")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 14
IL-25 Treatment Improves Metabolic Syndrome in High-Fat Diet and Genetic Models of Obesity
Authors Smith AD, Fan A, Qin B, Desai N, Zhao A, Shea-Donohue T
Received 28 August 2021
Accepted for publication 23 November 2021
Published 21 December 2021 Volume 2021:14 Pages 4875—4887
DOI https://doi.org/10.2147/DMSO.S335761
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Juei-Tang Cheng
Allen D Smith,1 Anya Fan,2 Bolin Qin,1 Neemesh Desai,2 Aiping Zhao,2 Terez Shea-Donohue3
1Diet, Genomics, and Immunology Laboratory, Beltsville Human Nutrition Research Center, Agricultural Research Service, U.S. Department of Agriculture, Beltsville, MD, USA; 2Department of Radiation Oncology University of Maryland School of Medicine, Baltimore, MD, USA; 3Division of Digestive Diseases and Nutrition, National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), National Institutes of Health, Bethesda, MD, USA
Correspondence: Allen D Smith
Diet, Genomics, and Immunology Laboratory, Beltsville Human Nutrition Research Center, Agricultural Research Service, U.S. Department of Agriculture, Beltsville, MD, USA
Tel +1 301-504-8577
Fax +1- 301 504-9062
Email [email protected]
Introduction: Endemic obesity is considered the driving force for the dramatic increase in incidence of type 2 diabetes (T2D). There is mounting evidence that chronic, low-grade inflammation driven by Th1/Th17 cells and M1 macrophages, is a critical link between obesity and insulin resistance. IL-25 promotes development of a Th2 immune response and M2 macrophages that counteract the inflammation associated with obesity and T2D.
Methods: Mice were fed a high-fat diet (HFD) for 16 weeks and then treated with IL-25 or BSA as a control for 21 days. Body weight, blood glucose levels, intraperitoneal glucose tolerance, and gene expression were evaluated in mice treated with BSA or IL-25. Ob/ob mice fed a normal control diet were also treated with BSA or IL-25 and body weight and blood glucose levels were measured. Transepithelial electrical resistance and sodium-linked glucose absorption were determined in muscle-free small intestinal tissue and glucose absorption assessed in vitro in intestinal epithelial and skeletal muscle cell lines.
Results: Administration of IL-25 to HFD fed mice reversed glucose intolerance, an effect mediated in part by reduction in SGLT-1 activity and Glut2 expression. Importantly, the improved glucose tolerance in HFD mice treated with IL-25 was maintained for several weeks post-treatment indicating long-term changes in glucose metabolism in obese mice. Glucose intolerance was also reversed by IL-25 treatment in genetically obese ob/ob mice without inducing weight loss. In vitro studies demonstrated that glucose absorption was inhibited by IL-25 treatment in the epithelial IPEC-1 cells but increased glucose absorption in the L6 skeletal muscle cells. This supports a direct cell-specific effect of IL-25 on glucose metabolism.
Conclusion: These results suggest that the IL-25 pathway may be a useful target for the treatment of metabolic syndrome.
Keywords: metabolic syndrome, glucose, ob/ob, rodent
Introduction
Endemic obesity is considered the driving force for the dramatic increase in incidence of type 2 diabetes (T2D). Immunometabolism is an emerging field of investigation, and a major focus has been the contribution of pro-inflammatory cytokines and mediators to obesity and the disruption of metabolic homeostasis. There is mounting experimental and clinical evidence showing that chronic, low-grade inflammation is a critical link between obesity and insulin resistance.1–3 This low-grade inflammation in obesity is found systemically, as indicated by increased circulating levels of pro-inflammatory cytokines including TNF-α, Macrophage Migration Inhibitory Factor (MIF), IL-64,5 and leukocytes expressing a pro-inflammatory cytokine profile in obese individuals.5 Plasma levels of IL-12 are elevated in obese patients and recent evidence highlights IL-12 family cytokines as potential regulators linking obesity to nutrient intake and insulin resistance.6,7 Th17 cells are associated with T2D, and IL-17A and IL-17F remain strong markers of Th17 responses.8 In addition, low-grade inflammation is found in metabolically important tissues, such as liver and adipose tissue, and features accumulation of various types of inflammatory cells (Th1/Th17 cells, macrophages, etc.).9
Anti-inflammatory agents are a major target for insulin-sensitizing therapies and have been validated in animal studies.10,11 Recent clinical trials targeting pro-inflammatory cytokines/mediators (TNFα, IL-1β, Inhibitor of Nuclear Factor Kappa B Kinase Subunit Beta (IKKβ)), however, were only modestly successful in treating diabetes.12–17 IL-22, an IL-10 family member generated by type 3 innate lymphoid cells (ILC3), Th17 and Th22 cells, improved metabolic syndrome parameters in mice fed a high-fat diet (HFD) and genetically obese leptin-receptor-deficient (db/db) mice.18 A limiting factor in these studies is that targeting a single inflammatory cytokine or mediator may not yield robust therapeutic benefits. Other factors include insufficient drug concentrations and concerns about adverse effects. These issues demonstrate the need for a different strategy and approach in the development of anti-inflammatory biological treatments for diabetes.
IL-25 is a member of the IL-17 cytokine family that plays a major role in the promotion and initiation of type 2 immunity, downregulation of Th1/Th17 pro-inflammatory cytokines, such as IL17A and IL-12, and development of alternatively activated macrophages (M2).19–21 The GI epithelium produces IL-25, and the relatively rare doublecortin-like kinase 1 (DCLK1)+ tuft cells are the sole epithelial source.22 Tuft cell hyperplasia occurs during nematode infection-induced upregulation of Th2 cytokines.23 Of interest, we and others showed that hepatocytes also produce IL-25.24,25 In addition, we demonstrated that the receptor for IL-25 (IL-25R) is a heterodimer consisting of IL-17RB and IL-17RA,26 which is expressed by various cells such as structural cells (enterocytes, adipocytes, skeletal muscle), immune cells including macrophages20,21 and ILC2 that mediate Th2 immunity.27 In addition, IL-25-responsive innate cells, including IL-13-producing ILC2, regulate weight and glucose homeostasis in mice fed a HFD via the induction and maintenance of eosinophils and M2 in visceral adipose tissue.28 Thus, IL-25 represents a viable immunometabolic target for the treatment of obesity and associated T2D. Although there are a number of studies that have evaluated IL-25 administration in mice fed a HFD, whether the beneficial effects of IL-25 treatment are retained after treatment is unknown. In addition, there is less information on its effects on genetically obese mice or on the effects of IL-25 on intestinal epithelial and skeletal muscle metabolism. The goal of the present study was to determine the effects of IL-25 administration on diet- and genetically induced changes in glucose metabolism and explore the potential mechanisms of these effects.
Materials and Methods
General Husbandry
All mice were maintained in a specific pathogen-free facility with a 12-h light/dark cycle (6PM/6AM) with free access to food and water. These studies were conducted with institutional approval from both the University of Maryland, Baltimore and the USDA Beltsville Area Animal Care and Use Committees, in accordance with principles set forth in the Guide for Care and Use of Laboratory Animals, Institute of Laboratory Animal Resources, National Research Council, Health and Human Services Publication.
High-Fat Diet (HFD) Model of Obesity
The murine model of HFD-induced T2D is commonly used for evaluating efficacy of anti-diabetic drugs. Mice were fed either the normal control diet (NCD, 10% of kcal from fat) or the HFD (60% of kcal from fat) (Research Diet, New Brunswick, NJ) ad libitum unless indicated. Male C57BL/6 mice were fed the NCD or HFD for 16 weeks and were weighed weekly. After feeding the HFD for 16 weeks, mice were fasted overnight, weighed, and fasting glucose measured (One Touch Ultra Meter). Based on these data, mice were individually randomized to achieve matched (BW and fasting blood glucose) groups. Mice were injected ip with IL-25 (mouse rIL-25, R&D Systems, Minneapolis, MN) at a dose of 2 μg/kg three times weekly or 5 μg/kg twice weekly for 21 days. To serve as control groups, mice fed the HFD and NCD were treated with bovine serum albumin (BSA) using the 3 times weekly schedule. During the IL-25 or BSA treatment, all mice were weighed daily for the first week and then every three days thereafter. Following the 21-day treatment period, body weight and blood glucose levels were monitored periodically in groups of mice from the BSA and IL-25 treatment.
