")
Back to Journals » International Journal of General Medicine » Volume 14
Identification of the Potential Prognosis Biomarkers in Hepatocellular Carcinoma: An Analysis Based on WGCNA and PPI
Authors Huang J, Zhan Y, Jiang L, Gao Y, Zhao B, Zhang Y, Zhang W, Zheng J , Yu J
Received 15 September 2021
Accepted for publication 25 November 2021
Published 10 December 2021 Volume 2021:14 Pages 9555—9565
DOI https://doi.org/10.2147/IJGM.S338500
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Junting Huang,1,* Yating Zhan,1,* Lili Jiang,1,* Yuxiang Gao,1,* Binyu Zhao,1,* Yuxiao Zhang,1 Wenjie Zhang,1 Jianjian Zheng,1 Jinglu Yu2,3
1Key Laboratory of Diagnosis and Treatment of Severe Hepato-Pancreatic Diseases of Zhejiang Province, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, 325000, People’s Republic of China; 2Department of Laboratory Medicine, Lishui Municipal Central Hospital, Lishui, People’s Republic of China; 3The Fifth Affiliated Hospital of Wenzhou Medical University, Lishui, Zhejiang, 323000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jinglu Yu
Department of Laboratory Medicine, Lishui Municipal Central Hospital, The Fifth Affiliated Hospital of Wenzhou Medical University, No. 289 Kuocang Road, Lishui, Zhejiang, 323000, People’s Republic of China
Email [email protected]
Jianjian Zheng
Key Laboratory of Diagnosis and Treatment of Severe Hepato-Pancreatic Diseases of Zhejiang Province, The First Affiliated Hospital of Wenzhou Medical University, No. 2 Fuxue Lane, Wenzhou, Zhejiang, People’s Republic of China
Email [email protected]
Aim: This study was done to determine biomarkers for the prognostic prediction of hepatocellular carcinoma (HCC).
Materials and Methods: In the Gene Expression Omnibus, the gene expression profiles of HCC were downloaded. Biomarkers were identified by weighted gene co-expression network analysis and protein–protein interaction network analysis.
Results: There were 24 modules, which were characterized by the high correlation with HCC. Meanwhile, through enrichment analysis, differentially expressed genes were largely participated in the ubiquitination and autophagy processes. Moreover, PRC1, TOP2A and CKAP2L may be the hub genes involved in HCC tumorigenesis, and their biomarker roles were further demonstrated via Gene Expression Profiling Interactive Analysis (GEPIA) and Oncomine databases. In addition, the levels of PRC1, TOP2A and CKAP2L were obviously up-regulated in the sera of HCC patients.
Conclusion: PRC1, TOP2A and CKAP2L may serve as biomarkers for the prognostic prediction of HCC patients.
Keywords: hepatocellular carcinoma, biomarker, WGCNA, PPI, prognosis
Introduction
According to the cancer statistics reported in 2020, hepatocellular carcinoma (HCC) is the main type of Primary Carcinoma of the Liver and the second leading causes of cancer-related death globally, with a five-year survival rate < 20%.1 Currently, surgical resection, a standard therapy for HCC, contributes to the prognosis of HCC.2 It is known that most HCC patients are diagnosed at the middle and/or advanced stages, which may lead to delayed treatment and poor prognosis.3 Due to the reason that the current detection techniques including diagnostic imaging and biomarkers still need to be improved, there is an urgent need for accurately diagnosing HCC at an early stage.4 Moreover, the identification of reliable prognostic molecular targets for HCC is imperative.5
Weighted gene co-expression network analysis (WGCNA) is a method that divides differentially expressed genes (DEGs) with closely relevant stemness index into gene modules (GMs).6 According to function enrichment analysis of DEGs, disease characteristics and cancer-associated biomarkers can be identified. In this study, a microarray dataset (GSE76427) was obtained from the Gene Expression Omnibus (GEO) database, then WGCNA as well as protein-protein interaction (PPI) network analysis was used to find HCC-related hub genes.7 Interestingly, there may be a positive correlation between HCC development and hub genes including PRC1, TOP2A and CKAP2L, which was confirmed by the datasets from Gene Expression Profiling Interactive Analysis (GEPIA) and Oncomine databases. Notably, the protein levels of these hub genes were increased in the liver tissues from HCC patients in the Human Protein Atlas (THPA) database. The results of this study demonstrate that the HCC-related hub genes such as PRC1, TOP2A and CKAP2L may be used as promising prognostic biomarkers for HCC, which may provide us with fresh insights into the molecular mechanisms of HCC.
Materials and Methods
Data Collection
From the GEO database (https://www.ncbi.nlm.nih.gov/), the mRNA expression profile and the relevant clinical data of human HCC were downloaded. The dataset GSE76427 including 115 HCC and 52 adjacent normal tissue samples was extracted from the platforms GPL10558 (Illumina HumanHT-12 v4.0 Expression BeadChip). Then the mRNA expression profile was utilized to structure co-expression networks and confirm hub genes.
Identification of DEGs
The “limma” R package was utilized to calculate fold change (FC) value of gene expression level between HCC and adjacent normal tissues.8 In addition, the method of Benjamin-Hochberg was applied to obtain the false discovery rate (FDR).9 All DEGs were screened by using the criteria of FDR < 0.05 and |log FC| > 1.5.
