")
Back to Journals » Lung Cancer: Targets and Therapy » Volume 10
Immune Checkpoint Inhibitors for the Treatment of Unresectable Stage III Non–Small Cell Lung Cancer: Emerging Mechanisms and Perspectives
Received 2 November 2019
Accepted for publication 20 December 2019
Published 3 January 2020 Volume 2019:10 Pages 161—170
DOI https://doi.org/10.2147/LCTT.S184380
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sai-Hong Ignatius Ou
Hiroyuki Inoue,1,2 Isamu Okamoto1
1Research Institute for Diseases of the Chest, Graduate School of Medical Sciences, Kyushu University, Fukuoka, Japan; 2Center for Clinical and Translational Research, Kyushu University Hospital, Fukuoka, Japan
Correspondence: Isamu Okamoto
Research Institute for Diseases of the Chest, Graduate School of Medical Sciences, Kyushu University, 3-1-1 Maidashi, Higashi-Ku, Fukuoka 812-8582, Japan
Tel +81-92-642-5378
Fax +81-92-642-5390
Email [email protected]
Abstract: There has been no improvement in outcome for patients with unresectable locally advanced (stage III) non–small cell lung cancer (NSCLC) for more than 10 years. The standard treatment for these patients is definitive concurrent chemotherapy and radiation (CCRT). Although the goal of treatment in this setting is to achieve a cure, most patients progress and their prognosis is poor, with a 5-year survival rate of 15–30%. There is thus an urgent need for the development of novel anticancer treatments in this patient population. Recent advances in cancer immunotherapy have led to a marked improvement in clinical outcome for advanced NSCLC. Such immunotherapy mainly consists of the administration of immune checkpoint inhibitors (ICIs) such as antibodies to cytotoxic T lymphocyte–associated protein–4 (CTLA-4) or to either programmed cell death–1 (PD-1) or its ligand PD-L1. Durvalumab (MEDI4736) is a high-affinity human immunoglobulin G1 monoclonal antibody that blocks the binding of PD-L1 on tumor cells or antigen-presenting cells to PD-1 on T cells. The PACIFIC study recently evaluated consolidation immunotherapy with durvalumab versus placebo administered after concurrent chemoradiotherapy (CCRT) in patients with unresectable stage III NSCLC. It revealed a significant improvement in both progression-free and overall survival with durvalumab, and this improvement was associated with a favorable safety profile. This achievement has made durvalumab a standard of care for consolidation after CCRT in patients with unresectable stage III NSCLC, and it has now been approved in this setting by regulatory agencies in the United States, Canada, Japan, Australia, Switzerland, Malaysia, Singapore, India, and the United Arab Emirates. In this review, we briefly summarize the results of the PACIFIC trial, including those of post hoc analysis, and we address possible molecular mechanisms, perspectives, and remaining questions related to combined treatment with CCRT and ICIs in this patient population.
Keywords: durvalumab, PD-L1, immunogenic cell death, lung cancer
Introduction
Non–small cell lung cancer (NSCLC) is the leading cause of cancer-related mortality worldwide, being one of the most common neoplasms in developed countries and having a poor prognosis.1 Early stages (I and II) account for ~20% of lung cancer diagnoses, with affected individuals having a 5-year survival rate of 40% to 70% after standard surgical treatment (lobectomy with systemic lymph node resection). Approximately 20% to 25% of NSCLC cases are diagnosed after the disease has progressed to clinical stage III.
Although locally advanced (stage III) NSCLC is heterogeneous, it is defined as having spread locoregionally through primary tumor extension into extrapulmonary structures (T3 or T4) and involving hilar or mediastinal lymph nodes (N1–N3), but without distant metastases (M0). At this stage, even if the cancer is considered unresectable, the treatment strategy should be to attain a cure. At the time of initial diagnosis, it is imperative for medical oncologists to deliberately choose the best treatment strategy for each patient through assembly of a multidisciplinary treatment team including thoracic surgeons and radiation oncologists, although the indication for surgical treatment of clinical N2 stage III NSCLC may vary across institutions.
For more than a decade, concomitant chemoradiotherapy (CCRT) has remained the standard treatment for unresectable stage III NSCLC, irrespective of tumor histology or molecular characteristics. The expected survival at 5 years for such patients is only 15% to 30%, however,2–4 highlighting the fact that most are not cured by CCRT5,6 and undergo relapse, with nearly 40% manifesting locoregional recurrence and ~50% developing distant metastases.7,8 This situation clearly calls for the development of novel anticancer treatments to augment the rate of cure or to improve clinical outcome. Given the high risk of metastasis and short progression-free survival (PFS) after CCRT, consolidation therapy defined as treatment administered after a defined number of chemotherapy cycles with or without radiotherapy9 has been considered a possible strategy to improve clinical outcome. Whereas the development of molecularly targeted therapy has improved clinical outcome in advanced NSCLC, it has not affected the management of stage III NSCLC patients. Indeed, there have been no substantial advances in the treatment of unresectable stage III NSCLC for more than a decade despite the performance of numerous randomized Phase III trials including those of induction or consolidation therapy with chemotherapeutic agents, biologics, or a cancer vaccine.8,10−12
In contrast to the failure to develop new therapies for unresectable stage III NSCLC, much progress has been made in our understanding of the underlying mechanisms of tumor immunology in particular, with regard to the role of immune checkpoints, which contribute to suppression of the tumor-associated antigen (TAA)–specific antitumor immune response, also referred to as T cell exhaustion.13
The extent of T cell activation is coordinately determined by interaction of the T cell receptor (TCR) with the antigen on antigen-presenting cells (APCs) as well as by costimulatory or co-inhibitory interactions of CD28 on T cells with CD80 or CD86 on APCs and of PD-1 (programmed cell death–1) on T cells with PD-L1 (programmed cell death–ligand 1) on APCs. Tumor cells also express PD-L1 as a co-inhibitory ligand, which contributes to evasion of the protective antitumor immune response and thereby promotes tumor growth.14 Immune checkpoint inhibitors (ICIs) that target the interaction between PD-L1 on tumor cells and PD-1 on exhausted T cells can reinvigorate the host immune system and allow it to mediate the cytolytic destruction of tumor cells.15 Such inhibitors including nivolumab, pembrolizumab, and atezolizumab have shown a high antitumor efficacy and have revolutionized the treatment of advanced (incurable) NSCLC, and they are now the standard second-line treatment as monotherapy as well as a standard first-line treatment in combination with chemotherapy, irrespective of the PD-L1 status of the tumor tissue.16–18 Given the broad clinical benefit of these ICI-based immunotherapies for subsets of patients with metastatic NSCLC, PD-1/PD-L1 inhibitors have been considered a potential option to improve curative treatment regimens for stage III NSCLC.
Durvalumab (MEDI4736), a fully human monoclonal antibody to PD-L1 that blocks the binding of PD-L1 to PD-1 (as well as that to CD80), was evaluated in the pivotal PACIFIC study19,20 and was approved in July 2018 in Japan as a consolidation therapy after CCRT for unresectable stage III NSCLC. We here discuss the results of this practice-changing trial including Japanese patients and address remaining questions regarding the clinical implementation of ICIs in this stage setting.
PACIFIC Study: Main Results
The PACIFIC study was conducted in patients with unresectable stage III NSCLC who were not selected on the basis of tumor histology or PD-L1 expression level. This phase III study compared the safety and efficacy of consolidation therapy with durvalumab versus placebo in patients who did not experience disease progression after induction therapy with of CCRT. This global study enrolled 713 patients at 234 participating centers. Patients randomly assigned to the experimental arm received durvalumab at 10 mg/kg intravenously every 2 weeks for up to 1 year, beginning 1 to 42 days after the completion of CCRT. Key exclusion criteria were as follows; previous exposure to anti–PD-1 or PD-L1 antibodies; receipt of immunotherapy or an investigational drug within 4 weeks before the first dose (6 weeks for monoclonal antibodies); active or previous autoimmune disease (within the past 2 years) or a history of primary immunodeficiency; evidence of uncontrolled, concurrent illness or ongoing or active infections; unresolved toxic effects of grade 2 or higher (according to the Common Terminology Criteria for Adverse Events [CTCAE]); and grade 2 or higher pneumonitis from previous chemoradiotherapy. The primary end points of the study were PFS and overall survival (OS). PFS, as assessed by a blinded independent central review, was significantly improved with durvalumab versus placebo (17.2 versus 5.6 months; hazard ratio [HR], 0.51; P<0.001). Durvalumab treatment also conferred a significant improvement in OS rate at 2 years, with an absolute difference of 10.7% (66.3%, with a 95% confidence interval [CI] of 61.7–70.4%, in the durvalumab arm versus 55.6% [95% CI, 48.9–61.8%] in the placebo arm; P=0.005). After a median follow-up time of 25.2 months, the median OS had not been reached with durvalumab and was 28.7 months with placebo (HR, 0.68; P=0.0025).20 As reported at the annual meeting of the American Society of Clinical Oncology in 2019, updated OS data were consistent with those previously reported (stratified HR of 0.69, with a 95% CI of 0.55–0.86), with the median still not having been reached (95% CI, 38.4 months to not reached) with durvalumab compared with a value of 29.1 months (95% CI, 22.1–35.1 months) with placebo. The 3-years OS rate was 57.0% with durvalumab and 43.5% with placebo, with an absolute difference of 13.5%, which is larger than that seen for the 2-year OS rate. The development of brain metastasis occurred at a lower frequency in the durvalumab arm than in the placebo arm (6.3% versus 11.8%). With regard to toxicity, adverse events (AEs) of grade 3 or 4 of any cause occurred in 30.5% and 26.1% of patients who received durvalumab and placebo, respectively. Frequent AEs leading to discontinuation of the trial regimen included pneumonitis (4.8% and 2.6% in the durvalumab and placebo arms, respectively), radiation pneumonitis (1.3% each), and pneumonia (1.1% and 1.3%, respectively). Safety outcomes were similar regardless of PD-L1 status. Collectively, these results showed that durvalumab therapy conferred a significantly longer OS and PFS compared with placebo and was associated with a well-tolerated safety profile. On the basis of this impressive clinical benefit, the US Food and Drug Administration as well as regulatory agencies in Canada, Japan, Australia, Switzerland, Malaysia, Singapore, India, and the United Arab Emirates approved durvalumab as consolidation therapy after CCRT for nonselected patients with unresectable stage III NSCLC. In contrast to negative outcomes in prior six randomized phase II
PACIFIC Study: Post Hoc Analysis
The OS and PFS benefit of durvalumab was apparent in all prespecified subgroups of the trial, including those based on tumor histology, smoking history, clinical stage, and level of PD-L1 expression in tumor tissue (defined as high versus low or negative). Exploratory post hoc analysis showed an improved PFS and OS with durvalumab regardless of chemotherapy or radiation dose administered or of the time from radiation or randomization to study treatment. Post hoc analysis of OS in patients with different levels of PD-L1 expression in archived tumor samples that were obtained before CCRT and retrospectively stained with the Ventana SP263 immunohistochemistry assay revealed that, among the 63% of patients with evaluable PD-L1 expression, those with a PD-L1 tumor proportion score (TPS) of <1% (PD-L1–negative tumors) might not have benefited from durvalumab (HR of 1.36, with a 95% CI of 0.79–2.34), in contrast to those with higher levels of PD-L1 expression (PD-L1–positive tumors). On the basis of this failure to clearly establish an OS benefit in individuals with PD-L1–negative tumors, who constituted a relatively small subset of patients, the European Medicines Agency made the decision to limit the use of durvalumab to patients with PD-L1–expressing tumors, even though the subgroup of patients of unknown PD-L1 status showed a strong survival benefit, consistent with that of the intention-to-treat (ITT) population. It is possible that this apparent lack of an OS benefit in patients with a PD-L1 TPS of <1% was the result both of uncertainties associated with unplanned post hoc analysis and of differences in patient characteristics between the durvalumab arm and the placebo arm, given that the OS curves for patients treated with placebo differed substantially between those with a PD-L1 TPS of <1% and those with a PD-L1 TPS of ≥ 1%. In addition, it may be of note that the archived tumor tissue assayed for PD-L1 was obtained only before CCRT, and not after. Given that preclinical and clinical studies have shown that CCRT can up-regulate PD-L1 expression in tumors,21–23 the PD-L1 expression level apparent in tumors before CCRT may change during the interval before initiation of consolidation therapy and may thus be unsuitable as a predictive biomarker for durvalumab. The identification of novel and reliable predictive biomarkers for ICI consolidation therapy after CCRT should thus be a priority.
Immunogenic Cell Death (ICD) as a Molecular Mechanism of the Antitumor Effect of CCRT Followed by ICIs
Evidence that radiotherapy or chemotherapy, or the combination thereof, can induce ICD24–26 suggests that this phenomenon might contribute to the clinical benefit of combination immunotherapy. Indeed, several preclinical studies have shown that chemotherapy and radiotherapy sensitize tumor cells to ICIs-based immunotherapy.27–29 Although the dichotomy of ICD versus non-ICD has not been clearly defined, the immunogenic potential in tumor lesions is dependent on the initiating stimulus, such as the type of chemotherapy and the radiation dose, as well as on the genetic background of the host, the cellular origin of the tumor, and the type of cancer cell death including apoptosis, necroptosis, and pyroptosis as forms of programmed cell death (PCD) and necrosis as a type of non-PCD (Figure 1).25,30–33 On induction of ICD, the dying cancer cells release or expose on their surface a range of molecules that can act as either adjuvants or danger signals for the innate immune system (Figure 2).34 These danger signals, referred to as damage-associated molecular patterns (DAMPs),35 are thought to contribute to the potential immunogenicity of stressed or dying uninfected cells. DAMPs released from dying cells can serve as an “eat me” signal to stimulate the uptake of TAAs by APCs, especially dendritic cells (DCs), as well as the maturation of these cells. The mature DCs then migrate to local tumor-draining lymph nodes, where they effectively prime TAA-specific T cells and generate secondary CD4+ helper T (TH) cell– and CD8+ cytotoxic T lymphocyte (CTL)–mediated immunity directed toward primary TAAs as well as the newly released TAAs, including neoantigens that arise from cancer-specific mutations.36 This broadening of the secondary immune response due to the increased accessibility of new antigens released from collapsing tumor cells is known as epitope spreading. The CD4+ TH1 cells amplify and sustain the differentiation of CD8+ CTLs into effector and memory phenotypes. The generated effector CTLs traffic back to the residual and micrometastatic tumor lesions through the systemic circulation and eventually reside in and mediate the cytolytic destruction of these lesions through the release of cytotoxic granules.
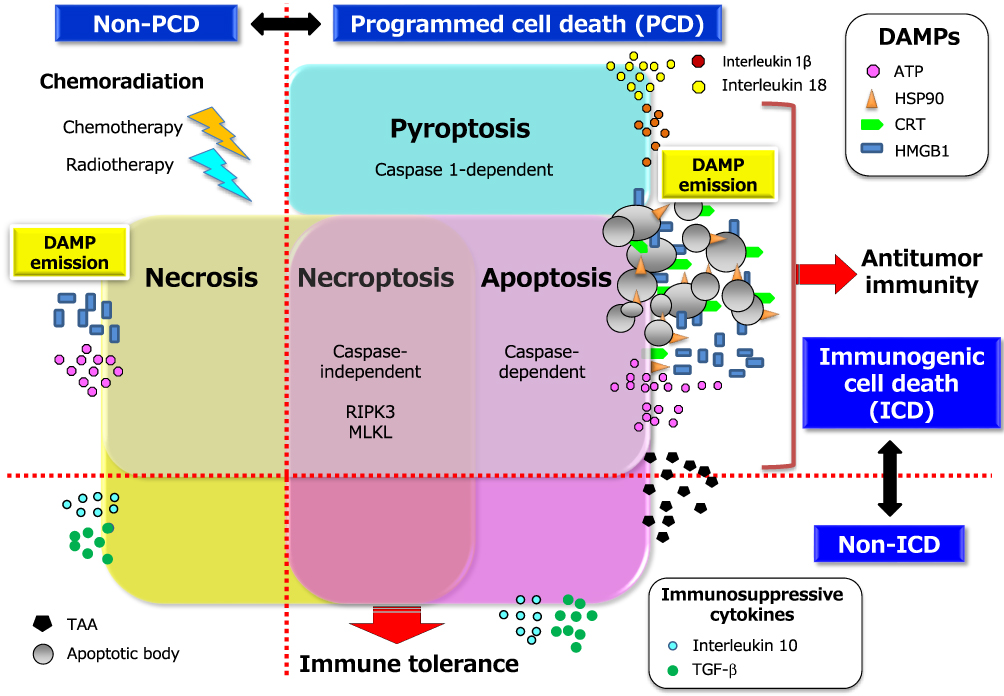 |
Figure 1 Two conceptual dichotomies among multiple forms of cancer cell death: ICD versus non-ICD, and PCD versus non-PCD. The multiple forms of cancer cell death include apoptosis, necroptosis, pyroptosis, and necrosis. The first three of these types of cell death are classified as PCD, and the last as non-PCD. PCD is defined as the death of a cell mediated by an active intracellular program, and is therefore also referred to as cellular suicide. Cell death can also be classified as ICD or non-ICD depending on the extent of DAMP induction or release from dying cells in response to chemoradiation. Representative DAMPs, as hallmarks of ICD, include CRT and HSP90 exposed on the outer surface of the cell membrane as well as ATP and HMGB1 released into the extracellular space. CRT, which promotes the engulfment of TAAs derived from cancer cells by DCs, is exposed only on cells undergoing immunogenic apoptosis, being absent on cells dying in an immunologically silent manner. Non-ICD can be accompanied by the release of immunosuppressive cytokines such as transforming growth factor–β (TGF-β) and interleukin 10 that are abundant in tumor tissue as well as by enrichment of regulatory T cells, both of which may promote tumor tolerance. The precise expression patterns of DAMPs and the balance between these two dichotomies in cell death types may determine the fate of the subsequent adaptive immune response namely, immune tolerance versus antitumor immunity. Abbreviations: RIPK3, receptor-interacting kinase 3; MLKL, mixed lineage kinase domain-like. |
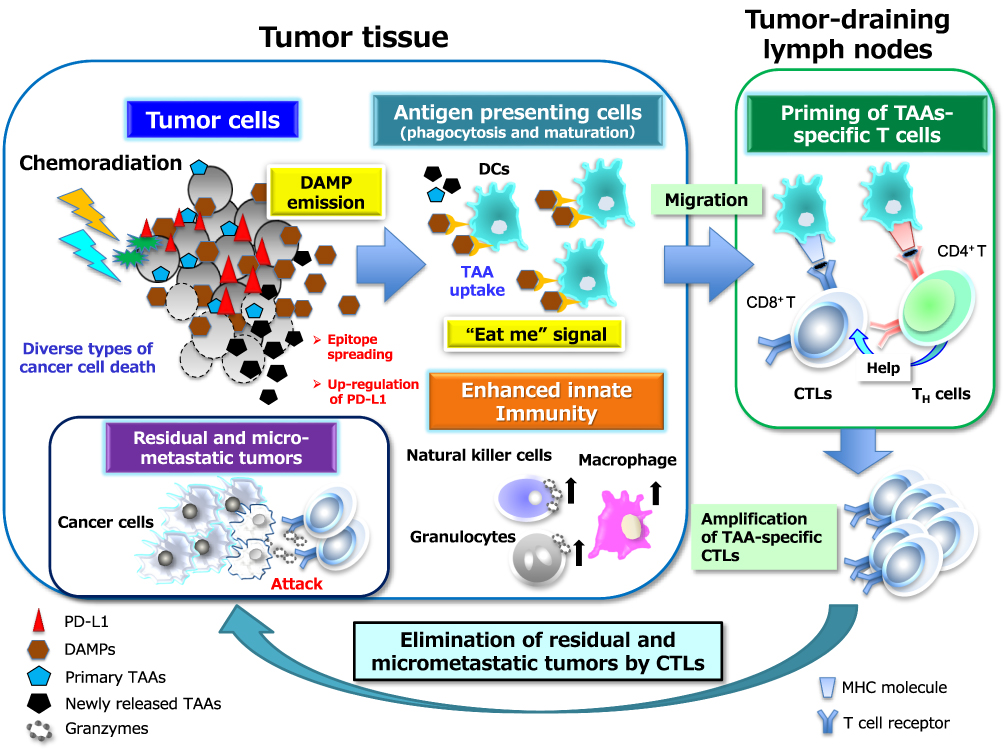 |
Figure 2 Chemoradiation-induced ICD and DAMP-mediated secondary antitumor immunity. The death of tumor cells induced by chemoradiation can be accompanied by the release or exposure of various DAMPs including ATP, HMGB1, and CRT. The interaction of these DAMP molecules with their cognate receptors including P2X7R, TLR4 or RAGE (receptor for advanced glycation end-products), and CD91, respectively can then facilitate both phagocytosis of primary and newly released TAAs by acting as an “eat me” signal as well as the maturation of APCs, particularly DCs. The mature DCs migrate to local tumor-draining lymph nodes to initiate priming of T cells mediated by the specific binding of antigenic peptide–MHC complexes on DCs to cognate TCRs. This binding elicits a signaling cascade from the TCR complex that ultimately triggers gene expression programs that induce the transition of the T cells from their resting state to a state of activation and proliferation. This transition also requires the presence of costimulatory signals elicited by the binding of the ligands CD80 or CD86 on APCs to their receptor CD28 on T cells as well as by that of the ligands OX40 and 4-1BB on APCs to their receptors on T cells. In general, the expression of costimulatory ligands is suppressed on APCs present in tumor tissue, but this suppression can be overcome by endogenous adjuvants such as certain DAMPs that activate pattern recognition receptors on APCs. Appropriate DAMP induction, acting through DC activation, may thus contribute to effective priming and generation of secondary CD4+ TH cell– or CD8+ CTL–mediated immunity directed toward primary TAAs and newly released TAAs including neoantigens (a phenomenon known as epitope spreading). TH1-skewing cytokines released from CD4+ TH1 cells amplify and sustain the expansion of CD8+ CTLs and their differentiation into effector and memory phenotypes. These multistep processes may eventually lead to the durable control of residual and micrometastatic tumor lesions by effector CTLs in a manner dependent on the induction of tumor cell death by directed exocytosis of cytolytic effectors such as granzymes. All abbreviations are defined in the text. |
DAMPs include both secreted or released molecules such as high mobility group protein B1 (HMGB1) and components of polynucleotides such as adenosine triphosphate (ATP), as well as molecules exposed on the outer leaflet of the plasma membrane such as calreticulin (CRT) and heat shock protein 90 (HSP90). A preclinical study showed that radiotherapy induced ICD in a dose-dependent manner as well as enhanced the induction of such death by chemotherapeutic agents (carboplatin or paclitaxel), as evidenced by the up-regulation of three DAMPs molecules, in mouse breast cancer cells.37 In the clinical setting, preoperative chemoradiotherapy in patients with esophageal squamous cell carcinoma was shown to induce tumor antigen–specific T cell responses, and the increase in the serum HMGB1 concentration was found to positively correlate with clinical outcome.38 These findings provide support for the notion that the induction of ICD by CCRT regimens may contribute at least in part to the fostering of a peritumoral proimmunogenic milieu.
Effector CTLs with an expanded TCR repertoire generated as a result of ICD in primary tumors must migrate to and infiltrate the tumor tissue in order to control tumor growth. However, tumors deploy numerous countermeasures to evade such attack, including activation of the PD-1–PD-L1 checkpoint,39 physical barriers formed by abnormal vasculature and stromal cells, and disruption of chemokine-mediated guidance of T cells to the tumor tissue.40 The binding of PD-L1 on tumor cells to PD-1 on T cells suppresses T cell migration and proliferation as well as the secretion of cytotoxic mediators, induces apoptosis of CTLs, and restricts tumor cell killing. However, inhibitors of PD-L1 such as durvalumab disrupt the PD-1–PD-L1 axis and thereby reverse T cell suppression and enhance secondary endogenous antitumor immunity, resulting in a durable antitumor response in cancer patients (Figure 3). DAMPs may therefore play an essential role in linking the antitumor effect of CCRT to promotion of a response to ICIs that target the PD-1–PD-L1 axis, although the most effective molecules among DAMPs and the optimal timing of their emission from tumor lesions during chemoradiation remain to be determined. Quantification of DAMPs by serial measurements during therapy might prove useful for the establishment of a novel predictive biomarker for CCRT combined with ICI immunotherapy.
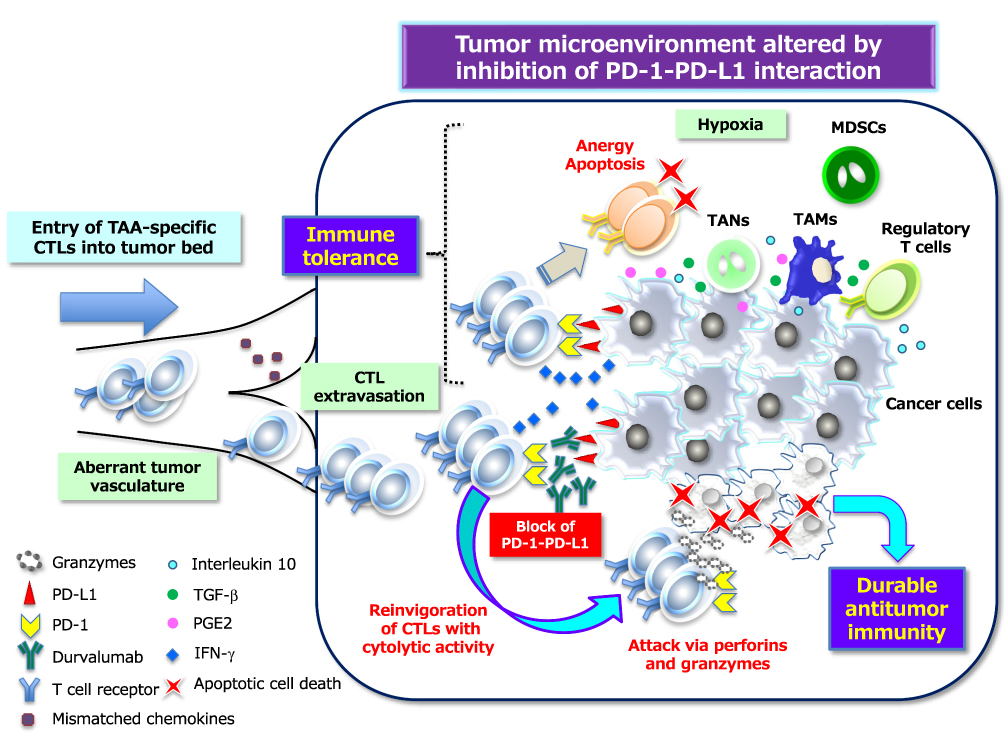 |
Figure 3 PD-1–PD-L1 interaction–dependent barrier to effector CTL activity in solid tumors. TAA-specific effector CTLs that can overcome chemokine mismatches and the aberrant vasculature of residual or metastatic tumor beds are able to attack cancer cells in a manner dependent on the interaction between peptide-MHC complexes and TCRs. The activated T cells secrete interferon-γ (IFN-γ), which induces the up-regulation of immunosuppressive mechanisms such as the expression of PD-L1 on cancer cells. PD-L1–mediated signaling via PD-1 displayed on effector CTLs renders them exhausted, anergic, or apoptotic, and thus no longer able to destroy cancer cells. However, inhibitors of PD-L1 such as durvalumab that interrupt the PD-1–PD-L1 interaction reinvigorate exhausted CTLs and restore them to the effector phase so that they are able to exert long-lasting antitumor effects in cancer patients. Immunosuppressive immune cells can also be attracted to tumor tissue via chemotaxis in response to chemokines induced by hypoxia. Inhibitory immune infiltrates include myeloid-derived suppressor cells (MDSCs), regulatory T cells, tumor-associated neutrophils (TANs), and tumor-associated macrophages (TAMs), all of which can produce immunosuppressive molecules such as interleukin 10, TGF-β, and prostaglandin E2 (PGE2). All abbreviations have been described previously. |
PACIFIC Study: Results for the Japanese Subpopulation
According to the interim analysis performed in 2018, for the 112 Japanese patients in the ITT population of the PACIFIC study, the median PFS in the durvalumab arm (n=72) was longer than that in the placebo arm (n=40), with values of not evaluable (NE), with a 95% CI of 10.9 months to NE, versus 7.2 months, with a 95% CI of 2.0 to 18.6 months, respectively (HR, 0.49 [95% CI, 0.26–0.89]). The median OS was NE (95% CI, NE–NE) versus NE (95% CI, NE–NE), with an HR of 0.96 (95% CI, 0.48–1.92). The apparent lack of an OS benefit in the Japanese subpopulation may be attributable to several factors including (1) the relatively small numbers of patients in the two arms, which rendered the analysis underpowered to detect a significant difference; (2) ethnic differences in tumor biology, histology, driver mutations (EGFR and BRAF mutations as well as ALK and, ROS1 translocations), and immunology, such as differential expression of major histocompatibility complex (MHC) class I molecules; and (3) the fact that the OS curve in the placebo group was superior to that seen historically in unresectable stage III NSCLC patients, likely as a result of more careful and intensive management of subsequent treatments such as molecularly targeted therapy and other ICIs by the Japanese medical oncologists.
A comparative overview of the safety profile for durvalumab between Japanese and non-Japanese patients is summarized in Table 1. Rates of grade ≥3 AEs of any cause, treatment-related death, severe AEs, and AEs leading to treatment discontinuation, were lower in Japanese patients than in non-Japanese patients. With regard to immune-related AEs (irAEs), those with a rate >10 percentage points higher and those of grade ≥3 with a rate >2 percentage points higher in Japanese patients than in non-Japanese patients included pneumonitis (54.2% versus 14.1% and 5.6% versus 1.0%, respectively), suggesting that it may be difficult in many cases for medical oncologists to determine whether to continue or discontinue durvalumab treatment after the appearance of pneumonitis. The increased frequency of pneumonitis in Japanese patients might reflect differences in the immune system between Asian and white ethnicities. Indeed, ethnic differences in Toll-like receptor 2 (TLR2) and TLR4 gene polymorphisms may underlie differential innate immunity to specific pathogens such as Mycobacterium tuberculosis.41–43 Differential activation of innate immunity mediated by TLRs on APCs in tumor lesions might affect the host response to CCRT and ultimately lead to enhanced adaptive antitumor immunity in Japanese patients, with TAA-specific T cells destroying tumor cells as on-target effect or damaging normal lung tissue and causing pneumonitis as an off-target effect.
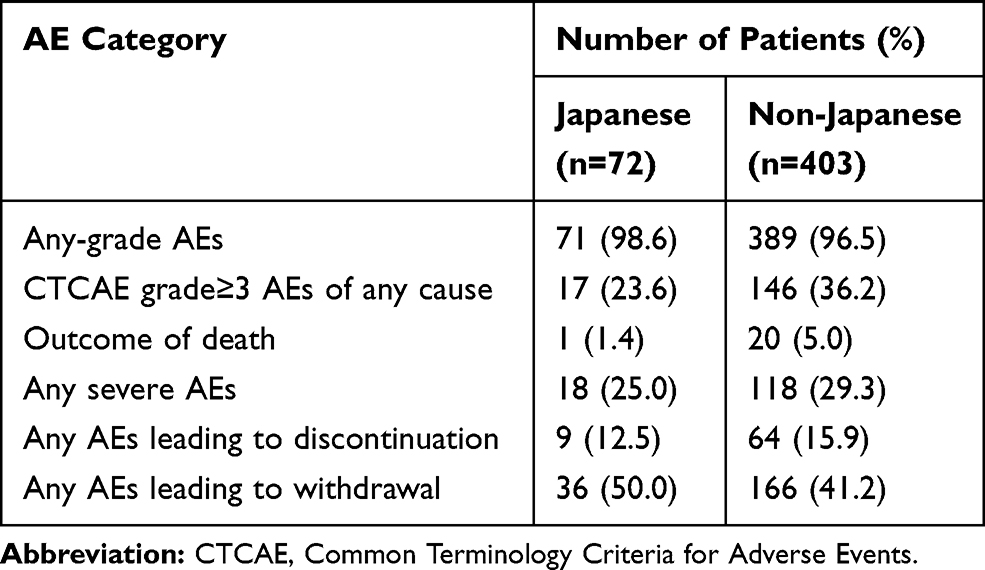 |
Table 1 Summary of Safety Findings for Japanese and Non-Japanese Subpopulations Treated with Durvalumab in the PACIFIC Study |
Notwithstanding the lack of an OS benefit for durvalumab in the Japanese subpopulation of the PACIFIC study, the marked improvement in PFS and relatively safe profile with a lower rate of severe AEs have resulted in Japanese patients with unresectable stage III NSCLC receiving consolidation therapy with durvalumab following CCRT as a new standard of care, irrespective of PD-L1 status and without use of the Ventana SP263 immunohistochemistry assay.
Perspectives and Remaining Questions After the PACIFIC Study
The use of durvalumab as consolidation therapy after CCRT has resulted in a clinically meaningful retardation of the development of local relapse and distant metastasis in patients with unresectable stage III NSCLC. However, the ultimate goal of any novel therapy for such patients is to improve the cure rate. Although long-term follow-up data, such as the 5-year OS rate, which has generally been accepted as the likelihood of cure, are still required, the fact that durvalumab improved the 2-year and 3-year OS rates (66.3% and 57.0%, respectively) in the PACIFIC study is suggestive that it might improve the cure rate in these patients. Indeed, the fact that the Kaplan-Meier curve of PFS for durvalumab showed a plateau at ~40% survival suggests that these remaining patients might have attained a cure. Intriguingly, the OS curve for the durvalumab arm appears to be diverging from that of the placebo arm with time, with an absolute improvement in OS rate of 10.7% at 2 years and 13.5% at 3 years. This divergence might reflect the development of long-lasting antitumor T cell responses unleased as a result of PD-L1 inhibition by durvalumab. It is thus possible that durvalumab therapy may achieve a long-term improvement in OS and become more firmly established as a practice-changing consolidation therapy for unresectable stage III NSCLC.
There are still unaddressed clinical questions with regard to the addition of durvalumab in this stage setting. First, strict eligibility criteria in the PACIFIC trial restricted the enrolled subjects to very selected subpopulation. It was recently reported that in clinical practice, approximately 70% of patients with unresectable stage III NSCLC treated with definitive chemoradiotherapy could be eligible to receive consolidation therapy with durvalumab.44 No information is available with regard to the safety and efficacy of durvalumab for patient populations that did not meet the exam eligibility requirements, including those with multiple comorbidities or impaired lung function, those with a poor performance status, and those who required recovery beyond 6 weeks after the completion of CCRT. Such information is also limited for patients with a PD-L1 TPS of <1%. Consideration of a different treatment for such patients may be inappropriate until definitive data are available. Such a situation requires a deliberative discussion including the patient and a multidisciplinary team regarding the uncertain and unprecedented outcomes brought about by this treatment.
Second, it remains unclear which induction chemotherapy regimen is optimal for subsequent ICI therapy. In the PACIFIC study, among such chemotherapy regimens, cisplatin-based combination chemotherapy was associated with a lower HR for durvalumab monotherapy compared with carboplatin-based chemotherapy (0.59 [95% CI, 0.43–0.81] versus 0.86 [95% CI, 0.60–1.23]), suggestive of a superior contribution of cisplatin to the survival prolongation. Clinical and preclinical studies have suggested mechanisms by which cisplatin might confer a better survival in this setting. For example, platinum-based therapy was found to increase PD-L1 expression levels in tumor tissue.45,46 In addition, cisplatin may confer a more immunogenic tumor microenvironment. Indeed, a preclinical study of human papilloma virus–associated cancer showed that cisplatin-induced tumor cell death and the release of DAMPs resulted in the infiltration of inflammatory APCs with increased levels of costimulatory molecule expression, eventually leading to the induction of tumor-specific CD8+ T cells.47 Qualitative and quantitative differences in the induction of PD-L1 expression and DAMPs release between cisplatin and other chemotherapeutic regimens may thus influence the clinical benefit afforded by durvalumab, although there are currently no data for carboplatin in comparison with cisplatin in this regard. Further studies are warranted to investigate the potential interaction of chemotherapy with radiation with regard to immunological effects and thereby to optimize induction chemotherapy regimens.
Third, the optimal timing of durvalumab therapy relative to radiotherapy also remains to be determined. Subset analysis of the PACIFIC study showed that patients treated with durvalumab within 14 days of the last radiation dose had a significantly longer OS compared with those for whom this interval was 14 to 42 days (HR for <14 days, 0.42 [95% CI, 0.27–0.67]; HR for 14–42 days, 0.81 [95% CI, 0.62–1.06]). This finding suggests that the earlier onset of durvalumab treatment maximized the antitumor immune response. The PACIFIC 2 trial (NCT03519971), a randomized, double-blind, placebo-controlled, multicenter, international phase III study is currently ongoing to assess the efficacy and safety of durvalumab given concurrently with platinum-based chemoradiation (durvalumab plus standard-of-care CCRT) in patients with unresectable stage III NSCLC. In Japan, a Phase II trial (DORPHIN study, WJOG11619L) to investigate the efficacy and safety of durvalumab plus concurrent radiation therapy as well as an interventional phase II study (DATE study, TORG1937) assessing the efficacy and safety of durvalumab administered immediately after completion of CCRT are both under way for such patients.
Fourth, the optimal modality of ICIs partnered with CCRT, given either concurrently or as consolidation therapy, remains unknown. Results from three phase II trials recently demonstrated promising antitumor activities of other ICIs including pembrolizumab, atezolizumab, and nivolumab administered either as consolidation therapy following CCRT (LUN 14–179 [NCT02343952] and Part I DETERRED [NCT02525757]) or concurrently with CCRT (Part II DETERRED [NCT02525757] and NICOLAS [NCT02434081]),48 although these activities require confirmation in randomized phase III trials.
Conclusion
Patients with unresectable stage III NSCLC have a dismal prognosis despite the administration of CCRT with curative intent. There has been no improvement in patient outcome or management in this setting for more than a decade. However, clinical data from the recent PACIFIC trial support the potential of ICIs administered as consolidation therapy after CCRT to improve both local and systemic disease control in such patients with an acceptable safety profile, although the results of long-term follow-up for OS of this trial and others are still awaited. Given the apparent lack of an OS benefit and relatively higher incidence of pneumonitis as an irAE in Japanese patients treated with durvalumab after CCRT in the PACIFIC study, careful monitoring of the clinical course for longer periods of time is warranted in such individuals. The development of more precise biomarkers such as PD-L1 expression, tumor mutation burden, and extent of ICD and determination of the optimal timing between chemoradiation and immunotherapy are important goals for further study. The PACIFIC trial has suggested that other therapeutic modalities, including induction immunotherapy, radiation dose adaptation, and consolidation therapy with other ICIs also warrant investigation.
Disclosure
Dr Isamu Okamoto reports grants, personal fees from AstraZenaca, during the conduct of the study; grants from Boehringer Ingelheim, Astellas Pharma, Novartis, and AbbVie; grants, personal fees from Taiho Pharmaceutical, Ono Pharmaceutical, MSD, Eli Lilly, Bristol-Myers Squibb, Chugai Pharma; personal fees from Pfizer, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi:10.3322/caac.21387
2. Nogami N, Takigawa N, Hotta K, et al. A phase II study of cisplatin plus S-1 with concurrent thoracic radiotherapy for locally advanced non-small-cell lung cancer: the Okayama Lung Cancer Study Group Trial 0501. Lung Cancer. 2015;87:141–147. doi:10.1016/j.lungcan.2014.11.001
3. Aoe K, Takigawa N, Hotta K, et al. A phase II study of S-1 chemotherapy with concurrent thoracic radiotherapy in elderly patients with locally advanced non-small-cell lung cancer: the Okayama Lung Cancer Study Group Trial 0801. Eur J Cancer. 2014;50:2783–2790. doi:10.1016/j.ejca.2014.07.024
4. Kuyama S, Hotta K, Tabata M, et al. Impact of HER2 gene and protein status on the treatment outcome of cisplatin-based chemoradiotherapy for locally advanced non-small cell lung cancer. J Thorac Oncol. 2008;3:477–482. doi:10.1097/JTO.0b013e31816e2ea3
5. Hotta K, Nagio T, Matsuo K, Kiura K. Cure- or care-oriented regimen for stage III non-small-cell lung cancer? J Clin Oncol. 2011;29:e320. doi:10.1200/JCO.2010.32.6025
6. Segawa Y, Kiura K, Takigawa N, et al. Phase III trial comparing docetaxel and cisplatin combination chemotherapy with mitomycin, vindesine, and cisplatin combination chemotherapy with concurrent thoracic radiotherapy in locally advanced non-small-cell lung cancer: OLCSG 0007. J Clin Oncol. 2010;28:3299–3306. doi:10.1200/JCO.2009.24.7577
7. Senan S, Brade A, Wang L-H, et al. PROCLAIM: randomized phase III trial of pemetrexed-cisplatin or etoposide-cisplatin plus thoracic radiation therapy followed by consolidation chemotherapy in locally advanced nonsquamous non-small-cell lung cancer. J Clin Oncol. 2016;34:953–962. doi:10.1200/JCO.2015.64.8824
8. Bradley JD, Paulus R, Komaki R, et al. Standard-dose versus high-dose conformal radiotherapy with concurrent and consolidation carboplatin plus paclitaxel with or without cetuximab for patients with stage IIIA or IIIB non-small-cell lung cancer (RTOG 0617): a randomised, two-by-two factorial Phase 3 study. Lancet Oncol. 2015;16:187–199. doi:10.1016/S1470-2045(14)71207-0
9. Thatcher N, Heighway J. Maintenance and consolidation therapy in patients with unresectable stage III/IV non-small cell lung cancer. Oncologist. 2010;15:1034–1042. doi:10.1634/theoncologist.2009-0292
10. Hanna N, Neubauer M, Yiannoutsos C, et al. Phase III study of cisplatin, etoposide, and concurrent chest radiation with or without consolidation docetaxel in patients with inoperable stage III non-small-cell lung cancer: the Hoosier Oncology Group and U.S. Oncology. J Clin Oncol. 2008;26:5755–5760. doi:10.1200/JCO.2008.17.7840
11. Ahn JS, Ahn YC, Kim J-H, et al. Multinational randomized phase III trial with or without consolidation chemotherapy using docetaxel and cisplatin after concurrent chemoradiation in inoperable stage III non-small-cell lung cancer: KCSG-LU05-04. J Clin Oncol. 2015;33:2660–2666. doi:10.1200/JCO.2014.60.0130
12. Butts C, Socinski MA, Mitchell PL, et al. Tecemotide (L-BLP25) versus placebo after chemoradiotherapy for stage III non-small-cell lung cancer (START): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2014;15:59–68. doi:10.1016/S1470-2045(13)70510-2
13. Santini FC, Hellmann MD. PD-1/PD-L1 axis in lung cancer. Cancer J. 2018;24:15–19. doi:10.1097/PPO.0000000000000300
14. Swaika A, Hammond WA, Joseph RW. Current state of anti-PD-L1 and anti-PD-1 agents in cancer therapy. Mol Immunol. 2015;67:4–17. doi:10.1016/j.molimm.2015.02.009
15. Granier C, De Guillebon E, Blanc C, et al. Mechanisms of action and rationale for the use of checkpoint inhibitors in cancer. Esmo Open. 2017;2:e000213. doi:10.1136/esmoopen-2017-000213
16. Herbst RS, Baas P, Kim D-W, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet. 2016;387:1540–1550. doi:10.1016/S0140-6736(15)01281-7
17. Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373:1627–1639. doi:10.1056/NEJMoa1507643
18. Rittmeyer A, Barlesi F, Waterkamp D, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet. 2017;389:255–265. doi:10.1016/S0140-6736(16)32517-X
19. Antonia SJ, Villegas A, Daniel D, et al. Durvalumab after chemoradiotherapy in stage III non-small-cell lung cancer. N Engl J Med. 2017;377:1919–1929. doi:10.1056/NEJMoa1709937
20. Antonia SJ, Villegas A, Daniel D, et al. Overall survival with durvalumab after chemoradiotherapy in stage III NSCLC. N Engl J Med. 2018;379:2342–2350. doi:10.1056/NEJMoa1809697
21. Derer A, Spiljar M, Bäumler M, et al. Chemoradiation increases PD-L1 expression in certain melanoma and glioblastoma cells. Front Immunol. 2016;7:610. doi:10.3389/fimmu.2016.00610
22. Hecht M, Büttner-Herold M, Erlenbach-Wünsch K, et al. PD-L1 is upregulated by radiochemotherapy in rectal adenocarcinoma patients and associated with a favourable prognosis. Eur J Cancer. 2016;65:52–60. doi:10.1016/j.ejca.2016.06.015
23. Yoneda K, Kuwata T, Kanayama M, et al. Alteration in tumoural PD-L1 expression and stromal CD8-positive tumour-infiltrating lymphocytes after concurrent chemo-radiotherapy for non-small cell lung cancer. Br J Cancer. 2019;121:490–496. doi:10.1038/s41416-019-0541-3
24. Golden EB, Apetoh L. Radiotherapy and immunogenic cell death. Semin Radiat Oncol. 2015;25:11–17. doi:10.1016/j.semradonc.2014.07.005
25. Inoue H, Tani K. Multimodal immunogenic cancer cell death as a consequence of anticancer cytotoxic treatments. Cell Death Differ. 2014;21:39–49. doi:10.1038/cdd.2013.84
26. Krysko DV, Garg AD, Kaczmarek A, et al. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12:860–875. doi:10.1038/nrc3380
27. Azad A, Yin Lim S, D’Costa Z, et al. PD-L1 blockade enhances response of pancreatic ductal adenocarcinoma to radiotherapy. EMBO Mol Med. 2017;9:167–180. doi:10.15252/emmm.201606674
28. Dovedi SJ, Adlard AL, Lipowska-Bhalla G, et al. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res. 2014;74:5458–5468. doi:10.1158/0008-5472.CAN-14-1258
29. Deng L, Liang H, Burnette B, et al. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest. 2014;124:687–695. doi:10.1172/JCI67313
30. Brix N, Tiefenthaller A, Anders H, Belka C, Lauber K. Abscopal, immunological effects of radiotherapy: narrowing the gap between clinical and preclinical experiences. Immunol Rev. 2017;280:249–279. doi:10.1111/imr.12573
31. Lauber K, Brix N, Ernst A, et al. Targeting the heat shock response in combination with radiotherapy: sensitizing cancer cells to irradiation-induced cell death and heating up their immunogenicity. Cancer Lett. 2015;368:209–229. doi:10.1016/j.canlet.2015.02.047
32. Wang Y-J, Fletcher R, Yu J, Zhang L. Immunogenic effects of chemotherapy-induced tumor cell death. Genes Dis. 2018;5:194–203. doi:10.1016/j.gendis.2018.05.003
33. Galluzzi L, Vitale I, Aaronson SA, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018;25:486–541. doi:10.1038/s41418-017-0012-4
34. Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi:10.1146/annurev.iy.12.040194.005015
35. Garg AD, Nowis D, Golab J, et al. Immunogenic cell death, DAMPs and anticancer therapeutics: an emerging amalgamation. Biochim Biophys Acta. 2010;1805:53–71. doi:10.1016/j.bbcan.2009.08.003
36. Casares N, Pequignot MO, Tesniere A, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med. 2005;202:1691–1701. doi:10.1084/jem.20050915
37. Golden EB, Frances D, Pellicciotta I, et al. Radiation fosters dose-dependent and chemotherapy-induced immunogenic cell death. Oncoimmunology. 2014;3:e28518. doi:10.4161/onci.28518
38. Suzuki Y, Mimura K, Yoshimoto Y, et al. Immunogenic tumor cell death induced by chemoradiotherapy in patients with esophageal squamous cell carcinoma. Cancer Res. 2012;72:3967–3976. doi:10.1158/0008-5472.CAN-12-0851
39. Juneja VR, McGuire KA, Manguso RT, et al. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J Exp Med. 2017;214:895–904. doi:10.1084/jem.20160801
40. Lanitis E, Dangaj D, Irving M, Coukos G. Mechanisms regulating T-cell infiltration and activity in solid tumors. Ann Oncol. 2017;28:xii18–xii32. doi:10.1093/annonc/mdx238
41. Lazarus R, Vercelli D, Palmer LJ, et al. Single nucleotide polymorphisms in innate immunity genes: abundant variation and potential role in complex human disease. Immunol Rev. 2002;190:9–25. doi:10.1034/j.1600-065x.2002.19002.x
42. Smirnova I, Mann N, Dols A, et al. Assay of locus-specific genetic load implicates rare Toll-like receptor 4 mutations in meningococcal susceptibility. Proc Natl Acad Sci U S A. 2003;100:6075–6080. doi:10.1073/pnas.1031605100
43. Randhawa AK, Shey MS, Keyser A, et al. Association of human TLR1 and TLR6 deficiency with altered immune responses to BCG vaccination in South African infants. PLoS Pathog. 2011;7:e1002174. doi:10.1371/journal.ppat.1002174
44. Sakaguchi T, Ito K, Furuhashi K, et al. Patients with unresectable stage III non-small cell lung cancer eligible to receive consolidation therapy with durvalumab in clinical practice based on PACIFIC study criteria. Respir Investig. 2019;57:466–471. doi:10.1016/j.resinv.2019.03.011
45. Ock C-Y, Kim S, Keam B, et al. Changes in programmed death-ligand 1 expression during cisplatin treatment in patients with head and neck squamous cell carcinoma. Oncotarget. 2017;8:97920–97927. doi:10.18632/oncotarget.18542
46. Song Z, Yu X, Zhang Y. Altered expression of programmed death-ligand 1 after neo-adjuvant chemotherapy in patients with lung squamous cell carcinoma. Lung Cancer. 2016;99:166–171. doi:10.1016/j.lungcan.2016.07.013
47. Beyranvand Nejad E, van der Sluis TC, van Duikeren S, et al. Tumor eradication by cisplatin is sustained by CD80/86-mediated costimulation of CD8+ T cells. Cancer Res. 2016;76:6017–6029. doi:10.1158/0008-5472.CAN-16-0881
48. Melosky B, Juergens R, McLeod D, et al. Immune checkpoint-inhibitors and chemoradiation in stage III unresectable non-small cell lung cancer. Lung Cancer. 2019;134:259–267. doi:10.1016/j.lungcan.2019.05.027
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.