Genetic Model of Obesity
After HFD-induced obesity, the ob/ob mouse is the next most well-documented murine model of T2D. The ob/ob mouse is genetically deficient in leptin and displays metabolic abnormalities like those observed in obese humans with T2D. Male lean WT mice and ob/ob (7-week-old, Jackson Laboaratory, Bar Harbor, ME) were fed the NCD, weighed every three days, and fasting blood glucose tested every two weeks until fasting levels in ob/ob mice were >150 g/L. At that time, WT and ob/ob mice were randomized to achieve matched (BW and fasting blood glucose) groups and both WT and ob/ob mice were injected ip with BSA or IL-25 at a dose of 5 μg/kg three times/week for 21 days. At the end of the treatment period, tissues were weighed and processed for subsequent analyses.
Food Intake
Food intake was determined in all groups of mice in all studies by weighing the food every three days to determine intake/cage, and then calculated for intake/mouse. In some of the HFD diet groups, food intake in cages of mice receiving BSA was matched to that of mice receiving IL-25. The remaining food was weighed daily and every three days the amount of food available to the BSA-treated HFD group was matched by weight to the previous 3-day intake of the corresponding IL-25 treated HFD group.
Intraperitoneal Glucose Tolerance Test (IPGTT)
After an overnight (∼16 hrs) fast, glucose was administered (1.5 g/kg body weight) intraperitoneally (ip) in PBS. The levels of glucose in blood taken from the tail were measured using a OneTouch Ultra glucometer (LifeScan, Milpitas, CA) just before glucose administration and then 30, 60, and 120 minutes later.25
Tissue Weights
At euthanasia spleen, liver, subcutaneous fat (left side), epididymal fat was excised, rinsed, blotted dry, and weighed.
Molecular Analyses
At the time of euthanasia, sections of small intestine, liver, and visceral fat were processed and stored for subsequent real-time PCR analysis. As per manufacturer’s instructions, total RNA was extracted from full thickness sections of upper (jejunum) small intestine with TRIzol reagent (Invitrogen, Carlsbad, CA). RNA integrity, quantity, and genomic DNA contamination were assessed using the Agilent Bioanalyzer 2100 and RNA 6000 Nano kit (Agilent Technologies, Palo Alto, CA). Only those RNA samples with 28S/18S ratios between 1.5 and 2 and no DNA contamination were studied further. RNA samples (2 µg) were reverse-transcribed to cDNA using the First Strand cDNA Synthesis Kit (MBI Fermentas, Hanover, MD) with random hexamer primer. Amplification reactions were performed on an iCycler detection system (Bio-Rad, CA). Primer sequences were designed using Beacon Designer 5.0 (Premier Biosoft International, Palo Alto, CA) and synthesized by the Biopolymer Laboratory of the University of Maryland. Primer sequences used for SGLt1 and Glut2 were published previously.29,30
Functional Analyses
Muscle-free sections of small intestinal mucosae were taken from mice fed the HFD or ob/ob mice and mounted in modified microsnapwells to determine changes in transepithelial electrical resistance (TEER).25,29 TEER was measured at 30-min intervals using a planar electrode (Endohm Snap electrode) connected to an EVOM-G WPI analyzer (World Precision Instruments) and expressed as ohms per square centimeter. In mice fed the HFD, muscle-free samples of small intestine were also mounted in Ussing chambers that exposed 0.126 cm2 of tissue to 5 mL of Krebs buffer and allowed to equilibrate for 25 min. Glucose (0.625 to 40 mM) was added to the mucosal side of the mounted tissue to assess sodium-linked glucose absorption.
Cell Cultures
The IPEC-1 cell line (obtained from Dr Harry Dawson, USDA/ARS/BHNRC) was used for the in vitro studies. Undifferentiated IPEC-1 cells were maintained by serial passage in growth medium (GM): Dulbecco’s modified Eagle’s medium/F12 medium (Invitrogen) supplemented with 5% fetal bovine serum (Invitrogen), insulin (5 μg/mL), transferrin (5 μg/mL), selenium (5 ng/mL) (ITS Premix®, BD Biosciences, Bedford, MA), epidermal growth factor (5 μg/liter) (BD Biosciences), penicillin (100 U/mL), and streptomycin (100 μg/mL) (Invitrogen). To induce differentiation, undifferentiated cells were harvested by trypsinization, and 0.25 × 106 cells/well were plated on 35-mm diameter collagen-coated dishes (Costar, Corning, Inc.). Cells were maintained in serum containing GM for 48 h and then switched to the same medium containing 10–7 M dexamethasone (Sigma), but without FBS. Medium was then changed every 2 days. After 10 days, IPEC-1 cells exhibit enterocytic features, including polarization with well-defined microvilli facing the apical medium.31 The cells were used for immunofluorescence cell staining or glucose uptake. Rat L6 skeletal muscle cells were maintained at sub-confluent conditions in growth media containing DMEM with 0.045 g/mL glucose, 100 U/mL penicillin, 100 μg/mL streptomycin, and 10% fetal bovine serum. Cells were maintained in a humidified 37°C incubator with ambient oxygen and 5% CO2. The use of mammalian cell lines has been approved by the Institutional Biosafety Committee.
2-NBD Glucose Uptake Assays
To study the cellular glucose uptake, a glucose uptake cell-based assay kit was used following manufacturer’s instructions (Cayman Chemical, Ann Arbor, MI). The kit employs 2-NBDG, a fluorescent-labeled deoxyglucose analog, as a probe for the detection of glucose uptake by cultured cells. Differentiated iPEC-1 cells or rat L6 skeletal muscle cells were incubated with IL-25 at two concentrations (50, 100 ng/mL) for 2 hours and then with the fluorescent glucose analog 2-NBDG for 2 hours to measure cellular glucose uptake. 2-NBDG uptake was measured using an Eclipse TE2000-S microscope with filters for detecting fluorescein, and the fluorescence intensity was quantified by ImageJ software (NIH Image).
Statistical Analyses
Data are expressed as means ± SEM. Significance was determined using a one-way analysis of variance (ANOVA) followed by Bonferroni test to compare differences among two or more means. A p value of <0.05 was considered significant.
Results
IL-25 Treatment Resulted in Significant Weight Loss in HFD Mice
BSA-treated mice fed the HFD gained an average of 1.87 gm over the 21-day treatment (Figure 1A and Supplemental Figure 1A). In contrast, mice fed the HFD and treated with 2 µg/kg of IL-25 three times per week lost an average of 14.2 gms or 24.9% of their initial weight. Similarly, mice fed the HFD and treated with 5 µg/kg of IL-25 twice weekly lost an average of 14.8 gms or 25.8% of their initial weight (Figure 1A and B, supplemental Figure 1 B-C). Despite the significant weight reduction in IL-25-treated mice fed the HFD all mice fed the HFD weighed significantly more than the mice feed the NCD. Other than transient hypophagia in both the HFD and ob/ob mice, no other adverse effects of IL-25 treatment were noted.