Construction of the Gene Co-Expression Network
WGCNA package was utilized to construct a co-expression network for the identified DEGs in GSE76427 in this study.10 According to Pearson’s correlation matrices, we constructed a weighted adjacency matrix by a power function am=|cmn|® (cmn=Pearson’s correlation between gene m and gene n). To emphasize the weak correlations and strong correlations between genes, the soft threshold power β was set as 4. Then, we transferred the adjacencies to the topological overlap matrix (TOM), and used the gene dendrogram with a minimum module size of 20 to perform average linkage hierarchical clustering. Finally, genes classified to the same module tend to exhibit similar expression patterns and functions.11
Protein-Protein Interaction (PPI)
PPI network information with a combined score of 0.4 was obtained from the Search Tool for the Retrieval of Interacting Genes (STRING) database (http://string-db.org) based on uploaded DEGs.12 By using a tool of Network Analyzer in Cytoscape software 3.6.1 (https://cytoscape.org), the degree score of each gene was obtained. Then, genes that have a degree higher than 5 were defined as hub genes in this study.
Identification of Hub Genes
The key module genes were composed of highly interconnected nodes and had been demonstrated to have important functions.13 In this study, key module genes in the co-expression network were considered to have high module membership (MM) measured by Pearson’s correlation, and were correlated with the clinical trait.14 The common hub genes in PPI network and the co-expression network were defined as “true” hub genes for subsequent analysis.
Hub Genes Validation
To validate the hub genes, GEPIA database (http://gepia.cancer-pku.cn) was used to compare expression levels and survival analysis data between HCC and adjacent normal tissues.15 Additionally, Oncomine database (http://www.oncomine.org) was also utilized to verify the expression values of the hub genes. All the results with P<0.05 were considered of statistically significance.16 Furthermore, the Human Protein Atlas (http://www.proteinatlas.org) offered immunohistochemistry validation to the hub genes.
Function Enrichment Analysis
We used the R package “cluster Profiler” to perform gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis on the DEGs identified in the 24 modules, respectively.17 Gene sets were considered significantly enriched under a threshold of P<0.05.
The Human Protein Atlas (THPA)
The Human Protein Atlas (https://www.proteinatlas.org/) was composed of Tissue Atlas, Pathology Atlas and Cell Atlas. This database provided proteomics data and cell-specific localization information across certain normal tissues and organs and 20 of the most common types of cancer. In this study, this database was utilized to study the protein expression and cell-specific localization of the PCR1, TOP2A and CKAP2L gene in normal liver tissues and cancer tissues.
Validation of qRT-PCR
Sera samples were obtained from 20 HCC patients and 20 healthy controls from Lishui Municipal Central Hospital. The study was approved by the ethics committee of Lishui Municipal Central Hospital and the informed consents were received from all subjects in this study. Next, TRIzol LS reagent was used to extract total RNA from the sera of HCC patients and healthy controls. Then we used the ribo SCRIPTTM reverse transcription kit to reversely transcribe the mRNA into cDNA. The mRNA expression level was calibrated with glyceraldehyde-3-phosphate dehydrogenase (GAPDH). SYBR Green master mix was added, and then real-time PCR was performed by using a 7500 rapid quantitative PCR system (Applied Biosystems, USA). We recorded the CT value of each well, and the relative quantification of the amplified products was carried out by using the 2−ΔCt method.
Statistical Analysis
In this study, R version 3.6.1 (http://www.R-project.org) was used to perform statistical analysis. A paired t-test was utilized to check for differences in gene expression between tumor and normal tissues. The P value of less than 0.05 was considered of statistically significance.
Results
Identification of DEGs Between HCC Patients and Normal Controls
The workflow of this study is shown in Figure 1.
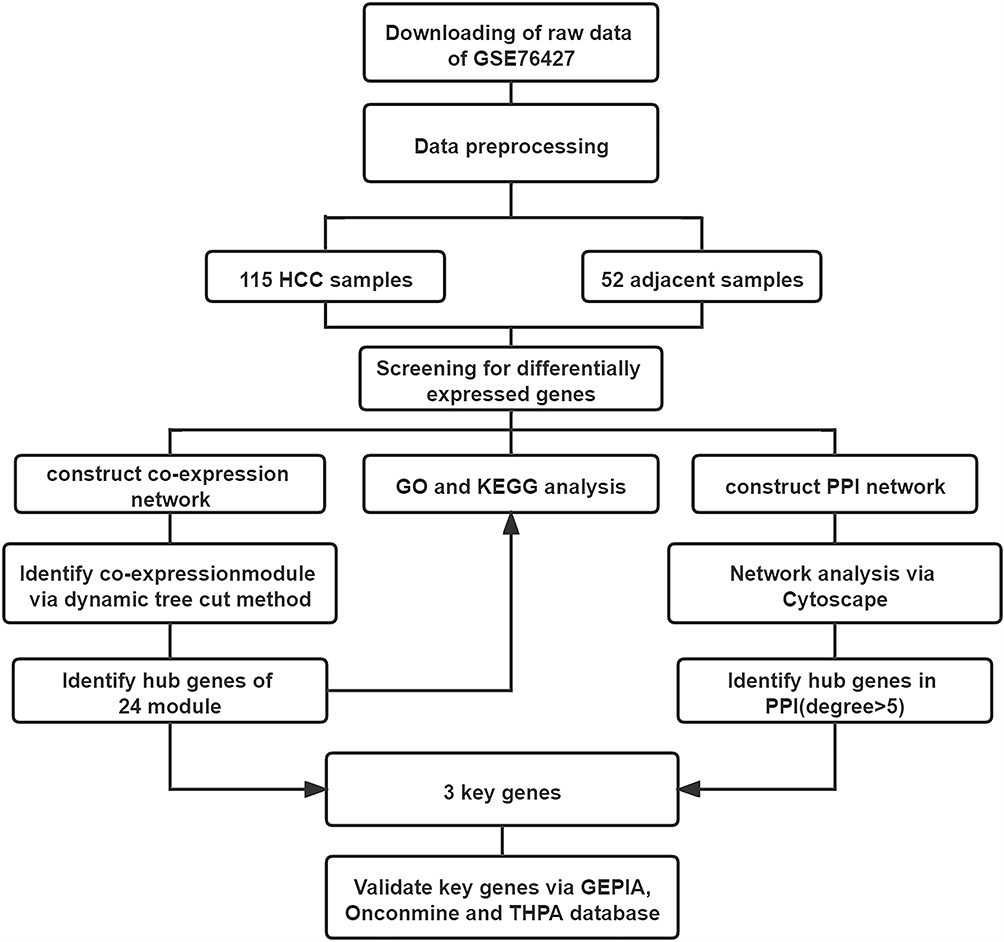 |
Figure 1 Study design. Flowchart of data collection, preprocessing, analysis and validation. |
The aim of this study was to identify the roles of DEGs in the development and progression of HCC. The expression data of GSE76427 was downloaded from the GEO database. All patients were included into subsequent analysis after the data preprocessing and quality evaluation of the WGCNA R package. Using the threshold of FDR <0.05 and the adjusted P value <0.05, over 13648 DEGs were identified for further analysis, and the minimum log |FC| was greater than 1.5. Among these, 6922 were up-regulated, while 6726 were down-regulated.
Construction of a Weighted Co-Expression Network for Key Modules
Co-expression analysis was performed on all clinical samples, and no significant outliers were found (Figure 2A). The weighted co-expression network in this study was constructed with a weighted parameter (soft-threshold β = 4) that ensured the adjacency function of the scale-free network (Figure 2B and C). Here, the dynamic tree was used to cut and merge similar modules, and 24 modules were identified and visualized through the clustering dendrogram (Figure 3A and B). Interestingly, all modules were related to certain clinical features (Figure 3C).
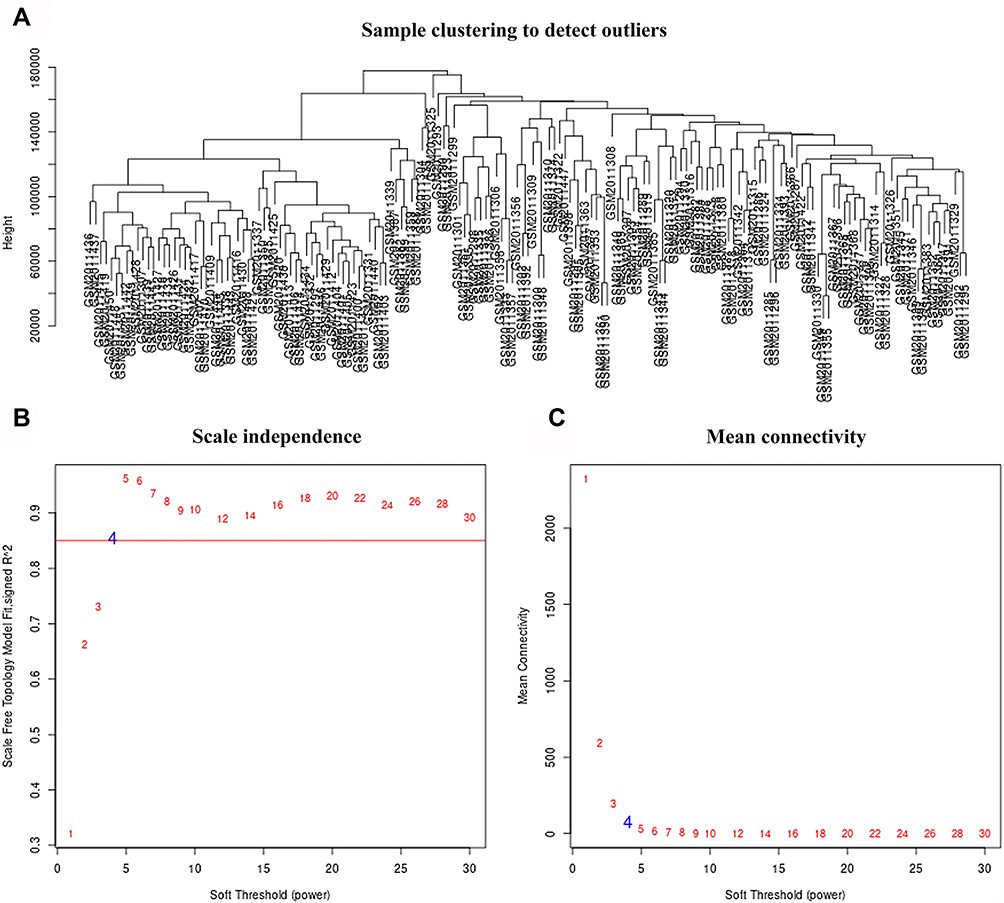 |
Figure 2 Use Weighted Gene Co-expression Network Analysis (WGCNA) to determine the soft threshold power (β). (A) Clustering’s expression data was from the differentially expressed genes between HCC (n = 115) and adjacent normal (n = 52) tissues. (B) Scale-free topology model fitting index (R2, y-axis). (C) Average connectivity of diverse soft-thresholding powers, where the red Arabic numerals indicate the soft thresholds. We determine β = 4, so as to balance the maximizing R2 and maintaining a high average number of connections. |
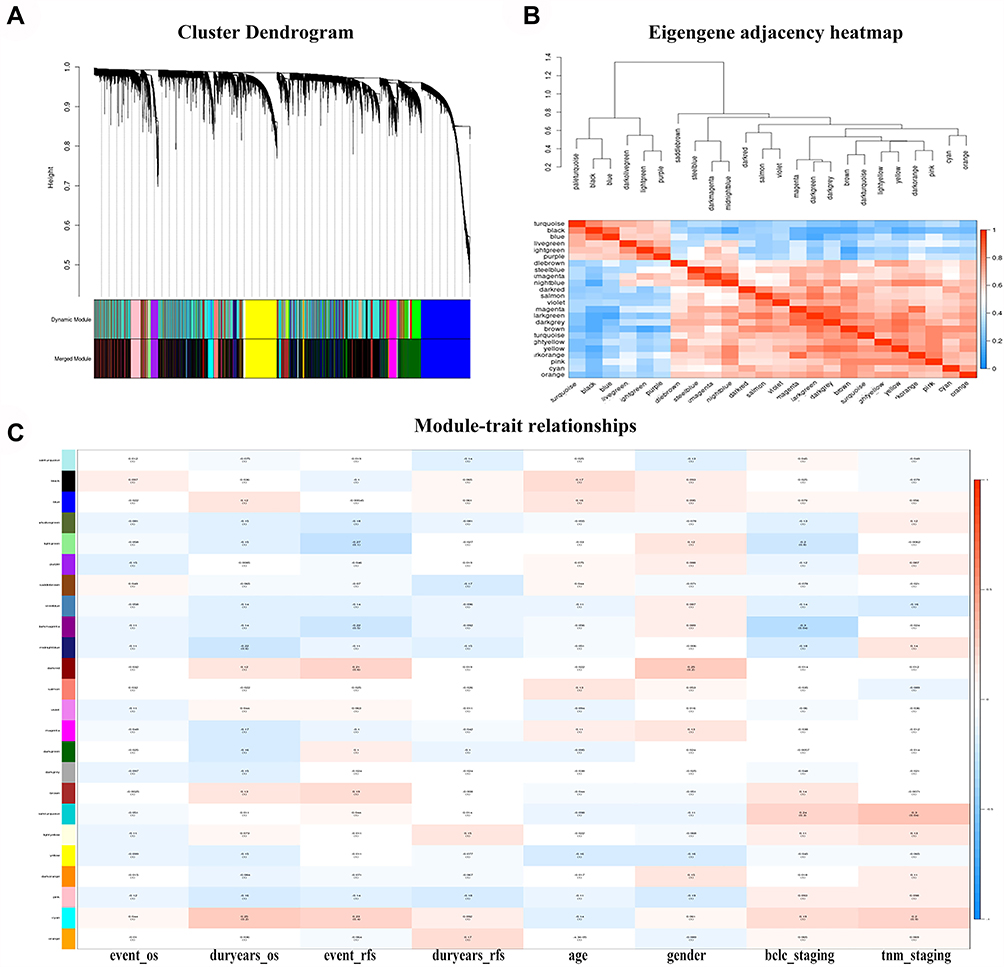 |
Figure 3 Identify modules related to HCC tumorigenesis. (A) Dendrogram of differentially expressed genes clustered based on a dissimilarity measure (1-TOM). (B) Adjacent heatmap of characteristic genes of different modules. (C) Heatmap of the correlation between the characteristic genes of the module and the clinical characteristics of HCC. |
GO and KEGG Pathway Analysis of DEGs
All DEGs were classified into clusters with the same GO biological process (BP) terms and KEGG pathway functions using the R package “cluster Profiler”. As indicated by the analysis of GO term results, the upregulated DEGs in tumors were mainly enriched in ubiquitin-like protein transferase activity (Figure 4A), whereas the downregulated DEGs were enriched in coenzyme binding and carboxylic acid binding (Figure 4C). Moreover, the results of KEGG pathway function analysis revealed that the upregulated genes in tumors were mainly enriched in ribosome, cell cycle, spliceosome and ubiquitin mediated proteolysis (Figure 4B), whereas the downregulated DEGs were enriched in complement and coagulation cascades (Figure 4D). Next, GO and KEGG pathway enrichment analysis were performed in the 24 modules, respectively. Intriguingly, cell cycle was highly associated with 2 modules (black and yellow) (Figure 4F and H), and the genetic compositions of the above two modules were completely different, since the black module consisted of downregulated DEGs, whereas the yellow module was composed of upregulated DEGs.
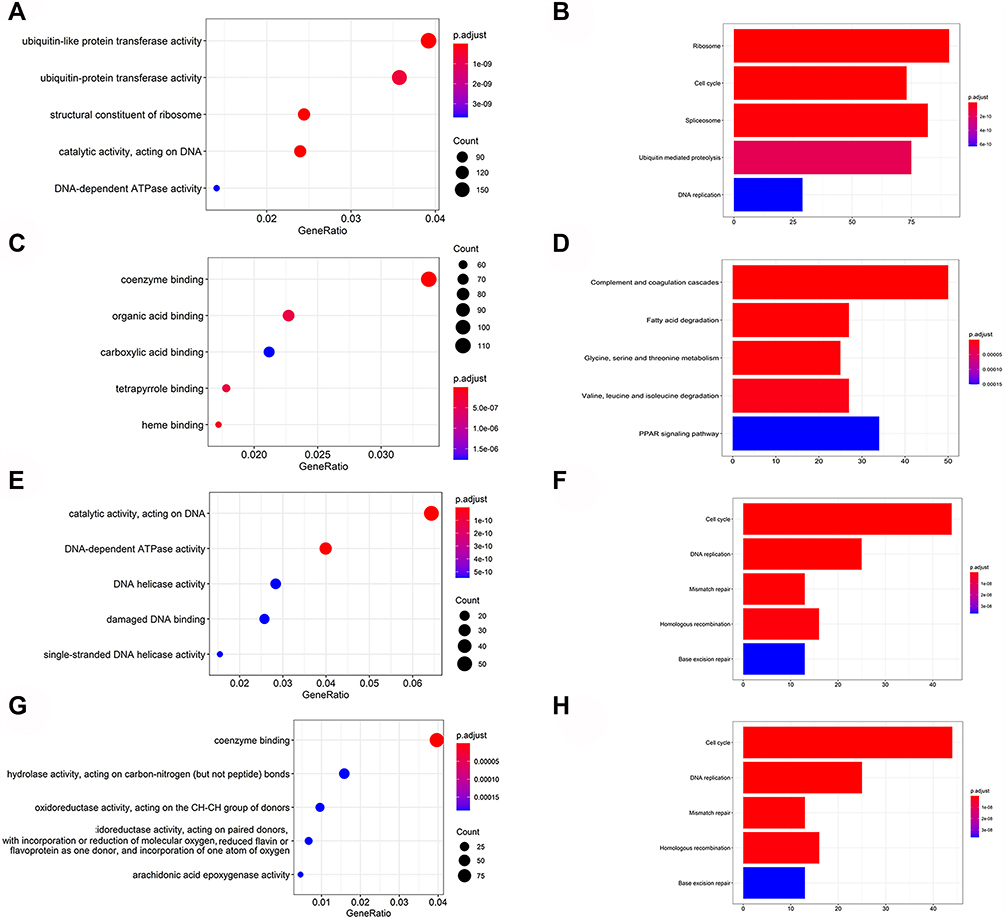 |
Figure 4 GO and KEGG enrichment analysis of genes in the total module. (A and B) Enrichment results of all up-regulated genes via GO and KEGG analysis. (C and D) Enrichment results of all down-regulated genes via GO and KEGG analysis. GO and KEGG analysis of the 24 identified modules showed that the yellow modules (E and F) and black modules (G and H) were highly correlated with the cell cycle. The enrichment score is represented by the size of the bubble or the length of the column, and the enrichment meaning is represented by the color. |
Hub Gene Identification Through PPI and WGCNA
Next, the hub genes of DEGs were explored in PPI network via STRING analysis and only 161 genes reached the cutoff criterion (degree>5) in Cytoscape. Moreover, it was found that 162 hub genes were identified by WGCNA analysis. Further studies showed that only 3 hub genes including PRC1, TOP2A and CKAP2L, were confirmed by both PPI and WGCNA, as shown in Venn diagram (Figure 5A). Notably, the above 3 genes were involved in the same cluster in the PPI network using Markov Clustering (MCL) algorithm (inflation parameter=5) (Figure 5B).
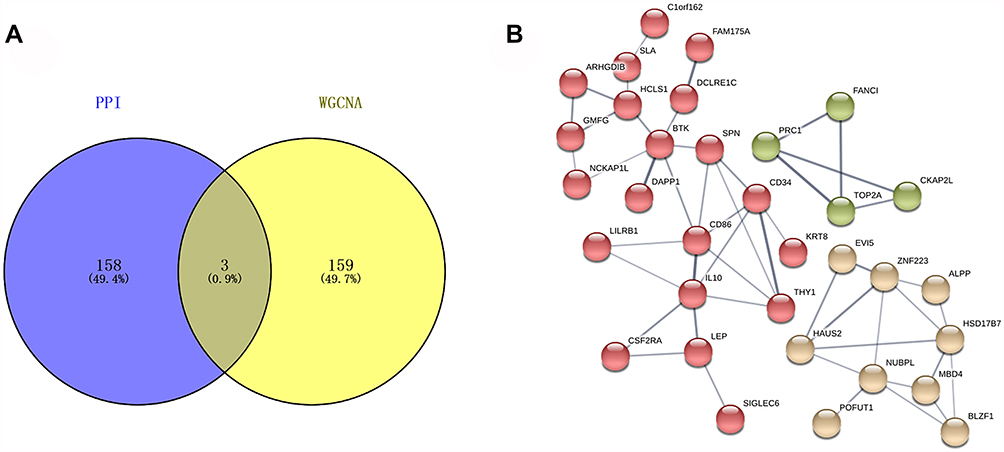 |
Figure 5 Common hub genes in co-expression network and PPI network. (A) Venn diagram showed the hub genes in the co-expression network and the PPI network, in which three common network genes were further analyzed and verified. (B) Three selected genes were involved in the same cluster in the PPI network using Markov Clustering (MCL) algorithm (inflation parameter=5). |
Hub Genes Validation in the GEPIA, Oncomine and THPA Database
It was found that the expressions of PRC1, TOP2A and CKAP2L were increased in HCC tissues via GEPIA (Figure 6A–C). In addition, HCC patients with low expressions of these genes exhibited prolonged survival (Figure 6D–F). Furthermore, the data from Oncomine database confirmed higher expression levels of PRC1, TOP2A and CKAP2L in tumor tissues in comparison with adjacent normal tissues (Figure 7A–C). Additionally, the results of immunohistochemistry staining from THPA database indicated that PRC1, TOP2A and CKAP2L may be oncogenes (Figure 7D–F).
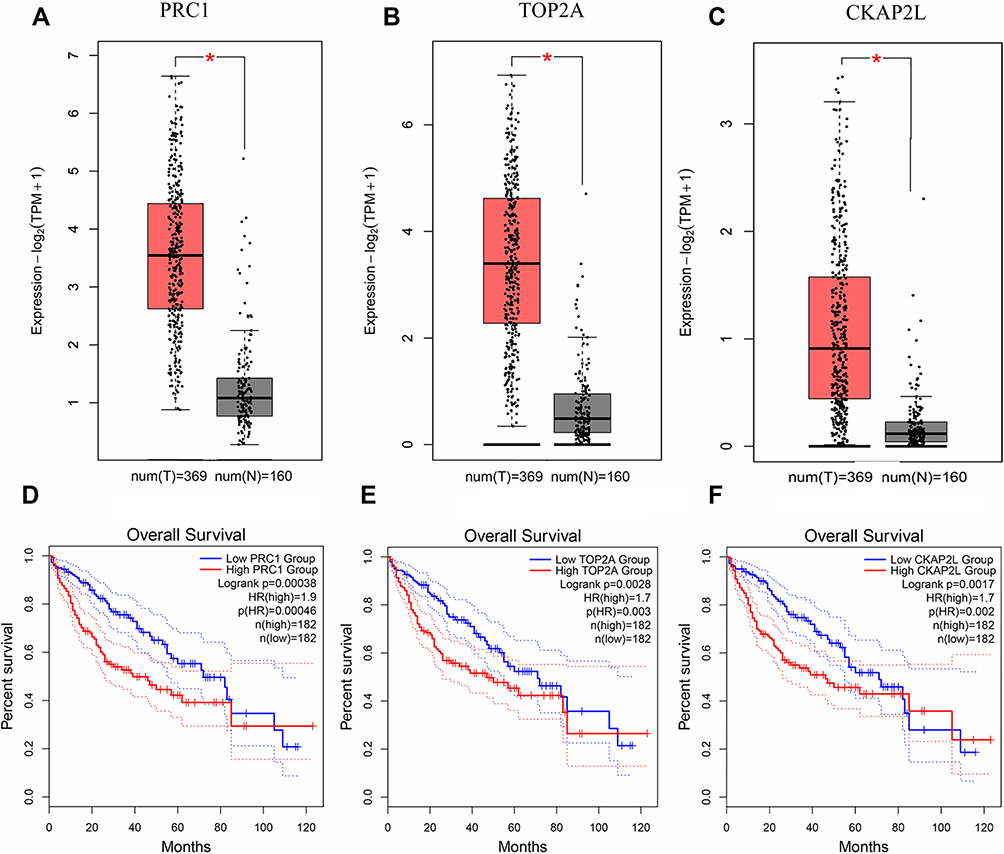 |
Figure 6 Hub gene verification in GEPIA. (A–C) Gene expression levels of the hub genes between tumors and normal tissues. (A) PRC1, (B) TOP2A, (C) CKAP2L. (D–F) Survival analysis of the hub genes in HCC. (D) PRC1, (E) TOP2A, (F) CKAP2L. The red line indicates high gene expression, and the blue line represents low gene expression. *P < 0.05. |
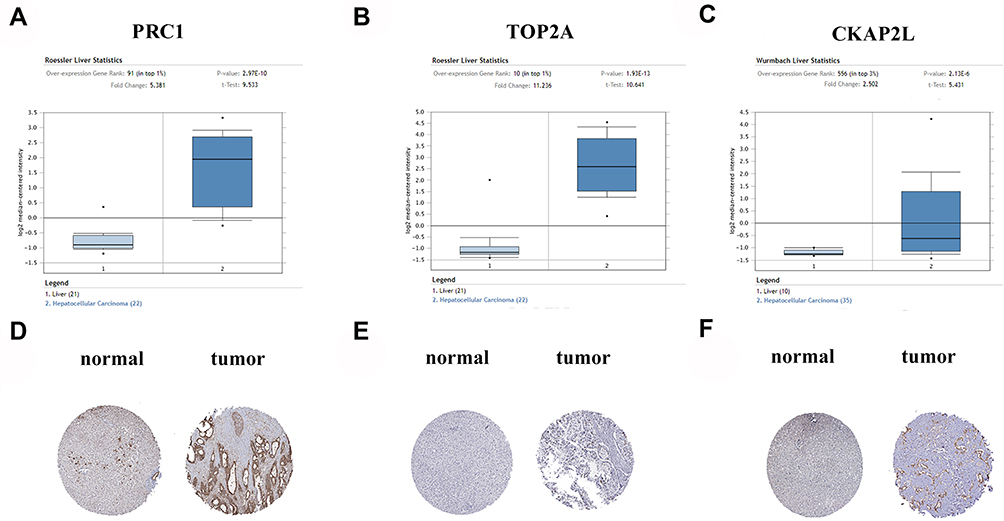 |
Figure 7 The hub genes in Oncomine and THPA database. (A–C) Expression level of the hub genes in tumor tissue and paired normal tissue based on Oncomine database. (A) PRC1, (B) TOP2A, (C) CKAP2L. (D–F) Expression of three hub genes in HCC samples and normal tissues in THPA. (D) PRC1, (E) TOP2A, (F) CKAP2L. |
Hub Genes Validation in Sera of HCC Patients
The expressions of PRC1, TOP2A and CKAP2L were also examined in the sera of HCC patients. Notably, it was found that serum levels of PRC1, TOP2A and CKAP2L were up-regulated in HCC patients in comparison with the healthy controls (Figure 8). Our results suggest that PRC1, TOP2A and CKAP2L may be promising HCC biomarkers.
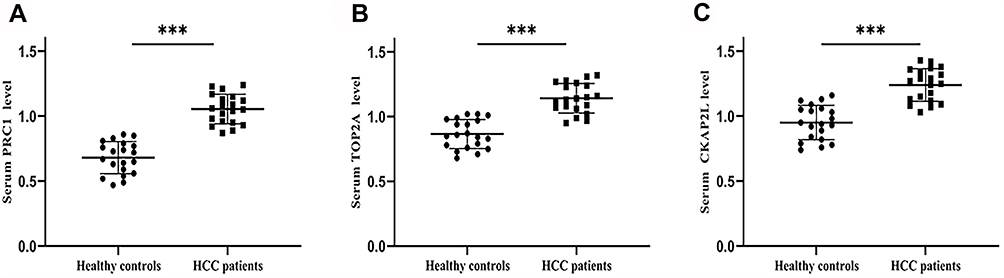 |
Figure 8 Validation of qRT-PCR. (A) Serum PRC1 level, (B) Serum TOP2A level, (C) Serum CKAP2L level. ***P < 0.001. |
Discussion
Currently, there is an urgent need to improve the diagnosis of HCC, which may promote the survival rate of HCC patients.18 We explored the hub biomarkers and molecular mechanisms of HCC via GSE76427. Our findings contribute to the development of new predictive models and biomarkers for the detection of patients with HCC. Our data also revealed the involvement of the pathways of ubiquitination and autophagy in HCC. Finally, using WGCNA and PPI, we identified the key hub genes in HCC.19
Interestingly, our results indicated that PRC1, TOP2A, and CKAP2L were highly correlated with the progression and prognosis of HCC. Currently, this is the first report to screen the key oncogenes (PRC1, TOP2A and CKAP2L) via WGCNA analysis and PPI network in HCC, which may provide promising prognosis biomarkers for HCC.
Herein, all DEGs were divided into two groups: upregulated genes and downregulated genes. The enrichment analysis of GO biological process revealed that many upregulated genes were enriched in the ubiquitinating process, while the downregulated ones were enriched in the autophagy process. Further studies were performed in the 24 modules via GO (BP) and KEGG pathway enrichment analysis. As a result, 2 modules (black and yellow) were found to be associated with cell cycle. Notably, the black module was consisted of downregulated DEGs, whereas the yellow module was composed of upregulated DEGs, indicating that cell cycle may be one of the major processes for HCC-associated ubiquitination and autophagy programming. For example, recent studies have reported that ubiquitylation could contribute to the development of new processes related to DNA replication and mitosis.20 Besides, Zheng et al found that selective autophagy is a mechanism that maintains orderly DNA repair and genome integrity by degrading specific cell cycle proteins, regulating cell division, and promoting DNA damage repair.21 Combined with these, targeting ubiquitination and autophagy may be a promising treatment strategy for HCC.
Three hub genes including PRC1, TOP2A and CKAP2L were identified in the intersection of WGCNA and PPI network analysis. Among them, PRC1 is a key regulator of cytokinesis.22 As an oncogene, PRC1 has been demonstrated to be a substrate of several cyclin-dependent kinases, as well as an exhibitor of E3 ubiquitin-ligase activity.23 The ubiquitin pathway has been reported to be involved in cell cycle regulation.24 Previously, de-ubiquitin enzyme have been found to reverse the process of protein ubiquitin degradation, and then affect the occurrence and prognosis of HCC, including tumor signal pathway, apoptosis, autophagy, and cell cycle regulation.25 Currently, tazemetostat (EPZ-6438), a competitive inhibitor of PRC1, has been applied to some advanced malignant tumors, including lymphoma, sarcoma and mesothelioma,26 indicating that PRC1 may be a promising target of HCC.
TOP2A is significantly enriched in the nucleotide binding, nuclear chromosome and magnesium ion binding in GO biological processes. It is known that TOP2A encodes an enzyme, DNA topoisomerase, which controls the topological state of DNA during the transcription process and is often used as a target of anti-cancer drugs.27 Further studies have revealed that TOP2A contributes to the activation of the PI3K/Akt/mTOR signaling pathway in patients with gallbladder cancer (GBC), resulting in the promotion of cell proliferation, migration and invasion in GBC.28 Taken together, since we still do not know the role of TOP2A in HCC, this may be a new idea of the molecular mechanism of HCC.
Recently, CKAP2L, participating in the cell division of neural progenitor cell, has been found to be involved in spindle organization defects, such as lagging chromosomes, mitotic spindle defects and chromatin bridges.11 Increasing evidence has demonstrated that CKAP2L could be induced by the Mitogen Activated Protein Kinase (MAPK) pathway, leading to the invasion of lung cancer.29 Furthermore, the MAPK signaling pathway plays a key role in the progression of autophagy, associated with the sensitivity to drug treatment.30 Therefore, higher CKAP2L may be associated with poor prognosis in tumors. Interestingly, all the genes (PRC1, TOP2A, and CKAP2L) were involved in the same cluster (cell cycle) in the PPI network, which was consistent with the previous functional enrichment analysis.
However, this study still has many limitations. The underlying molecular mechanisms of PRC1, TOP2A and CKAP2L promoting HCC development remain unclear, which could be explored in the future. In addition, larger sample size studies are needed to verify the clinical values of PRC1, TOP2A and CKAP2L in HCC prognosis.
Conclusion
Our results categorize the significance of many HCC-related biological advances including ubiquitination and autophagy. Our findings also suggest that PRC1, TOP2A and CKAP2L may be novel prognostic biomarkers in HCC.
Abbreviations
HCC, hepatocellular carcinoma; WGCNA, weighted gene co-expression network analysis; DEGs, differentially expressed genes; PPL, protein-protein interaction; GMs, gene modules; GEO, Gene Expression Omnibus; GEPIA, Gene Expression Profiling Interactive Analysis; THAT, The Human Protein Atlas; FC, fold change; FDR, false discovery rate; TOM, topological overlap matrix; STRING, Search Tool for the Retrieval of Interacting Genes; MM, module membership; GEPIA, Gene Expression Profiling Interactive Analysis; GO, gene ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; BP, biological process; MCL, Markov Clustering; GBC, gallbladder cancer; MAKE, mitogen activated protein kinase.
Future Perspective
PRC1, TOP2A and CKAP2L may serve as biomarkers for the prognostic prediction of HCC patients in the future.
Data Sharing Statement
Publicly available datasets were analyzed in this study. This data can be found here: https://www.ncbi.nlm.nih.gov/geo/, http://gepia.cancer-pku.cn/ and https://www.proteinatlas.org/.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Ethical Disclosure
The authors state that they have obtained the Ethics Committee of Lishui Municipal Central Hospital approval and that this study conforms to the provisions of the Declaration of Helsinki (as revised in Seoul, Korea, October 2008).
Funding
The Major Research and Development Project of Lishui (2019ZDYF11), Chinese National Training Program of Innovation and Entrepreneurship for Undergraduates (No.202010343027) and Zhejiang Provincial Research Center for Cancer Intelligent Diagnosis and Molecular Technology (JBZX-202003). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Disclosure
The authors declare that they have no competing interests.
References
1. Siegel R, Miller K, Jemal AJ. Cancer statistics, 2016. CA Cancer J Clin. 2020;70(1):7–30. doi:10.3322/caac.21590
2. Bray F, Ferlay J, Soerjomataram I, Siegel R, Torre L, Jemal AJ. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
3. Wang W, Wei CJG. Advances in the early diagnosis of hepatocellular carcinoma. Genes Dis. 2020;7(3):308–319. doi:10.1016/j.gendis.2020.01.014
4. Yang N, Ekanem N, Sakyi C, Ray SJ. Hepatocellular carcinoma and microRNA: new perspectives on therapeutics and diagnostics. Adv Drug Deliv Rev. 2015;81:62–74. doi:10.1016/j.addr.2014.10.029
5. Bai K, He S, Shu L, et al. Identification of cancer stem cell characteristics in liver hepatocellular carcinoma by WGCNA analysis of transcriptome stemness index. