")
Back to Journals » OncoTargets and Therapy » Volume 12
Immune-Mediated Antitumor Effect By VEGFR2 Selective Inhibitor For Gastric Cancer
Authors Yang J, Yan J, Shao J, Xu Q, Meng F, Chen F, Ding N, Du S, Zhou S, Cai J, Wang Q, Liu B
Received 5 October 2019
Accepted for publication 31 October 2019
Published 15 November 2019 Volume 2019:12 Pages 9757—9765
DOI https://doi.org/10.2147/OTT.S233496
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Federico Perche
Ju Yang, Jing Yan, Jie Shao, Qiuping Xu, Fanyan Meng, Fangjun Chen, Naiqing Ding, Shiyao Du, Shujuan Zhou, Juan Cai, Qin Wang, Baorui Liu
The Comprehensive Cancer Centre of Drum Tower Hospital, Medical School of Nanjing University, Clinical Cancer Institute of Nanjing University, Nanjing 210008, People’s Republic of China
Correspondence: Baorui Liu
The Comprehensive Cancer Centre of Drum Tower Hospital, Medical School of Nanjing University, Clinical Cancer Institute of Nanjing University, Nanjing 210008, People’s Republic of China
Email [email protected]
Background: It was previously reported that targeting vascular epithelial growth factor (VEGF)/VEGFR could modulate the antitumor immunity. VEGFR2 inhibitor YN968D1 is a highly selective VEGFR2 inhibitor and was approved for the treatment of late-stage gastric cancer in 2014, but its role in antitumor immunity remains unknown.
Materials and methods: In this study, we investigated the effects of YN968D1 on the function of T cells in vitro by testing the cytotoxicity and cytokine production. Next, we constructed peritoneal dissemination and subcutaneous gastric cancer mouse model to assess the cytotoxicity of YN968D1-treated T cells in vivo, respectively.
Results: We found that the use of YN968D1 in CD8+ T cells could reduce the expression levels of inhibitory checkpoints, such as Lag-3, PD-1, and Tim3, escalate the production of IFN-γ and IL-2 and promote the cytotoxicity of T cells dramatically in vitro. The transfer of YN968D1-treated T cells achieved better tumor control compared to DMSO-treated T cells or control in both peritoneal dissemination and subcutaneous gastric cancer mouse models.
Conclusion: Our results indicate that YN968D1 can enhance the T cell-mediated antitumor immunity.
Keywords: YN968D1, gastric cancer, T cells, cytotoxicity, anti-tumor immunity
Background
T cell exhaustion is a state of T cell dysfunction that arises during chronic infections and cancer because of persistent pathogens or cancer cells. Exhausted T cells lose their ability to secrete IL-2, TNF-α, and IFN-γ1 Phenotypic changes in T cells occur in exhausted T cells, including the expression of the immune checkpoints such as programmed cell death receptor-1 (PD-1), cytotoxic T lymphocyte-associated molecule-4 (CTLA-4), lymphocyte activation gene-3 (LAG-3), and T cell immunoglobulin (TIM-3). The most commonly used strategy of immunotherapy is targeting the inhibitory checkpoints. Beyond checkpoint inhibitors, engineered T cells such as chimeric antigen receptor (CAR) T cells and T cell receptor (TCR) modified T cells also show promising results in clinical immunotherapy. Both checkpoint inhibitors and engineered T cells aim to harness the immune system to generate an anti-tumor response by boosting the function of T cells.2
Vascular endothelial growth factor (VEGF) and its receptors play a crucial role in the angiogenesis required for tumor growth. VEGF receptor2 (VEGFR2) is the main signaling VEGFR in blood vascular endothelial cells. In addition to the well-defined role of VEGF in the development of vessels, VEGF can also dampen anti-tumor immunity, resulting in immune escape and tumor development. VEGF promotes the recruitment and proliferation of immunosuppressive regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs), and inhibits the differentiation and activation of dendritic cells (DCs).3,4 Importantly, VEGF can further frustrate antitumor immunity by interfering with the quantity and function of effector T cells. Firstly, VEGF hinders the differentiation of T cells from early hematopoietic progenitor cells5 Secondly, VEGF reduces the cytotoxic activity of T cells derived from the peripheral blood and inhibits the proliferation of T cells in a VEGF receptor2 (VEGFR2) dependent manner.6,7 Finally, VEGF induces T cell exhaustion via VEGFR2 in the tumor microenvironment by increasing the expression levels of inhibitory checkpoints, such as PD-1, CTLA-4, Tim-3, and Lag-3.8
Given the suppressive role of VEGF/VEGFR in T cells, there are some clinical and preclinical studies attempting to restore the function and activation of T cells by targeting VEGF/VEGFR. It was reported that bevacizumab, a humanized anti-VEGF monoclonal antibody, could increase T cell compartments in patients receiving a bevacizumab-based first-line therapy for metastatic colorectal cancer9 In patients with metastatic non-small cell lung cancer, bevacizumab enhanced cytotoxic T-lymphocytes responses10 Sunitinib is a multi-target tyrosine kinase inhibitor blocking VEGFR1, VEGFR 2, and VEGFR3 et al The use of sunitinib in a colon cancer-bearing mouse model resulted in a shift of cytokine and costimulatory molecule expression profiles that could favor T-cell activation and Th1 responses11 Dual angiopoietin-2 and VEGFA inhibition increased the proportion of CD8+ T cells expressing an activated IFN-γ or CD69+ phenotype in both transgenic and transplanted mammary tumor models.12
YN968D1 is a tyrosine kinase inhibitor that selectively inhibits VEGFR2. Previous studies demonstrated that YN968D1 improved progression-free survival (PFS) and overall survival (OS) in patients with refractory gastric or gastroesophageal junction cancer (NCT00970138).13,14 Clinical trials are ongoing for other types of cancer, such as breast cancer and hepatocellular carcinoma to evaluate the efficacy of YN968D1. YN968D1 inhibits tumor angiogenesis by blocking VEGFR2-mediated signaling pathway and exhibits effective antitumor activity15 Though VEGFR2 also plays a key role in suppressing the antitumor immunity, it remains unclear whether it can also modulate the function of T cells like other antiangiogenic agents.
To address this gap, in this study we investigated the effects of YN968D1 on the activation and function of T cells in vitro. We also constructed mice models bearing gastric cancer peritoneal dissemination or subcutaneous tumor to study whether YN968D1 can improve the efficacy of the existing adoptive T cell transfer treatment in vivo.
Materials And Methods
Ethics Statement
All the experiments were performed under approved guidelines. The blood collection procedure was carried out under the guidelines verified and approved by the Ethics Committee of Drum Tower Hospital. All blood donors signed informed consents. The animal experiment was performed under the guidelines verified and approved by the Ethics Committee of Drum Tower Hospital.
Cell Isolation, Culture, And Reagents
Peripheral blood samples were collected from healthy donors in heparinized tubes. Peripheral blood mononuclear cells (PBMCs) were separated using Ficoll-Paque (TBD science, China) and suspended in culture medium consisting of X-VIVO (Lonza, Switzerland) and 10% heat-inactivated FBS (Lonza, Switzerland) at a concentration of 5×106 cells/mL. PBMCs were incubated for 2 hrs at 37 °C under 5% CO2 in 6-well plates. Then, non-adherent cells were removed and suspended in culture medium. We used human anti-CD3 antibody (R&D systems, Minneapolis) at a final concentration of 25ng/mL and recombinant human interleukin-2 (rhIL-2, R&D systems) at a final concentration of 200IU/mL on day 1 for the stimulation and proliferation of T cells. RhIL-2 was added every 3 days for a total of 14 days to obtain the activated T cells. The YN968D1 was provided by Shanghai Hengrui Pharmaceutical Co Ltd (China). The YN968D1 was dissolved in DMSO and later diluted in PBS. The YN968D1 was added to T cells at a final concentration of 20nM.
Cell Counting Kit 8 (CCK8) Assay
The CCK-8 assay was used to test the proliferation of T cells after the treatment with YN968D1. T cells were plated into 96-well plates at a density of 2×103 cells per well in 100μL serum-free X-VIVO. T cells were treated with YN968D1 at various concentrations ranging from 1.5nM to 50000nM with an exponential gradient for 48 hrs. Then the CCK8 assay was performed according to the user manual.
Cytotoxicity Assay
We used the Roche (lactase dehydrogenase) LDH cytotoxicity detection kit to detect the cytotoxicity of the T cells. The assay was based on the measurement of LDH released into the media when the cell membrane was not intact. Tumor cells MKN-45 (104 cells per well) were plated in 96-well plates. Next, the T cells were added at different effector: target ratios (10:1; 5:1; 3:1) for 18–20 hrs. Finally, the LDH release was detected and the results were analyzed according to the manufacturer’s instructions. All tests were done in triplicate.
Flow Cytometry
Cells were harvested and stained with antibodies labeled by fluorescence for 30 mins at 4°C in the dark. The antibodies used in this study included: CD3-FITC (BD Bioscience), CD8-APC (BD Bioscience), PD-1-PE (BD Bioscience), CTLA-4-PE (BD Bioscience), Lag-3-PE (BD Bioscience). Human Th1/Th2 Cytokine Kit (Cytometric Bead Array) was applied to determine the expression level of IL-2, IL-4, IL-6, TNF-α, and IFN-γ. Gating strategy: Firstly, we gated lymphocytes and then CD3+ T cells were gated, at last, CD8+ T cells were gated to determine the expression levels of the inhibitory checkpoint molecules.
ELISPOT Assay
We estimated the frequency of IFN-γ-producing T cells after co-incubation with gastric cancer cell MKN-45 for 18 hrs by IFN-γ ELISPOT kit (Dakewei, China). T cells and MKN-45 were co-incubated as the cytotoxicity assay. The protocol and data analyses have been described previously.16
Human Xenograft Tumor Models
BALB/c nude mice were intraperitoneally or subcutaneously injected with 1×107 human gastric cancer cells MKN-45. Six mice were used for each group. Mice bearing tumors were treated by adoptive T cells transfer on day 10, day 13, and day 17 after tumor planting. For each mouse, approximately 1×107 T cells suspended in 200μl saline were transferred by intraperitoneal injection or intravenous tail injection for each treatment. Tumor volumes were recorded every other day for the subcutaneous model. The volume calculations were obtained using the formula V = (W2 × L)/2, where W was tumor width, L was tumor length and V was tumor volume. Peripheral blood serum was collected to detect cytokine production. The mice were killed and the tumors were excised and weighed on the 14th day after the first adoptive T cells transfer. For the survival analysis, ten mice were used for each group and the endpoint was the death of the mice.
