")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 17
IL-17A Promotes Epithelial ADAM9 Expression in Cigarette Smoke-Related COPD
Authors Li D, Wang T, Ma Q, Zhou L, Le Y, Rao Y, Jin L, Pei Y, Cheng Y, Huang C, Gai X, Sun Y
Received 15 June 2022
Accepted for publication 30 September 2022
Published 14 October 2022 Volume 2022:17 Pages 2589—2602
DOI https://doi.org/10.2147/COPD.S375006
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Russell
Danyang Li,1 Tong Wang,2 Qianli Ma,3 Lu Zhou,1 Yanqing Le,1 Yafei Rao,1 Liang Jin,1 Yuqiang Pei,1 Yaning Cheng,4 Chen Huang,5 Xiaoyan Gai,1 Yongchang Sun1
1Department of Respiratory and Critical Care Medicine, Peking University Third Hospital, Beijing, 100191, People’s Republic of China; 2Department of Thoracic Surgery, Peking University Third Hospital, Beijing, 100191, People’s Republic of China; 3Department of Thoracic Surgery, China-Japan Friendship Hospital, Beijing, 100029, People’s Republic of China; 4School of Basic Medical Sciences, Peking University Health Science Center, Beijing, 100191, People’s Republic of China; 5Center of Basic Medical Research, Peking University Third Hospital, Beijing, 100191, People’s Republic of China
Correspondence: Xiaoyan Gai; Yongchang Sun, Department of Respiratory and Critical Care Medicine, Peking University Third Hospital, No. 49 North Garden Road, Haidian District, Beijing, 100191, People’s Republic of China, Email [email protected]; [email protected]
Background: It has been reported that a disintegrin and metalloproteinase 9 (ADAM9) is involved in the pathogenesis of cigarette smoke (CS)-associated chronic obstructive pulmonary disease (COPD). But how CS exposure leads to upregulation of ADAM9 remains unknown.
Methods: Patients who underwent lobectomy for a solitary pulmonary nodule were enrolled and divided into three groups: non-smokers with normal lung function, smokers without COPD and smoker patients with COPD. Immunoreactivity of interleukin (IL)-17A and ADAM9 in small airways and alveolar walls was measured by immunohistochemistry. Wild-type and Il17a−/− C57BL/6 mice were exposed to CS for six months, and ADAM9 expression in the airway epithelia was measured by immunoreactivity. In addition, the protein and mRNA expression levels of IL-17A and ADAM9 were assessed in CS extract (CSE) and/or IL-17A-treated human bronchial epithelial (HBE) cells.
Results: The immunoreactivity of ADAM9 was increased in the airway epithelia and alveolar walls of patients with COPD compared to that of the controls. The expression of IL-17A was also upregulated in airway epithelial cells of patients with COPD and correlated positively with the level of ADAM9. The results from the animal model showed that Il17a−/− mice were protected from emphysema induced by CS exposure, together with a reduced level of ADAM9 expression in the airway epithelia, suggesting a possible link between ADAM9 and IL-17A. Consistently, our in vitro cell model showed that CSE stimulated the expression of ADAM9 and IL-17A in HBE cells in a dose- and time-dependent manner. Recombinant IL-17A induced ADAM9 upregulation in HBE cells and had a synergistic effect with CSE, whereas blocking IL-17A inhibited CSE-induced ADAM9 expression. Further analysis revealed that IL-17A induced c-Jun N-terminal kinase (JNK) phosphorylation, thereby increasing ADAM9 expression.
Conclusion: Our results revealed a novel role of IL-17A in CS-related COPD, where IL-17A contributes to ADAM9 expression by activating JNK signaling.
Keywords: chronic obstructive pulmonary disease, a disintegrin and metalloproteinase 9, interleukin-17A, airway epithelium
Introduction
Chronic obstructive pulmonary disease (COPD) is a chronic inflammatory disease characterized by a progressive decline of lung function resulting from alveolar destruction and airway remodeling. It has become a leading cause of morbidity and mortality worldwide and results in a substantial economic and social burden.1 Cigarette smoking is a major risk factor for COPD.1 Inhalation of cigarette smoke (CS) activates the innate and adaptive immune systems, leading to the release of a range of inflammatory mediators, including cytokines, chemokines, and proteases.2 Uncontrolled protease secretion contributes to the degradation of the extracellular matrix (ECM) components, and hence airspace enlargement (emphysema), a hallmark of COPD.3
A variety of proteases, such as serine proteinases, cysteine proteinases, and matrix metalloproteases (MMPs), have been reported to participate in the development of emphysema,3 whereas a disintegrin and metalloproteinases (ADAMs), such as ADAM8,4 ADAM9,5,6 ADAM15,7 and ADAM33,8 as a less studied subgroup of metalloproteinases, were found to be involved in COPD pathogenesis in recent years. ADAMs are type I transmembrane proteins containing six important domains, among which the metalloproteinase and disintegrin domains endow them with proteolytic and adhesive functions, respectively.9 ADAM9, one of the ADAM proteins, is widely expressed in a variety of cells, such as neutrophils, monocytes, macrophages, T cells, epithelial cells, endothelial cells, fibroblasts, and many types of tumor cells, and is demonstrated to play crucial roles in many inflammatory and neoplastic diseases.10 Elevated ADAM9 expression could promote cancer progression by increasing cell proliferation, invasion and migration.10 For example, ADAM9 is involved in lung cancer metastasis to the brain through facilitating CUB-domain–containing protein 1 cleavage.11 Moreover, polymorphonuclear neutrophils-derived ADAM9 contributes to acute lung injury development by degrading ECM proteins and promoting alveolar-capillary barrier injury.12 ADAM9 was also shown to be associated with COPD pathogenesis. Patients with COPD had a higher ADAM9 expression in lung epithelial cells and alveolar macrophages than non-smokers and smokers without COPD.5,6 In addition, ADAM9 deficiency could reduce lung inflammation and emphysema in a CS-exposed mouse model.5 But how CS exposure leads to enhanced expression of ADAM9 in COPD remains unknown.
Interleukin (IL)-17A is a multifunctional pro-inflammatory cytokine involved in COPD pathogenesis. Increased IL-17A+ T cells in the peripheral blood and lung tissues have been observed and associated with the severity of lung function in COPD.13 In addition, IL-17A signaling is implicated in the formation of tertiary lymphatic tissues in patients with end-stage COPD and long-term CS-exposed mice.14,15 Besides its role in chronic airway inflammation, IL-17A is required for emphysema development. Il17a−/− mice are protected against emphysema induced by elastase or CS exposure partly through regulating neutrophilic inflammation and macrophage recruitment.16–19 Furthermore, IL-17A can mediate protease-antiprotease imbalance by promoting MMP expression, leading to the development of emphysema and airway inflammation. For instance, IL-17A promoted MMP12 expression in CS-exposed models,19,20 and MMP-12 overexpression was the major cause of alveolar wall destruction.21 Moreover, intranasal stimulation with IL-17A increased the concentration of MMP-9 and its precursor proMMP-9 in mouse airways,22 whereas IL-17A neutralization reduced the expression of MMP-9 and other pro-inflammatory cytokines and attenuated Pseudomonas aeruginosa infection in a COPD mouse model.23 Considering the function of IL-17A in mediating MMP expression and that ADAM9 is also a metalloprotease, we speculate that IL-17A may be responsible for enhanced ADAM9 expression in CS-related COPD.
In this study, we firstly evaluated the expression of ADAM9 and IL-17A in lung tissue samples from smokers with or without COPD and from non-smokers and found that the expression of ADAM9 and IL-17A was elevated in COPD patients. In a well-established model of COPD induced by long-term CS exposure, we demonstrated that ADAM9 expression was reduced in Il17a−/− mice compared with WT mice. In an in vitro model, CS exposure induced expression of ADAM9 and IL-17A in HBE cells, and IL-17A promoted ADAM9 expression by activating c-Jun N-terminal kinase (JNK) signaling. Taken together, our results revealed a novel role of IL-17A in the regulation of ADAM9 in CS-associated COPD.