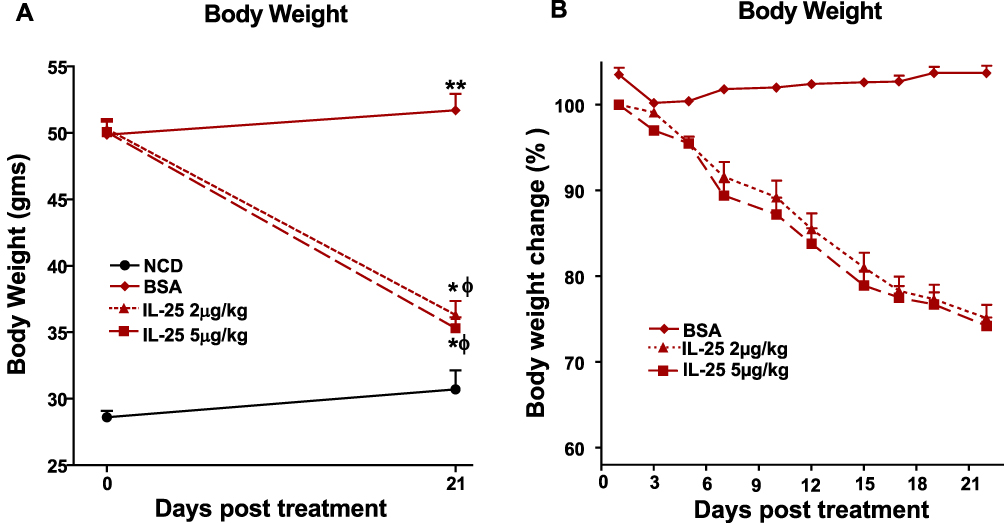 |
Figure 1 IL-25 treatment induces body weight loss in mice fed the HFD. Mice were fed the NCD or HFD for 16 weeks followed by ip injection with IL-25 at a dose of 2 μg/kg three times weekly or 5 μg/kg twice weekly for 21 days and periodically weighed. Panel (A) change in body weight in grams over 21 days in mice fed the NCD or HFD and treated with IL-25. (B) percent changes in body weight over time in BSA or IL-25-treated mice fed the HFD (% of pretreatment weight); mean±SEM, n=7–8 *p<0.05, **p<0.01 versus NCD; ф p<0.05 versus BSA treated HFD. |
IL-25 Treatment Resulted in Significant Changes in Organ Weights in HFD Mice
To determine if IL-25 induced reductions in body weight were associated with corresponding alterations in tissue weights, we collected and weighed the metabolically sensitive tissues, liver, epididymal fat and subcutaneous fat. Liver weight was increased significantly in BSA-treated mice fed the HFD and treatment with either the dosing regimen of 2 or 5 µg/kg of IL-25 normalized liver weights (Supplemental Figure 2A). Both epididymal (Supplemental Figure 2B) and subcutaneous (Supplemental Figure 2C) fat expressed as a percent of body weight were increased significantly in mice fed the HFD and this increase was attenuated similarly by both dosing regimens of IL-25. The spleen acts as a secondary lymphoid organ and spleen weight was unaltered in mice fed the HFD alone but was increased significantly by both IL-25 dosing regimens indicating IL-25 elicited a systemic immune response (Supplemental Figure 2D).
IL-25 Improved Glucose Tolerance in HFD Mice
To determine if IL-25 induced weight loss was associated with improved glucose tolerance, we determined glucose levels in mice fed the HFD diet before and after treatment with BSA or IL-25. Fasting blood glucose levels were increased significantly by feeding mice the HFD and remained elevated in BSA-treated mice over the 21-day period. In contrast, both dosing regimens of IL-25 significantly lowered fasting glucose (Figure 2A and Supplemental Figure 3A–C). At the end of the 21-day treatment period, all mice underwent an IPGTT. Peak glucose levels were significantly higher in BSA-treated mice fed the HFD when compared to mice fed the NCD and remained elevated even at 120 minutes (Figure 2B). In contrast, treatment with either the 2 or 5 µg/kg dosing regimens normalized the IPGTT (Figure 2B) as well as the calculated area under the curve (Figure 2C).
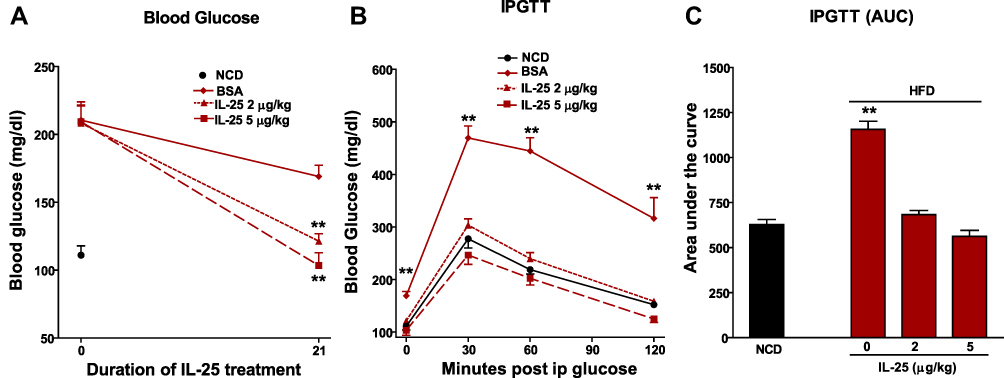 |
Figure 2 IL-25 treatment reduces fasting blood glucose levels and improves glucose tolerance in mice fed the HFD. Mice were fed the NCD or HFD for 16 weeks. (A) Fasting blood glucose levels measured before and after IL-25 treatment for 21 days, **p<0.01 versus pretreatment value (day 0); (B) intraperitoneal glucose tolerance test (IPGTT) administered 21 days after IL-25 treatment; and (C) calculated area under the IPGTT curve. Mean±SEM, n=7–8 **p<0.01 versus NCD. |
Dependence of IL-25 Effects on Glucose Tolerance on Weight Loss in Mice Fed the HFD
To determine the contribution of IL-25 induced weight loss on the normalized glucose tolerance in IL-25-treated mice, we matched the HFD food intake in a group of BSA-treated mice to that of a group of mice treated with 5 µg/kg of IL-25 three times weekly. The matched group’s food intake closely paralleled that of the IL-25 treated group and food intake was consistently less than in the BSA treated group fed ad libitum (Figure 3A). When food intake/cage was corrected to body weight/cage, there was a significant difference in food intake between the BSA treated HFD group fed ad libitum over the entire treatment period (46.81 ± 0.83 vs 37.18 ± 2.78 mg/g bw/day; p < 0.05) and the BSA treated food matched group; however, there was no difference between the IL-25 treated group (36.09 ± 2.66 mg/g bw/day) and the BSA treated feed matched group . Despite the near identical food intake, the IL-25 treated group lost a greater percentage of body weight over the treatment period than the BSA treated food intake matched group (Figure 3B). In addition, body weight in the BSA treated food matched group, but not in the IL-25 treated cohort, showed signs of recovery by the end of the 3-week treatment period. Changes in IPGTT (Figure 3C) and in the area under the curve (Figure 3D) in these groups mirrored the changes in body weight in that the BSA treated feed matched HFD cohort had intermediate values between the IL-25 and BSA treated ad libitum groups.
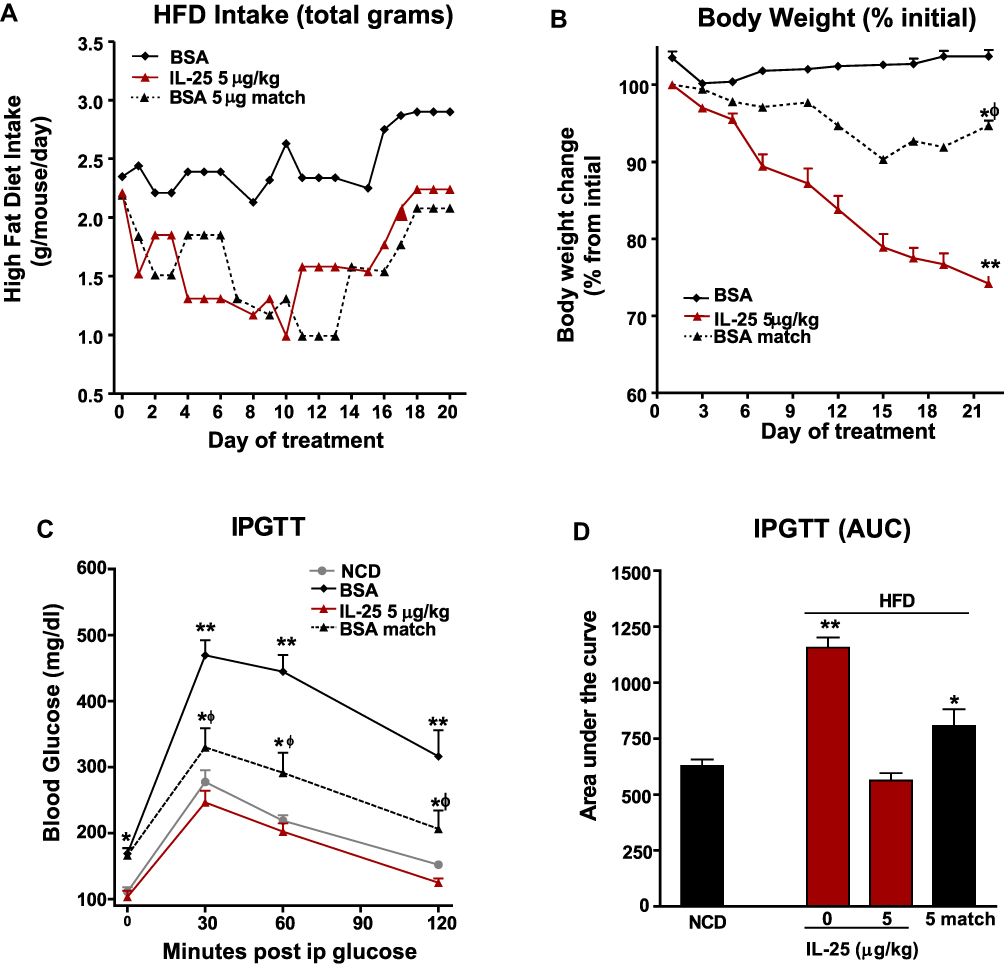 |
Figure 3 Body weight loss and improvement in glucose tolerance is not simply the results of decreased food consumption. Mice were fed the HFD for 16 weeks and treated with BSA or IL-25 and feed intake measured. One group of mice receiving BSA had their feed restricted to levels consumed by the IL-25 treated mice. (A) Daily feed intake in BSA, IL-25 treated and BSA-feed matched groups; (B) changes in body weight in BSA, IL-25 treated and BSA-feed matched groups, *p<0.05, **p<0.01 vs BSA-treated HFD group, ф p<0.05 vs IL-25 group; (C) glucose levels during an intraperitoneal glucose tolerance test (IPGTT) in NCD or HFD fed mice treated with BSA, with or without feed restriction, or IL-25; and (D) calculated area under the curve (AUC) of the IPGTT curves in (C) mean±SEM, n=7–8 *p<0.05, **p<0.01 vs NCD group, ф p<0.05 vs NCD. |
IL-25 Affects Intestinal Glucose Absorption in HFD Mice
We previously showed that nematode infection as well as exogenous administration of IL-4, IL-13 or IL-25 induced a stereotypic inhibition of glucose absorption in mice fed a traditional diet.21,32,33 This effect was attributed to a decrease in the sodium linked glucose transporter −1 (SGLT1) activity, rather than expression.29 In the present study, sodium linked glucose transport was reduced significantly in BSA-treated mice fed the HFD and was inhibited further by both doses of IL-25 (Figure 4A). SGLT1 expression also was reduced in the BSA-treated mice fed the HFD, but not in the IL-25 treated mice fed the HFD (Figure 4B). These data indicate that the HFD affected nutrient absorption and that IL-25 has additive effects on inhibition of glucose absorption that are independent of SGLT1 expression.
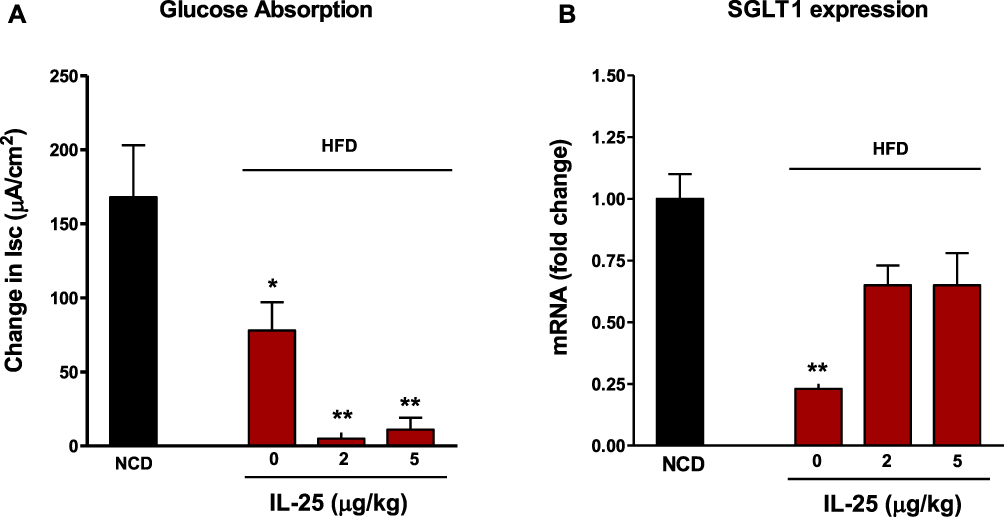 |
Figure 4 Feeding mice the HFD reduces sodium linked glucose transport and SGLT1 expression that are further changed by IL-25 treatment. (A) Sodium-linked glucose transport was reduced significantly in BSA-treated mice fed the HFD compared to mice fed a NCD and was inhibited further by both doses of IL-25. (B) SGLT1 expression was significantly reduced in mice fed the HFD compared to mice fed a NCD but was reversed by IL-25 treatment. Mean±SEM, n=5 *p<0.05, **p<0.01 vs NCD. |
The Metabolic Effects of IL-25 Extend Beyond the Treatment Period in HFD Mice
To determine if the beneficial effects of IL-25 persisted beyond the treatment period, cohorts of IL-25-treated mice fed the HFD were monitored after cessation of therapy. Body weight and fasting glucose levels were determined weekly. Mice steadily gained weight over a six-week follow-up period (Figure 5A); however, fasting glucose levels remained steady throughout (Figure 5B). These data indicate that IL-25 is an effective therapy against HFD-induced hyperglycemia, and that the beneficial effects of IL-25 continue for an extended time post-therapy.
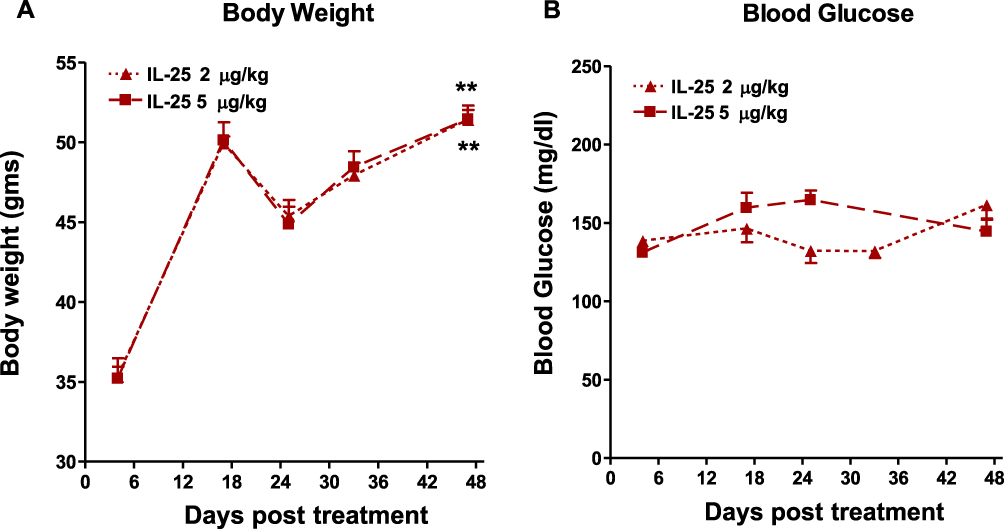 |
Figure 5 Body weight is regained by post IL-25 treated mice fed the HFD but the beneficial effects on glucose tolerance were maintained. (A) Body weight in HFD-fed mice was monitored after cessation of IL-25 treatment for 48 days; (B) fasting blood glucose post cessation of IL-25 treatment in HFD fed mice. mean±SEM, n=7 **p<0.01 vs 0 days post treatment. |
IL-25 Effects on Body Weight and Glucose Tolerance in Genetically Obese Mice
At the end of the 21-day treatment period, the 5 µg/kg IL-25 dosing regimen did not alter body weight in either the WT (WT/BSA 26.5 ± 0.6 vs WT/IL-25 26.4 ± 0.6) or ob/ob mice (ob/ob/BSA 63.3 ± 1.9 vs ob/ob/IL-25 60.5 ± 1.8) fed the NCD diet. Fasting glucose levels were significantly higher in BSA treated ob/ob mice when compared to BSA treated WT. Fasting glucose levels were reduced by treating ob/ob mice twice weekly with 5 µg/kg IL-25 but had no effect on WT mice when compared to BSA treatment. IGPTT was also performed in cohorts of mice from each group. Peak glucose levels occurred at 30 minutes in all groups and were significantly higher in the BSA-treated ob/ob group when compared to the corresponding WT group (Figure 6A). IL-25 therapy significantly reduced peak glucose levels in ob/ob mice, while IL-25 had no effect on WT mice. The effect of IL-25 on glucose tolerance was confirmed by a significant reduction in the AUC (Figure 6B). The intestinal expression of SGLT1 was similar in all groups (Supplemental Figure 4A) but Glut2 expression was reduced in WT, but not in ob/ob mice (Supplemental Figure 4B).