Statistical Analyses
All the results were displayed as mean ± SEM. Students t-test was applied to evaluate the effects of YN968D1 as compared to the DMSO or control. Results were considered statistically significant when p-value <0.05 (two-tailed).
Results
YN968D1 Improved The Cytotoxicity Of T Cells In Vitro
A previous study reported that the anti-VEGFR2 antibody could reverse the increased expressions of PD-1 and other inhibitory checkpoints induced by VEGF produced in the tumor microenvironment8 T cell activation was associated with increased expressions of VEGFR and checkpoints.6,17 Thus, we studied whether VEGFR2 inhibitor YN968D1 could reverse the enhanced expression of checkpoints in activated T cells. The T cell viability was not affected, when the concentration of YN968D1 was within 100nM (Figure 1A). And we found that in vitro YN968D1 did not affect the proportion of CD3+CD8+ T cells either in peripheral blood mononuclear cells (PBMC) or in activated T cells (Figure 1B). The reduction of Lag-3 was most obvious when the concentration of YN968D1 was 20nM (data not shown). As shown in Figure 2, expression levels of inhibitory checkpoints including PD-1, Tim-3, and CTLA-4, increased in activated CD3+CD8+ T cells, compared to PBMC. YN968D1 significantly reduced the expression levels of Lag-3, PD-1, and Tim-3 in both PBMC and CD3+CD8+ T cells in vitro. The decreases were more distinct in activated CD3+CD8+ T cells than PBMC, especially for Lag-3 (P<0.05) (Figure 2B–D). YN968D1 reduced the expression of Lag-3 by approximately 15% in activated CD3+CD8+ T cells (Figure 2B). However, our results indicate that YN968D1 did not affect the expression of CTLA-4 in activated CD3+CD8+ T cells (Figure 2E).
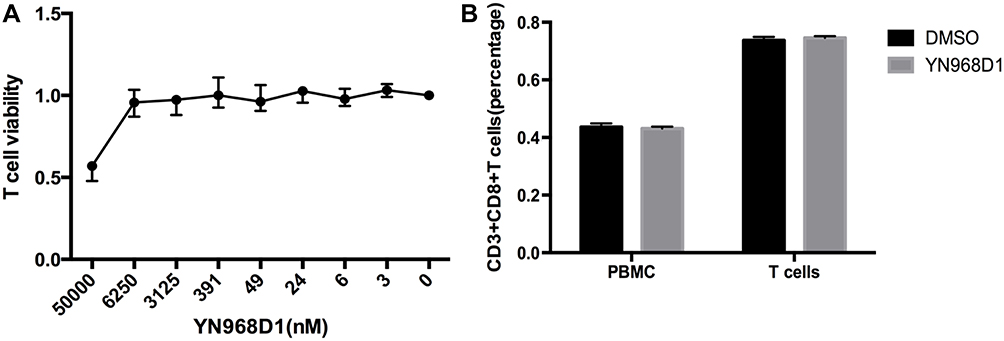 |
Figure 1 (A) The proliferation of activated T cells treated by YN968D1 at various concentrations in vitro for 48hours; (B) The effects of YN968D1 (20nM) on the proportion of CD3+CD8+ T cells in PBMC and activated T cells in vitro. |
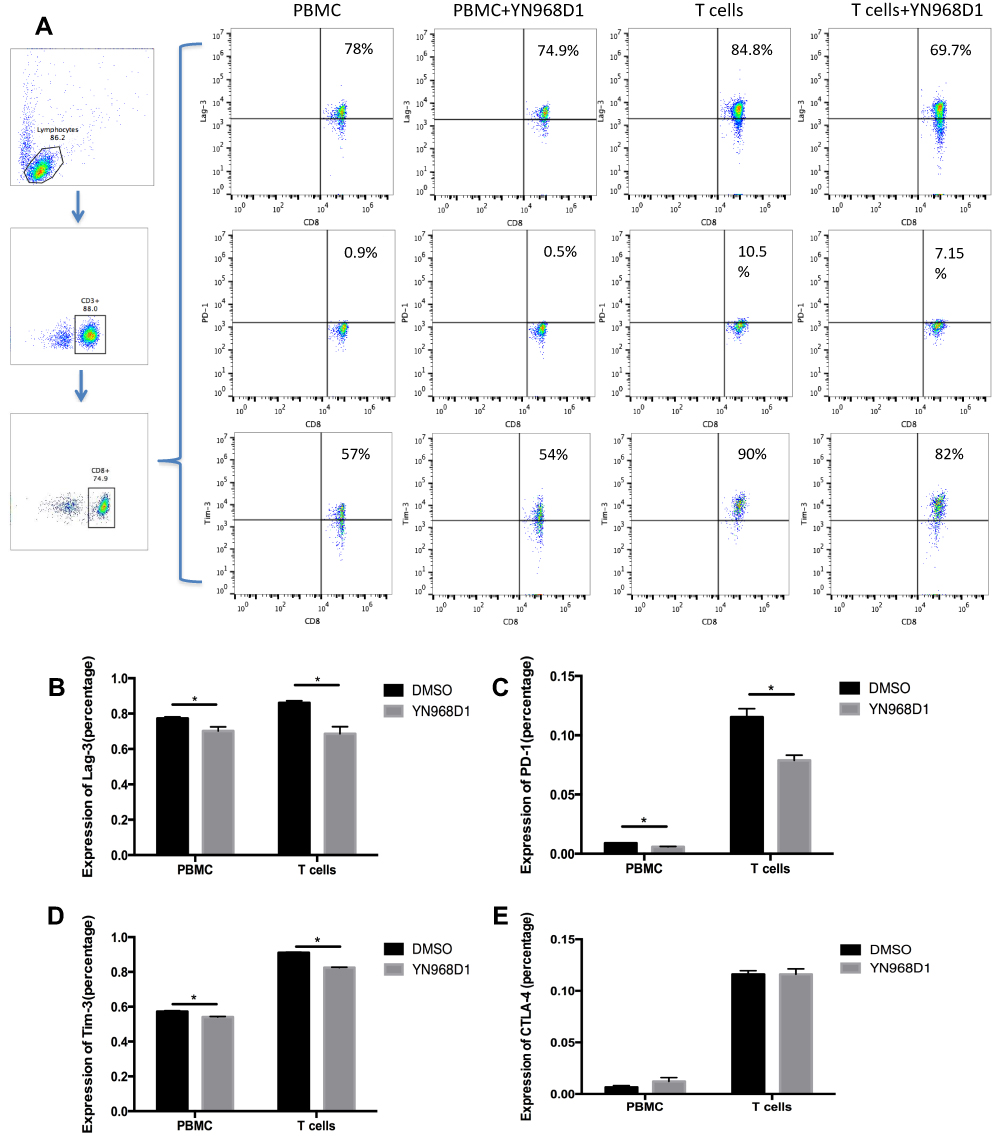 |
Figure 2 PBMC was isolated and T cells were activated as described in materials and methods. PBMC or activated T cells were treated with YN968D1 at a final concentration of 20nM for 48h. (A) Expressions of Lag-3, PD-1, and CTLA-4 in PBMC or activated T cells before and after the use of YN968D1. (B) Expression of Lag-3 in PBMC and activated T cells before and after the use of YN968D1 (20nM). (C) Expression of PD-1 in PBMC and activated T cells before and after the use of YN968D1 (20nM). (D) Expression of Tim-3 in PBMC and activated T cells before and after the use of YN968D1 (20nM). (E) Expression of CTLA-4 in PBMC and activated T cells before and after the use of YN968D1 (20nM). * means P value <0.05. |
As we know, inhibitory checkpoints are highly involved in CD8+ T cell exhaustion and diminish the cytotoxicity of CD8+ T cells. Regarding the role of YN968D1 in lowering expressions of inhibitory checkpoints, we investigated the effects of YN968D1 on the cytotoxicity of activated T cells. The cytotoxicity of immune cells was detected by the measurement of lactate dehydrogenase (LDH) at various effector: target ratios (E: T). The effector and target refer to T cells and MKN-45 gastric cancer cells, respectively. YN968D1 notably enhanced the cytotoxicity of activated T cells at different E: T ratios, particularly when the E: T ratio was 10: 1 (P<0.005) (Figure 3A). Activated T cells elicited enhanced excretions of IL-2 and IFN-γ compared to PBMC and the enhancement was significantly promoted after the addition of YN968D1 (Figure 3C). Though YN968D1 also increased the excretions of IL-4 and TNF-α and decreased IL-6 in activated T cells, the changes were modest and not statistically significant (Figure 3C). The IFN-γ ELISPOT assay was applied to quantify T cell response to MKN-45 gastric cancer cells. The addition of YN968D1 boosted the cytotoxicity of activated T cells to MKN-45 dramatically at different E: T ratios, as shown in Figure 3B and D (P<0.005).
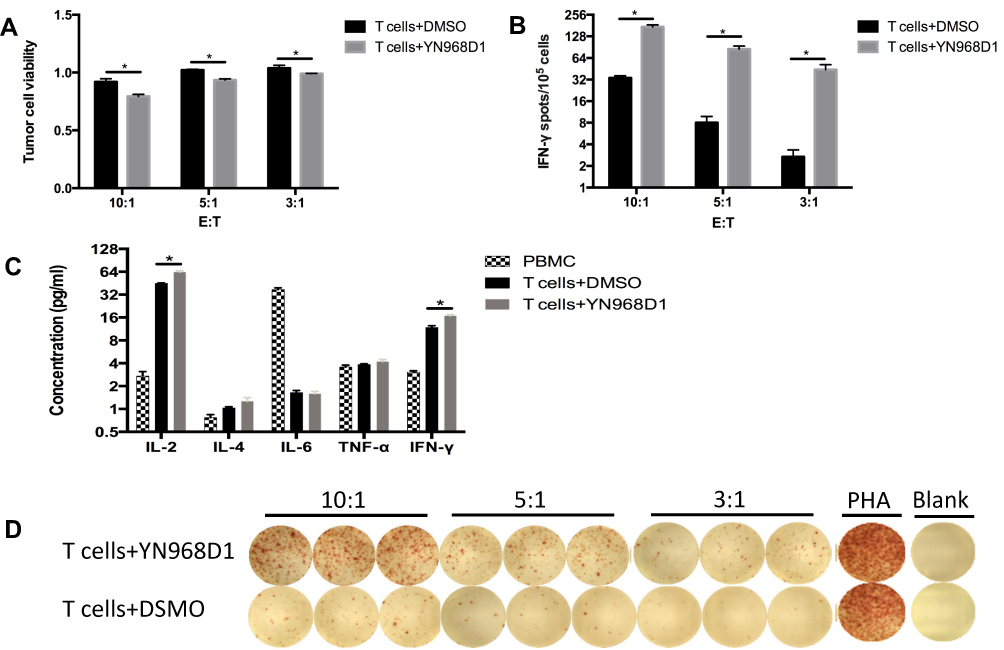 |
Figure 3 (A) The cytotoxicity of T cells was evaluated by the LDH assay at different (E) T ratios. (C) Human Th1/Th2 Cytokine Kit determined the expression level of IL-2, IL-4, IL-6, TNF-α, and IFN-γ of DMSO or YN968D1-treated T cells. (B, D) The frequency of IFN-γ-producing T cells after co-incubation with gastric cancer cell MKN-45 for 18 hrs was evaluated by IFN-γ ELISPOT kit (Dakewei, China). T cells and MKN-45 were co-incubated at different E: T ratios (10:1, 5:1, 3:1). * means P value <0.05. |
YN968D1 Improved The Cytotoxicity Of T Cells In Vivo
MKN-45 cells are gastric cancer cells with high potential for intraperitoneal dissemination. Firstly, we established a xenograft model with intraperitoneal dissemination by intraperitoneal injection of MKN-45 cells. All the intraperitoneal dissemination tumor lesions were resected on the 14th day after the first adoptive T cells transfer. The schematic illustration of the treatment process of peritoneal metastasis tumor model was shown in Figure 4A. As illustrated in Figure 4B, YN968D1-treated T cells significantly lowered the total tumor burden compared with DMSO-treated T cells or control for the intraperitoneal dissemination model. YN968D1-treated T cells decreased the overall weight of lesions with diameters larger than 3mm (Figure 4C) or less than 3mm (Figure 4D) and the number of metastases (Figure 4E) compared with DMSO-treated T cells (P<0.05) or control. YN968D1-treated T cells also prolonged the survival of mice bearing intraperitoneal dissemination compared with DMSO-treated T cells (P<0.05) or control, as shown in Figure 4F. Cytokine release syndrome (CRS) should be taken into account during T cell immunotherapy. IL-6, as an inflammatory mediator and a treatment target for CRS18 was tested in our study to monitor the treatment-related toxicities. As indicated in Figure 4G, expressions of IL-6 in peripheral blood serum were relatively low and similar for each group.
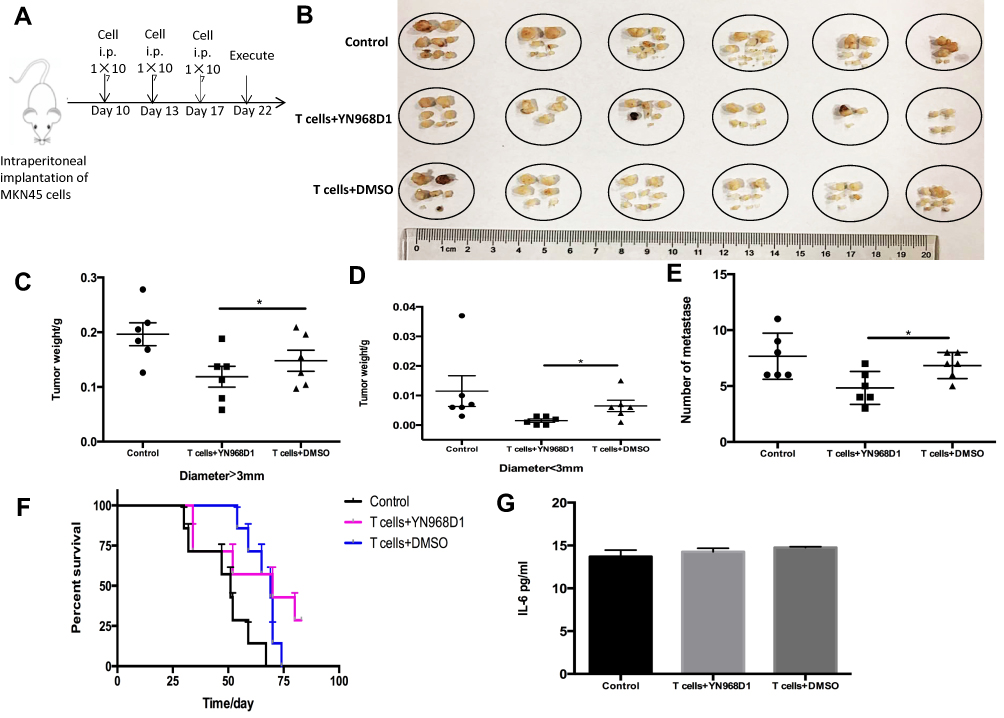 |
Figure 4 (A) A xenograft model with intraperitoneal dissemination or was established by intraperitoneal injection of MKN-45 cells. Schematic illustration of treatment process of peritoneal metastasis tumor model. (B) The mice were killed and the tumors were excised and weighed on the 14th day after the first adoptive T cells transfer. (C) The weight of tumors with a diameter >3mm. (D) The weight of tumors with a diameter <3mm. (E) Number of metastases. (F) The overall survival of mice with intraperitoneal dissemination treated with saline (control), DMSO-treated T cells or YN968D1-treated T cells. (G) Expressions of IL-6 in peripheral blood serum of mice with intraperitoneal dissemination. * means P value <0.05. |
Next, we constructed a subcutaneous xenograft model using the MKN-45 cell line to observe tumor growth. The schematic illustration of treatment process was shown in Figure 5A. On the 14th day after the first adoptive T cells transfer through intravenous tail injection, all the subcutaneous tumors were harvested and weighed. As shown in Figure 5C, YN968D1-treated T cells apparently slowed the subcutaneous tumor growth rate, compared to the DMSO-treated T cells (P<0.05) or control. Similar to the findings in the intraperitoneal dissemination xenograft mouse model, YN968D1-treated T cells resulted in a significantly lower tumor weight (P<0.05), as indicated in Figure 5B and D.
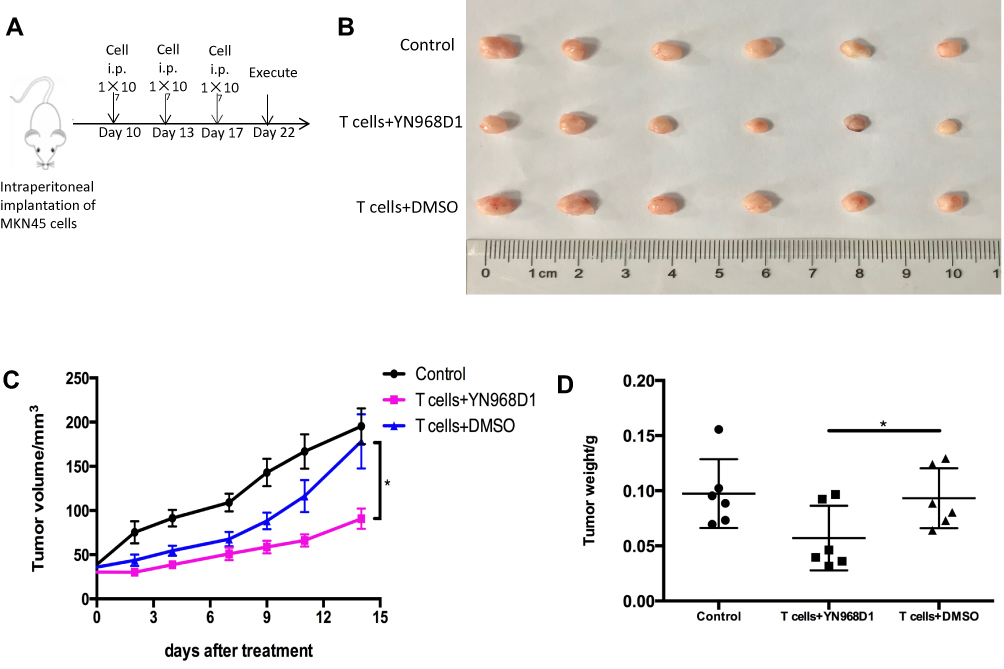 |
Figure 5 BALB/c nude mice were subcutaneously injected with human gastric cancer cells MKN-45. The tumor-bearing mice were treated by saline (control), DMSO-treated T cells or YN968D1-treated T cells, respectively. (A) Schematic illustration of treatment process. (B) Mice bearing tumor were treated by adoptive T cells transfer on day 10, day 13, and day 17 after tumor planting. The mice were killed and the subcutaneous tumors were harvested on the 14th day after the first adoptive T cells transfer. (C) Tumor volumes were recorded every other day for the subcutaneous model. (D) Weights of subcutaneous tumors on the 14th day after the first adoptive T cells transfer. * means P value<0.05. |
Discussion
The preclinical data demonstrated that YN968D1 could partially reverse the expressions of inhibitory checkpoints induced by the activation of T cells, and improve the cytotoxicity of T cells in vitro and in vivo. Our study could pave the way for developing an effective combination of YN968D1 with immunotherapeutic strategies for patients with cancer.
This study found that YN968D1, a novel antiangiogenic agent, could reduce the expression levels of PD-1, Lag-3, and Tim-3 and increase the production of IFN-γ in activated T cells. Our findings are supported by several studies. Voron et al found that anti-VEGFR2 antibody could revert the increased expressions of PD-1, Tim-3, Lag-3, and CTLA-4 induced by VEGF produced in the tumor microenvironment8 Ozao-Choy et al.’s findings indicated that sunitinib could reduce the expression of PD-1 and CTLA-4 while increasing the production of IFN-γ in tumor-infiltrating lymphocytes in a colon cancer-bearing mouse model11 However, our study found that YN968D1 did not exert an influence on the expression of CTLA-4 in activated T cells. This is possible because we investigated T cells activated by CD3 antibody and IL-2 in vitro but not tumor-infiltrating lymphocytes in the VEGF-producing microenvironment. Another reason was the use of different agents. YN968D1 used in our study is a small molecule that elicits a selective inhibitory activity against VEGFR signaling,19,20 while Voron et al used anti-VEGFR2 antibody8 and sunitinib, investigated by Ozao-Choy et al.11 is a multi-target inhibitor that can block VEGFR1, VEGFR 2, and VEGFR3, platelet-derived growth factor receptors α and β, stem cell factor receptor, and Flt3.21
Antiangiogenic agents can inhibit tumor growth by shutting down the blood supply and also confer immune-modulatory effects, but the efficacy of using antiangiogenic agents alone is still limited in clinical practice. Our study suggests the absolute reduction of PD-1 expression induced by YN968D1 was only around 3–5%, which is relatively low, though the difference was statistically significant, as shown in Figure 2. One reason might be the upregulation of PD-L1 was dependent on IFN-γ produced by activated CD8+ T cells22 and further elevated after the use of YN968D1 (Figure 3C). The results indirectly indicated that it might be promising to combine the antiangiogenic therapy with a checkpoint inhibitor. A preclinical study by Schmittnaegel et al suggested that PD-1 blockade could improve tumor control by a dual angiopoietin-2 and VEGF inhibitor.12 A recent study demonstrated that anti-PD-L1 therapy could sensitize tumors to antiangiogenic therapy and prolong its efficacy, and conversely, antiangiogenic therapy could improve anti-PD-L1 treatment by facilitating enhanced CTL infiltration, activity, and tumor cell destruction.23 In addition to the preclinical studies, there are phase I/II clinical trials assessing the safety and tolerability of combining drugs targeting VEGF/VEGFRs with immune checkpoints antibodies/inhibitors in patients with various types of solid cancer (NCT01472081; NCT02348008; NCT02039674; NCT02400385; NCT02337491; NCT02443324; NCT01633970)3 NCT00790010 is a phase I clinical trial to investigate the effect of ipilimumab (anti-CTLA-4) plus bevacizumab (anti-VEGF) in patients with metastatic melanoma. In this trial, patients undergoing combination therapy showed a great advantage in OS (median OS, ipilimumab plus bevacizumab vs. ipilimumab: 25.1 vs. 10.1 months) compared with the results of previous studies.24
In our study, we used YN968D1 at a low concentration to promote the cytotoxicity of activated T cells in vitro and in vivo, indicating high doses might be not appropriate to enhance anti-tumor immunity. Vascular normalization and hypoxia correction has been assumed to be the general mechanism of anti-angiogenic agents and can result in increasing infiltration and activity of T cells in the tumor microenvironment. Anti-angiogenic agents may at first improve the structure and the function of tumor vessels by normalization, however, continued or increased administration can produce vessel pruning, resulting in a more hypoxic environment.25,26 This aspect has been supported by a few studies. Huang et al found that lower doses of anti-VEGFR2 antibody could reprogram the tumor microenvironment toward potentiation of cancer vaccine therapies.27 Shi et al found that a high dose of anti-VEGF agent could obviously diminish the oxygenation in vivo, which was observed throughF-MRI and multispectral analysis.28,29
The strengths of this study include the fact that it is the first to study the effects of YN968D1 on T cells and we have used different models to validate our findings, in addition to the data in vitro. Our study is limited by the use of immunocompromised mouse models. Therefore, we did not investigate the influence of YN968D1 on other immune cells, such as Tregs and MDSCs in the tumor microenvironment. In the future, we will study the effects of YN968D1 on the tumor immune microenvironment using immunocompetent tumor-bearing mouse model.
Conclusion
In conclusion, immunity was involved in the treatment response of YN968D1. Conversely, YN968D1 can promote the cytotoxicity of activated T cells for gastric cancer in vivo and in vitro. It might be promising to combine YN968D1 with immunotherapies for patients with gastric cancer.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12:492–499. doi:10.1038/ni.2035
2. Marin-Acevedo JA, Soyano AE, Dholaria B, Knutson KL, Lou Y. Cancer immunotherapy beyond immune checkpoint inhibitors. J Hematol Oncol. 2018;11:8. doi:10.1186/s13045-017-0552-6
3. Lapeyre-Prost A, Terme M, Pernot S, et al. Immunomodulatory activity of VEGF in cancer. Int Rev Cell Mol Biol. 2017;330:295–342.
4. Yang J, Yan J, Liu B. Targeting VEGF/VEGFR to modulate antitumor immunity. Front Immunol. 2018;9:978. doi:10.3389/fimmu.2018.00978
5. Ohm JE, Gabrilovich DI, Sempowski GD, et al. VEGF inhibits T-cell development and may contribute to tumor-induced immune suppression. Blood. 2003;101:4878–4886. doi:10.1182/blood-2002-07-1956
6. Ziogas AC, Gavalas NG, Tsiatas M, et al. VEGF directly suppresses activation of T cells from ovarian cancer patients and healthy individuals via VEGF receptor type 2. Int J Cancer. 2012;130:857–864. doi:10.1002/ijc.v130.4
7. Gavalas NG, Tsiatas M, Tsitsilonis O, et al. VEGF directly suppresses activation of T cells from ascites secondary to ovarian cancer via VEGF receptor type 2. Br J Cancer. 2012;107:1869–1875. doi:10.1038/bjc.2012.468
8. Voron T, Colussi O, Marcheteau E, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med. 2015;212:139–148. doi:10.1084/jem.20140559
9. Manzoni M, Rovati B, Ronzoni M, et al. Immunological effects of bevacizumab-based treatment in metastatic colorectal cancer. Oncology. 2010;79:187–196. doi:10.1159/000320609
10. Martino EC, Misso G, Pastina P, et al. Immune-modulating effects of bevacizumab in metastatic non-small-cell lung cancer patients. Cell Death Discov. 2016;2:16025. doi:10.1038/cddiscovery.2016.25
11. Ozao-Choy J, Ma G, Kao J, et al. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. 2009;69:2514–2522. doi:10.1158/0008-5472.CAN-08-4709
12. Schmittnaegel M, Rigamonti N, Kadioglu E, et al. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci Transl Med. 2017;9:eaak9670. doi:10.1126/scitranslmed.aak9670
13. Li J, Qin S, Xu J, et al. Apatinib for chemotherapy-refractory advanced metastatic gastric cancer: results from a randomized, placebo-controlled, parallel-arm, phase II trial. J Clin Oncol. 2013;31:3219–3225. doi:10.1200/JCO.2013.48.8585
14. Li J, Qin S, Xu J, et al. Randomized, double-blind, placebo-controlled phase III trial of apatinib in patients with chemotherapy-refractory advanced or metastatic adenocarcinoma of the stomach or gastroesophageal junction. J Clin Oncol. 2016;34:1448–1454. doi:10.1200/JCO.2015.63.5995
15. Tian S, Quan H, Xie C, et al. YN968D1 is a novel and selective inhibitor of vascular endothelial growth factor receptor-2 tyrosine kinase with potent activity in vitro and in vivo. Cancer Sci. 2011;102:1374–1380. doi:10.1111/cas.2011.102.issue-7
16. Su S, Zou Z, Chen F, et al. CRISPR-Cas9-mediated disruption of PD-1 on human T cells for adoptive cellular therapies of EBV positive gastric cancer. Oncoimmunology. 2017;6:e1249558. doi:10.1080/2162402X.2016.1249558
17. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. doi:10.1038/nrc3239
18. Tanaka T, Narazaki M, Kishimoto T. Immunotherapeutic implications of IL-6 blockade for cytokine storm. Immunotherapy. 2016;8:959–970. doi:10.2217/imt-2016-0020
19. Hu X, Zhang J, Xu B, et al. Multicenter phase II study of apatinib, a novel VEGFR inhibitor in heavily pretreated patients with metastatic triple-negative breast cancer. Int J Cancer. 2014;135:1961–1969. doi:10.1002/ijc.28829
20. Peng H, Zhang Q, Li J, et al. Apatinib inhibits VEGF signaling and promotes apoptosis in intrahepatic cholangiocarcinoma. Oncotarget. 2016;7:17220–17229. doi:10.18632/oncotarget.7948
21. Sun L, Liang C, Shirazian S, et al. Discovery of 5-[5-fluoro-2-oxo-1,2- dihydroindol-(3Z)-ylidenemethyl]-2,4- dimethyl-1H-pyrrole-3-carboxylic acid (2-diethylaminoethyl)amide, a novel tyrosine kinase inhibitor targeting vascular endothelial and platelet-derived growth factor receptor tyrosine kinase. J Med Chem. 2003;46:1116–1119. doi:10.1021/jm0204183
22. Spranger S, Spaapen RM, Zha Y, et al. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med. 2013;5:200ra116. doi:10.1126/scitranslmed.3006504
23. Allen E, Jabouille A, Rivera LB, et al. Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci Transl Med. 2017;9:eaak9679. doi:10.1126/scitranslmed.aak9679
24. Yi M, Jiao D, Qin S, et al. Synergistic effect of immune checkpoint blockade and anti-angiogenesis in cancer treatment. Mol Cancer. 2019;18:60. doi:10.1186/s12943-019-0974-6
25. Rapisarda A, Melillo G. Overcoming disappointing results with antiangiogenic therapy by targeting hypoxia. Nat Rev Clin Oncol. 2012;9:378–390. doi:10.1038/nrclinonc.2012.64
26. Bueno MJ, Mouron S, Quintela-Fandino M. Personalising and targeting antiangiogenic resistance: a complex and multifactorial approach. Br J Cancer. 2017;116:1119–1125. doi:10.1038/bjc.2017.69
27. Huang Y, Yuan J, Righi E, et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci U S A. 2012;109:17561–17566. doi:10.1073/pnas.1215397109
28. Shi Y, Oeh J, Eastham-Anderson J, et al. Mapping in vivo tumor oxygenation within viable tumor by 19F-MRI and multispectral analysis. Neoplasia. 2013;15:1241–1250. doi:10.1593/neo.131468
29. Shi Y, Oeh J, Hitz A, et al. Monitoring and targeting anti-VEGF induced hypoxia within the viable tumor by 19F-MRI and multispectral analysis. Neoplasia. 2017;19:950–959. doi:10.1016/j.neo.2017.07.010
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.