Materials and Methods
Study Participants
Patients undergoing lobectomy for solitary pulmonary nodules (including benign and malignant nodules) at Peking University Third Hospital and China-Japan Friendship Hospital from 2018 to 2022 were recruited and divided into three groups: age-matched non-smokers with normal lung function (NS), smokers without COPD (S), and smoker patients with COPD (COPD). COPD was diagnosed by a post-bronchodilator forced expiratory volume in one second/forced vital capacity (FEV1/FVC) < 0.70 and the patients did not have exacerbations for at least three months before the study. Patients with a smoking history of ≥ 10 packs/year and a post-bronchodilator FEV1/FVC > 0.70 were defined as smokers without COPD. Only male patients were enrolled, because female smokers with COPD were less common in the Chinese population. Individuals with other chronic pulmonary diseases, such as asthma, bronchiectasis, and interstitial lung disease, autoimmune diseases, acute infection, a history of thoracic surgery, and long-term use of oral glucocorticoids or immunosuppressants were excluded. The study was approved by the Ethics Committee of Peking University Third Hospital [batch number: (2018)208-01], and conducted following the Declaration of Helsinki. Informed consent was obtained from all participants. Demographic data are presented in Table 1.
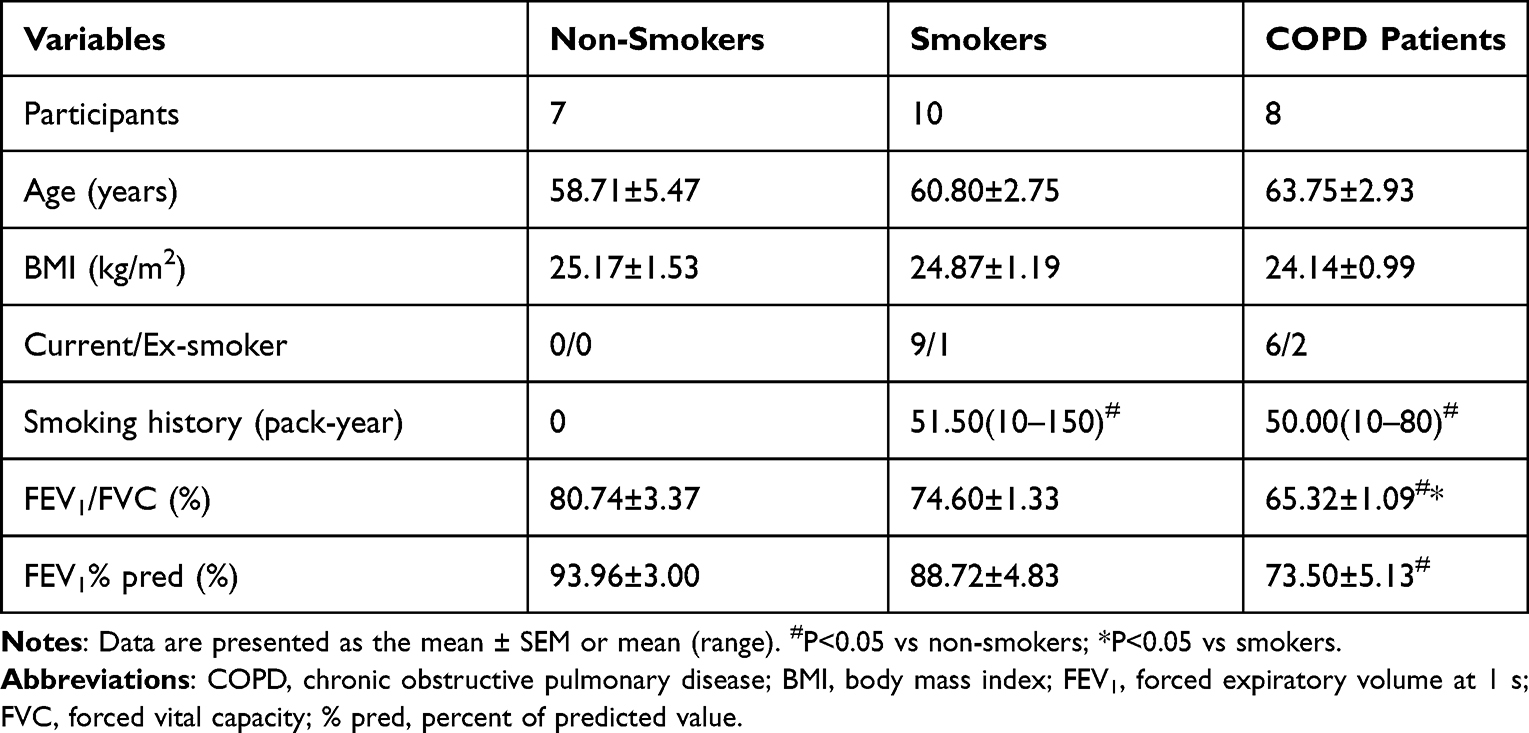 |
Table 1 General Characteristics of All Participants |
Human Lung Tissue Collection
Lung sections were obtained from lobectomy specimens from patients with solitary pulmonary nodules. Lung tissues were taken > 2 cm from the nodule, and specimens were taken > 10 cm from the tumor margin in cases with malignant tumors.
Animal Model of COPD
Six-to-eight-week-old female WT C57BL/6 mice were purchased from Beijing Vital River Experimental Animal Company (Beijing, China). Il17a−/− mice with a C57BL/6 background were generously donated by Dr. Huanzhong Shi (Capital Medical University, Beijing, China). Animals were housed under specific pathogen-free conditions with a light-dark cycle of 12 h. All animal protocols were approved by the Ethics Committee of Peking University Third Hospital [batch number: (2018)208-01] and followed the “Laboratory animal—Guideline for ethical review of animal welfare (GB/T 35892-2018)” and the “Guide for the Care and Use of Laboratory Animals: Eighth Edition (2011)”. WT and Il17a−/− mice were exposed to CS for six months to develop a COPD mouse model, as previously described.24 Briefly, mice were exposed to cigarette smoke (Baisha cigarettes, Hunan, China; tar: 11 mg, nicotine: 0.9 mg, CO:12 mg) for 50 min, twice a day, five days per week using a nose-only smoke exposure system (SG-300; SIBATA, Tokyo, Japan). The smoke was suctioned at a speed of 20 mL per 8 s, mixed with room air at a ratio of 1:9, and injected into the inhalation tower. The control groups were exposed to room air for six months. Hematoxylin and eosin (H&E) staining was performed to assess the severity of emphysema in the CS-exposed mice. The mean linear intercept (MLI) and destructive index (DI) were used to quantify the degree of airspace enlargement and alveolar wall destruction, respectively.
Immunohistochemistry
Lung tissues were fixed with 4% paraformaldehyde and paraffin-embedded after lung resection. Paraffin-embedded tissues were cut into 4 μm-thick sections. After dewaxing, hydration, and antigen retrieval, the sections were blocked with goat serum (ZSGB-Bio, Beijing, China) at room temperature for 40 min and immunostained with rabbit anti-ADAM9 (ab186833, Abcam, Cambridge, MA, USA) or rabbit anti-IL-17A (ab79056, Abcam, Cambridge, MA, USA) antibodies overnight. Subsequently, sections were incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (ZSGB-Bio, Beijing, China). Immunoreactivity was visualized using a DAB Detection System kit (ZSGB-Bio, Beijing, China). Images were captured using a NanoZommer-SQ Digital slide scanner (Hamamatsu, Japan) and analyzed using ImageJ software (National Institutes of Health, MD, USA). Two to three images per lung sections were randomly selected to measure the mean optical density of ADAM9 and IL-17A in epithelial cells. The average was taken for further statistical analysis.
Cigarette Smoke Extract (CSE) Preparation and Cell Proliferation Assessment
CSE was prepared as previously described.25 Briefly, smoke from five cigarettes (Baisha, Hunan, China) was slowly bubbled into a tube containing 10 mL Dulbecco’s modified Eagle medium (DMEM) (Biological Industries Ltd, Kibbutz Beit Haemek, Israel). Solutions with an absorbance value of approximately 4.0 were considered acceptable and were defined as 100% CSE solution. The CSE solution was filtered through a 0.22 µm filter (Millipore, Sigma, Billerica, MA, USA) to remove bacteria. The cytotoxic effect of CSE at different concentrations (0%, 1%, 3%, 5%, 7%, 10%) on HBE cells was detected using a Cell Counting Kit-8 (CCK-8; Yeasen Biotech, Shanghai, China).
Cell Culture
The HBE cell line was purchased from the American Type Culture Collection (ATCC, VA, USA). HBE cells were cultured in DMEM medium (Biological Industries Ltd, Kibbutz Beit Haemek, Israel) supplemented with 10% fetal bovine serum (Gibco, Thermo Fisher Scientific, Waltham, MA, USA), 100 U/mL penicillin and 100 μg/mL streptomycin (Hyclone, Global Life Sciences Solutions USA LLC, Marlborough, MA, USA) at 37 °C in 5% CO2-humidified atmosphere. Different concentrations of CSE and/or recombinant human IL-17A (200-17, PeproTech, Rocky Hill, NJ, USA) were added to the medium when cells grew to 70% confluence. The cells were collected after 0 to 48 h for further analysis. IL-17A neutralizing antibody (69021-1-Ig, Proteintech, Rosemont, IL, USA; 200ng/mL) was used to neutralize cell-intrinsic IL-17A. To investigate the effect of JNK signaling on IL-17A and/or CSE-induced ADAM9 expression in HBE cells, cells were pretreated for 1 h with 10 µM JNK inhibitor SP600125 (MedChemExpress, Monmouth Junction, NJ, USA).
Polymerase Chain Reaction (PCR)
Total RNA was extracted from HBE cells by using the TRIzol reagent (Life, Thermo Fisher Scientific, Waltham, MA, USA). Reverse transcription-polymerase chain reaction (RT-PCR) was performed using the QuantStudio 5 Real-Time PCR system (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA) and One Step TB Green® PrimeScript™ RT-PCR Kit II (Perfect Real Time) (Takara, Tokyo, Japan). Relative gene expression was analyzed using the ΔΔCt method. The PCR-specific primer sequences used for analysis were as follows: human ADAM9 forward primer, 5′-ATAGTGCTCCCTCCTGTGGT-3′; human ADAM9 reverse primer, 5′-ACAGGTACTTCCTTCGCAGC-3′; human IL-17A forward primer, 5′- CGGACTGTGATGGTCAACCT-3′; human IL-17A reverse primer, 5′- TCCTCATTGCGGTGGAGATT-3′; human GAPDH forward primer, 5′-AAATCAAGTGGGGCGATGCTG-3′; human GAPDH reverse primer, 5′-GCAGAGATGATGACCCTTTTG-3′.
Immunoblotting
Cells were lysed in RIPA lysis buffer (Applygen, Beijing, China) containing protease inhibitors (Applygen, Beijing, China) and phosphatase inhibitors (Applygen, Beijing, China). Protein concentrations were measured using a BCA kit (Applygen, Beijing, China). After sodium dodecyl sulfate-polyacrylamide gel electrophoresis for 45 min at a constant voltage of 80 V and 45 min at 120 V, the separated proteins were transferred to PVDF membranes (Millipore, Sigma, Billerica, MA, USA). The membranes were blocked with 5% skimmed milk powder at room temperature for 1.5 h and then incubated overnight at 4 °C with primary antibodies: rabbit anti-ADAM9 mAb (1:1000, 4151, Cell Signaling Technology, Danvers, MA, USA), mouse-anti-IL-17A mAb (1:1000, 66148-1-lg, Proteintech, Rosemont, IL, USA), rabbit anti-JNK mAb (ab208035, Abcam, Cambridge, MA, USA), and rabbit anti-phospho-JNK mAb (ab76572, Abcam, Cambridge, MA, USA). HRP-labeled goat anti-rabbit or goat anti-mouse lgG (ZSGB-Bio, Beijing, China) was used as secondary antibodies. Immunoreactive bands were detected using Immobilon Western Chemiluminescent HRP Substrate (Millipore, Sigma, Billerica, MA, USA) and analyzed using ImageJ software (National Institutes of Health, MD, USA).
Statistical Analyses
Data are presented as mean ± SEM or mean (range). Comparisons between two groups were analyzed using the Student’s t-test after normality and lognormality tests. Statistical differences among multiple groups were determined using a one-way analysis of variance (ANOVA) test followed by Tukey’s multiple comparisons test or Kruskal–Wallis test with Dunn’s multiple comparisons test. Correlations were determined using Pearson’s correlation analysis. All statistical data were analyzed using GraphPad Prism 8 software (GraphPad Software, La Jolla, CA, USA). Statistical significance was set at p < 0.05.
Results