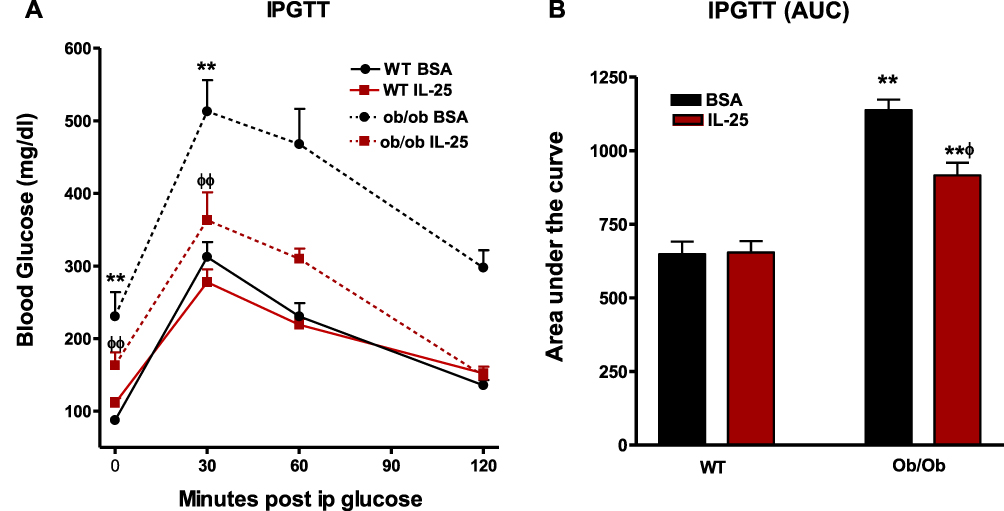 |
Figure 6 IL-25 treatment improves glucose tolerance in genetically obese ob/ob mice. WT or ob/ob mice were treated with BSA or IL-25 for 21 days and the effect of IL-25 treatment on glucose levels during intraperitoneal glucose tolerance test (IPGTT), (A) and the area under the curve (AUV) of the IPGTT curves determined, (B) mean±SEM, n=8–10 **p<0.01 vs WT BSA; ф p<0.05, ф ф p<0.01 vs ob/ob BSA. |
IL-25 Effects on TEER in HFD-Induced and Genetic Obesity
There is evidence that obesity and hyperglycemia are associated with increased intestinal permeability.34,35 To determine if intestinal permeability was increased in the small intestine in diet-induced obesity and/or in genetically obese mice TEER was measured. We confirmed previous reports of enhanced permeability in the small intestine of mice fed a HFD and show that IL-25 did not further reduce TEER (Supplemental Figure 5A). In contrast, IL-25 reduced TEER in WT mice but not in ob/ob mice (Supplemental Figure 5B).
Acute Effects of IL-25 on Glucose Absorption in IPEC-1 and L-6 Skeletal Muscle Cells in vitro
Receptors for IL-25 are present in both immune and structural cells. Therefore, IL-25 may have direct beneficial effects on structural cells, such as enterocytes, adipocytes, and skeletal muscle. To better understand how IL-25 may be affecting glucose metabolism in vivo, we initiated experiments to evaluate the effect of IL-25 on glucose absorption by gut epithelial and skeletal muscle cells. To investigate the acute inhibitory effects of IL-25 on glucose uptake, IPEC-1 cells were co-incubated with IL-25 at two concentrations (50, 100 ng/mL) and then with the fluorescent glucose analog 2-NBDG for 120 min. As shown in Figure 7A, glucose uptake was significantly reduced by 50, 100 ng/mL IL-25, respectively (p < 0.05). In contrast, IL-25 treatment enhanced glucose uptake in skeletal muscle L6 cells. Treatment with 100 ng/mL of IL-25 significantly stimulated glucose uptake compared to control and was comparable to the increase seen with 100 nM insulin treatment (Figure 7B).
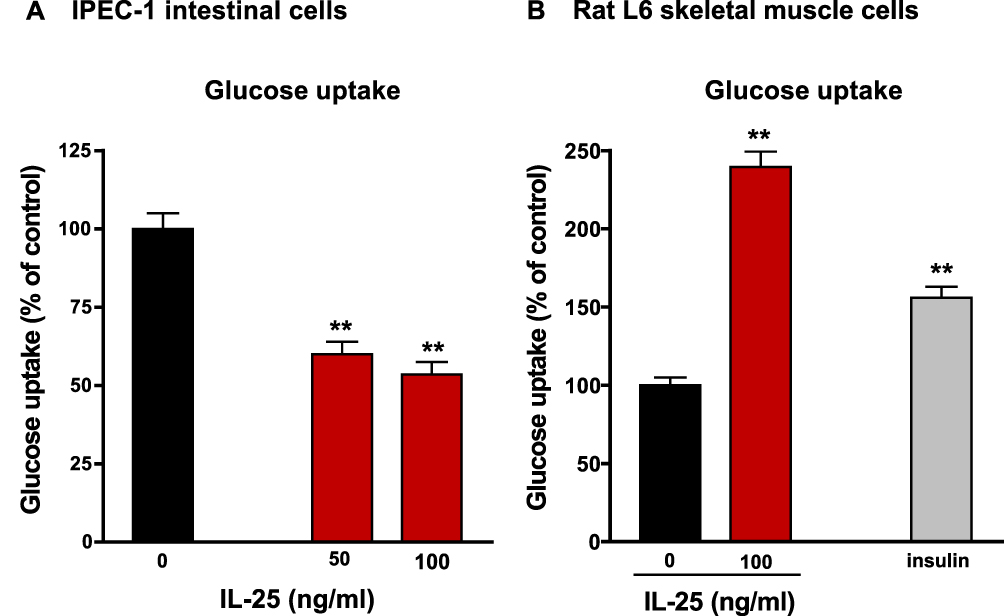 |
Figure 7 IL-25 has a direct effect on glucose uptake in iPEC-1 cells and skeletal smooth muscle cells. Differentiated iPEC-1 cells (A) or L6 skeletal cells (B) were incubated with IL-25 at two concentrations (50, 100 ng/mL) for two hours and then with the fluorescent glucose analog 2-NBDG for 2 hours to measure cellular glucose uptake. 2-NBDG uptake was measured using an Eclipse TE2000-S microscope and the fluorescence intensity was quantified by ImageJ software (NIH Image). Mean±SEM, n=5–6 **p<0.01 vs 0. |
Discussion
There is increasing recognition of the contribution of immune cells, such as eosinophils, tuft cells, ILC2, and most recently γδ T cells to the maintenance of the lean gut phenotype. IL-25 is unique in that its primary source in the gut is the epithelial tuft cell, which has chemosensing properties conferred by the gustatory G protein α-gustducin that sense pathogens.36 Although tuft cells comprise only 0.4% of epithelial cells,37 they are increased significantly during Th2-driven responses. IL-25-responsive innate cells, including IL-13-producing ILC2, regulate weight and glucose homeostasis in mice fed a HFD via the induction and maintenance of eosinophils in visceral adipose tissue.40 In addition, IL-25 promotes macrophage polarization towards the M2 phenotype that is characteristic of lean tissues.3,29,38,39 In the present study, we demonstrate that IL-25 treatment can positively impact glucose metabolism in both diet-induced and in genetically obese mice. Importantly, the beneficial effects of IL-25 against HFD-induced hyperglycemia extend beyond cessation of therapy indicating long-term IL-25 induced changes in glucose metabolism.
Feeding a HFD has multiple effects on gut function that contribute to obesity-induced inflammation. Mice fed a HFD had increased small intestinal permeability with reduced expression of tight junction proteins. This facilitates translocation of bacteria or bacterial products leading to dysbiosis and metabolic endotoxemia and can be reversed by antibiotic treatment.34 Germ-free mice were also protected against metabolic syndrome induced by a HFD,40 further implicating the microbiota in the HFD-induced changes in barrier function. In the present study, both IL-25 alone and the HFD reduced small intestinal TEER, consistent with previous findings, but IL-25 did not exacerbate the effects of the HFD indicating perhaps a limit to lower levels of barrier function as measured by TEER. IL-25 also did not decrease TEER in ob/ob mice. Both ob/ob mice and nematode infected mice, which upregulates IL-25, have low levels of leptin. Leptin is known to increase gut permeability and to be pro-inflammatory, and the latter effect may be offset by treatment with IL-25 by promoting a M2 phenotype in macrophages. This is supported by the observation that IL-25 inhibited monocyte-derived cytokines and prevented LPS induced lethal endotoxemia in mice.41 Taken together, these data show diet rather than genetic-induced hyperglycemia negatively impacts barrier function. This may be attributed to hyperglycemia and/or dysbiosis, but the absence of a further reduction by IL-25 is important when considering its therapeutic potential.
Caloric intake is an important factor in obesity, and IL-25 treatment had different effects on weight loss in the HFD and hyperphagia-induced obesity in ob/ob mice. Mice on the HFD experienced a transient reduction in food intake; however, when food intake in a BSA-treated HFD group was matched to that in the IL-25 treated HFD group, only about 10% of the weight loss in the IL-25 treated group could be attributed to reduced food intake. Moreover, BSA matched mice gained weight from day 14–21 coincident with increased food intake. In contrast to the results obtained with mice fed the HFD, IL-25 treatment did not result in weight loss in the ob/ob mice. IL-25 is thought to be part of the immune-mediated hypophagia associated with enteric nematode infection39,42 that is linked to increased release of CCK, a hormone that inhibits food intake.43 Mice fed a HFD and subjected to a nematode infection also exhibited increased levels of IL-25 and decreased expression of the immunoregulatory adipokine leptin.20 Importantly, both the HFD and genetic obesity significantly increased fasting blood glucose as well as peak IPGT levels and both parameters were lowered significantly by IL-25 irrespective of weight loss.
Carbohydrate absorption is an active process that involves cotransport of sugar with sodium ions through the SGLT-1, the activity of which is enhanced by GLP44 and reduced by CCK and leptin.45,46 There is also a growing recognition of the importance of the insulin-sensitive bidirectional glucose transporter, GLUT2. On the basolateral aspect, GLUT2 facilitates glucose exit from the enterocyte and GLUT2 is also sensitive to metabolic changes, with translocation to the luminal side of enterocytes in obesity47 thus, further enhancing glucose uptake. The content and amount of luminal nutrients in the diet drive the changes in intestinal morphology and function that distinguish the intestine in lean and obese individuals.48 Carbohydrate transporter expression is modulated by luminal availability of carbohydrates49 such that diet high in starch or sucrose upregulates the expression of SGLT1 and GLUT2.50 Trafficking of GLUT2 to the apical membrane is regulated by SGLT1 activity resulting in enhanced glucose absorption, a mechanism that matches transport to dietary intake. Reports that glucose absorption is more rapid and efficient in obese than in lean subjects predisposing to weight gain and T2D51 have been attributed, in part, to the upregulated expression of SGLT1 and GLUT2 in T2D patients.52 In the present study, the HFD decreased both SGLT1 activity and expression consistent with previous observations53,54 and supporting the opposing effects of carbohydrate versus fat on glucose transporter function and expression. That the transporters are not regulated by hyperphagia in mice fed a normal diet is reinforced by the absence of changes in SGLT1 or GLUT2 expression in ob/ob mice. IL-25 treatment further reduced glucose absorption in mice fed the HFD, consistent with our previous findings of inhibited SGLT1 activity and decreased GLUT2 expression induced by Th2 cytokines, IL-25, and nematode infection.21,32,33,55 These data provide further support for an effect of IL-25 on glucose metabolism in both models that is independent of weight loss. This idea is consistent with previous studies demonstrating that cytokines induced by nematode infection, which include IL-25, induce a “lean enterocyte” phenotype.29 Intestinal epithelial cells express receptors for IL-25 and as observed in vivo,29 IL-25 treatment inhibited glucose uptake into the porcine epithelial cell-line IPEC-1 demonstrating a direct effect on glucose transport.
In contrast to the intestine, the major insulin-regulated glucose transporter in mammalian skeletal muscle and adipose tissue is glucose transporter protein 4 (GLUT4).56 Indeed, transport of glucose into the skeletal muscle cells is mediated primarily by GLUT4 and is the rate-limiting step in whole-body glucose metabolism under normoglycemic conditions. Skeletal muscle accounts for ~75% of whole-body insulin-stimulated glucose uptake and T2D impaired skeletal muscle handling of glucose.57 GLUT4 is downregulated in adipose tissue and in skeletal muscle in obesity. Herein, IL-25 treatment increased glucose uptake into L6 skeletal muscle cells in vitro comparable to that induced by insulin, suggesting that improved glucose metabolism in IL-25 treated mice may be due in part to increased glucose uptake in skeletal muscle.
The long-term changes in adipose and other tissues that continue even after weight loss have immune underpinnings and are termed obesogenic memory.58 IL-25 mediated changes in host-protective immunity and gut function are dependent on IL-13 and STAT6.21 IL-25 promotes development of M2 macrophages in HFD mice,18,59 an effect associated with reduced lipolysis and hepatic steatosis.59 In the present study, the beneficial effects of IL-25 treatment on glucose metabolism persisted for weeks after cessation of treatment. Obesity-induced inflammation reduces the number of ILC2 cells in adipose tissue.60 Exogenous IL-25 stimulates ILC2 release of IL-5 and IL-13 that can restore the Th2 bias in immune cells, including macrophages, thus opposing the pro-inflammatory effects of obesity that can lead to metabolic syndrome. IL-25 administration increased ILC2 cells and M2 in VAT of obese mice,28 and this may contribute to the persistence of a Th2 environment with improved glucose intolerance and insulin sensitivity even after cessation of IL-25 treatment. This is consistent with our previous findings showing the decrease in SGLT1 activity, but not protein, in response to enteric nematode infection was dependent on IL-4/IL-13 mediated STAT6 activation of alternatively activated M2 macrophages.
The long-term changes in glucose metabolism may also be due to epigenetic changes in enterocytes. Hyperglycemia reprogrammed the colonic epithelial transcriptome to alter mucosal barrier function by a mechanism that involves GLUT2,34 which is expressed in the colon, albeit at low levels. Epigenetic regulation of stem cells plays a role in intestinal homeostasis and can be modulated by dietary metabolites from gut microbiota.61 IL-25 simulates ILC2 release of IL-13 which in turn acts in a feed-forward fashion on intestinal stem cells to regulate the self-renewal of Lgr5+ stem cells62 as well as to promote differentiation into tuft and goblet cells.23 There is a constitutive expression of IL-25 receptors on ILC2 in the small intestine suggesting an important role for IL-25 in intestinal homeostasis. Previous studies demonstrated that exogenous IL-25 can bypass the need for tuft cells and remodel the intestinal stem cell niche to enhance immune responses.63 Data from the present study suggest that this niche remodeling may also contribute to the persistence of beneficial effects of IL-25 on glucose metabolism post-treatment.
Conclusion
The results presented here identify IL-25 as a potential target for strategies designed to improve obesity-induced metabolic syndrome and T2D. IL-25 plays a major role in the promotion and initiation of type 2 immunity, downregulation of Th1/Th17 pro-inflammatory cytokines, such as IL17A and IL-12, and development of M2 macrophages19–21 that can counteract the pro-inflammatory environment associated with T2D (Supplemental Figure 6). This approach offers distinct advantages over targeting individual pro-inflammatory cytokines and IL-25 treatment was well tolerated over a three-week treatment period with no major adverse events. IL-25 normalized glucose tolerance tests in T2D induced by feeding a HFD or in genetically induced obesity (ob/ob). The beneficial immunometabolic actions on glucose metabolism post-IL-25 treatment in the present study may be attributed to the ability of M2 macrophages to maintain insulin sensitivity. Importantly, effects of IL-25 are maintained for a period-of-time post treatment, suggesting that therapeutic dosing could be similar to that of other biologics with only intermittent self-injections. Thus, the IL-25 pathway is an attractive target for the development of strategies to improve obesity-induced metabolic syndrome and T2D that needs to be explored further.
Disclosure
This work was prepared while Aiping Zhao was employed at University of Maryland School of Medicine and his current address is Center for Scientific Review, National Institutes of Health, Bethesda, MD, USA. The opinions expressed in this article are the author’s own and do not reflect the view of the National Institutes of Health, the Department of Health and Human Services, or the United States government. Dr Aiping Zhao reports a patent US9724392B2 issued to University of Maryland Baltimore. The authors report no other conflicts of interest in this work.
References
1. Odegaard JI, Chawla A. Pleiotropic actions of insulin resistance and inflammation in metabolic homeostasis. Science. 2013;339(6116):172–177. doi:10.1126/science.1230721
2. Osborn O, Olefsky JM. The cellular and signaling networks linking the immune system and metabolism in disease. Nat Med. 2012;18(3):363–374. doi:10.1038/nm.2627
3. Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. 2010;72:219–246. doi:10.1146/annurev-physiol-021909-135846
4. Katsuki A, Sumida Y, Murashima S, et al. Serum levels of tumor necrosis factor-α are increased in obese patients with noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab. 1998;83(3):859–862.
5. Ghanim H, Aljada A, Hofmeyer D, Syed T, Mohanty P, Dandona P. Circulating mononuclear cells in the obese are in a proinflammatory state. Circulation. 2004;110(12):1564–1571. doi:10.1161/01.CIR.0000142055.53122.FA
6. Nam H, Ferguson BS, Stephens JM, Morrison RF. Impact of Obesity on IL-12 Family Gene Expression in Insulin Responsive Tissues. Biochim Biophys Acta. 2013;1832(1):11–19. doi:10.1016/j.bbadis.2012.08.011
7. Winkler G, Dworak O, Salamon F, Salamon D, Speer G, Cseh K. Increased interleukin-12 plasma concentrations in both, insulin-dependent and non-insulin-dependent diabetes mellitus. Diabetologia. 1998;41(4):488. doi:10.1007/s001250050935
8. Ip B, Cilfone N, Belkina AC, et al. Th17 cytokines differentiate obesity from obesity-associated type 2 diabetes and promote TNFα production. Obesity. 2016;24(1):102–112. doi:10.1002/oby.21243
9. Winer S, Chan Y, Paltser G, et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med. 2009;15(8):921–929. doi:10.1038/nm.2001
10. Brooks-Worrell B, Narla R, Palmer JP. Biomarkers and immune-modulating therapies for Type 2 diabetes. Trends Immunol. 2012;33(11):546–553. doi:10.1016/j.it.2012.07.002
11. Donath MY. Targeting inflammation in the treatment of type 2 diabetes: time to start. Nat Rev Drug Discov. 2014;13(6):465–476. doi:10.1038/nrd4275
12. Bernstein L, Berry J, Kim S, Canavan B, Grinspoon SK. EFfects of etanercept in patients with the metabolic syndrome. Arch Intern Med. 2006;166(8):902–908. doi:10.1001/archinte.166.8.902
13. Dominguez H, Storgaard H, Rask-Madsen C, et al. Metabolic and Vascular Effects of Tumor Necrosis Factor-α Blockade with Etanercept in Obese Patients with Type 2 Diabetes. J Vasc Res. 2005;42(6):517–525. doi:10.1159/000088261
14. Faghihimani E, Aminorroaya A, Rezvanian H, Adibi P, Ismail-Beigi F, Amini M. Salsalate improves glycemic control in patients with newly diagnosed type 2 diabetes. Acta Diabetol. 2013;50(4):537–543. doi:10.1007/s00592-011-0329-2
15. Goldfine AB, Fonseca V, Jablonski KA, Pyle L, Staten MA, Shoelson SE. The Effects of Salsalate on Glycemic Control in Patients With Type 2 DiabetesA Randomized Trial. Ann Intern Med. 2010;152(6):346–357. doi:10.7326/0003-4819-152-6-201003160-00004
16. Rissanen A, Howard CP, Botha J, Thuren T. for the Global I. Effect of anti-IL-1β antibody (canakinumab) on insulin secretion rates in impaired glucose tolerance or type 2 diabetes: results of a randomized, placebo-controlled trial. Diabetes Obes Metab. 2012;14(12):1088–1096. doi:10.1111/j.1463-1326.2012.01637.x
17. Sloan-Lancaster J, Abu-Raddad E, Polzer J, et al. Double-Blind, Randomized Study Evaluating the Glycemic and Anti-inflammatory Effects of Subcutaneous LY2189102, a Neutralizing IL-1β Antibody, in Patients With Type 2 Diabetes. Diabetes Care. 2013;36(8):2239–2246. doi:10.2337/dc12-1835
18. Wang X, Ota N, Manzanillo P, et al. Interleukin-22 alleviates metabolic disorders and restores mucosal immunity in diabetes. Nature. 2014;514(7521):237–241. doi:10.1038/nature13564
19. Iwakura Y, Nakae S, Saijo S, Ishigame H. The roles of IL-17A in inflammatory immune responses and host defense against pathogens. Immunol Rev. 2008;226:57–79. doi:10.1111/j.1600-065X.2008.00699.x
20. Yang Z, Grinchuk V, Urban JF
21. Zhao A, Urban JF
22. Gerbe F, van Es JH, Makrini L, et al. Distinct ATOH1 and Neurog3 requirements define tuft cells as a new secretory cell type in the intestinal epithelium. J Cell Biol. 2011;192(5):767–780. doi:10.1083/jcb.201010127
23. von Moltke J, Ji M, Liang H-E, Locksley RM. Tuft-cell-derived IL-25 regulates an intestinal ILC2–epithelial response circuit. Nature. 2016;529(7585):221–225. doi:10.1038/nature16161
24. Sarra M, Cupi ML, Bernardini R, et al. IL-25 prevents and cures fulminant hepatitis in mice through a myeloid-derived suppressor cell-dependent mechanism. Hepatology. 2013;58(4):1436–1450. doi:10.1002/hep.26446
25. Wang AJ, Yang Z, Grinchuk V, et al. IL-25 or IL-17E Protects against High-Fat Diet-Induced Hepatic Steatosis in Mice Dependent upon IL-13 Activation of STAT6. J Immunol. 2015;195(10):4771–4780. doi:10.4049/jimmunol.1500337
26. Rickel EA, Siegel LA, Yoon B-RP, et al. Identification of Functional Roles for Both IL-17RB and IL-17RA in Mediating IL-25-Induced Activities. J Immunol. 2008;181(6):4299–4310. doi:10.4049/jimmunol.181.6.4299
27. Huang Y, Paul WE. Inflammatory group 2 innate lymphoid cells. Int Immunol. 2016;28(1):23–28. doi:10.1093/intimm/dxv044
