kirkwood JM, Strawderman MH, Ernstoff MS, Smith TJ, Borden EC, Blum RH. Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684. J Clin Oncol. 1996;14(1):7–17. | |
Kirkwood JM, Manola J, Ibrahim J, et al; Eastern Cooperative Oncology Group. A pooled analysis of eastern cooperative oncology group and intergroup trials of adjuvant high-dose interferon for melanoma. Clin Cancer Res. 2004;10(5):1670–1677. | |
Sullivan RJ, Lorusso PM, Flaherty KT. The intersection of immune-directed and molecularly targeted therapy in advanced melanoma: where we have been, are, and will be. Clin Cancer Res. 2013;19(19):5283–5291. | |
O’Day S, Boasberg P. Management of metastatic melanoma 2005. Surg Oncol Clin N Am. 2006;15(2):419–437. | |
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. | |
Schwartz RH. Costimulation of T lymphocytes: the role of CD28, CTLA-4, and B7/BB1 in interleukin-2 production and immunotherapy. Cell. 1992;71(7):1065–1068. | |
Egen JG, Allison JP. Cytotoxic T lymphocyte antigen-4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity. 2002;16(1):23–35. | |
Parry RV, Chemnitz JM, Frauwirth KA, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. 2005;25(21):9543–9553. | |
Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271(5256):1734–1736. | |
Chambers CA, Kuhns MS, Egen JG, Allison JP. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol. 2001;19:565–594. | |
Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–2465. | |
Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027–1034. | |
Shin T, Yoshimura K, Shin T, et al. In vivo costimulatory role of B7-DC in tuning T helper cell 1 and cytotoxic T lymphocyte responses. J Exp Med. 2005;201(10):1531–1541. | |
Barber DL, Wherry EJ, Masopust D, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439(7077):682–687. | |
Dong H, Strome SE, Salomao DR, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8(8):793–800. | |
Rosenwald A, Wright G, Leroy K, et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med. 2003;198(6):851–862. | |
Taube JM, Anders RA, Young GD, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4(127):127ra37. | |
Marzec M, Zhang Q, Goradia A, et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc Natl Acad Sci U S A. 2008;105(52):20852–20857. | |
Atefi M, Avramis E, Lassen A, et al. Effects of MAPK and PI3K pathways on PD-L1 expression in melanoma. Clin Cancer Res. 2014;20(13):3446–3457. | |
Parsa AT, Waldron JS, Panner A, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007;13(1):84–88. | |
Taube JM, Klein A, Brahmer JR, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res. Epub April 8, 2014. | |
Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. | |
Robert, C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364(26):2517–2526. | |
Wolchok JD, Weber JS, Maio M, et al. Four-year survival rates for patients with metastatic melanoma who received ipilimumab in phase II clinical trials. Ann Oncol. 2013;24(8):2174–2180. | |
Wolchok JD, Joos A, O’Day S, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. 2009;15(23):7412–7420. | |
Ott PA, Hodi FS, Robert C. CTLA-4 and PD-1/PD-L1 blockade: new immunotherapeutic modalities with durable clinical benefit in melanoma patients. Clin Cancer Res. 2013;19(19):5300–5539. | |
Brahmer JR, Drake CG, Wollner I, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28(19):3167–3175. | |
Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. | |
Topalian SL, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32(10):1020–1030. | |
Weber JS, Kudchadkar RR, Yu B, et al. Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or -naive melanoma. J Clin Oncol. 2013;31(34):4311–4318. | |
Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369(2):122–133. | |
Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369(2):134–144. | |
Berger R, Rotem-Yehudar R, Slama G, et al. Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic malignancies. Clin Cancer Res. 2008;14(10):3044–3051. | |
Armand P, Nagler A, Weller EA, et al. Disabling immune tolerance by programmed death-1 blockade with pidilizumab after autologous hematopoietic stem-cell transplantation for diffuse large B-cell lymphoma: results of an international phase II trial. J Clin Oncol. 2013;31(33):4199–4206. | |
Westin JR, Chu F, Zhang M, et al. Safety and activity of PD1 blockade by pidilizumab in combination with rituximab in patients with relapsed follicular lymphoma: a single group, open-label, phase 2 trial. Lancet Oncol. 2014;15(1):69–77. | |
Hamid O, Sosman JA, Lawrence DP, et al. Clinical activity, safety, and biomarkers of MPDL3280A, an engineered PD-L1 antibody in patients with metastatic melanoma. J Clin Oncol. 2013;31 (Suppl) [abstr 9010]. | |
Spigel DR, Gettinger SN, Horn L, et al. Clinical activity, safety and biomarkers of MPDL3208A, an engineered PD-L1 antibody in patients with locally advanced or metastatic non-small cell lung cancer (NSCLC). J Clin Oncol. 2013;31 (Suppl) [abstr 8008]. | |
Cho DC, Sosman JA, Sznol K, et al. Clinical activity, safety and biomarkers of MPDL3208A, an engineered PD-L1 antibody in patients with metastatic renal cell carcinoma (RCC). J Clin Oncol. 2013;31 (Suppl) [abstr 4505]. | |
Lutzky J, Antonia SJ, Blake-Haskins A, et al. A phase I study of MEDI4736, an anti-PD-L1 antibody, in patients with solid tumors. J Clin Oncol. 2014;32:5s (Suppl) [abstr 3001]. | |
Segal NH, Antonia SJ, Brahmer JR, et al. Preliminary data from a multi-arm expansion study of MEDI4736, an anti-PD-L1 antibody. J Clin Oncol. 2014;32:5s (Suppl) [abstr 3002]. | |
Heery CR, O’Sullivan Coyne GH, Madan RA, et al. Phase I open-label, multiple ascending dose trial of MSB0010718C, an anti-PD-L1 monoclonal antibody, in advanced solid malignancies. J Clin Oncol. 2014;32:5s (Suppl) [abstr 3064]. | |
Infante JR, Powderly JD, Burris HA, et al. Clinical and pharmacodynamic (PD) results of a phase I trial with AMP-224 (B7-DC Fc) that binds to the PD-1 receptor. J Clin Oncol. 2014;31 (Suppl) [abstr 3044]. | |
Robert, C, Schadendorf D, Messina M, Hodi FS, O’Day S; MDX010-20 investigators. Efficacy and safety of retreatment with ipilimumab in patients with pretreated advanced melanoma who progressed after initially achieving disease control. Clin Cancer Res. 2013;19(8):2232–2239. | |
Lipson, E.J, Sharfman WH, Drake CG, et al. Durable cancer regression off-treatment and effective reinduction therapy with an anti-PD-1 antibody. Clin Cancer Res. 2013;19(2):462–468. | |
Hino, R, Kabashima K, Kato Y, et al. Tumor cell expression of programmed cell death-1 ligand 1 is a prognostic factor for malignant melanoma. Cancer. 2010;116(7):1757–1766. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.