Expression of ADAM9 and IL-17A Was Increased and Correlated in Lung Tissues from COPD
To explore whether IL-17A is associated with ADAM9 expression in COPD, we performed immunostaining to assess the distribution and expression levels of ADAM9 and IL-17A in lung tissue samples from smokers with or without COPD and non-smokers. As shown in Figures 1A and 2A, both ADAM9+ and IL-17A+ cells were detected in the small airways, alveolar walls, and infiltrating immune cells. The immunoreactivity of ADAM9 was significantly increased in the bronchiolar (Figure 1B) and alveolar epithelial cells (Figure 2B) in COPD compared with non-smokers. Increased expression of IL-17A was also observed in the airway epithelia of COPD patients (Figure 1C), whereas no difference in IL-17A expression was detected in the alveolar epithelia among the three groups (Figure 2C). Furthermore, the expression of ADAM9 in airway epithelial cells was correlated positively with IL-17A expression in all three groups (Figure 1D). A similar pattern was observed in the alveolar epithelia (Figure 2D). In addition, the expression of IL-17A in the airway epithelia showed a negative correlation with FEV1/FVC (Figure 1E). However, we did not find a relationship between the expression level of ADAM9 and FEV1/FVC or FEV1% pred in the airway epithelia or the alveolar epithelia in smokers with or without COPD.
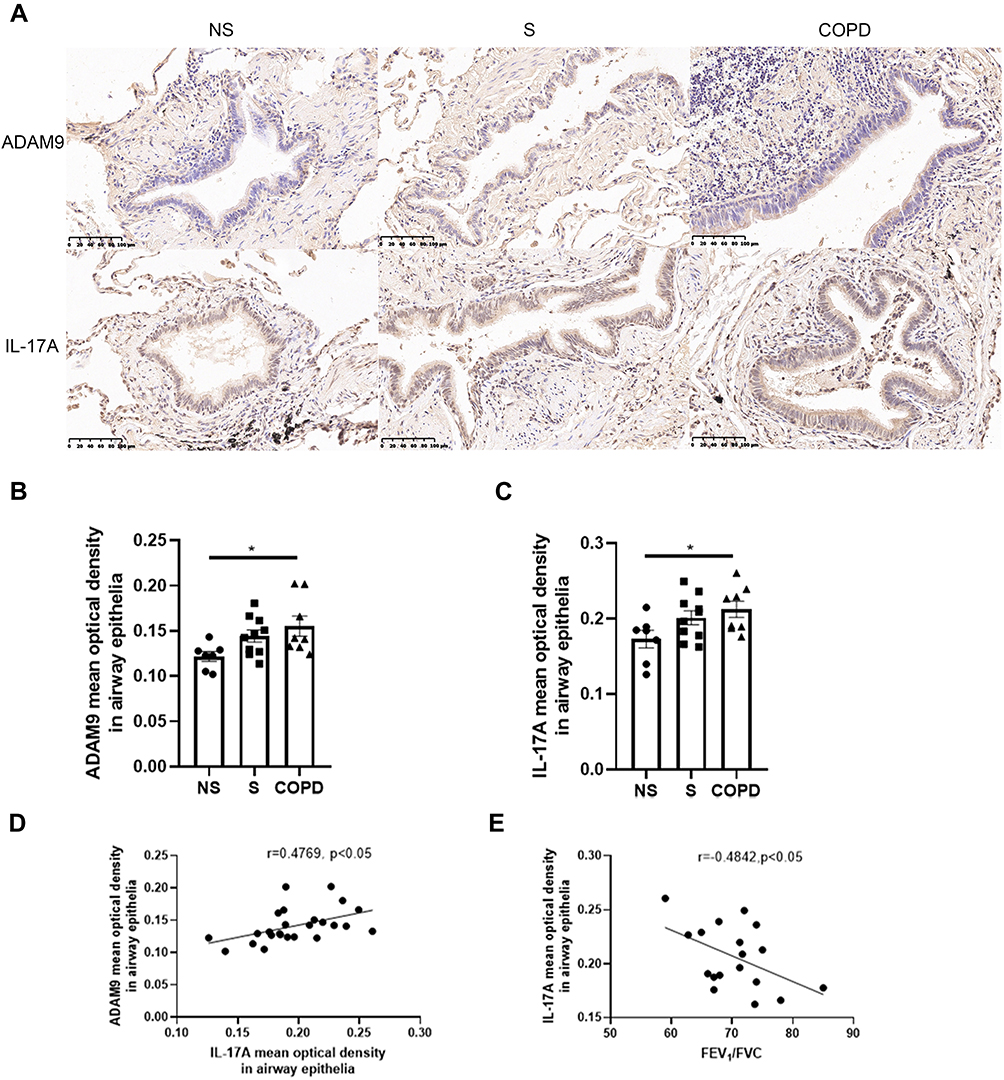 |
Figure 1 Elevated expression of ADAM9 and IL-17A in the airway epithelia of patients with chronic obstructive pulmonary disease (COPD). (A) Bright-field micrographs of the airway epithelia in non-smokers (NS, n = 7), smokers (S, n = 10), and COPD patients (COPD, n = 8). Positive immunoreactivity of ADAM9 and IL-17A was visualized by immunohistochemistry and 3,3′-diaminobenzidine (DAB) detection chromogen (brown). Scale bar = 100 μm. Quantitative analysis of ADAM9 (B) and IL-17A (C) was performed using mean optical density. (D) Correlation of the mean optical density between ADAM9 and IL-17A in the airway epithelia of three groups of people (n = 25). (E) Correlation between the mean optical density of IL-17A in airway epithelia and FEV1/FVC in smokers and patients with COPD (n = 18). *p < 0.05. |
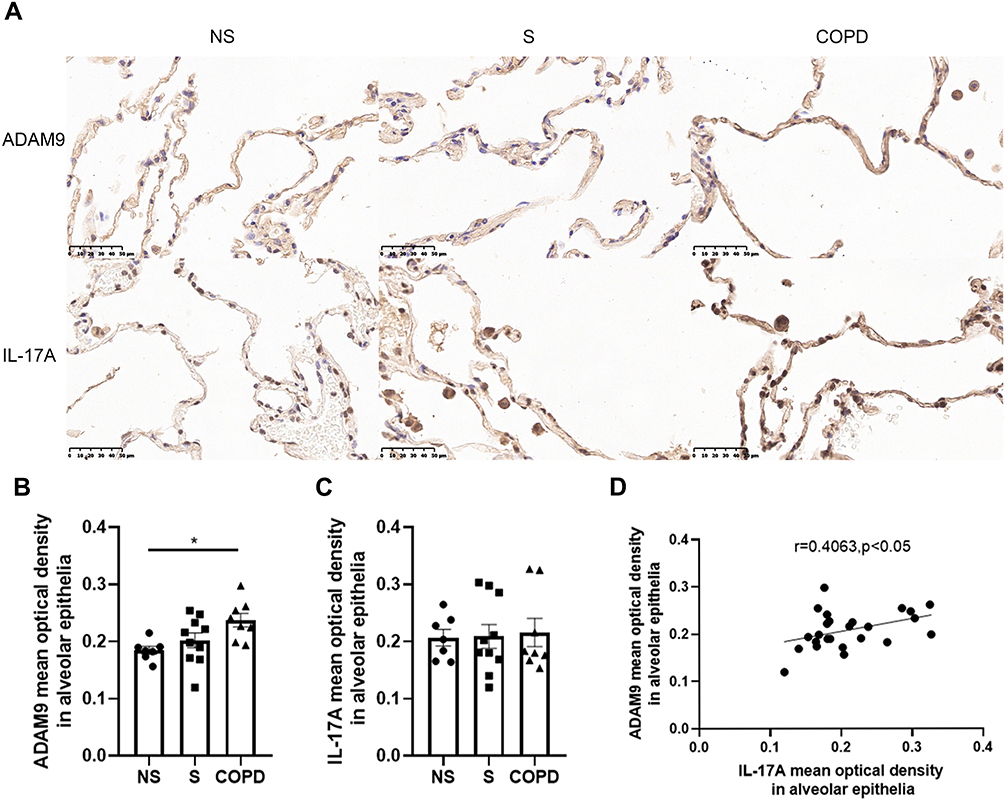 |
Figure 2 Increased expression of ADAM9 in the alveolar epithelia of patients with chronic obstructive pulmonary disease (COPD). (A) Representative images of ADAM9 and IL-17A in the alveolar epithelia of non-smokers (NS, n = 7), smokers (S, n = 10), and COPD patients (COPD, n = 8). Scale bar = 50 μm. Quantitative analysis of ADAM9 (B) and IL-17A (C) was performed using mean optical density. (D) Correlation of the mean optical density between ADAM9 and IL-17A in the alveolar epithelia of three groups of people (n = 25). *p < 0.05. |
CS-Induced Emphysema and ADAM9 Expression Were Dependent on IL-17A
After six months of CS exposure, the mice were evaluated for emphysema and expression of ADAM9. As expected, WT mice receiving chronic CS exposure displayed a significant increase in MLI and DI in lung tissue slices compared to that in WT mice exposed to room air, indicating that a COPD-like phenotype was established in CS-exposed mice (Figure 3A, C and D). In contrast, alveolar enlargement was ameliorated in Il17a−/− CS-exposed mice (Figure 3A, C and D), which was consistent with previous findings that IL-17A contributed to the development of COPD.16,17 Notably, ADAM9 expression was reduced in Il17a−/− CS-exposed mice compared to that in WT mice receiving the same amount of CS exposure (Figure 3B and E). Considering the previous report that Adam9−/− mice were protected from emphysema development,5 we propose that the role of IL-17A in COPD pathology may be partly attributed to its induction of ADAM9 expression.
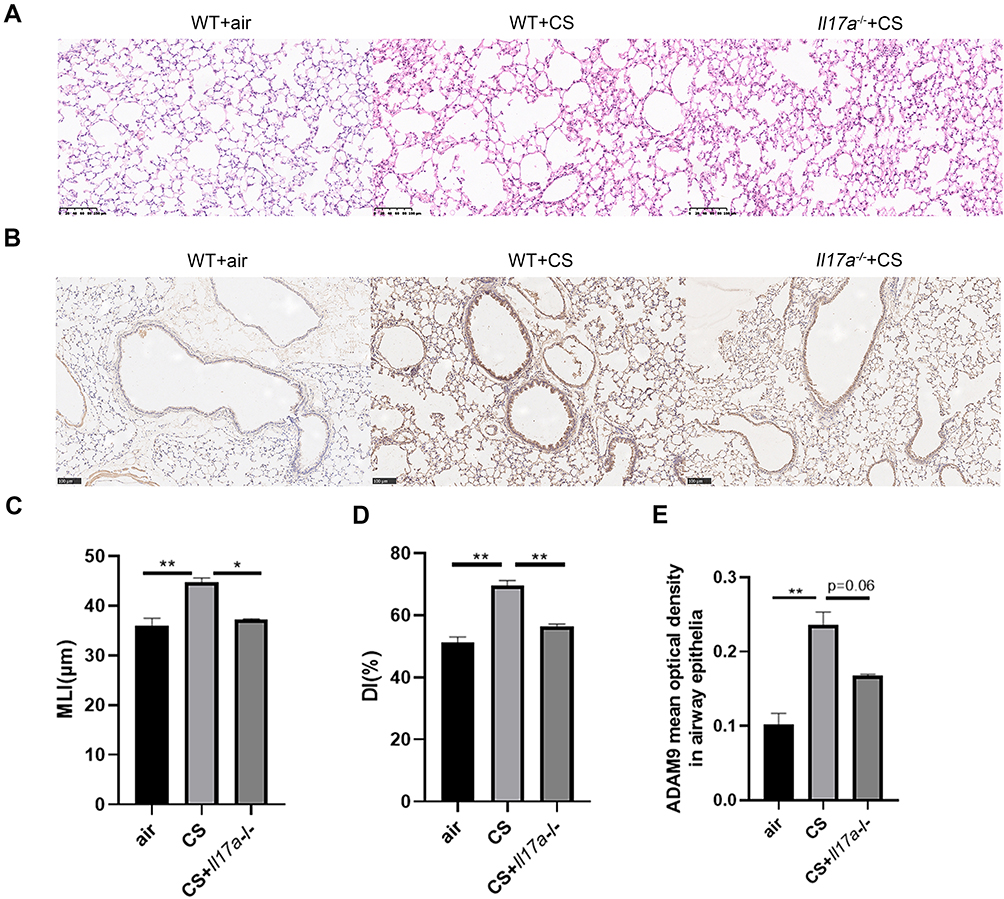 |
Figure 3 Expression of ADAM9 is down-regulated by Il17a knockout in CS-exposed mice. Wild-type (WT) mice and Il17a−/− mice were exposed to cigarette smoke (CS) or room air for six months. (A) Representative H&E staining images of the lung tissue sections in room air-exposed WT mice (n = 3), CS-exposed WT mice (n = 3), and CS-exposed Il17a−/− mice (n = 2). Scale bar = 100 μm. Mean linear intercept (MLI) (C) and destructive index (DI) (D) were measured to evaluate the degree of emphysema. (B) Immunohistochemical staining of ADAM9 in the airway epithelia of room air-exposed WT mice (n = 3), CS-exposed WT mice (n = 3), and CS-exposed Il17a−/− mice (n = 2). Scale bar = 100 μm. (E) Quantitative analysis of ADAM9 was performed using mean optical density. *p < 0.05, **p < 0.01. |
CSE Upregulated the Expression of ADAM9 and IL-17A in Airway Epithelial Cells
CCK-8 assay was performed to evaluate the cytotoxicity of CSE to HBE cells, and a concentration of 5% was found to start inhibiting cell activity (Figure 4A). Therefore, HBE cells were cultured with a supplement of different concentrations of CSE (from 0% to 5%) for 24 or 48 h. As the concentration of CSE increased, the expression level of Adam9 mRNA showed a tendency to increase (Figure 4B and C), as did the expression of Il17a mRNA (Figure 4E). Moreover, transcription of Adam9 was upregulated by CSE over time (Figure 4D). The CSE-induced upregulation of ADAM9 and IL-17A was further confirmed at the protein levels (Figure 4F–H). In addition, we observed an earlier upregulation of IL-17A, which was significantly increased at 6 h, whereas the expression of ADAM9 was increased at 12 h after CSE treatment (Figure 4I–K).
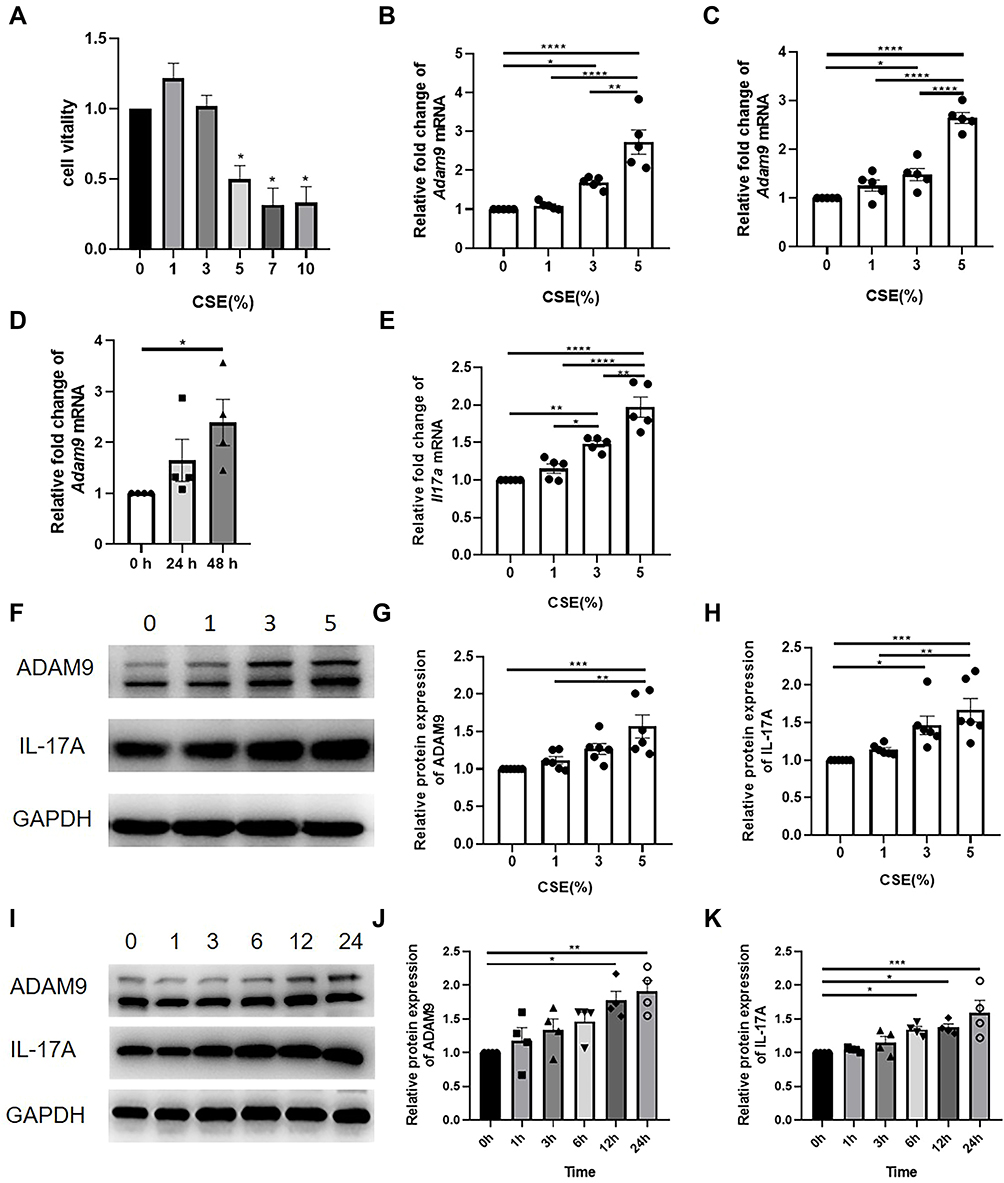 |
Figure 4 Cigarette smoke exposure induces the expression of ADAM9 and IL-17A in human bronchial epithelial (HBE) cells. (A) The cytotoxicity of the cigarette smoker extract (CSE) was measured by the CCK-8 kit (n = 4). HBE cells were cultured with different concentrations of CSE for 24 to 48 h. Gene expression of Adam9 was measured in HBE cells after 24 h (n = 5) (B) or 48 h (n = 5) (C). (D) Gene expression of Adam9 in HBE cells treated with 5% CSE (n = 4). (E) Gene expression of Il17a in HBE cells after 24 h CSE treatment (n = 4). (F–H) Immunoblot and densitometric analysis of ADAM9 and IL-17A in CSE-treated HBE cells (n = 6). (I–K) Immunoblot and densitometric analysis of ADAM9 and IL-17A at 0 to 24 h after CSE treatment (n = 4). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. |
IL-17A Induced ADAM9 Expression in Airway Epithelial Cells
We next investigated the effect of IL-17A on CSE-induced ADAM9 upregulation in HBE cells. Treatment with recombinant human IL-17A alone led to a dose-dependent increase in Adam9 transcription, with no effect on cell vitality confirmed by CCK-8 assay (Figure 5A and B). A similar result was obtained by Western blot, although ADAM9 only exhibited a mild upregulation at the protein level (Figure 5D and E). However, co-treatment with IL-17A and CSE significantly increased ADAM9 production in vitro (Figure 5C, F and G), suggesting a synergistic effect of IL-17A and CS exposure in the induction of ADAM9 expression. In addition, IL-17A neutralizing antibody significantly reduced CSE-induced ADAM9 expression in HBE cells (Figures 5H, I and S1A). These results indicate that HBE cell-intrinsic IL-17A is involved in CSE-induced ADAM9 expression.
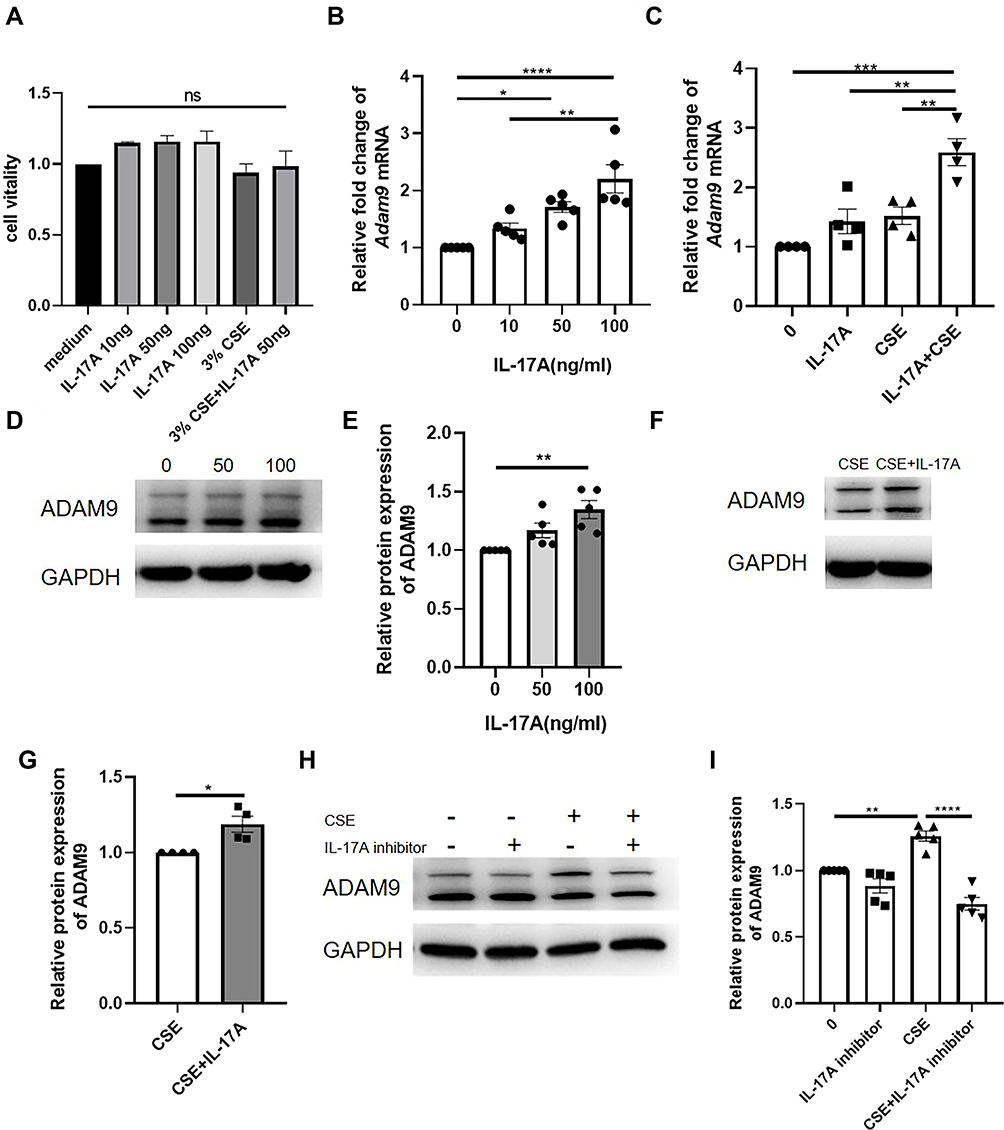 |
Figure 5 ADAM9 expression is upregulated by IL-17A in human bronchial epithelial (HBE) cells. (A) The cytotoxicity of different concentrations of recombinant human IL-17A with or without cigarette smoker extract (CSE) to HBE cells was measured by CCK-8 assay (n = 3). (B) Transcription of Adam9 was measured in HBE cells treated with different concentrations of IL-17A for 24 h (n = 5). (C) Transcription of Adam9 was increased with the supplement of 50 ng/mL recombinant human IL-17A cytokine with or without 3% CSE (n = 4). (D–G) Representative Western blot analysis of ADAM9 in IL-17A (n = 5) and/or CSE-treated HBE cells (n = 4). (H and I) Representative Western blot analysis of ADAM9 in IL-17A neutralizing antibody and/or CSE-treated HBE cells (n = 5). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns, no significant difference. |
IL-17A Induced ADAM9 Expression Through Activating JNK Signaling
Several studies have demonstrated that JNK signaling is involved in COPD pathogenesis, including airway inflammation, mucus secretion, EMT, and airway remodeling.26–28 Therefore, we investigated whether the JNK signaling pathway also participated in the IL-17A-mediated regulation of ADAM9 expression in CSE-treated HBE cells. We found that both IL-17A and CSE induced JNK phosphorylation in HBE cells, whereas total JNK was not affected by IL-17A or CSE (Figure 6A–C). The combined use of IL-17A and CSE synergistically induced a higher level of JNK phosphorylation, which was blocked by the addition of the JNK inhibitor SP600125 (Figures 6A, B, D and S1B). Moreover, decreased ADAM9 expression was observed in SP600125-treated cells, which could be reversed by adding IL-17A and/or CSE (Figure 6E and F).
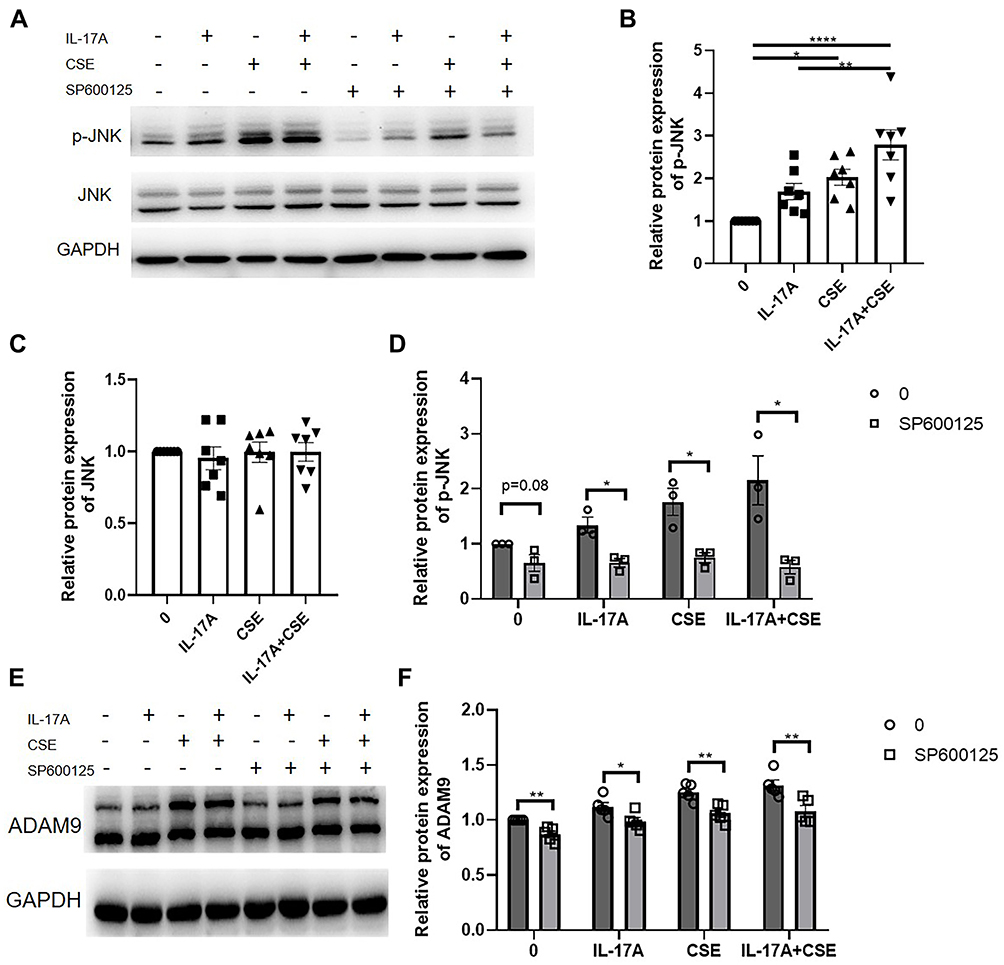 |
Figure 6 Activation of JNK signaling by IL-17A induces ADAM9 expression in human bronchial epithelial (HBE) cells. (A–D) Protein expression of JNK and p-JNK were examined in HBE cells treated with 50 ng/mL IL-17A with or without 3% cigarette smoker extract (CSE) for 10 min (n = 7). JNK inhibitor (SP600125, 10 µM) was added to inhibit JNK phosphorylation 1 h before the addition of IL-17A or CSE (n = 3). (E and F) HBE cells were treated with SP600125 for 1 h and then treated with IL-17A and/or CSE for 24 h. The expression of ADAM9 was examined by Western blot analysis (n = 5). *p < 0.05, **p < 0.01, ****p < 0.0001. |
Discussion
Excess ECM degradation is involved in COPD-associated emphysema, and abnormal metalloprotease activity participates in this process.29 A recent study demonstrated that ADAM9, which reportedly promotes acute lung injury by degrading the ECM and damaging the alveolar-capillary barrier,12 was also involved in the development of COPD.5 ADAM9 has been suggested to degrade lung elastin and induce alveolar cell death by shedding EGFR and VEGFR2, thus promoting emphysema development.5 However, the mechanism by which ADAM9 production is induced in COPD was unclear. In this study, we provided evidence that IL-17A, a crucial contributor to COPD pathogenesis, was an upstream modulator of ADAM9.
Multiple studies have explored IL-17A expression in COPD. The number of IL-17A+ inflammatory cells was higher in the small airway subepithelia in patients with COPD than in healthy people with or without smoking history.30–33 In contrast, the results regarding epithelial IL-17A immunoreactivity were conflicting. Some studies reported higher levels of immunoreactivity of IL-17A in the airway epithelia and alveolar walls in COPD,31,34 while others observed no differences.30,33 In this study, we observed increased IL-17A expression in the small airways, but not in the alveolar walls. The inconsistencies in different studies could be due to sample differences, as IL-17A expression was significantly increased in severe to extremely severe COPD compared to that in controls,14 or due to different methods used to measure immunoreactivity.
In line with a previous report,5 we observed enhanced ADAM9 expression in small airways and alveolar walls of COPD patients compared with controls. Notably, we found a positive correlation between ADAM9 and IL-17A expression in the airway epithelia as well as in the alveolar walls. To confirm the effect of IL-17A on ADAM9 expression in COPD, we performed an experiment using Il17a−/− mice exposed to long-term CS. After six months of CS exposure, Il17a−/− mice exhibited less ADAM9 expression in small airways than WT mice. Moreover, Il17a knockout attenuated the severity of emphysema, similar to the results obtained by Adam9 deletion in a previous study.5 These findings provide evidence that IL-17A induces upregulation of ADAM9 in COPD.
To elucidate the effect of IL-17A on ADAM9 expression, we further demonstrated, for the first time to our knowledge, that ADAM9 was increased in HBE cells in response to CSE stimulation in a time- and concentration-dependent manner. Recombinant IL-17A also induced ADAM9 expression in HBE cells. It is well known that IL-17A expression is increased in COPD patients as well as in mouse models.13 CS could induce the expansion of Th17 and Tc17 cells in the lungs, which are the primary source of IL-17A.13 Moreover, epithelial cells could express IL-17A in an autocrine manner.35 Consistent with other studies, we observed that IL-17A was expressed at a relatively low level in unstimulated HBE cells but was upregulated by CS exposure. The increase of IL-17A was earlier than ADAM9 after CSE treatment. In addition, IL-17A neutralization inhibited CSE-induced ADAM9 expression. Hence, apart from IL-17A that originates from infiltrating immune cells, epithelial cell-derived IL-17A is likely to contribute to ADAM9 expression in lung epithelial cells of smokers with COPD.
Furthermore, we explored IL-17A-mediated signaling in the regulation of ADAM9 expression. We revealed that both IL-17A and CSE induced JNK phosphorylation, whereas inhibiting JNK phosphorylation suppressed ADAM9 upregulation by IL-17A with or without CSE, indicating that IL-17A induced ADAM9 expression through JNK activation in CS-related COPD.
Our study had several limitations. We did not perform experiments to explore the role of ADAM9 in COPD-associated emphysema, although it has been suggested that ADAM9 could induce alveolar epithelial cell death.5 Whether IL-17A neutralization could reduce CSE-induced epithelial cell death through inhibiting ADAM9 expression remains to be clarified. IL-17A and ADAM9 are reportedly implicated in EMT in some diseases.36,37 IL-17A has been demonstrated to synergize with CS to induce EMT in bronchial epithelial cells by activating nuclear factor-kappa B (NF-κB) and CCAAT/enhancer-binding protein beta (CEBPβ) signaling.38,39 Similarly, Adam9 deletion was shown to protect CS-exposed C57BL/6 mice from airway fibrosis.5 Adam9−/− mice had fewer α- spinal muscular atrophy+ (a-SMA+) myofibroblasts around the small airways after six months of CS exposure than WT mice.5 Further experiments should be performed to examine whether IL-17A induces EMT through ADAM9 signaling in COPD. In addition, Umeda et al reported that ADAM9 could be exclusively expressed in Th17 cells and enhance their differentiation, thus contributing to the development of systemic lupus erythematosus, indicating that a reciprocal connection may exist between ADAM9 and IL-17A.40 As increased Th17 cells and ADAM9 expression in epithelial cells can be simultaneously observed in COPD, there may be a positive feedback between ADAM9 and IL-17A in COPD, which also needs further investigation.
Acknowledgments
The authors appreciate the Center of Basic Medical Research of Peking University Third Hospital for their technical support. We also thank all the patients for their contributions to this research.
Funding
This study was supported by the National Natural Science Foundation of China Youth Fund Project (No.81800038).
Disclosure
Danyang Li and Tong Wang are joint first authors. The authors report no conflicts of interest in this work.