28. Hams E, Locksley RM, McKenzie ANJ, Fallon PG. Cutting Edge: IL-25 Elicits Innate Lymphoid Type 2 and Type II NKT Cells That Regulate Obesity in Mice. J Immunol. 2013;191(11):5349–5353. doi:10.4049/jimmunol.1301176
29. Notari L, Riera DC, Sun R, et al. Role of macrophages in the altered epithelial function during a type 2 immune response induced by enteric nematode infection. PLoS One. 2014;9(1):e84763. doi:10.1371/journal.pone.0084763
30. Zhao A, Urban JF
31. Gonzalez-Vallina R, Wang H, Zhan R, et al. Lipoprotein and apolipoprotein secretion by a newborn piglet intestinal cell line (IPEC-1). Am J Physiol. 1996;271(2 Pt 1):G249–259. doi:10.1152/ajpgi.1996.271.2.G249
32. Madden KB, Whitman L, Sullivan C, et al. Role of STAT6 and mast cells in IL-4- and IL-13-induced alterations in murine intestinal epithelial cell function. JImmunol. 2002;169(8):4417–4422. doi:10.4049/jimmunol.169.8.4417
33. Madden KB, Yeung KA, Zhao A, et al. Enteric nematodes induce stereotypic STAT6-dependent alterations in intestinal epithelial cell function. JImmunol. 2004;172(9):5616–5621. doi:10.4049/jimmunol.172.9.5616
34. Thaiss CA, Levy M, Grosheva I, et al. Hyperglycemia drives intestinal barrier dysfunction and risk for enteric infection. Science. 2018;359(6382):1376–1383. doi:10.1126/science.aar3318
35. Ahmad R, Rah B, Bastola D, Dhawan P, Singh AB. Obesity-induces Organ and Tissue Specific Tight Junction Restructuring and Barrier Deregulation by Claudin Switching. Sci Rep. 2017;7(1):5125. doi:10.1038/s41598-017-04989-8
36. Howitt MR, Lavoie S, Michaud M, et al. Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science. 2016;351(6279):1329–1333. doi:10.1126/science.aaf1648
37. Steensels S, Depoortere I. Chemoreceptors in the Gut. Annu Rev Physiol. 2018;80:117–141. doi:10.1146/annurev-physiol-021317-121332
38. Caslin HL, Bhanot M, Bolus WR, Hasty AH. Adipose tissue macrophages: unique polarization and bioenergetics in obesity. Immunol Rev. 2020;295(1):101–113. doi:10.1111/imr.12853
39. Worthington JJ, Samuelson LC, Grencis RK, McLaughlin JT. Adaptive immunity alters distinct host feeding pathways during nematode induced inflammation, a novel mechanism in parasite expulsion. PLoS Pathog. 2013;9(1):e1003122. doi:10.1371/journal.ppat.1003122
40. Rabot S, Membrez M, Bruneau A, et al. Germ-free C57BL/6J mice are resistant to high-fat-diet-induced insulin resistance and have altered cholesterol metabolism. FASEB J. 2010;24(12):4948–4959. doi:10.1096/fj.10-164921
41. Caruso R, Stolfi C, Sarra M, et al. Inhibition of monocyte-derived inflammatory cytokines by IL-25 occurs via p38 Map kinase-dependent induction of Socs-3. Blood. 2009;113(15):3512–3519. doi:10.1182/blood-2008-08-172767
42. McDermott JR, Leslie FC, D’Amato M, Thompson DG, Grencis RK, McLaughlin JT. Immune control of food intake: enteroendocrine cells are regulated by CD4+ T lymphocytes during small intestinal inflammation. Gut. 2006;55(4):492–497. doi:10.1136/gut.2005.081752
43. Moran TH. Cholecystokinin and satiety: current perspectives. Nutrition. 2000;16(10):858–865. doi:10.1016/S0899-9007(00)00419-6
44. Au A, Gupta A, Schembri P, Cheeseman CI. Rapid insertion of GLUT2 into the rat jejunal brush-border membrane promoted by glucagon-like peptide 2. Biochem J. 2002;367(Pt 1):247–254. doi:10.1042/bj20020393
45. Ducroc R, Guilmeau S, Akasbi K, Devaud H, Buyse M, Bado A. Luminal Leptin Induces Rapid Inhibition of Active Intestinal Absorption of Glucose Mediated by Sodium-Glucose Cotransporter 1. Diabetes. 2005;54(2):348–354. doi:10.2337/diabetes.54.2.348
46. Hirsh AJ, Cheeseman CI. Cholecystokinin Decreases Intestinal Hexose Absorption by a Parallel Reduction in SGLT1 Abundance in the Brush-Border Membrane. J Biol Chem. 1998;273(23):14545–14549. doi:10.1074/jbc.273.23.14545
47. Ait-Omar A, Monteiro-Sepulveda M, Poitou C, et al. GLUT2 Accumulation in Enterocyte Apical and Intracellular Membranes. Diabetes. 2011;60(10):2598–2607. doi:10.2337/db10-1740
48. Dailey MJ. Nutrient-induced intestinal adaption and its effect in obesity. Physiol Behav. 2014;136:74–78. doi:10.1016/j.physbeh.2014.03.026
49. Ferraris RP, Diamond JM. Specific regulation of intestinal nutrient transporters by their dietary substrates. Annu Rev Physiol. 1989;51:125–141. doi:10.1146/annurev.ph.51.030189.001013
50. Honma K, Mochizuki K, Goda T. Inductions of histone H3 acetylation at lysine 9 on SGLT1 gene and its expression by feeding mice a high carbohydrate/fat ratio diet. Nutrition. 2009;25(1):40–44. doi:10.1016/j.nut.2008.07.006
51. Zhang X, Young RL, Bound M, et al. Comparative Effects of Proximal and Distal Small Intestinal Glucose Exposure on Glycemia, Incretin Hormone Secretion, and the Incretin Effect in Health and Type 2 Diabetes. Diabetes Care. 2019;42(4):520–528. doi:10.2337/dc18-2156
52. Dyer J, Wood IS, Palejwala A, Ellis A, Shirazi-Beechey SP. Expression of monosaccharide transporters in intestine of diabetic humans. Am J Physiol Gastrointest Liver Physiol. 2002;282(2):G241–248. doi:10.1152/ajpgi.00310.2001
53. Wiśniewski JR, Gizak A, Rakus D. Integrating Proteomics and Enzyme Kinetics Reveals Tissue-Specific Types of the Glycolytic and Gluconeogenic Pathways. J Proteome Res. 2015;14(8):3263–3273. doi:10.1021/acs.jproteome.5b00276
54. Losacco MC, de Almeida CFT, Hijo AHT, et al. High-fat diet affects gut nutrients transporters in hypo and hyperthyroid mice by PPAR-a independent mechanism. Life Sci. 2018;202:35–43. doi:10.1016/j.lfs.2018.03.053
55. Shea-Donohue T, Sullivan C, Finkelman FD, et al. The Role of IL-4 in Heligmosomoides polygyrus-Induced Alterations in Murine Intestinal Epithelial Cell Function. J Immunol. 2001;167(4):2234–2239. doi:10.4049/jimmunol.167.4.2234
56. Chang L, Chiang S-H, Saltiel AR. Insulin Signaling and the Regulation of Glucose Transport. Mol Med. 2004;10(7–12):65–71. doi:10.2119/2005-00029.Saltiel
57. Huang S, Czech MP. The GLUT4 Glucose Transporter. Cell Metab. 2007;5(4):237–252. doi:10.1016/j.cmet.2007.03.006
58. Blaszczak AM, Bernier M, Wright VP, et al. Obesogenic Memory Maintains Adipose Tissue Inflammation and Insulin Resistance. Immunometabolism. 2020;2:3.
59. Feng J, Li L, Ou Z, et al. IL-25 stimulates M2 macrophage polarization and thereby promotes mitochondrial respiratory capacity and lipolysis in adipose tissues against obesity. Cell Mol Immunol. 2018;15(5):493–505. doi:10.1038/cmi.2016.71
60. Brestoff JR, Kim BS, Saenz SA, et al. Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature. 2015;519(7542):242–246. doi:10.1038/nature14115
61. Bhat MI, Kapila R. Dietary metabolites derived from gut microbiota: critical modulators of epigenetic changes in mammals. Nutr Rev. 2017;75(5):374–389. doi:10.1093/nutrit/nux001
62. Zhu P, Zhu X, Wu J, et al. IL-13 secreted by ILC2s promotes the self-renewal of intestinal stem cells through circular RNA circPan3. Nat Immunol. 2019;20(2):183–194. doi:10.1038/s41590-018-0297-6
63. Schneider C, O’Leary CE, von Moltke J, et al. A Metabolite-Triggered Tuft Cell-ILC2 Circuit Drives Small Intestinal Remodeling. Cell. 2018;174(2):271–284.e214. doi:10.1016/j.cell.2018.05.014
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.