")
Back to Journals » Infection and Drug Resistance » Volume 15
Immunity Cell Responses to RSV and the Role of Antiviral Inhibitors: A Systematic Review
Authors Churiso G , Husen G, Bulbula D, Abebe L
Received 27 August 2022
Accepted for publication 23 November 2022
Published 14 December 2022 Volume 2022:15 Pages 7413—7430
DOI https://doi.org/10.2147/IDR.S387479
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Suresh Antony
Gemechu Churiso,1 Gose Husen,2 Denebo Bulbula,2 Lulu Abebe3
1Department of Medical Laboratory Sciences, Dilla University, Dilla, Ethiopia; 2Department of Orthopedic Surgery, Dilla University, Dilla, Ethiopia; 3Department of Psychiatry, Dilla University, Dilla, Ethiopia
Correspondence: Gemechu Churiso, Email [email protected]
Abstract: Antigen-presenting cells recognize respiratory syncytial virus antigens, and produce cytokines and chemokines that act on immune cells. Dendritic cells play the main role in inflammatory cytokine responses. Similarly, alveolar macrophages produce IFN-β, IFN-α, TNF-α, IL-6, CXCL10, and CCL3, while alternatively activated macrophages differentiate at the late phase, and require IL-13 or IL-4 cytokines. Furthermore, activated NKT cells secrete IL-13 and IL-4 that cause lung epithelial, endothelial and fibroblasts to secrete eotaxin that enhances the recruitment of eosinophil to the lung. CD8+ and CD4+T cells infection by the virus decreases the IFN-γ and IL-2 production. Despite this, both are involved in terminating virus replication. CD8+T cells produce a larger amount of IFN-γ than CD4+T cells, and CD8+T cells activated under type 2 conditions produce IL-4, down regulating CD8 expression, granzyme and IFN-γ production. Antiviral inhibitors inhibit biological functions of viral proteins. Some of them directly target the virus replication machinery and are effective at later stages of infection; while others inhibit F protein dependent fusion and syncytium formation. TMC353121 reduces inflammatory cytokines, TNF-α, IL-6, and IL-1β and chemokines, KC, IP-10, MCP and MIP1-α. EDP-938 inhibits viral nucleoprotein (N), while GRP-156784 blocks the activity of respiratory syncytial virus ribonucleic acid (RNA) polymerase. PC786 inhibits non-structural protein 1 (NS-1) gene, RANTES transcripts, virus-induced CCL5, IL-6, and mucin increase. In general, it is an immune reaction that is blamed for the disease severity and pathogenesis in respiratory syncytial virus infection. Anti-viral inhibitors not only inhibit viral entry and replication, but also may reduce inflammatory cytokines and chemokines. Many respiratory syncytial virus inhibitors are proposed; however, only palivizumab and ribavirin are approved for prophylaxis and treatment, respectively. Hence, this review is focused on immunity cell responses to respiratory syncytial virus and the role of antiviral inhibitors.
Keywords: immune cells, respiratory syncytial virus, antiviral inhibitors
Introduction
Respiratory syncytial virus (RSV) is an enveloped negative sense RNA virus that has trans membrane surface proteins like F, G and SH, and non-glycosylated virion matrix protein (M), proteins associated with RNA to form viral nucleocapsid (P, N, L and M2 open reading frame 1), and other proteins that are non-structural viral products (NS1 and NS2).1,2 It causes lower respiratory tract infection affecting mostly elder people and children.3,4 The RSV virus has 2 main subtypes; subtype A and B.5 Luminal columnar cells, especially ciliated cells, can be affected by RSV, and the virus can be shed solely from the apical surface of ciliated cells and spread to neighbouring cells by the cilial beats.6 RSV G protein interaction with C-X-C Motif Chemokine Receptor 1 (CXCR1) on the cell surface of human amniotic epithelial cells (HAECs) is very important to the pathogenesis of the virus. It contributed to around 50% of RSV infectivity in these cells. CXCR1 is predominantly detected on the ciliated cells, and the percentage of these receptors is varied from person to person.7 Infected people show symptoms like cough, sneezing, runny nose, fever, decrease in appetite, and wheezing within 4 to 6 days of infection.8 The virus can cause severe infection that includes bronchitis, pneumonia, inflammation of the small airways in the lung, and is common in children younger than one year of age. Older adults and infants earlier than six months of age may need hospitalization and additional oxygen or intubation.9
Innate and adaptive immunities are induced during RSV infection. Innate immune cells and other virally infected cells secret inflammatory cytokines, which attract and activate immune cells that induce acute inflammation that helps virus clearance.3 Cellular immunities are very important to clear the RSV infection;10 unlike virus-neutralizing antibodies that are induced and suboptimal during initial infection and wanes over time. Despite this, re-infection with the virus is common. Even though cellular immunities are very important for the clearance of the virus, they can cause serious lower respiratory tract bronchitis immune pathogenesis.11 Furthermore, the treatment for RSV infection is generally supportive care, and sometimes hospital care may also be required.12 Currently, however, palivizumab is being used for prevention and treatment of RSV infection, in addition to ribavirin (FDA-approved therapeutic).13
Immunity Cell Responses to RSV
During RSV infection, antigen presenting (APC) cells recognize the virus by sensors and receptors found on the cell surface and in the endolysosome. Toll-like receptors (TLRs) that are essential for innate recognition of bacterial components are involved in antiviral response as well. They are TLR4 and cluster of differentiation 14 (CD14), a co-receptor for several Toll-like receptors. Persistent longer RSV infection has been observed in TLR4 deficient mice than normal mice, indicating that a shared receptor activation pathway can be used to induce innate immune responses in bacterial and viral infection.14,15 TLR2 and TLR6 signalling can also activate innate immune response against RSV, promoting IL-6, TNF-α, CCL2 (monocyte chemo attractant protein 1), and CCL5 production. Similarly, TLR2 interaction with RSV promotes neutrophil migration and DC activation in the lung.3 The immune response to RSV infection in the lung of TLR7 (-/-) mice is more pathogenic as inflammation and mucus production are increased. It is due to an alteration in T cell response with an increase in mucogenic cytokines like IL-17, IL-4, and IL-13 though no effect of TLR7 deficiency on type I interferons (IFNs). Dendritic cells (DCs) from these mice indicated a special activation of IL-23, a cytokine that promote Th17, and reduction in IL-12 production.1
Dendritic Cells
The number of DC cells increases following RSV infection. They play a great role in detecting the viral antigens and producing cytokines and chemokines that affect other immune cells. They also take viral antigens and move to the lung draining lymph nodes where they start T cell immune responses.16 Phenotypic characterization of DCs showed variation between donors for conventional type 1 dendritic cells (cDC1), cDC2 and CD14+ monocytes, with, much difference in plasmacytoid dendritic cells (pDc) frequencies. cDC1 has lower frequency in mucosal sites while the frequency of cDC2 is higher than cDC1 with higher frequency of cDC2 in lung and jejunum. The frequency of pDC is the highest in lungs, lymph nodes (LNs), bone marrow (BM), and spleen.17 The number of cDC is increased in RSV-BAL while the number of pDC is normal, and the number of both cDC and pDC decreases, and a higher number of cDCs express CD83, CD40, ICOS-ligand and PDL-1 in the peripheral blood of RSV cases.18 At the time of admission, the quantity of mDCs is higher in the follow-up patients than in healthy controls. However, the quantity of pDCs is decreased in patients than in healthy controls for RSV bronchitis. The mDCs/pDCs ratio is also higher at the time of admission.19 RSV infection enhances the expression of CD83, CD86, major histocompatibility complex II (MHC II) and MHC I on monocyte derived DCs (mDCs). However, T cells activated by RSV infected DCs showed reduced proliferation.20 There is a significant increase of mDCs in mucosal areas than in the blood of RSV infected patients. There are also increased concentrations of IL-6, IL-8, MIP-1, IP-10 and TNF-α in nasal wash samples of RSV infected patients. Monocyte chemoattractant protein-1 (MCP-1) has a vital role in lung inflammation by stimulating histamines and leukotriene release, enhancing Th2 polarization and attraction of monocytes to the lungs. RSV can also infect mDCs, decreasing the capacity to stimulate CD4+T cells.21
RSV-infected epithelial cells mainly produce the inflammatory cytokines FNF-α, IL-6 and IL-8 that intern activate DCs and other immune cells, and the level and pattern of these cytokines are similar for all of the RSV strains. In in vitro infection of peripheral blood mononuclear cells (PBMC) with RSV induced the production of IL-8, IL-6, IFN-α, and TNF-α and a low amount of IFN-β. The production of IL-6 is improved in the presence of autologous serum. Most importantly, CD14+ monocytes show a main role in the inflammatory cytokine response against RSV; however, pDCs are important for the secretion of IFN-α. In CD14+ cells depleted PBMC; pDCs produces IFN-α in the presence of autologous serum. TLR7 recognizes single stranded viral ribonucleic acid (RNA) brought to endosomal compartments in pDC can elicit an IFN-α response via cytoplasmic sensors. Hence, the way of the virus and the cell interaction and the way of cell entry will affect the consequence of the innate response.22 Stimulated pDCs cannot increase proliferation and maturation of antigen-specific T cells; they rather enhance antiviral activity by producing type I IFNs. But their depletion decreases viral clearance and exacerbation of pulmonary inflammation, airway hyper-responsiveness, and mucus production. Therefore, pDCs are important for initial type I IFN secretion, CD8+T cell response, and natural killer (NK) cell activation in systemic RSV infection. Most of inflammatory DCs expression CD11β, Ly6C, and intermediate levels of CD11c, and MHC class II. Monocyte-derived CD11c+DCs, that express CX3C chemokine receptor 1 (CX3CR1), can move through the pulmonary arterial vasculature and capture embolic materials. Therefore, these cells are important for priming naive T cells in the lung draining mediastinal lymph nodes.23