References
1. Christenson SA, Smith BM, Bafadhel M, Putcha N. Chronic obstructive pulmonary disease. Lancet. 2022;399(10342):2227–2242. doi:10.1016/S0140-6736(22)00470-6
2. Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2016;138(1):16–27. doi:10.1016/j.jaci.2016.05.011
3. Owen CA. Roles for proteinases in the pathogenesis of chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2008;3(2):253–268. doi:10.2147/copd.s2089
4. Oreo KM, Gibson PG, Simpson JL, Wood LG, McDonald VM, Baines KJ. Sputum ADAM8 expression is increased in severe asthma and COPD. Clin Exp Allergy. 2014;44(3):342–352. doi:10.1111/cea.12223
5. Wang X, Polverino F, Rojas-Quintero J, et al. A disintegrin and a metalloproteinase-9 (ADAM9): a novel proteinase culprit with multifarious contributions to COPD. Am J Respir Crit Care Med. 2018;198(12):1500–1518. doi:10.1164/rccm.201711-2300OC
6. Cui L, Li H, Xie M, et al. Relationship between proteinase with a disintegrin and a metalloproteinase domain-9 (ADAM9), inflammation, airway remodeling, and emphysema in COPD patients. Int J Chron Obstruct Pulmon Dis. 2020;15:3335–3346. doi:10.2147/COPD.S276171
7. Wang X, Zhang D, Higham A, et al. ADAM15 expression is increased in lung CD8(+) T cells, macrophages, and bronchial epithelial cells in patients with COPD and is inversely related to airflow obstruction. Respir Res. 2020;21(1):188. doi:10.1186/s12931-020-01446-5
8. Fachri M, Hatta M, Massi MN, et al. The strong correlation between ADAM33 expression and airway inflammation in chronic obstructive pulmonary disease and candidate for biomarker and treatment of COPD. Sci Rep. 2021;11(1):23162. doi:10.1038/s41598-021-02615-2
9. Seals DF, Courtneidge SA. The ADAMs family of metalloproteases: multidomain proteins with multiple functions. Genes Dev. 2003;17(1):7–30. doi:10.1101/gad.1039703
10. Chou CW, Huang YK, Kuo TT, Liu JP, Sher YP. An overview of ADAM9: structure, activation, and regulation in human diseases. Int J Mol Sci. 2020;21(20):7790. doi:10.3390/ijms21207790
11. Lin CY, Chen HJ, Huang CC, et al. ADAM9 promotes lung cancer metastases to brain by a plasminogen activator-based pathway. Cancer Res. 2014;74(18):5229–5243. doi:10.1158/0008-5472.CAN-13-2995
12. Roychaudhuri R, Hergrueter AH, Polverino F, et al. ADAM9 is a novel product of polymorphonuclear neutrophils: regulation of expression and contributions to extracellular matrix protein degradation during acute lung injury. J Immunol. 2014;193(5):2469–2482. doi:10.4049/jimmunol.1303370
13. Le Rouzic O, Pichavant M, Frealle E, Guillon A, Si-Tahar M, Gosset P. Th17 cytokines: novel potential therapeutic targets for COPD pathogenesis and exacerbations. Eur Respir J. 2017;50(4):1602434. doi:10.1183/13993003.02434-2016
14. Roos AB, Sandén C, Mori M, Bjermer L, Stampfli MR, Erjefält JS. IL-17A is elevated in end-stage chronic obstructive pulmonary disease and contributes to cigarette smoke-induced lymphoid neogenesis. Am J Respir Crit Care Med. 2015;191(11):1232–1241. doi:10.1164/rccm.201410-1861OC
15. Xiong J, Zhou L, Tian J, et al. Cigarette smoke-induced lymphoid neogenesis in COPD involves IL-17/RANKL pathway. Front Immunol. 2020;11:588522. doi:10.3389/fimmu.2020.588522
16. Kurimoto E, Miyahara N, Kanehiro A, et al. IL-17A is essential to the development of elastase-induced pulmonary inflammation and emphysema in mice. Respir Res. 2013;14(1):5. doi:10.1186/1465-9921-14-5
17. Chang Y, Al-Alwan L, Audusseau S, et al. Genetic deletion of IL-17A reduces cigarette smoke-induced inflammation and alveolar type II cell apoptosis. Am J Physiol Lung Cell Mol Physiol. 2014;306(2):132–143. doi:10.1152/ajplung.00111.2013
18. Roos AB, Sethi S, Nikota J, et al. IL-17A and the promotion of neutrophilia in acute exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2015;192(4):428–437. doi:10.1164/rccm.201409-1689OC
19. Bozinovski S, Seow HJ, Chan SP, et al. Innate cellular sources of interleukin-17A regulate macrophage accumulation in cigarette- smoke-induced lung inflammation in mice. Clin Sci (Lond). 2015;129(9):785–796. doi:10.1042/CS20140703
20. Chen K, Pociask DA, McAleer JP, et al. IL-17RA is required for CCL2 expression, macrophage recruitment, and emphysema in response to cigarette smoke. PLoS One. 2011;6(5):e20333. doi:10.1371/journal.pone.0020333
21. Baggio C, Velazquez JV, Fragai M, Nordgren TM, Pellecchia M. Therapeutic targeting of MMP-12 for the treatment of chronic obstructive pulmonary disease. J Med Chem. 2020;63(21):12911–12920. doi:10.1021/acs.jmedchem.0c01285
22. Prause O, Bozinovski S, Anderson GP, Lindén A. Increased matrix metalloproteinase-9 concentration and activity after stimulation with interleukin-17 in mouse airways. Thorax. 2004;59(4):313–317. doi:10.1136/thx.2003.008854
23. Ding F, Han L, Fu Q, et al. IL-17 aggravates pseudomonas aeruginosa airway infection in acute exacerbations of chronic obstructive pulmonary disease. Front Immunol. 2021;12:811803. doi:10.3389/fimmu.2021.811803
24. Zhou L, Le Y, Tian J, et al. Cigarette smoke-induced RANKL expression enhances MMP-9 production by alveolar macrophages. Int J Chron Obstruct Pulmon Dis. 2019;14:81–91. doi:10.2147/COPD.S190023
25. Xiong J, Le Y, Rao Y, et al. RANKL mediates muscle atrophy and dysfunction in a cigarette smoke-induced model of chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2021;64(5):617–628. doi:10.1165/rcmb.2020-0449OC
26. Zhou J, Zhao Y, Zhou H, et al. Autophagy plays an essential role in cigarette smoke-induced expression of MUC5AC in airway epithelium. Am J Physiol Lung Cell Mol Physiol. 2016;310(11):1042–1052. doi:10.1152/ajplung.00418.2015
27. Eurlings IM, Reynaert NL, van de Wetering C, et al. Involvement of c-Jun N-terminal kinase in TNF-α-driven remodeling. Am J Respir Cell Mol Biol. 2017;56(3):393–401. doi:10.1165/rcmb.2015-0195OC
28. Wagner C, Uliczka K, Bossen J, et al. Constitutive immune activity promotes JNK- and FoxO-dependent remodeling of drosophila airways. Cell Rep. 2021;35(1):108956. doi:10.1016/j.celrep.2021.108956
29. Turino GM. The origins of a concept: the protease-antiprotease imbalance hypothesis. Chest. 2002;122(3):1058–1060. doi:10.1378/chest.122.3.1058
30. Eustace A, Smyth LJC, Mitchell L, Williamson K, Plumb J, Singh D. Identification of cells expressing IL-17A and IL-17F in the lungs of patients with COPD. Chest. 2011;139(5):1089–1100. doi:10.1378/chest.10-0779
31. Montalbano AM, Riccobono L, Siena L, et al. Cigarette smoke affects IL-17A, IL-17F and IL-17 receptor expression in the lung tissue: ex vivo and in vitro studies. Cytokine. 2015;76(2):391–402. doi:10.1016/j.cyto.2015.07.013
32. Chang Y, Nadigel J, Boulais N, et al. CD8 positive T cells express IL-17 in patients with chronic obstructive pulmonary disease. Respir Res. 2011;12(1):43. doi:10.1186/1465-9921-12-43
33. Chang Y, Al-Alwan L, Alshakfa S, et al. Upregulation of IL-17A/F from human lung tissue explants with cigarette smoke exposure: implications for COPD. Respir Res. 2014;15(1):145. doi:10.1186/s12931-014-0145-7
34. Chu S, Zhong X, Zhang J, Lao Q, He Z, Bai J. The expression of Foxp3 and ROR gamma t in lung tissues from normal smokers and chronic obstructive pulmonary disease patients. Int Immunopharmacol. 2011;11(11):1780–1788. doi:10.1016/j.intimp.2011.06.010
35. Wu M, Lai T, Jing D, et al. Epithelium-derived IL17A promotes cigarette smoke-induced inflammation and mucus hyperproduction. Am J Respir Cell Mol Biol. 2021;65(6):581–592. doi:10.1165/rcmb.2020-0424OC
36. Li S, Cong X, Gao H, et al. Tumor-associated neutrophils induce EMT by IL-17a to promote migration and invasion in gastric cancer cells. J Exp Clin Cancer Res. 2019;38(1):6. doi:10.1186/s13046-018-1003-0
37. van Kampen JGM, van Hooij O, Jansen CF, et al. MiRNA-520f reverses epithelial-to-mesenchymal transition by targeting ADAM9 and TGFBR2. Cancer Res. 2017;77(8):2008–2017. doi:10.1158/0008-5472.CAN-16-2609
38. Ma L, Jiang M, Zhao X, Sun J, Pan Q, Chu S. Cigarette and IL-17A synergistically induce bronchial epithelial-mesenchymal transition via activating IL-17R/NF-κB signaling. BMC Pulm Med. 2020;20(1):26. doi:10.1186/s12890-020-1057-6
39. Chu S, Ma L, Wu Y, Zhao X, Xiao B, Pan Q. C-EBPβ mediates in cigarette/IL-17A-induced bronchial epithelial-mesenchymal transition in COPD mice. BMC Pulm Med. 2021;21(1):376. doi:10.1186/s12890-021-01738-6
40. Umeda M, Yoshida N, Hisada R, et al. ADAM9 enhances Th17 cell differentiation and autoimmunity by activating TGF-β1. Proc Natl Acad Sci U S A. 2021;118:18. doi:10.1073/pnas.2023230118
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.