")
Back to Journals » Journal of Blood Medicine » Volume 7
IDH1 and IDH2 mutations as novel therapeutic targets: current perspectives
Authors Mondesir J, Willekens C, Touat M, de Botton S
Received 15 March 2016
Accepted for publication 31 May 2016
Published 2 September 2016 Volume 2016:7 Pages 171—180
DOI https://doi.org/10.2147/JBM.S70716
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Martin H Bluth
Johanna Mondesir1,2 Christophe Willekens3–5 Mehdi Touat6,7 Stéphane de Botton3–5
1Service d’Immunopathologie Clinique, Hôpital Saint Louis, 2CNRS UMR8104, INSERM U1016, Institut Cochin, Université Paris Descartes, Paris, 3Gustave Roussy, Université Paris-Saclay, Service d’Hématologie Clinique, 4INSERM U1170, Gustave Roussy, Université Paris-Saclay, Villejuif, 5Faculté de médecine Paris-Sud, Kremlin-Bicêtre, 6AP-HP, Hôpitaux Universitaires La Pitié Salpêtrière – Charles Foix, Service de Neurologie 2-Mazarin, Paris, 7Gustave Roussy, Université Paris‑Saclay, Département d’Innovation Thérapeutique et d’Essais Précoces, Villejuif, France
Abstract: Isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) are key metabolic enzymes that convert isocitrate to α-ketoglutarate. IDH1/2 mutations define distinct subsets of cancers, including low-grade gliomas and secondary glioblastomas, chondrosarcomas, intrahepatic cholangiocarcinomas, and hematologic malignancies. Somatic point mutations in IDH1/2 confer a gain-of-function in cancer cells, resulting in the accumulation and secretion in vast excess of an oncometabolite, the D-2-hydroxyglutarate (D-2HG). Overproduction of D-2HG interferes with cellular metabolism and epigenetic regulation, contributing to oncogenesis. Indeed, high levels of D-2HG inhibit α-ketoglutarate-dependent dioxygenases, including histone and DNA demethylases, leading to histone and DNA hypermethylation and finally a block in cell differentiation. Furthermore, D-2HG is a biomarker suitable for the detection of IDH1/2 mutations at diagnosis and predictive of the clinical response. Finally, mutant-IDH1/2 enzymes inhibitors have entered clinical trials for patients with IDH1/2 mutations and represent a novel drug class for targeted therapy.
Keywords: tumor metabolism, epigenetic, oncogene, IDH1, IDH2, glioma, acute myeloid leukemia, 2-HG, targeted therapies
Introduction
Isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) are metabolic enzymes catalyzing the conversion of isocitrate to α-ketoglutarate (αKG), while reducing NADP to NADPH. Point mutations in IDH1/2 define distinct subsets of low-grade glioma and secondary glioblastoma (GBM), chondrosarcoma, intrahepatic cholangiocarcinomas, hematologic malignancies, as well as premalignant diseases and rare inherited metabolism disorders.1–9 Multiple preclinical models have provided evidence for the oncogenic potential of IDH1/2 mutations, which alter epigenetic regulation, cancer cell differentiation, and metabolism.10–14 Depending on the associated genomic aberrations and the cellular context, the oncogenic potential of IDH1/2 mutations ranges from an initiating event – promoting transformation – to a secondary oncogenic event conferring selective advantage to cancer cells. In vitro and in vivo preclinical studies have demonstrated that inhibition of IDH1/2-mutant enzymes decreases intracellular D-2-hydroxyglutarate (D-2HG) levels, reverses epigenetic dysregulation, and releases the differentiation block. These findings supported initiation of the ongoing clinical trials evaluating novel IDH1/2 inhibitors in IDH1/2-mutant cancers.
Normal functions of IDH enzymes
The IDH family of enzymes comprises three proteins located in the cytoplasm and peroxysomes (IDH1) and mitochondria (IDH2 and IDH3),15,16 which are involved in a number of cellular processes, including mitochondrial oxidative phosphorylation, glutamine metabolism, lipogenesis, glucose sensing, and regulation of cellular redox status.17 IDH3 forms a heterotetrameric complex (two alpha, one beta, and one gamma subunit) catalyzing the NAD+-dependent conversion of isocitrate to αKG in the tricarboxylic acid cycle (Figure 1).
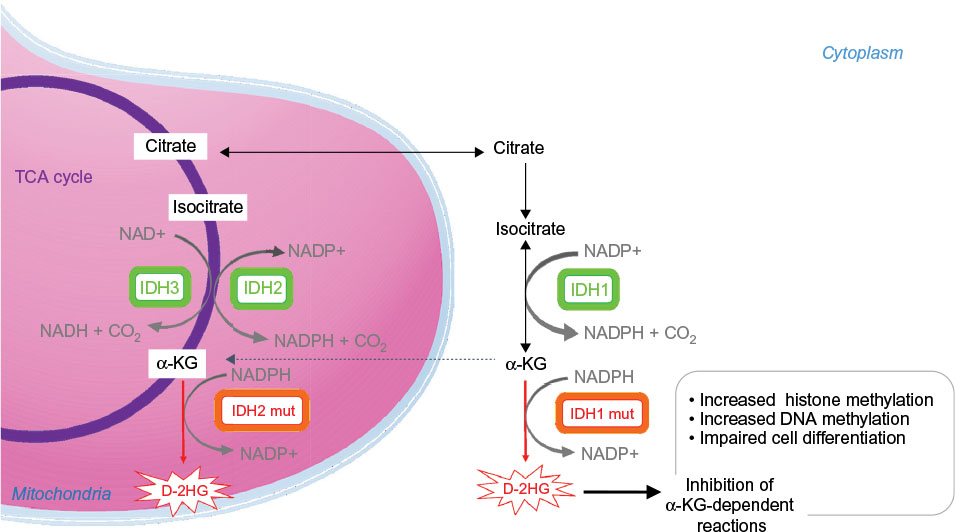 | Figure 1 Enzymatic activities of wild type and mutated IDH enzymes. Notes: The IDH family of enzymes comprises three proteins located in the cytoplasm and peroxysomes (IDH1), and mitochondria (IDH2 and IDH3). IDH1 and IDH2 catalyze the reversible NADP+-dependent oxidative decarboxylation of isocitrate to αKG. IDH3 catalyzes the NAD+-dependent conversion of isocitrate to αKG in the TCA cycle. IDH1 and IDH2 mutant enzymes gain neomorphic enzymatic activity, converting NADPH and αKG to NADP+ and D-2HG. D-2HG acts as a weak competitive inhibitor of αKG-dependent dioxygenases. αKG-dependent dioxygenases are involved in various cellular processes such as hypoxia, angiogenesis, maturation of collagens of the extracellular matrix, and regulation of epigenetics. Excess of D-2HG is associated with increased histone and DNA methylation, altering cancer cells differentiation. Abbreviations: αKG, alpha ketoglutarate; D-2HG, D-2-hydroxyglutarate; IDH, isocitrate dehydrogenase; DNA, deoxyribonucleic acid; mut, mutated; NAD, nicotinamide adenine dinucleotide; NADP, nicotinamide adenine dinucleotide phosphate; TCA cycle, tricarboxylic acid cycle. |
IDH1 and IDH2 are highly similar enzymes, forming homodimers and catalyzing the reversible NADP+-dependent oxidative decarboxylation of isocitrate to αKG (Figure 1). NADPH is a key cellular reducing agent required for detoxification processes through reduction of glutathione and thioredoxins and activation of catalase, which are all involved in the protection against the toxicity of reactive oxygen species and oxidative DNA damage.18 IDH1 is the main NADPH producer in the brain.19