In vitro infected human moDCs induce the production of pro-inflammatory cytokines IL-1β, IL-6, IL-12, TNF-α, IFN-γ, IFN-α and IFN-β. Infected moDCs also produce several chemokines, including CCL4, CCL2, CCL5, CCL3, CXCL10 and CXCL8. The production of IL-2, IFN-γ, TNF-α, and IL-4 decrease in the stimulation of naive T cells with RSV-infected DCs. In addition to this, infected DCs also exhibited a limited ability to stimulate cytokine production by polarized T cells.16
Macrophages
The percentage of monocytes and macrophages is also increased in BAL during RSV infection.18 Alveolar macrophages detect viral antigens, and release cytokines and chemokines early in the RSV infection.24 Ex vivo exposed alveolar macrophages to RSV produce IL-6, IFN-β, IFN-α, TNF-α, CCL3 and CXCL10 at 20 h after exposure, and increases with viral dose increases. However, these cytokines and chemokines are not produced by UV-RSV exposed alveolar macrophages. The in vivo study also indicated that alveolar macrophages together with other cells cause lung inflammation during viral infection. Type I IFNs can induce numerous IFN-stimulated genes (ISGs), which interfere with viral replication. Therefore, type I IFNs are inflammatory mediators that induce antiviral condition in infected cells and nearby cells, and for activation, and recruitment of immune cells.25
Alternatively activated macrophages are also the main cells that participate in RSV infection. They differentiate act at the late phase of the infection. Alternatively activated macrophages differentiation requires cytokines associated with Th2 immune responses, IL-4 or IL-13.26,27 RSV infection of WT purified murine primary peritoneal macrophages, bronchoalveolar lavage (BAL) macrophages, and RAW 264.7 macrophages induced IL-13 and IL-4 in vitro. Elevated levels of these cytokines were also shown in the lung tissue. In in vivo, RSV WT mice infection induced expression of arginase-1 in the majority of F4/80+ macrophages. This is also true for BAL cells taken from RSV-infected rats in which both FIZZ1 and arginase-1 mRNA highly increased at 4 days post infection. This indicates that both alveolar and interstitial macrophages are able to produce alternatively activated macrophage markers during RSV infection. Arginase 1 and FIZZ 1 are responsible for lung repair and remodelling after infection. Despite the production of pro-inflammatory cytokines and chemokines, IFN-β and cyclooxygenase 2 (COX-2) in RSV infection, counter regulatory cytokines, IL-13, IL-4, and IL-10 are also secreted during RSV infection. IL-14/IL-13 induced alternatively activated macrophages have a great role in the modification of lung pathology induced by RSV infection. Therefore, virus replication is controlled by early immune response, while lung pathology is controlled by later development of alternatively activated macrophages.27
Natural Killer Cells
NK cells are effector immune cells that control various viral infections, limiting their spread and pathology. However, these cells have a dual role in RSV infection since their initial responses are protective and followed by harmful responses that induce lung injury, because of the inhibition of the pro-inflammatory factors secretion and antibody responses.28 RSV modulated MHC I molecules on A549, BEAS-2B and HPBEC cells in vitro analysis. The level of these molecules increases on the surface of these cells at 24 h of infection in a dose, time and replication-dependent manner. And also, RSV infection increases the surface level of the major histocompatibility complex class I chain related gene A (MICA) and soluble MICA in BEAS-2B cells.29
The percentage of mature NK cells (CD3−DX5+) and T cells (CD3+DX5−) in the lungs are increased during RSV infection, and reach peak at day 3 of infection while T cells reach peak at day 7 of infection. Inducible surface marker for activated lymphocyte (CD69) on NK cells increases and reaches a peak on day 2, while it reaches a peak on day 7 on T cells. This indicates that NK cells are recruited and activated earlier than T cells that may be responsible for the lung injury at the early RSV infection. The percentage of CD27+ and NKG2D+NK cells increases and reaches a peak at 48 h. The level of IFNs in BALF elevated and reached a peak on day 3 of infection. RSV infection increases the production of IFNs by NK cells and the proportion of IFN positive NK cells increases in the lungs, and NK cell derived IFNs are responsible for the lung injury during RSV infection.30 RSV-infected neonatal NK cells produce more IFN-γ than uninfected cells, and CD107a, a marker of activated NK cells, also is up regulated in RSV-infected NK cells than un-infected cells. Similarly, adult NK cells can also produce IFN-γ up on RSV infection.31 In RSV infection, NK cells may produce IL-4 that regulates immune mediated cytokine production, which may also contribute to immune pathogenesis.32
Natural Killer T Cells
In RSV infection, natural killer T (NKT) cells are also activated and produce many cytokines, like IFN-γ, IL-13, IL-17, IL-5 and IL-4 in response to glycolipid antigens, α-GC. In addition to this, these cells interact with other immune cells mostly by producing IL-5 that attracts eosinophils to the lung, and then activation and differentiation. Activated NKT cells secrete IL-4 and IL-13 that cause lung epithelial, endothelial and fibroblasts to secrete eotaxin that increases the attraction of eosinophils to the lung.33
Neutrophils
The number of neutrophils in the airways and peripheral blood is higher in infants with RSV bronchitis.18 Experiments on the mouse model indicated that neutrophils are not the main participants to induce a pro-inflammatory environment, even though their number increases following RSV infection. Thus, they are not seen as an important component of response, since they are not seen limiting viral replication in the lungs during RSV infection.34 However, some studies indicate that neutrophils can prevent viral binding and spreading. Human neutrophils exposed to RSV showed web-like networks of DNA around the cell. These structures are covered with elastase and citH3 that indicate the establishment of neutrophil extracellular traps (NETs). The live cell imaging of neutrophils incubated with RSV also showed these structures within 30 minutes of exposure. The NETs trap RSV virions and inhibit viral binding and infection. Stimulated neutrophil formation of NETs reduces spread and consequent productive infection of RSV in lung.35,36 However, in RSV infection, Fas is up-regulated in neutrophils indicating the apoptosis of these cells due to the RSV.37
Gamma Delta T Cells
Gamma delta T cells are innate-like lymphocytes that respond to stress without clonal selection and differentiation. They have a very important role in immune response or regulation by recognizing antigens directly in an MHC unlimited manner.38 RSV positive bronchitis showed both TCR−CD3+ and γδTCR+T cells producing IFN-γ, but the number of IFN-γ secreting γδTCR+CD3+T cells is higher in acute infection, and IFN-γ response of γδT cells in the acute phase is lower in infants with recurrent wheezing. However, its number may return to normal in the convalescent phase. These cells are also the source of IL-4 in RSV infection.39 The level of γδT cells also increases in the lungs of both adult and neonate mice in RSV infection. However, the overall level of these cells is higher in neonates. The level of IFN-γ producing γδT cells and CD69 expressing activated γδT cells are increased only in the lungs of adult mice. Neutralization of these cytokines elevated lung viral number, bronchoalveolar lavage fluid (BALF) lymphocytes, neutrophils, and pulmonary pathology. During early infection in adults, γδT cells are the main producers of IL-17A, though some other cells like neutrophils are the sources of this cytokine. In neonates, however, both γδT cells and neutrophils are the main source of these cytokines in RSV infected mice. IL-17A increases infiltration of neutrophils into the virus-infected lungs. It potentiates the production of Il-8, a chemokine that attracts the neutrophil. Most importantly, the expression of IL-22 is also observed only in the lungs of RSV infected mice.40,41 Re-stimulated γδT cells taken from peripheral-blood of infants with severe disease failed to produce IFN-γ, and γδT-cells from neonatal mice had a reduced ability to produce IL-17. Thus, the defensive role of these cells in RSV infection is incompetent in an early life.42
Helper and Cytotoxic T Cells
CD8+ and CD4+T cells are efficiently infected by RSV like other cells, and their infection decreases IFN-γ and IL-2 production and the expression of Ki-67 and CD25 by activated CD4+T cells. The frequency of RSV positive CD4+T cells correlated with disease severity.43 Despite this, in the mice, both CD8+ and CD4+T cells are involved in terminating the virus replication after primary infection. Depletion of these cells prolongs the virus replication with no sign of illness, indicating that the immune response determines the disease in mice rather than the cytocidal effect of the virus.44 T cell mediated immune response has important roles in clearing RVS, and immune competent children stop releasing the virus within 21 days of infection. CD8+T cells are able to clear RVS infection in murine models though it may induce and enhance inflammatory response.45 Cytotoxic T lymphocytes (CTLs) response is started within 10 days of infection, and SH, M, NS2, F, N and M2 viral proteins are targets for CTLs.46 However, the failure of attachment protein (G protein) to initiate a CTL response may be a factor for RVS pathogenesis.47
Both CD8+ and CD4+T cells are present in the airways of RSV cases, but the percentage of CD8+T cells is higher than that of CD4+T cells. Moreover, there is an increase in the levels of IFN-γ, IL-2, IL-10, IL-13, and pro-inflammatory cytokines like IL-8, IL-1β, TNF-α and IL-6. The serum level of IFN-γ, TNF-α, IL-10 and IL-6 are also higher in RSV cases than in controls. Most importantly, the concentration of IL-6, TNF-α and IL-8 (innate pro inflammatory cytokines) and IL-4, IL-10, IL-2 and IL-13 (T cell derived cytokines) correlate with the number of BAL cDCs, while Il-10 concentration correlates with the number of pDCs.18 Th1 mediated cellular immunity is very important in RVS infection clearance. In mice, Th1 produce IL-2, IFN-γ, and induction of IgG2a antibody.48
In vitro stimulation of lung, mediastinal lymph nodes, and lung air (BAL) cells produced Il-10 and the production reached a peak on day 6 post infection, in the lung and mediastinal lymph nodes, and on day 8 post infection, in the BAL. Most of the Il-10 is produced by CD4+T cells. However, IL-10 secreting CD8+T cells are very low in any of these tissues. In vivo analysis also showed that the majority of IL-10 cytokines are produced by CD4+T cells during early RSV infection. However, production of IL-10 by CD4+T cells may be suppressed by CD8+T cells; and the elimination of CD8+T cells increases the production of this cytokine. There is a higher amount of IFN-γ production from CD8+T cells than CD4+T cells in RSV infection; which indicates that CD4+T cells modulation of the immune response towards anti-inflammatory cytokine release. Hence, both conventional and regulatory CD4+T cells are responsible for IL-10 production, but regulatory CD4+T cells account for more production. IL-10 has a vital role in limiting disease severity in enhancing the adaptive immunity to RSV infection.49 Furthermore, IL-10 plays a vital role in controlling the severity of RSV related pulmonary disease by limiting inflammation and immunopathology.50
Granzyme and perforin producing cells are increased in RSV infection. The production of granzyme and perforin by CD8+T cells indicates the lytic potential of these cells, while Ki-67 indicates proliferating or newly dividing cells. The peak level of T cell response varies from patient to patient. However, the kinetics of the response is similar in most patients. When disease severity and the viral load increased, however, the CD8+T cells percentage is highly low and similar to the percentage found in healthy controls.51 In in vitro RSV stimulation, the airway epithelial cells IL-15 production is up regulated through activation of transcription factor NF-kB, virus replication and up regulation of the IL-15 gene. IL-15 has a vital role in the survival of effector and memory CD8+T cells.29
The proportion of CD8+T cells is higher in severe patients than patients with moderate disease of infants hospitalized with RSV. But, patients with severe disease have a lower number of CD4+T cells, a higher CD8+/CD4+T cell ratio, an elevated frequency of T cytotoxic 2 (Tc2) cells, and a reduced frequency of T cytotoxic 1 (Tc1) cells compared with patients with mild disease. Increased frequency of T helper 2 (Th2) cells is also seen in patients with severe disease than in patients with moderate disease, but they have a similar frequency of other CD4+T cells. Patients with a lower viral load have a greater frequency of T cytotoxic 17 (Tc17) and T helper 17 (Th17) cells, and Tc1 cells, and a lower ratio of IL-4 producing T cells/IFN-γ1 producing T cells. But, patients with severe disease have a greater ratio of Tc2/Tc1 cells compared to those with moderate disease. However, there is no difference in the ratio of Th2/T helper 1 (Th1) cells in these cases. Most of Tc1 cells express high amount of CD8, while the majority of Tc17 and Tc2 cells express low CD8 molecules. CD8+T cells activated under type 2 conditions produce IL-14, down regulating granzyme, CD8 expression and IFN-γ in vitro. Tc1, Tc17 and Th17 and their cytokines IL-17 and IFN-γ confer protection to RSV disease. Almost all CD8+T cell populations express IL-4 (Tc2), IFN-γ (Tc1), and IL-17 (Tc17). Thus, cells expressing high amount of CD8 mainly produce IFN-γ while those expressing low levels mainly produce IL-4 and IL-17 (Figure 1).10 RSV-infected severe patients had elevated CD8+T cells (Tc2) level expressing IL-4; decreased proportion of CD8+T cells (Tc1) producing IFN-γ, and reduced concentration of IL-17. Patients with higher frequency of Tc1, Tc17 and Th17 (CD4+T cells expressing IL-17) have a shorter duration of hospitalization.52 In the mouse model, an over-time pulmonary increase of IL-23, IL-6 and IL-17 expression was observed, and anti-IL-17 treatment limited inflammation, and minimised viral load, and elevated RSV specific CD8+T cells.53
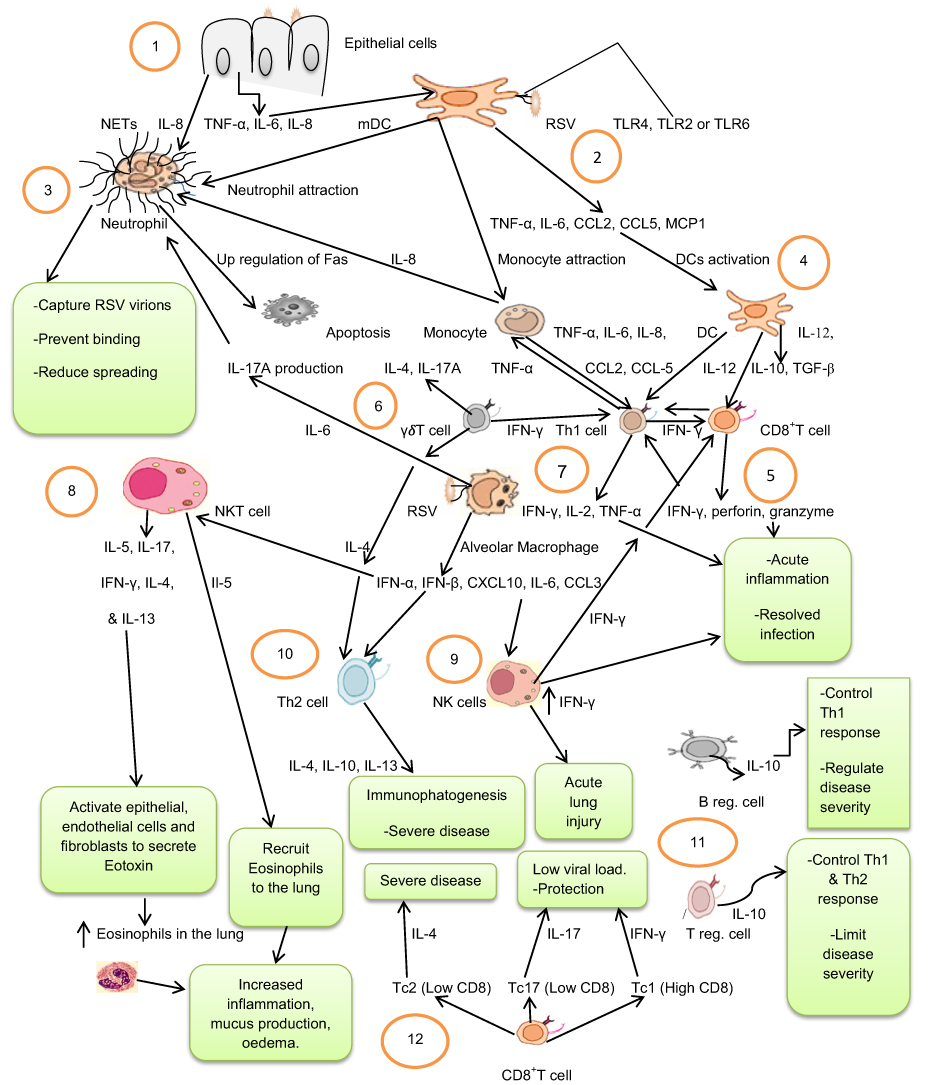 |
Figure 1 Immunity cell responses to Respiratory Syncytial Virus: 1. Virus-infected epithelial cells produce TNF-α, IL-6, and IL-8 that in turn activate and attract DCs and neutrophils. 2. Activated myeloid dendritic cells (mDCs) detect viral antigens and produce inflammatory cytokines and chemokines (TNF-α, IL-6, CCL2, CCL5, MCP1) that activate and attract monocytes, DCs, and neutrophils. 3. Activated neutrophils capture respiratory syncytial virus (RSV) virions by neutrophil extracellular traps (NETs) and prevent binding, and reduce spreading of the virus, or the virus may induce up-regulation of Fas on the neutrophils and induce apoptosis. 4. Activated DCs and monocytes produce IL-12, IL-10, TGF-β and TNF-α, IL-6, IL-8, CCL2, CCL-5, respectively, and activate Th1 and CD8+T cells. 5. Activated Th1 cells produce IFN-γ, IL-2 and TNF-α, while CD8+T cells produce IFN-γ, perforin, and granzyme that collectively induce acute inflammation that resolves the infection. 6. γδT cells are activated and produce IL-4, IL-17A and IFN-γ cytokines activating both Th1 and Th2 cells. 7. Alveolar Macrophages detect viral antigens and produce IFN-α, IFN-β, CXCL10, IL-6, and CCL3 that activate NK, Th2, and NKT cells. 8. Activated NKT cells produce IL-5, IL-17, IFN-γ, IL-4, and IL-13 cytokines. IL-5 acts as chemo-attractant of eosinophils while IL-4 and IL-13 activate eotoxin secretion that recruits eosinophils to the lung, increasing inflammation, mucus production, and oedema. 9. Activated NK cells produce a higher amount of IFN-γ and cause acute lung injury. 10. Th2 cells produce IL-4, IL-10 and IL-13, which cause immunephatogenesis and severe disease. 11. Neonatal regulatory B cells (B reg. cells), and regulatory T cells (Treg. cells) produce IL-10 and control Th1 and Th2 responses, and regulate disease severity. 12. CD8+T cells with low CD8 markers differentiate into T cytotoxic 2 cells (Tc2), or T cytotoxic 17 cells (Tc17) that produce IL-4 and IL-17, respectively. CD8+T cells with high CD8 markers differentiate into T cytotoxic 1 cells (Tc1) that produce IFN-γ. |
Viral factors induce Th2 predominated immune profiles at the mucosal level. RSV evades adaptive immunity by shifting the Th1/Th2 cytokine balance towards the increased level of Th2 cytokines. The levels of IL-4, IL-13, IL-6 and IL-10 and increase in nasopharyngeal aspirate (NPAs) of RSV infected patients.54 Similarly, the level of IL-10, IL-4 and IFN-γ increased, while the level of IL-2 decreased in RSV pneumonia.55 IFN-γ is higher in the early phase than the convalescent phase for severe and mild cases, while IL-10 is higher only in severe cases.56 IL-5 and IL-4 have also increased RSV-infected infants. These cytokines stimulate an immunoglobulin E (IgE) synthesis and then activate Eosinophils to induce type I hypersensitivity and late phase inflammation that indicates an allergic mechanism may contribute to the pathogenesis of wheezing acute bronchitis in RSV infection.57
PBMCs from acute illness and hospitalized subjects stimulated with live virus show no age-related difference in CD69+CD4+T cells that secrete IFN-γ, IL-2, IL-10, IL-4, IL-13, IL-17 and TNF-α, and CD69+CD8+T cells that secrete IFN-γ after stimulation by live or peptide pools in mild and severe patients.58 PBMCs from severe RSV cases have greater numbers of IFN-γ and TNF-α secreting CD69+CD4+T cells than PBMCs from mild cases after live virus stimulation. Severely ill patients have increased levels of IL-4 though the overall number of IL-4 secreting T cells is relatively low. Thus, an effector Th1-like CD4+T cell response may be high in severely ill RSV patients with no sign of a defective regulatory response (IL-10), and also give signs of a modest Th2 response as seen by IL-4 secretion. Severely ill patients have an increased number of IFN-γ producing CD69+CD8+T cells in the acute phase when stimulated by live RSV and NP peptides.58
Regulatory T Cells
Following an acute RSV disease, Foxp3+CD4+T regulatory (T regs) cells rapidly gather in the lung-draining mediastinal lymph nodes and lungs. They modify CD25 expression and express activated T reg molecules, such as CD44high, CD11ahigh, ICOS+, CTLA-4+, and CD43glyco+. T regs diminish the CD8+T cell response in prolonged viral infection.59 Depletion of CD4+Foxp3+CD25+ natural T regs in the BAL and lung by anti-CD25 antibody showed increased weight loss and prolonged recovery from the disease, and weight loss was followed by rise in innate immune cells in both tissues on day 8 post infection that indicates CD4+Foxp3+CD25+natural T reg limit either attraction, proliferation, or clearance of innate cells following RSV infection. Most importantly, there were increased numbers of CD8+ and CD4+T cells releasing IFN-γ, and CD4+T cells releasing IFN-γ and TNF-α. Therefore, the role of CD4+Foxp3+CD25+nT reg is controlling pro inflammatory antiviral responses, CD8+ and CD4+T cell responses.60
In another study in mice, T reg depleted mice showed a significant loss of weight than wild-type that indicates increased disease with increased cellularity in the BALF and lung eosinophilia, that followed by many CD8+ and CD4+T cells experiencing activated phenotype and stimulation of GATA3 and IL-13 expressing Th2 cells that were present in the airway until 14 days after infection. This indicates that Treg cells control pathogenic T cell responses and prevent lung eosinophilia.61 Tregs not only suppress exacerbated and harmful T cell responses but also control Th2 immune responses (IL-13 and Gata3 expressing T cells, and inflow of eosinophils into the airways). Therefore, Tregs play a vital role in preventing pathology in bronchitis. Acute Treg diminution in mice leads to the appearance of a mixed Th1/Th2 type immunity in primary infection, and both Th2 cells and eosinophils prolong beyond the peak of the immune response and viral clearance, indicating that these cells directly participate in prolonged disease. Thus, T regs induce a proper antiviral immune response and promote a resolution of the disease.61
Regulatory B Cells
B cell subsets with different phenotypes that highly express CD10 or CD5 are identified. Mature naive CD19+B cells are CD5−CD10−CD1chiCD21hiCD45RAhiCD23loCD24intCD38intIgDhiIgMlo/hi phenotype, though there is heterogeneity with some markers. Immature transitional B cells corresponding to CD24hiCD38hi B cells are CD5loCD10+CD1c−CD21−CD45RAintCD23−CD24hiCD38hiIgDintIgMhi phenotype. The previously undefined subset of CD5hi B cells is CD5hiCD10−CD1cloCD21intCD45RAintCD23hiCD24loCD38loIgDloIgMlo phenotype. Adult blood immature transitional B cells, CD24hiCD38hiB cells, secrete IL-10 in response to CD40. The elevation of these cells in cord blood indicates that these cells are the major source of IL-10 in new-borns. These cells are called neonatal Breg (nBregs) cells. Culturing of activated nBregs with activated CD4+Th1 cells inhibit IL-22 and IFN-γ but not TNF-α release from the CD4+T cells. Activated Breg cells are able to hinder the ability of pDCs to initiate IFN-γ producing T cell response by producing IL-10 cytokine. This is due to decreased APC functions of pDCs but not IFN-α response. Thus, the Bregs can be distinctly activated by RSV and can regulate the Th1 cell response by the IL-10 production.62
Thus, the main suppressive molecules that are associated with Breg cell function are TGF-β, IL-10 and IL-35, and aryl hydrocarbon receptor, programmed death ligand 1, CD39 and CD73. Bregs can be stimulated by microbial products of TLR4 or TLR9 ligands, CD40 ligation and inflammatory cytokines like IL-1β, IL-6 and TNF-α.63 RSV-G protein also binds to a neonatal nBregs and induces immunosuppressive cytokine IL-10 production, inhibiting the protective immune responses.64 RSV induces IL-10 production induction in cord blood nBreg cells within 6h of infection. nBregs are specially infected by the virus as compared to mature naive or immature transitional B cells. However, the infection does not affect the viability of nBreg cells. nBregs containing RSV produce more IL-10 than others, and UV-treated RSV does not induce production of IL-10. RSV infects nBreg cells via receptor recognition, and induces CX3CR1 that allows viral interaction with G glycoprotein. In new-borns, the B cell receptor (BCR) is not adequate to activate nBreg cells in RSV infection, so a second receptor is required. F and G glycoproteins have a combined role in impairing the immune system in new-borns to weaken the viral clearance. The F protein interaction with BCR initiates nBreg cell activation and allows G-CX3CR1 mediated infection. Unbalanced and unregulated T cell response to RSV limits viral clearance and causes immunopathology in the respiratory tract. No detectable Th2 cells were found in the blood of the new born. Thus, activated nBreg cells control IFN-γ producing Th1 cells and its mediated viral clearance, even though it is not clear if these cells reach lymph nodes, and if they directly affect Th1 cell immune response initiation (Figure 1).62
The Role of Antiviral Inhibitors
Currently, the understanding of the molecular mechanisms helped with the discovery of new antiviral inhibitors targeting host factors and viral proteins. These inhibitors inhibit the biological functions of viral proteins including formation of viral replication machinery or enzymatic activities.65 Many of them are tested as important therapeutics for RSV infection by targeting viral entry and replication. However, only ribavirin and palivizumab are approved for the treatment and prophylaxis of RSV infection, respectively.66 Moreover, though RSV infection causes respiratory disease in paediatric, immune-compromised and elderly people, efficacious treatment options are very limited and no vaccine is available. Therefore, new, potent, and orally available RSV fusion inhibitors are being developed.67
EP-023938
EP-023938 is a novel, non-fusion inhibitor of RSV, and its inhibition directly targets the virus replication machinery, and it is effective at later stages of infection. It inhibits RSV-A, RSV-B and clinical isolates with a 50% effective concentration (EC50) <200nM, with a 3-fold change in the presence of human serum albumin (HAS). No cytotoxicity is seen, 50% cytotoxicity concentration (CC50) >25µM. GS-5806, ALS-8112 or AZ-27 has a synergistic effect when combined with it. It has a high barrier to resistance maintaining activity against GS-5806 resistant virus. This molecule is a strong inhibitor of RSV; hence, it is a promising therapeutic for the fight against the virus.68
RFI-641
This molecule strongly inhibits the clinical isolates of RSV-A and RSV-B strains both in vitro and in vivo. Its mechanism of action is blocking the viral F protein dependent fusion and syncytium formation.69 The average 50% inhibition concentrations (IC50s) against all tested subtypes of A and B are 0.055 and 0.018 µg/mL, respectively, while the concentration that inhibits 90% of the clinical isolate was 0.03µg/mL. It is a strong inhibits a new virion formation. The mean IC90s for A and B clinical isolates are 0.22µg/mL and 0.12µg/mL, respectively.70 RFI-641 has no activity against other viruses. IC50 against influenza strains and human Para influenza Virus-3 (hPIV3) are >10 µg/mL, and relatively poor activity is observed against human cytomegalovirus (hCMV) and herpes simplex virus (HSV). Additionally, this molecule does not act non-specifically. Hence, its anti-RSV activity is specific. RFI-641 blocks two F glycoprotein mediated fusions: fusion of its envelope with cellular plasma membrane and syncytium formation.71
TMC353121
TMC353121 is a potential fusion inhibitor. Its activity is dose-dependent in non-human primate model, reducing the viral load, and it may also completely inhibit the viral replication with low plasma exposure, 0.39µg/mL. A dose-dependent reduction of IFN-γ, IL-6 and macrophage inflammatory protein α (MIP1α) was also observed.72 TMC353121 reduced cellular infiltration of BAL in all tested doses in mice, especially macrophages and lymphocytes. For the dose, 1–10mg/kg, the level of total cell in BAL is not different from non-infected mice, while the total number of BAL cells folded in RSV infected untreated mice. It reduced cytokines and chemokines in the BAL. Substantial reduction in inflammatory cytokines like IL-6, IL-1β and TNF-α, and chemokines like keratinocyte chemo-attractant (KC), monocyte chemo-attractant protein (MCP), MIP1 α and interferon gamma-induced protein 10 (IP-10) is observed 24h later. Thus, reducing lung inflammation.66,72
RO-0529
RO-0529 is a potent as well as a selective RSV-F protein inhibitor. Its 50% effective concentration (EC50) is 3nM in the animal model.73 It revealed that a > 1 log unit of viral titre decrease in the lung is achieved at a dose 12.5 mg/kg. Its main target is RSV-F protein. RSV-F protein induces a cell fusion and generates syncytia formation in the absence of RO-0529. However, this effect of syncytial formation is completely inhibited in the presence of 0.1µM RO-0529.74
Dammarenolic Acid (Ign T1), Aglaiol (Dup T1) and Niloticin (Cuc T1)
Compounds from the genus Aglaia are screened for anti-RSV and cytotoxicity. Of 18 selected compounds, ign T1, dup T1 and cuc T1 exhibited potent and selective anti-RSV activity. IgnT1 exhibited the strongest activity, followed by dupT1 and cucT1 exhibited only moderate anti-RSV activity. They target the RSV replication at the post entry stage. Cell viability is not affected by dupT1, but reduced to 50% by ignT1 at the concentrations that exhibited post-entry inhibition. The ignT1 exhibits specific anti-RSV effect that is not cell mediated.75
Ribavirin
Ribavirin is a clinically approved antiviral drug used to treat RSV infection, hepatitis C virus (HCV) and other viral haemorrhagic fever, even though its safety and efficacy have not been recognized for the treatment of RSV disease. It is only used for high-risk patients.76–78 Ribavirin triphosphate can interact with different viral RNA polymerases, and this is most likely important in the mechanism of action of ribavirin. Ribavirin is considered as it has no one universal mechanism of action. However, it inhibits different viruses in different ways.79 The most important mechanisms of action of ribavirin are reduction of intracellular guanosine triphosphate (GTP) pools, inhibition of the cellular inosine monophosphate (IMP) dehydrogenase, viral polymerase activity, viral capping, and initiation of error tragedy due to the increase of mutations in the viral genome.80
Palivizumab (Synagis)
Synagis is a humanized monoclonal antibody that prevents RSV infection.81 It is a RSV-F glycoprotein inhibitor that is used to prevent RSV disease in children and other high-risk groups.76–78 It is a passive immunization used as a prophylaxis.82 It is provided by once-a-month intramuscular injection during the RSV infection season. It is indicated for prematurely born infants, and children with congenital heart disease to limit hospitalization. It can also prevent recurrent wheezing in the first year of life at the healthy preterm gestational age of 33–35 weeks.83
BMS-433771
This molecule is orally available F protein induced membrane fusion inhibitor. It has very good potency against many clinical and laboratory isolates with an average EC50 of 20Nm.84 BMS-433771 inhibits the fusion of lipid membranes during the virus entry and syncytium formation in the late stage.85 BMS-433771 inhibits at an early stage of infection as well as the stage where it remains susceptible to antibody neutralization. This molecule is active against both A and B groups of RSV, with an average of EC50 20nM (Table 1). It has a favourable toxicity profile.84–86
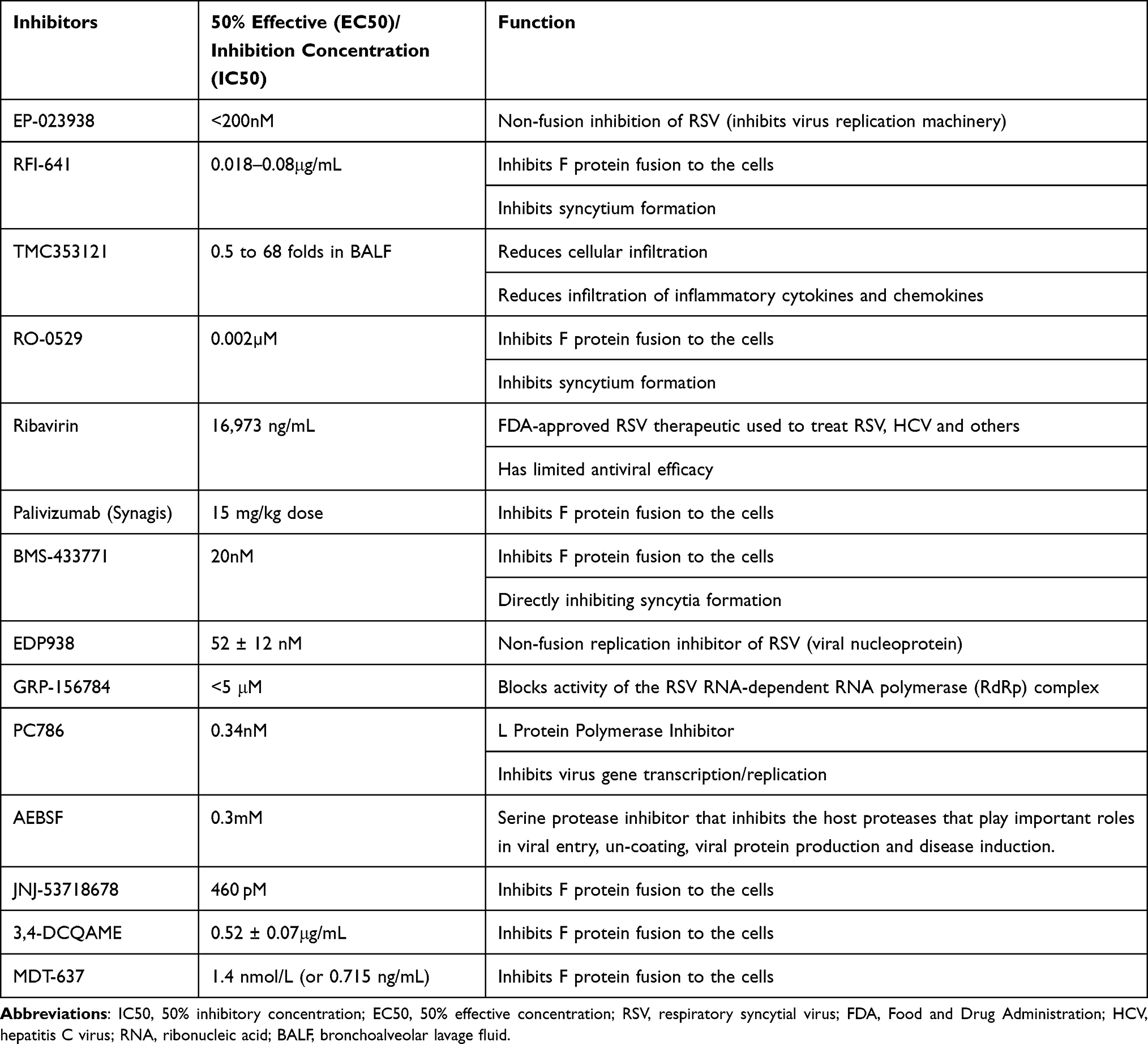 |
Table 1 Respiratory Syncytial Virus Inhibitors, 2022 |
EDP938
EDP-938 is a new post entry replication inhibitor. It inhibits RSV clinical isolate and laboratory strains in vitro in numerous cell-lines with EC50 of 21, 64, 23nM against Long A, VR-955B and M37A strains, respectively, in primary human bronchial epithelial cells (HBECs). In clinical trials, EDP938 taken orally for 5 days decreased both RSV clinical symptoms and viral load in healthy individuals challenged with RSV strains. The side effects were mild, and resolved. There is no dose relationship over EDP-938 regimens concerning efficacy measures. It has rapid absorption, and its half-life supports once or twice daily dosing. The window for intervention with EDP-938 may be longer, since this molecule inhibits the viral N protein, which is effective at the stages of entry into human cells, and after entry. In this human challenge trial, it has shown lowering of total symptom scores, viral load, and mucus weight. All adverse events occurring are headache, dizziness and diarrhoea, and they are mild.87,88 EDP-938 inhibition activity is induced by viral nucleoprotein (N). In in vitro investigation, EDP-938 presented a higher barrier to resistance compared to non-nucleoside L polymerase inhibitors or viral fusion with no cross-resistance.87
GRP-156784
GRP-156784 is a molecule that selectively blocks the activity of RSV RNA dependent RNA polymerase (RdRp). In the dose response assay, GRP-156784 returned EC50<5µM and 72h exposure CC50>µM. When tested on primary HPTECs, it showed unchanged antiviral activity. The previously characterized polymerase inhibitor JMN3-003 and RSV entry inhibitor BMS-433771 were added at the same point as reference. Inhibition results obtained for JMN3-003 and GRP-156784 were similar, whereas BMS-433771 showed a different time of addition profile. Reverse-transcription quantitative polymerase chain reaction (qPCR)-based quantitation of the copy numbers of RSV N protein encoding mRNAs produced in the presence of 10 µM GRP-156784 revealed 60% reduction, which is similar with that seen with the RSV-specific ribonucleoside analog polymerase inhibitor, ALIOS-8176. Its anti-viral activity is RSV specific, since influenza A virus strain (IAV-WSN), measles virus and vesicular stomatitis virus remained unaffected.89
PC786
PC786 is a potent non-nucleoside RSV-L protein polymerase inhibitor prepared for inhalation to treat RSV infection.90 In an in vitro cytopathic effect (CPE) assay, in Hep-2 cells, the PC786 antiviral profile is found to be a strong inhibitor of CPE induced by RSV-A and B strains with IC50 values of 0.40nM and 1.3nM, respectively, without causing detectable cytotoxicity. The average IC50 value of PC786 on CPE induced by clinical isolates is 0.34nM, which is 1940-fold more powerful than the known ant-RSV nucleotide inhibitor ALS-8112 (659nM). It does not inhibit CPE induced by other respiratory viruses like PIV3, influenza A and rhinovirus.91 PC786 also produced concentration-dependent and potent inhibition of the expression of luciferase driven by a mini-genome construct including of the P, L, N and M2-1 proteins of RSV A2 (IC50 value: 0.48 nM), and was 20- and 115-fold more powerful than the RSV L protein inhibitors AZ27 (Astra Zeneca) and Compound D (Boehringer Ingelheim), respectively, and over 430-fold more powerful than ALS-8112. This is evidence that PC786 inhibits virus gene transcription/replication by the RSV-A2 derived ribonucleo-protein (RNP) complex.91,92 In vivo, PC786 (2mg/mL) completely inhibited viral load in the lungs of BALB/c mice dosed by either intra-tracheal (20µL) or intranasal (40µL) routes. In cotton rats, PC786 dose dependently inhibited RSV titre in lung homogenates and significantly inhibited RSV virus titre at 3.3 and 10 mg/mL (50µL intra-nasally). PC786 also showed a dose-dependent inhibition of RANTES transcripts and RSV NS-1 gene transcripts in the lung.91 Late intervention with PC786 produced concentration dependent and potent inhibition of RSV replication with viral load dropping below the measurable limit after 3 days of treatment in human airway epithelium. It has also shown inhibitory activities over RSV induced double strand DNA, IL-6, CCL5 and mucin increase.93
AEBSF
This molecule is a serine protease inhibitor, which inhibits the host proteases that play vital roles in viral entry, un-coating, viral protein production and disease initiation. It showed a complete block in some of infected cells after 18 h of incubation, in BEAS-2B cells, A549 and HEp-2 cells, and the effect was dose-dependent, and it is mostly active during the entry phase.4,94 During the analysis of the effect of protease inhibitors on RSV infection in immortalized cells, the viral proteins spread over the whole cell cytoplasm after 12 h post infection. The number of RSV positive cells reached a plateau phase at 18h post infection. At a concentration of 0.3mM, AEBSF exhibited almost complete block of the RSV infection and a dose-dependent reduction of infection. Almost complete inhibition of RSV A2 infection is observed in all other cell lines of respiratory tract like BEASB-2B (transformed human bronchial epithelium), and A549 (human lung carcinoma) at this concentration. Treatment that combines the post-inoculation phase with either the peri or the pre-inoculation phase results in a greater significant reduction of RSV infection. This indicates that AEBSF treatment is more powerful after the attachment of the virion to the host cells and before the start of replication (during fusion).4
JNJ-53718678
This molecule binds to and stabilizes pre-fusion RSV F protein. It does not bind to the post-fusion RSV-F protein, indicating that the compound is specific for the pre-fusion conformation. The inhibitor may stabilize the pre-fusion conformation of RSV F protein and prevent triggering of the post-fusion state as indicated by the attachment of this compound to residues in the fusion peptide. The presence of this compound caused a concentration-dependent elevation of the fraction of pre-fusion F protein on the cells. In general, this compound antagonizes the refolding of the RSV-F protein from its pre-fusion to post-fusion structure, a process vital for RSV to enter the host cell effectively.67,95 Its concentration to reach EC50 in HeLa cells is 460pM. Its concentration to reach CC50 in HeLa cells is very high, showing a useful general selectivity index, SI = CC50/EC50 of >105. These indicate that the molecule shows a very strong antiviral activity with low cytotoxicity. The compound is also extremely active against a number of RSV strains. The inhibitor indicated no activity against human metapneumovirus (hMPV) or paramyxoviruses suggesting that great selectivity of the compound for RSV.67,96
3,4-DCQAME
3.4-DCQAME is a compound that can interact with the ectodomain of the RSV-F protein and inhibit viral fusion, causing inhibition of viral gene expression and infection. It strongly repressed the infection of RSV strains in HEp-2 cells without showing significant cytotoxicity. The CC50/EC50 of this molecule is more than 30-fold greater than that of ribavirin. This molecule may interfere with F protein function, which leads to blocking of viral entry. Blocking of the association between the cell membrane and F protein may inhibit fusion of RSV with the cell membrane but not attachment of the virus. In in vivo inhibition analysis, the lung tissue in RSV infected mice showed significant infection and pathology compared to non-infected mice. In mice treated with ribavirin and 3.4-DCQAME, the expression of F protein and viral load was decreased in a dose-dependent manner. The two inhibitors inhibited infiltration of inflammatory cells, lung inflammation, and pathologic changes of alveoli in mice. This molecule can block RSV infection both in vitro and in vivo. Toxicity effect assessment in BALB/c and SCID mice showed no difference in body weight in the treated both BALB/c and SCID mice compared to the untreated mice, showing that 3.4-DCQAME may not cause substantial weight loss.97
MDT-637
This molecule is RSV F protein inhibitor with antiviral activity against clinical and laboratory strains signifying broad RSV genetic diversity. This molecule is over 20,000-fold more powerful than ribavirin (the only FDA-approved RSV therapeutic). The concentration to reach EC50 in an RSV infection in in vitro assay is 1.4 nmol/L (or 0.715 ng/mL) (Table 1).98
Conclusion
During RSV infection, antigen presenting (APC) cells recognize the virus via sensors and receptors, and produce chemokines and cytokines that activate or recruit immune cells. CD14+ monocytes have a great role in the inflammatory cytokine production against RSV; however, pDCs are important for the IFN-α production. Alveolar macrophages exposed to RSV produce IFN-β, IFN-α, IL-6, TNF-α, CCL3 and CXCL10. Alternatively, activated macrophages differentiate at the late phase of the infection, and their differentiation requires cytokines related with strong Th2 mediated responses, IL-4 or IL-13. Despite the release of pro-inflammatory chemokine and cytokine cyclooxygenase 2 (COX-2) and IFN-β in RSV infection, counter regulatory cytokines, IL-10, IL-13 and IL-4 are also produced. NK cells have a dual role, as their initial responses are protective and followed by harmful responses that induce lung injury by inhibiting the production of pro-inflammatory factors. They are also the source of IFNs at the early stage of viral infection. It may also produce IL-4, which regulates immune mediated cytokine production, which may also contribute to immune pathogenesis. NKT cells interact with other immune cells mostly by producing IL-5 that attract eosinophils to the lung, and lead to cell activation and differentiation. These cells can also produce IL-13 and IL-4 that cause lung epithelial, endothelial and fibroblasts to secrete eotaxin that increases the attraction of eosinophils to the lung.
Gamma delta T cells are the main producers of IL-17A in early infection in adults. They are also the source of IL-4 and IFN-γ in RSV infection. IL-17A increases infiltration of neutrophils into lungs, and the release of Il-8, a neutrophil-attracting chemokine. Similarly, conventional T cell mediated immunity also has important role in clearing RSV. However, CD8+ and CD4+T cell infection by RSV decreases the IFN-γ and IL-2 release and the expression of Ki-67 and CD25 by activated CD4+T cells. Despite this, both CD8+ and CD4+T cells are involved in terminating the virus replication after primary infection. CD8+ cells are able to clear RVS infection in murine models though it may induce and enhance inflammatory response. There is also an elevation in the levels of IFN-γ, IL-2, IL-10, IL-13, IL-8, IL-1β, TNF-α and IL-6. Th1 mediated cellular immunity is also very essential in RVS infection clearance. In mice, Th1 has been characterized by the release of IL-2 and IFN-γ. The majority of Il-10 is produced by CD4+T cells. In contrast, the number of an IL-10 releasing CD8+T cells was very low in the infected tissues. However, these cells may decrease IL-10 release by CD4+T cells, as the elimination of CD8+T cells increases the production of this cytokine. CD8+T cells produce a higher amount of IFN-γ than CD4+T cells in RSV infection. Both the conventional and regulatory CD4+T cells are responsible for IL-10 secretion, but regulatory CD4+T cells account for more secretion. IL-10 has a vital role in regulating disease severity and in enhancing adaptive immune response. Increased frequency of Th2 cells is also seen in critical patients. Patients with a lower viral load have a greater frequency of Tc17, Th17 and Tc1 cells. But, patients with severe diseases have a larger ratio of Tc2/Tc1 cells. The majority of Tc1 cells express a greater level of CD8, while the majority of Tc17 and Tc2 cells express a lower level of CD8 marker. CD8+T cells activated in type 2 situations release IL-14; down regulating IFN-γ, CD8 expression, and granzyme in vitro. Tc1, Tc17 and Th17 and their cytokines IL-17 and IFN-γ confer protection against RSV disease. The number of CD8+T cells (Tc2) level expressing IL-4 increased in patients with severe disease; reduced concentration of IL-17, and decreased proportion of CD8+T cells (Tc1) producing IFN-γ. Patients with greater frequency of Tc1, Tc17 and Th17 (CD4+T cells expressing IL-17) have a shorter duration of hospitalization. Viral factors induceTh2 pre-dominated immune profiles at the mucosal level. IL-4 and IL-5 are also elevated in RSV infected infants. These cytokines activate IgE synthesis, activating Eosinophils that cause type I hypersensitivity, and late phase inflammation that may contribute to the pathogenesis of acute bronchitis. Severely ill patients have increased levels of IL-4 though the overall number of IL-4 secreting T cells is relatively low.
Following an acute infection, Foxp3+CD4+ T regs accumulate in the lung-draining lymph nodes. Tregs suppress the CD8+T cell mediated response in persistent viral infection. Therefore, the role of CD4+Foxp3+CD25+nT reg is controlling pro-inflammatory antiviral responses. Tregs reduce exacerbated and pathological T cell mediated responses, and also control Th2 mediated responses. Therefore, T regs play a central role in preventing pathology in bronchitis. The abundance of neonatal Breg (nBregs) cells in cord blood indicates that these cells are the chief producer of IL-10 in new-borns. Culturing RSV activated nBregs with activated CD4+Th1 cells repressed IL-22 and IFN-γ but not TNF-α secretion. pDCs priming of IFN-γ producing T cell response is inhibited by RSV activated Breg cells in an IL-10 dependent system. Thus, the Bregs can be activated by RSV, and may control the Th1 cell response. RSV infects nBreg cells, and induces CX3CR1, which allows viral interaction with G glycoprotein. F and G glycoproteins have a combined role in the new-borns to impair viral clearance, through hijacking the immune system.
Antiviral inhibitors inhibit the biological functions of viral proteins, including formation of viral replication machinery or enzymatic activities. Many inhibitors of RSV are tested as powerful therapeutics for RSV infection by targeting viral entry and replication. However, only palivizumab and ribavirin are approved for the prophylaxis and treatment of RSV infection, respectively. The main inhibitors of RSV are EP-023938, RFI-641, TMC353121, RO-0529, Dammarenolic acid (Ign T1), Aglaiol (Dup T1) and Niloticin (Cuc T1), Ribavirin, Palivizumab (Synagis), BMS-433771, EDP938, GRP-156784, PC786, AEBSF, JNJ-53718678, 3.4-DCQAME, and MDT-637. EP-023938 directly targets the virus replication machinery, and has shown a great barrier to resistance against virus in vitro, and it is effective at later stages of infection. RFI-641, RO-0529, BMS-433771, Synagis and 3.4-DCQAME inhibit clinical isolates of RSV strains with potent in vitro and in vivo inhibition activities. Their mechanism of action is blocking viral F protein mediated fusion and formation of syncytium. 3.4-DCQAME highly suppressed the infection of RSV strains in HEp-2 cells without causing significant cytotoxicity. TMC353121 is also a powerful RSV fusion inhibitor. Its antiviral activity is dose-dependent in non-human primates. A dose dependent decrease of MIP1α, IL-6 and IFN-γ was also observed. A substantial decrease in inflammatory cytokines, IL-6, IL-1β, and TNF-α, and chemokines, MCP, KC, IP-10 and MIP1 α is observed, reducing lung inflammation. Synagis is indicated for prevention of RSV in children and high vulnerable groups. It is a passive immunization used as a prophylaxis against RSV.
Compounds from the genus Aglaia, ign T1, dup T1 and cuc T1 exhibited potent and selective anti-RSV activity. They target the RSV replication at the post entry stage. Ribavirin’s safety and efficacy have not been recognized for the treatment of RSV disease. It is used only in high-risk groups. EDP-938 is a new non-fusion post entry replication inhibitor of RSV, inhibiting viral nucleoprotein (N). It inhibits RSV clinical isolate and laboratory strains in vitro in numerous cell lines. GRP-156784 blocks the activity of RSV RdRp. It showed unchanged antiviral activity on infected primary human bronchial epithelial cells. PC786 is a potent non-nucleoside RSV-L protein polymerase inhibitor prepared for inhalation. The PC786 antiviral profile is found to be a strong inhibitor of CPE caused by RSV-A and B strains in in vitro cytopathic effect assay in Hep-2 cells. PC786 also showed a dose-dependent inhibition of RANTES transcripts and RSV NS-1 gene transcripts in the lung, and a viral load decreasing below a measurable limit after 3 days of treatment in human airway epithelium. It has also shown inhibitory actions against RSV induced double strand DNA, IL-6, CCL5 and mucin increase. AEBSF inhibits the host proteases that have vital roles in viral entry, un-coating, viral protein production and disease induction. It has shown almost a total blockage in many of RSV infected cells after 18 h, in BEAS-2B, A549 and HEp-2 cells, and its effect was dose-dependent, and it is primarily active at entry stage. JNJ-53718678 however, binds and stabilizes a pre-fusion RSV-F protein.
In conclusion, it is an immune response that is accountable for the disease severity and pathogenesis in RSV infection. Th1 and CD8+T cells mediated immune responses induce acute inflammation, resolving the infection. NK cells have dual roles; their initial responses are protective, and followed by detrimental responses that induce lung injury. NKT cells produce cytokines that recruit eosinophils to the lung, increasing inflammation, mucus production, and oedema. Furthermore, Th2 cells produce counter regulatory cytokines that induce immunephatogenesis, severe disease, and prolonged infection. Cytokines produced by Th2 cells induce IgE synthesis and stimulate eosinophils, inducing type I hypersensitivity and late phase inflammation. CD8+T cells that have low CD8 markers (Tc2 cells) also produce IL-4, inducing severe disease. On the other hand, neonatal B reg cells and Treg cells produce IL-10 that controls Th1 mediated immune responses, limiting disease severity. Many inhibitors of RSV are tested as essential therapeutics for RSV infection targeting viral entry and replication. However, only ribavirin and palivizumab are accepted for the treatment and prophylaxis of RSV infection, respectively, even though they are used only in children and vulnerable groups. Anti-viral inhibitors not only inhibit viral entry and replication, but also may reduce inflammatory cytokines and chemokines TNF-α, IFN-γ, IL-1β, IL-6, MIP1α, CCL5, IP-10, and MCP limiting lung inflammation, as observed in some of inhibitors. Lung inflammation reduction is very important as immunopathology is the key factor for the lung injury and disease severity in RSV infection. Therefore, RSV inhibitors have dual roles, inhibiting viral entry and replication, and reducing inflammatory responses, which are very important for treating the patients, and for complete recovery from the infection.
Abbreviations
BAL – bronchoalveolar lavage; BALF – bronchoalveolar lavage fluid; BCR – B cell receptor; BM – bone marrow; CD14 – cluster of differentiation 14; cDC1 – conventional type 1 dendritic cells; CTLs – cytotoxic T lymphocytes; CXCR1 -C-X-C Motif Chemokine Receptor 1; DCs – dendritic cells; EC50 −50% effective concentration; GTP – guanosine triphosphate; HAECs – human amniotic epithelial cells; HAS – human serum albumin; HBECs – human bronchial epithelial cells; hCMV – human cytomegalovirus; HCV – hepatitis C virus; hMPV – human metapneumovirus; hPIV3 – human Para influenza virus-3; HSV-herpes simplex virus; IAV – influenza A virus; IC50s −50% inhibition concentrations; IFNs – interferons; IgE – immunoglobulin E; IMP – inosine monophosphate; IP-10 -interferon gamma-induced protein 10; ISGs – IFN- stimulated genes; KC – keratinocyte chemoattractant; LNs – lymph nodes; MCP – monocyte chemoattractant protein; MCP-1 -monocyte chemoattractant protein-1; MHC I – major histocompatibility complex I; MHC II – major histocompatibility complex II; MICA – major histocompatibility complex class I chain-related gene A; MIP1α -macrophage inflammatory protein α; N – nucleoprotein; nBregs – neonatal Breg; NETs – neutrophil extracellular traps; NK – natural killer cells; NKT – natural killer T cells; NPAs – nasopharyngeal aspirate; NS-1 -non-structural protein 1; PBMC – peripheral blood mononuclear cells; PBS -phosphate-buffered saline; pDc – plasmacytoid dendritic cells; qPCR -reverse-transcription quantitative polymerase chain reaction; RdRp -RNA dependent RNA polymerase; RNA – ribonucleic acid; RNP – ribonucleoprotein; RSV – respiratory syncytial virus; Tc1 – T cytotoxic 1 cells; Tc17 – T cytotoxic 17 cells; Tc2 – T cytotoxic 2 cells; Th1 – T helper 1 cells; Th17 – T helper 17 cells; Th2 – T helper 2 cells; Tregs – T regulatory cells.
Acknowledgment
We would like to thank Dilla University for technical support.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no competing interests.
References
1. Lukacs NW, Smit JJ, Mukherjee S, Morris SB, Nunez G, Lindell DM. Respiratory virus-induced TLR7 activation controls IL-17–associated increased mucus via IL-23 regulation. J Immunol. 2010;185(4):2231–2239. doi:10.4049/jimmunol.1000733
2. Heminway B, Yu Y, Tanaka Y, et al. Analysis of respiratory syncytial virus F, G, and SH proteins in cell fusion. Virology. 1994;200(2):801–805. doi:10.1006/viro.1994.1245
3. Murawski MR, Bowen GN, Cerny AM, et al. Respiratory syncytial virus activates innate immunity through Toll-like receptor 2. J Virol. 2009;83(3):1492–1500. doi:10.1128/JVI.00671-08
4. Van der Gucht W, Leemans A, De Schryver M, et al. Respiratory syncytial virus (RSV) entry is inhibited by serine protease inhibitor AEBSF when present during an early stage of infection. Virol J. 2017;14(1):1–10. doi:10.1186/s12985-017-0824-3
5. Jafri HS, Wu X, Makari D, Henrickson KJ. Distribution of respiratory syncytial virus subtypes A and B among infants presenting to the emergency department with lower respiratory tract infection or apnea. Pediatr Infect Dis J. 2013;32(4):335–340. doi:10.1097/INF.0b013e318282603a
6. Zhang L, Peeples ME, Boucher RC, Collins PL, Pickles RJ. Respiratory syncytial virus infection of human airway epithelial cells is polarized, specific to ciliated cells, and without obvious cytopathology. J Virol. 2002;76(11):5654–5666. doi:10.1128/JVI.76.11.5654-5666.2002
7. Chirkova T, Lin S, Oomens AG, et al. CX3CR1 is an important surface molecule for respiratory syncytial virus infection in human airway epithelial cells. J Gen Virol. 2015;96(Pt 9):2543. doi:10.1099/vir.0.000218
8. Orumie UC, Bartholomew DC. Respiratory syncytial virus infection in infants: a comparative study using discriminant. Probit Logistic Regression Analysis. 2022;1:654.
9. Pickles RJ, DeVincenzo JP. Respiratory syncytial virus (RSV) and its propensity for causing bronchiolitis. J Pathol. 2015;235(2):266–276. doi:10.1002/path.4462
10. Siefker D, Vu L, You D, et al. Respiratory syncytial virus disease severity is associated with distinct CD8+ T cell profiles. Am Assoc Immnol. 2019;2:8465.
11. Domachowske JB, Rosenberg HF. Respiratory syncytial virus infection: immune response, immunopathogenesis, and treatment. Clin Microbiol Rev. 1999;12(2):298–309. doi:10.1128/CMR.12.2.298
12. CDC 24/7: respiratory Syncytial Virus Infection (RSV). Available from: https://www.cdc.gov/rsv/about/symptoms.html.
13. WHO. Respiratory Syncytial Virus Surveillance. Available from: https://www.who.int/teams/global-influenza-programme/global-respiratory-syncytial-virus-surveillance.
14. Gagro A, Tominac M, Kršulović-Hrešić V, et al. Increased Toll-like receptor 4 expression in infants with respiratory syncytial virus bronchiolitis. Clin Exp Immunol. 2004;135(2):267–272. doi:10.1111/j.1365-2249.2004.02364.x
15. Kurt-Jones EA, Popova L, Kwinn L, et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol. 2000;1(5):398–401. doi:10.1038/80833
16. McDermott DS, Weiss KA, Knudson CJ, Varga SM. Central role of dendritic cells in shaping the adaptive immune response during respiratory syncytial virus infection. Future Virol. 2011;6(8):963–973. doi:10.2217/fvl.11.62
17. Granot T, Senda T, Carpenter DJ, et al. Dendritic cells display subset and tissue-specific maturation dynamics over human life. Immunity. 2017;46(3):504–515. doi:10.1016/j.immuni.2017.02.019
18. Kerrin A, Fitch P, Errington C, et al. Differential lower airway dendritic cell patterns may reveal distinct endotypes of RSV bronchiolitis. Thorax. 2017;72(7):620–627. doi:10.1136/thoraxjnl-2015-207358
19. Weng K, Zhang J, Mei X, et al. Lower number of plasmacytoid dendritic cells in peripheral blood of children with bronchiolitis following respiratory syncytial virus infection. Influenza Other Respi Viruses. 2014;8(4):469–473. doi:10.1111/irv.12242
20. de Graaff PM, de Jong EC, van Capel TM, et al. Respiratory syncytial virus infection of monocyte-derived dendritic cells decreases their capacity to activate CD4 T cells. J Immunol. 2005;175(9):5904–5911. doi:10.4049/jimmunol.175.9.5904
21. Gill MA, Long K, Kwon T, et al. Differential recruitment of dendritic cells and monocytes to respiratory mucosal sites in children with influenza virus or respiratory syncytial virus infection. J Infect Dis. 2008;198(11):1667–1676. doi:10.1086/593018
22. Schijf MA, Lukens MV, Kruijsen D, et al. Respiratory syncytial virus induced type I IFN production by pDC is regulated by RSV-infected airway epithelial cells, RSV-exposed monocytes and virus specific antibodies. PLoS One. 2013;8(11):e81695. doi:10.1371/journal.pone.0081695
23. Kim TH, Lee HK. Differential roles of lung dendritic cell subsets against respiratory virus infection. Immune Netw. 2014;14(3):128–137. doi:10.4110/in.2014.14.3.128
24. Pribul PK, Harker J, Wang B, et al. Alveolar macrophages are a major determinant of early responses to viral lung infection but do not influence subsequent disease development. J Virol. 2008;82(9):4441–4448. doi:10.1128/JVI.02541-07
25. Makris S, Bajorek M, Culley FJ, Goritzka M, Johansson C. Alveolar macrophages can control respiratory syncytial virus infection in the absence of type I interferons. J Innate Immun. 2016;8(5):452–463. doi:10.1159/000446824
26. Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32(5):593–604. doi:10.1016/j.immuni.2010.05.007
27. Shirey KA, Pletneva LM, Puche AC, et al. Control of RSV-induced lung injury by alternatively activated macrophages is IL-4Rα-, TLR4-, and IFN-β-dependent. Mucosal Immunol. 2010;3(3):291–300. doi:10.1038/mi.2010.6
28. Bhat R, Farrag MA, Almajhdi FN. Double-edged role of natural killer cells during RSV infection. Int Rev Immunol. 2020;39(5):233–244. doi:10.1080/08830185.2020.1770748
29. Zdrenghea M, Telcian AG, Laza-Stanca V, et al. RSV infection modulates IL-15 production and MICA levels in respiratory epithelial cells. Eur Respir J. 2012;39(3):712–720. doi:10.1183/09031936.00099811
30. Li F, Zhu H, Sun R, Wei H, Tian Z. Natural killer cells are involved in acute lung immune injury caused by respiratory syncytial virus infection. J Virol. 2012;86(4):2251–2258. doi:10.1128/JVI.06209-11
31. van Erp EA, Feyaerts D, Duijst M, et al. Respiratory syncytial virus infects primary neonatal and adult natural killer cells and affects their antiviral effector function. J Infect Dis. 2019;219(5):723–733. doi:10.1093/infdis/jiy566
32. Roduit C, Frei R, Ferstl R, et al. Late-Breaking Abstracts Presented At Scientific Sessions AAAAI Annual Meeting March 4-7, 2016. J Allergy Clin Immunol. 2016;137(2):89.
33. Lee SY, Noh Y, Goo JH, et al. Natural killer T cell sensitization during neonatal respiratory syncytial virus infection induces eosinophilic lung disease in re-infected adult mice. PLoS One. 2017;12(6):e0176940. doi:10.1371/journal.pone.0176940
34. Kirsebom F, Michalaki C, Agueda-Oyarzabal M, Johansson C. Neutrophils do not impact viral load or the peak of disease severity during RSV infection. Sci Rep. 2020;10(1):1–12. doi:10.1038/s41598-020-57969-w
35. Johnson JE, Gonzales RA, Olson SJ, Wright PF, Graham BS. The histopathology of fatal untreated human respiratory syncytial virus infection. Modern Pathology. 2007;20(1):108–119. doi:10.1038/modpathol.3800725
36. Cortjens B, De Boer OJ, De Jong R, et al. Neutrophil extracellular traps cause airway obstruction during respiratory syncytial virus disease. J Pathol. 2016;238(3):401–411. doi:10.1002/path.4660
37. Wang S-Z, Smith P, Lovejoy M, Bowden J, Alpers J, Forsyth K. The apoptosis of neutrophils is accelerated in respiratory syncytial virus (RSV)-induced bronchiolitis. Clin Exp Immunol. 1998;114(1):49–54. doi:10.1046/j.1365-2249.1998.00681.x
38. Sabbaghi A, Miri SM, Keshavarz M, Mahooti M, Zebardast A, Ghaemi A. Role of γδ T cells in controlling viral infections with a focus on influenza virus: implications for designing novel therapeutic approaches. Virol J. 2020;17(1):1–18. doi:10.1186/s12985-020-01449-0
39. Aoyagi M, Shimojo N, Sekine K, Nishimuta T, Kohno Y. Respiratory syncytial virus infection suppresses IFN-γ production of γδ T cells. Clin Exp Immunol. 2003;131(2):312–317. doi:10.1046/j.1365-2249.2003.02062.x
40. Stoppelenburg AJ, Salimi V, Hennus M, Plantinga M. Local IL-17A potentiates early neutrophil recruitment to the respiratory tract during severe RSV infection. PLoS One. 2013;8(10):e78461. doi:10.1371/journal.pone.0078461
41. Huang H, Saravia J, You D, Shaw AJ, Cormier SA. Impaired gamma delta T cell‐derived IL‐17A and inflammasome activation during early respiratory syncytial virus infection in infants. Immunol Cell Biol. 2015;93(2):126–135. doi:10.1038/icb.2014.79
42. Démoulins T, Brügger M, Zumkehr B, et al. The specific features of the developing T cell compartment of the neonatal lung are a determinant of respiratory syncytial virus immunopathogenesis. PLoS Pathog. 2021;17(4):e1009529. doi:10.1371/journal.ppat.1009529
43. Raiden S, Sananez I, Remes-Lenicov F, et al. Respiratory syncytial virus (RSV) infects CD4+ T cells: frequency of circulating CD4+ RSV+ T cells as a marker of disease severity in young children. J Infect Dis. 2017;215(7):1049–1058. doi:10.1093/infdis/jix070
44. Graham B, Bunton L, Wright P, Karzon D. Role of T lymphocyte subsets in the pathogenesis of primary infection and rechallenge with respiratory syncytial virus in mice. J Clin Invest. 1991;88(3):1026–1033. doi:10.1172/JCI115362
45. Anderson J, Norden J, Saunders D, Toms G, Scott R. Analysis of the local and systemic immune responses induced in BALB/c mice by experimental respiratory syncytial virus infection. J General Virol. 1990;71(7):1561–1570. doi:10.1099/0022-1317-71-7-1561
46. Cherrie AH, Anderson K, Wertz GW, Openshaw P. Human cytotoxic T cells stimulated by antigen on dendritic cells recognize the N, SH, F, M, 22K, and 1b proteins of respiratory syncytial virus. J Virol. 1992;66(4):2102–2110. doi:10.1128/jvi.66.4.2102-2110.1992
47. Srikiatkhachorn A, Braciale TJ. Virus-specific CD8+ T lymphocytes downregulate T helper cell type 2 cytokine secretion and pulmonary eosinophilia during experimental murine respiratory syncytial virus infection. J Exp Med. 1997;186(3):421–432. doi:10.1084/jem.186.3.421
48. Muñoz JL, McCarthy C, Clark M, Hall C. Respiratory syncytial virus infection in C57BL/6 mice: clearance of virus from the lungs with virus-specific cytotoxic T cells. J Virol. 1991;65(8):4494–4497. doi:10.1128/jvi.65.8.4494-4497.1991
49. Weiss KA, Christiaansen AF, Fulton RB, Meyerholz DK, Varga SM. Multiple CD4+ T cell subsets produce immunomodulatory IL-10 during respiratory syncytial virus infection. J Immunol. 2011;187(6):3145–3154. doi:10.4049/jimmunol.1100764
50. Sun J, Cardani A, Sharma AK, et al. Autocrine regulation of pulmonary inflammation by effector T-cell derived IL-10 during infection with respiratory syncytial virus. PLoS Pathog. 2011;7(8):e1002173. doi:10.1371/journal.ppat.1002173
51. Lukens MV, van de Pol AC, Coenjaerts FE, et al. A systemic neutrophil response precedes robust CD8+ T-cell activation during natural respiratory syncytial virus infection in infants. J Virol. 2010;84(5):2374–2383. doi:10.1128/JVI.01807-09
52. Siefker DT, Vu L, You D, et al. Respiratory syncytial virus disease severity is associated with distinct CD8+ T-cell profiles. Am J Respir Crit Care Med. 2020;201(3):325–334. doi:10.1164/rccm.201903-0588OC
53. Mukherjee S, Lindell DM, Berlin AA, et al. IL-17–induced pulmonary pathogenesis during respiratory viral infection and exacerbation of allergic disease. Am J Pathol. 2011;179(1):248–258. doi:10.1016/j.ajpath.2011.03.003
54. Bermejo-Martin JF, Garcia-Arevalo MC, Lejarazu ROD, et al. Predominance of Th2 cytokines, CXC chemokines and innate immunity mediators at the mucosal level during severe respiratory syncytial virus infection in children. Eur Cytokine Netw. 2007;18(3):163.
55. Ye Q, Shao WX, Shang SQ, Pan YX, Shen HQ, Chen XJ. Epidemiological characteristics and immune status of children with Respiratory Syncytial Virus. J Med Virol. 2015;87(2):323–329. doi:10.1002/jmv.24047
56. Fernández JA, Roine I, Vasquez A, Cáneo M. Soluble interleukin-2 receptor (sCD25) and interleukin-10 plasma concentrations are associated with severity of primary respiratory syncytial virus (RSV) infection. Eur Cytokine Netw. 2005;16(1):81–90.
57. Sung RYT, Hui SHL, Wong CK, Lam CWK, Yin J. A comparison of cytokine responses in respiratory syncytial virus and influenza A infections in infants. Eur J Pediatr. 2001;160(2):117–122. doi:10.1007/s004310000676
58. Roumanes D, Falsey A, Quataert S, et al. T-cell responses in adults during natural respiratory syncytial virus infection. J Infect Dis. 2018;218(3):418–428. doi:10.1093/infdis/jiy016
59. Fulton RB, Meyerholz DK, Varga SM. Foxp3+ CD4 regulatory T cells limit pulmonary immunopathology by modulating the CD8 T cell response during respiratory syncytial virus infection. J Immunol. 2010;185(4):2382–2392. doi:10.4049/jimmunol.1000423
60. Lee DC, Harker JA, Tregoning JS, et al. CD25+ natural regulatory T cells are critical in limiting innate and adaptive immunity and resolving disease following respiratory syncytial virus infection. J Virol. 2010;84(17):8790–8798. doi:10.1128/JVI.00796-10
61. Durant LR, Makris S, Voorburg CM, Loebbermann J, Johansson C, Openshaw PJ. Regulatory T cells prevent Th2 immune responses and pulmonary eosinophilia during respiratory syncytial virus infection in mice. J Virol. 2013;87(20):10946–10954. doi:10.1128/JVI.01295-13
62. Zhivaki D, Lemoine S, Lim A, et al. Respiratory syncytial virus infects regulatory B cells in human neonates via chemokine receptor CX3CR1 and promotes lung disease severity. Immunity. 2017;46(2):301–314. doi:10.1016/j.immuni.2017.01.010
63. Jansen K, Cevhertas L, Ma S, Satitsuksanoa P, Akdis M. Regulatory B cells, A to Z. Allergy. 2021;76(9):2699–2715. doi:10.1111/all.14763
64. Openshaw PJ. RSV takes control of neonatal Breg cells: two hands on the wheel. Immunity. 2017;46(2):171–173. doi:10.1016/j.immuni.2017.01.011
65. Lou Z, Sun Y, Rao Z. Current progress in antiviral strategies. Trends Pharmacol Sci. 2014;35(2):86–102. doi:10.1016/j.tips.2013.11.006
66. Olszewska W, Ispas G, Schnoeller C, et al. Antiviral and lung protective activity of a novel respiratory syncytial virus fusion inhibitor in a mouse model. Eur Respir J. 2011;38(2):401–408. doi:10.1183/09031936.00005610
67. Roymans D, Alnajjar SS, Battles MB, et al. Therapeutic efficacy of a respiratory syncytial virus fusion inhibitor. Nat Commun. 2017;8(1):1–15. doi:10.1038/s41467-017-00170-x
68. Rhodin M, McAllister N, Kim I, et al., EP-023938, A Novel Non-fusion Replication Inhibitor of Respiratory Syncytial Virus (RSV).
69. Razinkov V, Gazumyan A, Nikitenko A, Ellestad G, Krishnamurthy G. RFI-641 inhibits entry of respiratory syncytial virus via interactions with fusion protein. Chem Biol. 2001;8(7):645–659. doi:10.1016/S1074-5521(01)00042-4
70. Razinkov V, Huntley C, Ellestad G, Krishnamurthy G. RSV entry inhibitors block F-protein mediated fusion with model membranes. Antiviral Res. 2002;55(1):189–200. doi:10.1016/S0166-3542(02)00050-5
71. Huntley CC, Weiss WJ, Gazumyan A, et al. RFI-641, a potent respiratory syncytial virus inhibitor. Antimicrob Agents Chemother. 2002;46(3):841–847. doi:10.1128/AAC.46.3.841-847.2002
72. Ispas G, Koul A, Verbeeck J, et al. Antiviral activity of TMC353121, a respiratory syncytial virus (RSV) fusion inhibitor, in a non-human primate model. PLoS One. 2015;10(5):e0126959. doi:10.1371/journal.pone.0126959
73. Kocienski P. Synthesis of Ziresovir. Synfacts. 2019;15(10):1100.
74. Zheng X, Gao L, Wang L, et al. Discovery of Ziresovir as a Potent, Selective, and Orally Bioavailable Respiratory Syncytial Virus Fusion Protein Inhibitor. ACS Publications; 2019.
75. Esimone C, Eck G, Duong T, Überla K, Proksch P, Grunwald T. Potential anti-respiratory syncytial virus lead compounds from Aglaia species. Int J Pharmaceutical Sci. 2008;63(10):768–773.
76. Chu HY, Englund JA. Respiratory syncytial virus disease: prevention and treatment. In: Challenges and Opportunities for Respiratory Syncytial Virus Vaccines. Springer; 2013:235–258.
77. Maggon K, Barik S. New drugs and treatment for respiratory syncytial virus. Rev Med Virol. 2004;14(3):149–168. doi:10.1002/rmv.423
78. Krilov LR. Respiratory syncytial virus disease: update on treatment and prevention. Expert Rev Anti Infect Ther. 2011;9(1):27–32. doi:10.1586/eri.10.140
79. Parker WB. Metabolism and antiviral activity of ribavirin. Virus Res. 2005;107(2):165–171. doi:10.1016/j.virusres.2004.11.006
80. Leyssen P, Balzarini J, De Clercq E, Neyts J. The predominant mechanism by which ribavirin exerts its antiviral activity in vitro against flaviviruses and paramyxoviruses is mediated by inhibition of IMP dehydrogenase. J Virol. 2005;79(3):1943–1947. doi:10.1128/JVI.79.3.1943-1947.2005
81. Pollack PF, Groothuis JR, Barbarotto G. Development and use of palivizumab (Synagis): a passive immunoprophylactic agent for RSV. J Infection Chemother. 2002;8(3):201–206. doi:10.1007/s10156-002-0178-6
82. Narayan O, Bentley A, Mowbray K, et al. Updated cost-effectiveness analysis of palivizumab (Synagis) for the prophylaxis of respiratory syncytial virus in infant populations in the UK. J Med Econ. 2020;23(12):1640–1652. doi:10.1080/13696998.2020.1836923
83. Bont L. Palivizumab (Synagis®). Handbook of Therapeutic Antibodies; 2014:1825–1854.
84. Cianci C, Meanwell N, Krystal M. Antiviral activity and molecular mechanism of an orally active respiratory syncytial virus fusion inhibitor. J Antimicrobial Chemother. 2005;55(3):289–292. doi:10.1093/jac/dkh558
85. Cianci C, Yu K-L, Combrink K, et al. Orally active fusion inhibitor of respiratory syncytial virus. Antimicrob Agents Chemother. 2004;48(2):413–422. doi:10.1128/AAC.48.2.413-422.2004
86. Feng S, Hong D, Wang B, et al. Discovery of imidazopyridine derivatives as highly potent respiratory syncytial virus fusion inhibitors. ACS Med Chem Lett. 2015;6(3):359–362. doi:10.1021/acsmedchemlett.5b00008
87. Rhodin MH, McAllister NV, Castillo J, et al. EDP-938, a novel nucleoprotein inhibitor of respiratory syncytial virus, demonstrates potent antiviral activities in vitro and in a non-human primate model. PLoS Pathog. 2021;17(3):e1009428. doi:10.1371/journal.ppat.1009428
88. Ahmad A, Eze K, Noulin N, et al. EDP-938, a respiratory syncytial virus inhibitor, in a human virus challenge. N Eng J Med. 2022;386(7):655–666. doi:10.1056/NEJMoa2108903
89. Cox RM, Toots M, Yoon -J-J, et al. Development of an allosteric inhibitor class blocking RNA elongation by the respiratory syncytial virus polymerase complex. J Biol Chem. 2018;293(43):16761–16777. doi:10.1074/jbc.RA118.004862
90. Coates M, Brookes D, Kim Y-I, et al. Preclinical characterization of PC786, an inhaled small-molecule respiratory syncytial virus L protein polymerase inhibitor. Antimicrob Agents Chemother. 2017;61(9):e00737–17. doi:10.1128/AAC.00737-17
91. Coates M, Brookes D, Allen H, et al. Preclinical characterization of PC786, a potent antiviral inhibitor of respiratory syncytial virus replication. virus. 2016;1:2.
92. Zhang G-N, Li Q, Zhao J, et al. Design and synthesis of 2-((1H-indol-3-yl) thio)-N-phenyl-acetamides as novel dual inhibitors of respiratory syncytial virus and influenza virus A. Eur J Med Chem. 2020;186:111861. doi:10.1016/j.ejmech.2019.111861
93. Brookes DW, Coates M, Allen H, et al. Late therapeutic intervention with a respiratory syncytial virus L‐protein polymerase inhibitor, PC786, on respiratory syncytial virus infection in human airway epithelium. Br J Pharmacol. 2018;175(12):2520–2534. doi:10.1111/bph.14221
94. Van der Gucht W. Characterization of Clinical Respiratory Syncytial Virus Isolates and the Effect of Protease Inhibitors on Early Viral Entry. University of Antwerp; 2019.
95. Vendeville S, Tahri A, Hu L, et al. Discovery of 3-({5-Chloro-1-[3-(methylsulfonyl) propyl]-1 H-indol-2-yl} methyl)-1-(2, 2, 2-trifluoroethyl)-1, 3-dihydro-2 H-imidazo [4, 5-c] pyridin-2-one (JNJ-53718678), a Potent and Orally Bioavailable Fusion Inhibitor of Respiratory Syncytial Virus. J Med Chem. 2020;63(15):8046–8058. doi:10.1021/acs.jmedchem.0c00226
96. Xing Y, Proesmans M. New therapies for acute RSV infections: where are we? Eur J Pediatr. 2019;178(2):131–138. doi:10.1007/s00431-018-03310-7
97. Tang W, Li M, Liu Y, et al. Small molecule inhibits respiratory syncytial virus entry and infection by blocking the interaction of the viral fusion protein with the cell membrane. FASEB J. 2019;33(3):4287–4299. doi:10.1096/fj.201800579R
98. Kim YI, Pareek R, Murphy R, et al. The antiviral effects of RSV fusion inhibitor, MDT‐637, on clinical isolates, vs its achievable concentrations in the human respiratory tract and comparison to ribavirin. Influenza Other Respi Viruses. 2017;11(6):525–530. doi:10.1111/irv.12503
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.