In specific cellular contexts such as hypoxia/pseudohypoxia and/or altered oxidative metabolism, bidirectional αKG metabolism along oxidative and reductive pathways can be activated,20–22 thereby allowing generation of isocitrate/citrate from αKG and glutamine. Recent evidence indicated that both IDH1 and IDH2 enzymes play fundamental roles in these alternative metabolic pathways.22,23 Reductive αKG/glutamine metabolism allows cancer cells to maintain pools of biosynthetic precursors and suppress mitochondrial reactive oxygen species, resulting in the sustainment of rapid rates of proliferation, even when oxidative metabolism is impaired.24,25
IDH mutants produce D-2HG and inhibit αKG-dependent dioxygenases
IDH1/2 mutations are heterozygous, missense mutations, leading to the substitution of the amino acids arginine 132 in IDH1 and arginine 172 or 140 in IDH22–7 (Table 1). These residues play key role in substrate binding in the enzymes active site. Initial functional studies of IDH1/2-mutant cells revealed that IDH1/2 mutations decrease the ability of the mutant enzymes to convert isocitrate to αKG3 and that IDH1/2-mutant enzymes inhibit wild-type activity in a dominant-negative manner.26 Further functional analysis demonstrated that IDH1/2-mutant enzymes gain neomorphic enzymatic activity, converting NADPH and αKG to NADP+ and D-2HG.10,27,28 IDH1/2 mutant enzymes produce high levels of D-2HG in cells (50- to 100-fold higher than in normal tissues). 2HG is a chiral molecule that can exist as either a d-enantiomer or an l-enantiomer. IDH1/2 mutants exclusively produce D-2HG. Importantly, IDH1/2-mutant cells have normal αKG levels.10,28 In physiological condition, D-2HG intracellular concentration is low. D-2HG is not known to play any physiological metabolic role, and its production remains poorly understood.
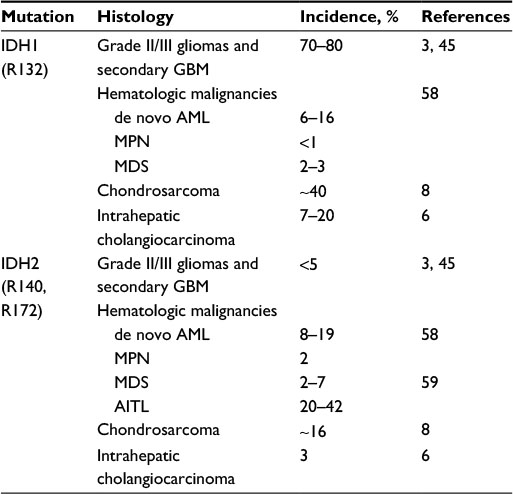 | Table 1 IDH mutations estimates in solid tumors and hematologic malignancies Abbreviations: IDH, isocitrate dehydrogenase; GBM, glioblastoma; AML, acute myeloid leukemia; MPN, myeloproliferative neoplasms; MDS, myelodysplastic syndrome; AITL, angioimmunoblastic T-cell lymphoma. |
IDH mutations are associated with epigenetic reprogramming, altering cancer cell differentiation
D-2HG and αKG are highly similar molecules, differing only by the presence of a C2 hydroxyl group in D-2HG instead of the C2 carbonyl of αKG. D-2HG can occupy the same binding pocket as α-KG and acts as a weak competitive inhibitor of αKG-dependent dioxygenases. αKG and Fe2+ are used as cofactors of the activity of >60 αKG-dependent dioxygenases,12,29 which are involved in a wide range of cellular processes such as hypoxia, angiogenesis, maturation of collagens of the extracellular matrix, and regulation of epigenetics. In vitro ectopic expression of IDH1/2 mutants produces high D-2HG levels that inhibit αKG-dependent dioxygenases, including histone demethylases Jumonji 2 (JMJD2) and Jmj C domain-containing histone demethylase-1 (JHDM1), and DNA demethylases ten-eleven translocation 2 (TET2) proteins, resulting in the impairment of key steps in histone and DNA demethylation. The epigenetic deregulation induced by IDH1/2 mutant enzymes translates into histone and DNA hypermethylation in a passage-dependent manner.10–13,30–35
Indeed, methylation profile of several human malignancies showed that IDH1/2-mutant tumors display a typical CpG island methylator phenotype characterized by high degree of DNA hypermethylation in CpG-rich domains. Hypermethylation is the dominant feature of IDH1/2-mutant acute myeloid leukemias (AMLs), and these mutants display similar DNA methylation profiles. Interestingly, TET2-mutant cells display an overlapping hypermethylation signature with IDH1/2-mutants cells.11,13 Gene expression profile of large cohorts of gliomas and AML has shown that IDH1/2-mutant tumors display a distinct gene expression profile enriched for genes expressed in progenitor cells.11,13,14,36–38 Importantly, such wide epigenetic modifications were associated with altered expression of genes involved in cellular differentiation, thereby resulting in a block to cellular differentiation, which can be reversed by pharmacological inhibition of the mutant enzymes.11,13,14,39–41 Consequently, as observed in TET2 mutants, increased expression of stem cell markers and impaired differentiation are seen in cells expressing IDH1/2 mutant enzymes.11,13,42 Hypermethylation can also compromise the binding of methylation-sensitive insulator proteins, which may result in the loss of insulation between topological DNA domains and aberrant gene activation, as recently demonstrated in IDH1-mutant gliomasphere models.43
Importantly, there is a correlation between intracellular concentrations of D-2HG and the epigenetic effects in IDH1/2-mutant tumors. Indeed, as D-2HG is a weak competitor of αKG,12 the phenotype of immature cell is only observed when a high level of accumulation of D-2HG is reached.44 Besides IDH1/2-mutant cancers, abnormal accumulation of D-2HG has been observed in D-2HG aciduria (D-2HGA), a rare inherited metabolic disorder characterized by extremely variable clinical presentations ranging from fatal neonatal encephalopathy and cardiomyopathy to asymptomatic cases. Mutations in D2HGDH and IDH2 are the molecular basis of this metabolic disorder, with a tendency to a more severe clinical phenotype in IDH2-mutant patients.45 Interestingly, although D-2HG levels are excessively high in patients with D-2HGA, no cancers have been reported so far in this population, which suggests that D-2HG accumulation is not sufficient alone to induce cancer. Indeed, while expression of mutant IDH1/2 enzymes was associated with increased progenitor cell marker expression and impaired cell differentiation, such epigenetics effects were not sufficient alone to drive oncogenesis. This suggests that other molecular alterations including FLT3 or NRAS are required to promote full transformation of IDH1/2-mutant clones in AML models.46,47
Finally, other metabolites are known to inhibit αKG-dependent dioxygenases. For example, excess of succinate is observed in case of mutations affecting genes coding for succinate dehydrogenase. Similar to IDH1/2 mutations, such alterations have been linked with DNA hypermethylation, decreased expression of key genes involved in neuroendocrine differentiation, and a block in cellular differentiation.48
IDH mutations are associated with metabolic reprogramming
Few investigations have questioned metabolic changes associated with IDH1/2 mutations. Recent metabolic flux analyses have shown that IDH1 mutations render tumor cells more dependent on mitochondrial oxidative tricarboxylic acid metabolism and compromise the conversion of glutamine to citrate and fatty acids under hypoxia as compared to IDH1-wild type cells.49 Interestingly, such compromised metabolic reprogramming resulted in decreased cell growth of IDH1-mutant cells upon hypoxia. Of note, D-2HG inhibits both ATP synthase and mTOR signaling in glioma cells, resulting in growth arrest and cell death under conditions of glucose limitation.50 Large-scale metabolic profiling of IDH1-mutant patient-derived glioma models showed that IDH1-mutant glioma cells display extreme vulnerability to depletion of the coenzyme nicotinamide adenine dinucleotide (NAD+),51 an essential metabolite involved in several fundamental cellular processes such as energetic metabolism, regulation of transcription, DNA repair, cell cycle progression, and apoptosis. Overall, IDH1/2 mutations are likely associated with wide metabolic reprogramming. Further deciphering of alterations in cellular metabolism associated with IDH1/2 mutations should highlight novel opportunities for therapeutic intervention and drug development.
Spectrum of cancer types with IDH mutations
First identified in colorectal cancer, IDH1/2 mutations affecting IDH1 (R132) or IDH2 (R140, R172) are found across a broad spectrum of cancer types (Table 1).
Brain tumors
IDH1 mutations were described in 2008, in exome-sequencing studies of GBM (WHO grade IV astrocytoma).2 Subsequent studies have shown that IDH1/2 mutations occur in a mutually exclusive manner in ~80% of WHO grade II/III oligodendrogliomas, astrocytomas, and oligoastrocytomas and secondary GBM (ie, GBM that had progressed from lower grade gliomas).3,52 Conversely, in primary GBM, IDH1/2 mutations are found in only 6% of patients, suggesting distinct mechanisms of tumorigenesis between GBM and lower grade gliomas.3,53 IDH1 R132H mutation represents 80% of all IDH mutations.52 Rarely, other mutations are found affecting either IDH1 at Arg132 (including R132S, R132C, R132G, and R132L substitutions) or IDH2 at Arg172 (R172K most frequently; Table 1). IDH1 R132H mutation can be diagnosed by immunohistochemistry or sequencing, while other mutations can be identified only by sequencing.54
IDH-mutant gliomas represent a distinct subset of gliomas, with specific clinical and molecular characteristics.36–38,55 In nearly all IDH1/2-mutant gliomas, the IDH1/2 mutation precedes other molecular alterations, suggesting that IDH1/2 mutation is an early causative event in the pathogenesis of this brain tumor subset.37,38,55–59 In line with this hypothesis, IDH1/2 mutation is the only molecular alteration that is almost constantly conserved at recurrence, and analysis of IDH1/2-mutant tumors showed that IDH1/2-mutant proteins are almost ubiquitously expressed in tumor cells.56–58,60 Accordingly, the IDH1/2 mutation is often referred to as a “trunk” initiating event in the clonal evolutionary tree of IDH1/2-mutant gliomas. Secondary genetic alterations occurring during the evolution of IDH1/2-mutant gliomas are often referred as “lineage-defining events”, as TP53 and ATRX mutations characterize tumors of astrocytic lineage, whereas hTERT promoter mutation and 1p/19q codeletion are associated with oligodendroglial tumors. Thereafter, as the disease progresses toward more aggressive tumors, tumor cells often acquire “tertiary” oncogenic alterations, frequently involving cell cycle regulation and growth control pathways, resulting in more malignant behavior.37,38,55,57
Importantly, IDH1/2 mutations have been associated with prognostic and predictive values as biomarkers in gliomas, and assessment of the IDH1/2 status is being implemented in routine clinical practice for patients with primary brain tumors.61 Natural history of IDH1/2-mutant glioma differs from the one of IDH1/2-wild type tumors. Recent studies have demonstrated that IDH1/2 mutations are associated with younger age, better prognosis, and better response to treatment. As an illustration, recent randomized Phase III trials have demonstrated that patients with IDH1/2-mutant gliomas had better overall survival after treatments with radiation therapy and chemotherapy.62–64 Among IDH1/2-mutant gliomas, patients with oligodendroglial tumors harboring 1p19q codeletion have the more favorable prognosis. The mechanisms underlying the relative chemo- and radiosensitive phenotypes associated with IDH1/2 mutations are not fully understood. Epigenetic silencing of the methyl-guanine methyl transferase (MGMT) – which encodes a DNA repair protein that counteracts the cytotoxic effect of alkylating agents – promoter gene by methylation is frequently observed in IDH1/2-mutant tumors and is associated with a partial inability of the tumor to repair the alkylating agent-induced DNA damage.
Hematologic malignancies
IDH1 (R132) or IDH2 (R140 and R172) mutations are found in myeloid malignancies, that is, myelodysplastic syndromes (MDS), AML, and myeloproliferative neoplasms, and also in angioimmunoblastic T-cell lymphoma (AITL).4,5,65,66 In myeloid malignancies, they are considered as an initiating event in 19% of patients with IDH1 mutations and 34% of patients with IDH2 mutations.67 However, IDH1 mutations are likely to be implicated in early stages of de novo AML as others are, that is, NPM1, DNMT3A, TET2, and ASXL1.68
In de novo AML, IDH1/2 mutations are associated with older age, normal karyotype, and NPM1 mutations. IDH1/2 and TET2 mutations are mutually exclusive.69 IDH1 mutations are found in 6%–16% of de novo AML and are associated with a poorer outcome in patients treated with intensive chemotherapy,70 even in patients with favorable prognosis as per European LeukemiaNet (ELN) classification.71–73
IDH2 mutations are found in 8%–19% of de novo AML. IDH2 R140Q mutation is the most frequent (75%–80%) whereas IDH2 R172K mutation is found in 20% of the cases. The prognostic impact of these mutations differs strongly. Depending on the mutational spectrum, IDH2 R140Q mutations confer favorable or no impact on overall survival.4,65,67,74,75 Conversely, patients with IDH2 R172K mutation have a worse prognosis, with lower complete remission rate, higher relapse rate, and lower overall survival.76,77
IDH1/2 mutations are found in 4% to 12% of MDS cases, with a higher incidence (up to 23%) in high-risk MDS. IDH1/2 mutations are associated with an older age, DNMT3A, ASXL1, SRSF2 mutations, and higher rate of transformation to AML.65,78–80
In myeloproliferative neoplasms, the incidence of IDH1/2 mutations range between 2% and 4%, rising up to 31% after transformation to AML. They are associated with older age and SRSF2 mutations. In patients with myelofibrosis, IDH1/2 mutations confer worse prognosis.81
Besides myeloid neoplasms, IDH2 mutations (mostly R172K) are found in 20%–45% of patients with AITL although its prognostic value is not yet known.66
Other malignancies associated with IDH mutations
IDH1/2 mutations have been detected in enchondromas and chondrosarcomas but rarely found in other mesenchymal tumors such as osteosarcomas. IDH1 mutations are the most frequent (40%–52%), and IDH2 mutations are present in 6%–11% of the cases.7,8
IDH1/2 mutations occur in up to 25% of intrahepatic cholangiocarcinomas. Again, IDH1 mutations are the most frequent (11%–24%) and IDH2 mutations are seen in 2%–6% of the cases.6,39,82
Finally, sporadic cases of IDH1/2 mutations have been reported in other types of solid cancers: thyroid cancer, melanoma, prostate carcinoma, lung cancer, breast adenocarcinoma, colorectal cancer, esophageal cancer, and bladder cancer.15,16,83
D-2HG as a predictive biomarker in IDH-mutant cancers
D-2HG released in the serum and/or urine by cancer cells harboring IDH1/2 mutations is a biomarker for IDH1/2 mutations, presumably reflecting the neomorphic enzymatic activity of the mutant enzymes. D-2HG levels are of interest for both the diagnosis and monitoring of patients with IDH1/2-mutant malignancies.84–89
At diagnosis, D-2HG is a strong predictive biomarker for the presence of IDH1/2 mutations in AML. High total 2HG concentration was highly predictive of the presence of an IDH1/2 mutation, although separation of the d- and l-enantiomers distinguished IDH1/2-mutant and -wild-type AML with greater specificity.86,87,90
Increased serum and/or urine D-2HG levels predict IDH1/2 mutation in AML and intrahepatic cholangiocarcinoma and may be used as predictive biomarker for tumor response/recurrence. Prospective evaluation of D-2HG levels during the treatment of newly diagnosed AML treated with standard chemotherapy revealed that both D-2HG level and mutated-IDH1/2 allele burden decreased with response to treatment. Failure to normalize D-2HG levels is associated with treatment failure, whereas elevated D-2HG levels at complete remission are associated with poorer outcome, suggesting that D-2HG is a biomarker predictive of clinical response to intensive chemotherapy in AML patients with IDH1/2 mutations.86,87 On the opposite, D-2HG levels are in the normal range in patients with IDH-mutant gliomas,91 suggesting that the brain–blood barrier prevents D-2HG from entering the circulation. Nevertheless, recent studies suggested that the urinary levels of D-2HG may increase the sensitivity and specificity for IDH1/2 mutation detection in glioma patients.92,93 Although the clinical value of noninvasive detection and monitoring of D-2HG levels has been well established in AML, the feasibility in glioma remains unclear. Most promising strategies are based on advanced imaging approaches, currently under investigation to determine their utility in clinical practice. Indeed, magnetic resonance spectroscopy (MRS) may detect and measure in vivo 2HG levels in patients harboring IDH1/2-mutant tumors.94–98 Recent pilot studies evaluating clinical applications of MRS in IDH1/2-mutant gliomas have shown that 2HG levels correlated with tumor volume and cellularity, and that cytotoxic therapy resulted in a decrease in 2HG levels, suggesting that MRS could assist as noninvasive tool for diagnosis and treatment follow-up.96,97
Targeting of IDH-mutant tumors
The discovery of IDH1/2 mutations has resulted in a number of novel therapeutic approaches (Table 2), which either restore normal IDH1/2 function or block production or downstream effects of D-2HG.
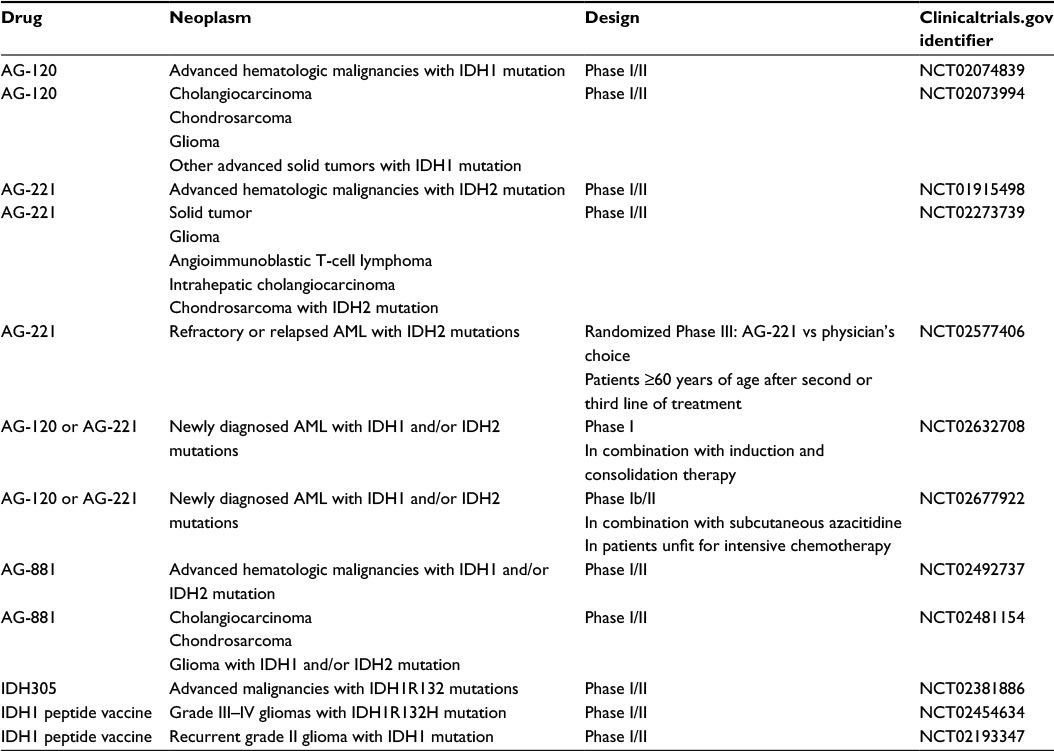 | Table 2 Ongoing clinical trials evaluating IDH inhibitors Abbreviations: IDH, isocitrate dehydrogenase; AML, acute myeloid leukemia. |
Hypomethylating agents
Hypomethylating agents (HMAs) may be of interest in the context of CpG island methylator phenotype induced by IDH1/2 mutations. Azacitidine and decitabine are DNA methyltransferase (DNMT) inhibitors that demonstrated significant clinical benefit not only in high-risk MDS but also in AML.99–101 The outcome of IDH1/2-mutant AML patients treated with HMAs has been retrospectively analyzed, although results are difficult to interpret due to the small number of patients (n=27). These series suggested the lack of association between IDH1/2 mutations and efficacy of HMAs102,103 or showed a better response to DNMT inhibitors among patients with IDH1/2-mutant AML.104
In gliomas, recent preclinical studies have reported that treatment with HMAs reduces DNA methylation of promoter loci of genes involved in glial differentiation. Treatment with HMAs resulted in reduction in cell proliferation and tumor regression in patient-derived IDH1-mutant glioma xenograft models.105,106 These approaches are currently evaluated in early phase trials (NCT02223052 and NCT02332889).
IDH mutant enzymes inhibitors
Preclinical in vitro and in vivo studies have validated the proof of concept that targeted inhibition of IDH1/2 mutants resulted in normalization in a dose-dependent manner of 2-HG, reversal of histone and DNA hypermethylation, and release of cellular differentiation block.39,41,107,108 AGI-5198 and AGI-6780 are selective inhibitors of mutant IDH1 and IDH2 enzymes, respectively. They normalized 2HG, reversed histone and DNA hypermethylation, and induced differentiation of not only TF-1 erythroleukemia cells but also primary human AML cells harboring IDH1/2 mutations.39,41 AGI-5198 in a dose-dependent manner reduced 2HG and in turn was associated with tumor growth inhibition in vitro and in vivo. In IDH1-mutant glioma models, AGI-5198 induced expression of genes associated with astrocytic and oligodendrocytic differentiation and reduced repressive histone trimethylation marks at these gene promoters.40 Together, these studies indicate that differentiation therapy may be achievable in cancers with IDH1/2 mutations, thereby supporting the initiation of clinical trials (Table 2). AG-120 and AG-221 are first-in-class, oral, potent, reversible, selective inhibitors of the IDH1 and IDH2 mutant enzymes, respectively. Separate first-in-human, Phase I, dose-escalation studies of AG-120 and AG-221 are underway in patients with IDH1/2-mutated hematologic malignancies (NCT02074839 and NCT01915498). Similar Phase I dose escalation studies of AG-120 in patients with IDH1-mutated gliomas and other solid tumors (NCT02073994) and of AG-221 in patients with IDH2-mutated gliomas, other solid tumors, and AITL (NCT02273739) are now open. The primary objective of these studies is to establish the safety and tolerability profile of AG-120 and AG-221, while secondary objectives are to characterize the pharmacokinetics, pharmacodynamics, and clinical efficacy. Preliminary unpublished clinical data from the ongoing Phase I trials for AG-120 and AG-221 indicates favorable safety profile, reduction of D-2HG levels, and finally encouraging response rate.109–111 These results support initiation of randomized Phase III and combination studies in AML (Table 2).
Immunotherapy
Recent studies have investigated vaccination-based immunotherapy to target IDH1 mutations. In principle, IDH1/2 mutants are ideal tumor-specific neoantigens due to their uniform occurrence at specific codons and ubiquitous expression throughout all tumor cells. Accordingly, recent preclinical studies showed that vaccination with IDH1 R132H-specific peptide elicited an MHC class II-specific antitumor response against IDH1 R132H-expressing tumor cells and reduced the growth of intracranial tumors.112,113 These preliminary results suggest that mutant IDH1-targeted immunotherapies can elicit potent antitumor immune responses. Clinical trials are ongoing to evaluate such strategies.
BCL-2 inhibition
Recent preclinical works in patient-derived models of IDH1/2-mutant AML have identified synthetic lethal interaction between the antiapoptotic gene BCL-2 and mutant-IDH1/2, showing that IDH1/2-mutant AML cells are more sensitive than their wild-type counterparts to the BCL-2 inhibitor ABT-199.114 ABT-199 was further tested in AML patients in a Phase II trial that has enrolled 32 patients.115 Interestingly, of the five patients who achieved complete remission with or without incomplete marrow recovery, three had IDH1/2 mutations. These preliminary data suggest that BCL-2 inhibition may be of interest in patients with IDH1/2-mutant AML.
Conclusion
The discovery of IDH1/2 mutations highlights the unique role of the “oncometabolite” D-2HG in oncogenesis. The druggable gain-of-function of the mutant enzymes has led to the generation of a new class of drugs. Relevant preclinical models and results of early Phase I trials in adults with hematologic malignancies demonstrate that targeting IDH1/2 mutant is a valid strategy. This is a new model of differentiation therapy that warrants combination strategies.
Disclosure
The authors report no conflicts of interest in this work.
References
Sjöblom T, Jones S, Wood LD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314(5797):268–274. | ||
Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–1812. | ||
Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765–773. | ||
Mardis ER, Ding L, Dooling DJ, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361(11):1058–1066. | ||
Kosmider O, Gelsi-Boyer V, Slama L, et al. Mutations of IDH1 and IDH2 genes in early and accelerated phases of myelodysplastic syndromes and MDS/myeloproliferative neoplasms. Leukemia. 2010;24(5):1094–1096. | ||
Borger DR, Tanabe KK, Fan KC, et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist. 2012;17(1):72–79. | ||
Amary MF, Damato S, Halai D, et al. Ollier disease and Maffucci syndrome are caused by somatic mosaic mutations of IDH1 and IDH2. Nat Genet. 2011;43(12):1262–1265. | ||
Amary MF, Bacsi K, Maggiani F, et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol. 2011;224(3):334–343. | ||
Kranendijk M, Struys EA, Salomons GS, Van der Knaap MS, Jakobs C. Progress in understanding 2-hydroxyglutaric acidurias. J Inherit Metab Dis. 2012;35(4):571–587. | ||
Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462(7274):739–744. | ||
Figueroa ME, Abdel-Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6):553–567. | ||
Xu W, Yang H, Liu Y, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19(1):17–30. | ||
Lu C, Ward PS, Kapoor GS, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483(7390):474–478. | ||
Turcan S, Rohle D, Goenka A, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483(7390):479–483. | ||
Yen KE, Bittinger MA, Su SM, Fantin VR. Cancer-associated IDH mutations: biomarker and therapeutic opportunities. Oncogene. 2010;29(49):6409–6417. | ||
Cairns RA, Mak TW. Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer Discov. 2013;3(7):730–741. | ||
Reitman ZJ, Yan H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: alterations at a crossroads of cellular metabolism. J Natl Cancer Inst. 2010;102(13):932–941. | ||
Lee SM, Koh HJ, Park DC, Song BJ, Huh TL, Park JW. Cytosolic NADP(+)-dependent isocitrate dehydrogenase status modulates oxidative damage to cells. Free Radic Biol Med. 2002;32(11):1185–1196. | ||
Bleeker FE, Atai NA, Lamba S, et al. The prognostic IDH1(R132) mutation is associated with reduced NADP+-dependent IDH activity in glioblastoma. Acta Neuropathol. 2010;119(4):487–494. | ||
Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB. Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev. 2008;18(1):54–61. | ||
Mullen AR, Wheaton WW, Jin ES, et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2011;481(7381):385–388. | ||
Mullen AR, Hu Z, Shi X, et al. Oxidation of alpha-ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell Rep. 2014;7(5):1679–1690. | ||
Jiang L, Shestov AA, Swain P, et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature. 2016;532(7598):255–258. | ||
Icard P, Poulain L, Lincet H. Understanding the central role of citrate in the metabolism of cancer cells. Biochim Biophys Acta. 2012;1825(1):111–116. | ||
Hiller K, Metallo CM. Profiling metabolic networks to study cancer metabolism. Curr Opin Biotechnol. 2013;24(1):60–68. | ||
Zhao S, Lin Y, Xu W, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science. 2009;324(5924):261–265. | ||
Ward PS, Patel J, Wise DR, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17(3):225–234. | ||
Gross S, Cairns RA, Minden MD, et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med. 2010;207(2):339–344. | ||
Loenarz C, Schofield CJ. Expanding chemical biology of 2-oxoglutarate oxygenases. Nat Chem Biol. 2008;4(3):152–156. | ||
Ng SS, Kavanagh KL, McDonough MA, et al. Crystal structures of histone demethylase JMJD2A reveal basis for substrate specificity. Nature. 2007;448(7149):87–91. | ||
Chowdhury R, Yeoh KK, Tian YM, et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011;12(5):463–469. | ||
Tsukada Y, Fang J, Erdjument-Bromage H, et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439(7078):811–816. | ||
Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013;502(7472):472–479. | ||
Pastor WA, Aravind L, Rao A. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol. 2013;14(6):341–356. | ||
Koivunen P, Lee S, Duncan CG, et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012;483(7390):484–488. | ||
Noushmehr H, Weisenberger DJ, Diefes K, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17(5):510–522. | ||
Brat DJ, Verhaak RG, Aldape KD, et al. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med. 2015;372(26):2481–2498. | ||
Ceccarelli M, Barthel FP, Malta TM, et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell. 2016;164(3):550–563. | ||
Wang F, Travins J, DeLaBarre B, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340(6132):622–626. | ||
Rohle D, Popovici-Muller J, Palaskas N, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340(6132):626–630. | ||
Kernytsky A, Wang F, Hansen E, et al. IDH2 mutation-induced histone and DNA hypermethylation is progressively reversed by small-molecule inhibition. Blood. 2015;125(2):296–303. | ||
Losman JA, Looper RE, Koivunen P, et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013;339(6127):1621–1625. | ||
Flavahan WA, Drier Y, Liau BB, et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature. 2016;529(7584):110–114. | ||
Lu C, Venneti S, Akalin A, et al. Induction of sarcomas by mutant IDH2. Genes Dev. 2013;27(18):1986–1998. | ||
Kranendijk M, Struys EA, van Schaftingen E, et al. IDH2 mutations in patients with D-2-hydroxyglutaric aciduria. Science. 2010;330(6002):336. | ||
Chen C, Liu Y, Lu C, et al. Cancer-associated IDH2 mutants drive an acute myeloid leukemia that is susceptible to Brd4 inhibition. Genes Dev. 2013;27(18):1974–1985. | ||
Kats LM, Reschke M, Taulli R, et al. Proto-oncogenic role of mutant IDH2 in leukemia initiation and maintenance. Cell Stem Cell. 2014;14(3):329–341. | ||
Letouzé E, Martinelli C, Loriot C, et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. 2013;23(6):739–752. | ||
Grassian AR, Parker SJ, Davidson SM, et al. IDH1 mutations alter citric acid cycle metabolism and increase dependence on oxidative mitochondrial metabolism. Cancer Res. 2014;74(12):3317–3331. | ||
Fu X, Chin RM, Vergnes L, et al. 2-Hydroxyglutarate inhibits ATP synthase and mTOR signaling. Cell Metab. 2015;22(3):508–515. | ||
Tateishi K, Wakimoto H, Iafrate AJ, et al. Extreme vulnerability of IDH1 mutant cancers to NAD+ depletion. Cancer Cell. 2015;28(6):773–784. | ||
Hartmann C, Meyer J, Balss J, et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol. 2009;118(4):469–474. | ||
Brennan CW, Verhaak RG, McKenna A, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462–477. | ||
Capper D, Weissert S, Balss J, et al. Characterization of R132H mutation-specific IDH1 antibody binding in brain tumors. Brain Pathol. 2010;20(1):245–254. | ||
Bai H, Harmancı AS, Erson-Omay EZ, et al. Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nat Genet. 2016;48(1):59–66. | ||
Watanabe T, Nobusawa S, Kleihues P, Ohgaki H. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol. 2009;174(4):1149–1153. | ||
Wakimoto H, Tanaka S, Curry WT, et al. Targetable signaling pathway mutations are associated with malignant phenotype in IDH-mutant gliomas. Clin Cancer Res. 2014;20(11):2898–2909. | ||
Johnson BE, Mazor T, Hong C, et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science. 2014;343(6167):189–193. | ||
Suzuki H, Aoki K, Chiba K, et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat Genet. 2015;47(5):458–468. | ||
Lass U, Nümann A, von Eckardstein K, et al. Clonal analysis in recurrent astrocytic, oligoastrocytic and oligodendroglial tumors implicates IDH1-mutation as common tumor initiating event. PLoS One. 2012;7(7):e41298. | ||
Stupp R, Brada M, van den Bent MJ, Tonn JC, Pentheroudakis G; Group EGW. High-grade glioma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2014;25(suppl 3):iii93–iii101. | ||
van den Bent MJ, Brandes AA, Taphoorn MJ, et al. Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. J Clin Oncol. 2013;31(3):344–350. | ||
Cairncross G, Wang M, Shaw E, et al. Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: long-term results of RTOG 9402. J Clin Oncol. 2013;31(3):337–343. | ||
Cairncross JG, Wang M, Jenkins RB, et al. Benefit from procarbazine, lomustine, and vincristine in oligodendroglial tumors is associated with mutation of IDH. J Clin Oncol. 2014;32(8):783–790. | ||
Im AP, Sehgal AR, Carroll MP, et al. DNMT3A and IDH mutations in acute myeloid leukemia and other myeloid malignancies: associations with prognosis and potential treatment strategies. Leukemia. 2014;28(9):1774–1783. | ||
Cairns RA, Iqbal J, Lemonnier F, et al. IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood. 2012;119(8):1901–1903. | ||
Molenaar RJ, Thota S, Nagata Y, et al. Clinical and biological implications of ancestral and non-ancestral IDH1 and IDH2 mutations in myeloid neoplasms. Leukemia. 2015;29(11):2134–2142. | ||
Welch JS, Ley TJ, Link DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150(2):264–278. | ||
Solary E, Bernard OA, Tefferi A, Fuks F, Vainchenker W. The Ten-Eleven Translocation-2 (TET2) gene in hematopoiesis and hematopoietic diseases. Leukemia. 2014;28(3):485–496. | ||
Feng JH, Guo XP, Chen YY, Wang ZJ, Cheng YP, Tang YM. Prognostic significance of IDH1 mutations in acute myeloid leukemia: a meta-analysis. Am J Blood Res. 2012;2(4):254–264. | ||
Döhner H, Estey EH, Amadori S, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115(3):453–474. | ||
Marcucci G, Maharry K, Wu YZ, et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a cancer and leukemia group B study. J Clin Oncol. 2010;28(14):2348–2355. | ||
Peterlin P, Renneville A, Ben Abdelali R, et al. Impact of additional genetic alterations on the outcome of patients with NPM1-mutated cytogenetically normal acute myeloid leukemia. Haematologica. 2015;100(5):e196–e199. | ||
Patel JP, Gönen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366(12):1079–1089. | ||
Abdel-Wahab O, Patel J, Levine RL. Clinical implications of novel mutations in epigenetic modifiers in AML. Hematol Oncol Clin North Am. 2011;25(6):1119–1133. | ||
Boissel N, Nibourel O, Renneville A, Huchette P, Dombret H, Preudhomme C. Differential prognosis impact of IDH2 mutations in cytogenetically normal acute myeloid leukemia. Blood. 2011;117(13):3696–3697. | ||
Green CL, Evans CM, Zhao L, et al. The prognostic significance of IDH2 mutations in AML depends on the location of the mutation. Blood. 2011;118(2):409–412. | ||
Lin CC, Hou HA, Chou WC, et al. IDH mutations are closely associated with mutations of DNMT3A, ASXL1 and SRSF2 in patients with myelodysplastic syndromes and are stable during disease evolution. Am J Hematol. 2014;89(2):137–144. | ||
Patnaik MM, Hanson CA, Hodnefield JM, et al. Differential prognostic effect of IDH1 versus IDH2 mutations in myelodysplastic syndromes: a Mayo Clinic study of 277 patients. Leukemia. 2012;26(1):101–105. | ||
Thol F, Weissinger EM, Krauter J, et al. IDH1 mutations in patients with myelodysplastic syndromes are associated with an unfavorable prognosis. Haematologica. 2010;95(10):1668–1674. | ||
Lasho TL, Jimma T, Finke CM, et al. SRSF2 mutations in primary myelofibrosis: significant clustering with IDH mutations and independent association with inferior overall and leukemia-free survival. Blood. 2012;120(20):4168–4171. | ||
Saha SK, Parachoniak CA, Ghanta KS, et al. Mutant IDH inhibits HNF-4α to block hepatocyte differentiation and promote biliary cancer. Nature. 2014;513(7516):110–114. | ||
Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. | ||
Rakheja D, Konoplev S, Medeiros LJ, Chen W. IDH mutations in acute myeloid leukemia. Hum Pathol. 2012;43(10):1541–1551. | ||
Fathi AT, Sadrzadeh H, Borger DR, et al. Prospective serial evaluation of 2-hydroxyglutarate, during treatment of newly diagnosed acute myeloid leukemia, to assess disease activity and therapeutic response. Blood. 2012;120(23):4649–4652. | ||
DiNardo CD, Propert KJ, Loren AW, et al. Serum 2-hydroxyglutarate levels predict isocitrate dehydrogenase mutations and clinical outcome in acute myeloid leukemia. Blood. 2013;121(24):4917–4924. | ||
Janin M, Mylonas E, Saada V, et al. Serum 2-hydroxyglutarate production in IDH1- and IDH2-mutated de novo acute myeloid leukemia: a study by the Acute Leukemia French Association Group. J Clin Oncol. 2014;32(4):297–305. | ||
Borger DR, Goyal L, Yau T, et al. Circulating oncometabolite 2-hydroxyglutarate is a potential surrogate biomarker in patients with isocitrate dehydrogenase-mutant intrahepatic cholangiocarcinoma. Clin Cancer Res. 2014;20(7):1884–1890. | ||
Willekens C, Micol J-B, Poinsignon V, et al. Serum 2-Hydroxyglutarate level can predict IDH2 mutation in myeloid sarcoma. Blood. 2015;126(23):3835–3835. | ||
Duran M, Kamerling JP, Bakker HD, van Gennip AH, Wadman SK. L-2-Hydroxyglutaric aciduria: an inborn error of metabolism? J Inherit Metab Dis. 1980;3(4):109–112. | ||
Capper D, Simon M, Langhans CD, et al. 2-Hydroxyglutarate concentration in serum from patients with gliomas does not correlate with IDH1/2 mutation status or tumor size. Int J Cancer. 2012;131(3):766–768. | ||
Lombardi G, Corona G, Bellu L, et al. Diagnostic value of plasma and urinary 2-hydroxyglutarate to identify patients with isocitrate dehydrogenase-mutated glioma. Oncologist. 2015;20(5):562–567. | ||
Fathi AT, Nahed BV, Wander SA, et al. Elevation of urinary 2-hydroxyglutarate in IDH-mutant glioma. Oncologist. 2016;21(2):214–219. | ||
Choi C, Ganji SK, DeBerardinis RJ, et al. 2-hydroxyglutarate detection by magnetic resonance spectroscopy in IDH-mutated patients with gliomas. Nat Med. 2012;18(4):624–629. | ||
Andronesi OC, Kim GS, Gerstner E, et al. Detection of 2-hydroxyglutarate in IDH-mutated glioma patients by in vivo spectral-editing and 2D correlation magnetic resonance spectroscopy. Sci Transl Med. 2012;4(116):116ra4. | ||
Andronesi OC, Loebel F, Bogner W, et al. Treatment response assessment in IDH-mutant glioma patients by non-invasive 3D functional spectroscopic mapping of 2-hydroxyglutarate. Clin Cancer Res. 2015;22(7):1632–1641. | ||
de la Fuente MI, Young RJ, Rubel J, et al. Integration of 2-hydroxyglutarate-proton magnetic resonance spectroscopy into clinical practice for disease monitoring in isocitrate dehydrogenase-mutant glioma. Neuro Oncol. 2016;18(2):283–290. | ||
Emir UE, Larkin SJ, de Pennington N, et al. Noninvasive quantification of 2-hydroxyglutarate in human gliomas with IDH1 and IDH2 mutations. Cancer Res. 2016;76(1):43–49. | ||
Fenaux P, Mufti GJ, Hellström-Lindberg E, et al. Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. J Clin Oncol. 2010;28(4):562–569. | ||
Kantarjian HM, Thomas XG, Dmoszynska A, et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J Clin Oncol. 2012;30(21):2670–2677. | ||
Dombret H, Seymour JF, Butrym A, et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood. 2015;126(3):291–299. | ||
DiNardo CD, Patel KP, Garcia-Manero G, et al. Lack of association of IDH1, IDH2 and DNMT3A mutations with outcome in older patients with acute myeloid leukemia treated with hypomethylating agents. Leuk Lymphoma. 2014;55(8):1925–1929. | ||
Benton CB, Ravandi F, Andreeff M, et al. Case series of patients with acute myeloid leukemia receiving hypomethylation therapy and retrospectively found to have IDH1 or IDH2 mutations. Leuk Lymphoma. 2014;55(6):1431–1434. | ||
Emadi A, Faramand R, Carter-Cooper B, et al. Presence of isocitrate dehydrogenase mutations may predict clinical response to hypomethylating agents in patients with acute myeloid leukemia. Am J Hematol. 2015;90(5):E77–E79. | ||
Borodovsky A, Salmasi V, Turcan S, et al. 5-azacytidine reduces methylation, promotes differentiation and induces tumor regression in a patient-derived IDH1 mutant glioma xenograft. Oncotarget. 2013;4(10):1737–1747. | ||
Turcan S, Fabius AW, Borodovsky A, et al. Efficient induction of differentiation and growth inhibition in IDH1 mutant glioma cells by the DNMT Inhibitor Decitabine. Oncotarget. 2013;4(10):1729–1736. | ||
Hansen E, Quivoron C, Straley K, et al. AG-120, an oral, selective, first-in-class, potent inhibitor of mutant IDH1, reduces intracellular 2HG and induces cellular differentiation in TF-1 R132H cells and primary human IDH1 mutant AML patient samples treated Ex Vivo. Blood. 2014;124(21):3734–3734. | ||
Quivoron C, David M, Straley K, et al. AG-221, an oral, selective, first-in-class, potent IDH2-R140Q mutant inhibitor, induces differentiation in a Xenotransplant Model. Blood. 2014;124(21):3735–3735. | ||
Stein EM, Altman JK, Collins R, et al. AG-221, an oral, selective, first-in-class, potent inhibitor of the IDH2 mutant metabolic enzyme, induces durable remissions in a phase I study in patients with IDH2 mutation positive advanced hematologic malignancies. Blood. 2014;124(21):115–115. | ||
Fan B, Le K, Manyak E, et al. Longitudinal pharmacokinetic/pharmacodynamic profile of AG-120, a potent inhibitor of the IDH1 mutant protein, in a phase 1 study of IDH1-mutant advanced hematologic malignancies. Blood. 2015;126(23):1310–1310. | ||
DiNardo C, de Botton S, Pollyea DA, et al. Molecular profiling and relationship with clinical response in patients with IDH1 mutation-positive hematologic malignancies receiving AG-120, a first-in-class potent inhibitor of mutant IDH1, in addition to data from the completed dose escalation portion of the phase 1 study. Blood. 2015;126(23):1306–1306. | ||
Schumacher T, Bunse L, Pusch S, et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature. 2014;512(7514):324–327. | ||
Pellegatta S, Valletta L, Corbetta C, et al. Effective immuno-targeting of the IDH1 mutation R132H in a murine model of intracranial glioma. Acta Neuropathol Commun. 2015;3:4. | ||
Chan SM, Thomas D, Corces-Zimmerman MR, et al. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat Med. 2015;21(2):178–184. | ||
Konopleva M, Pollyea DA, Potluri J, et al. A phase 2 study of ABT-199 (GDC-0199) in patients with acute myelogenous leukemia (AML). Blood. 2014;124(21):118–118. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.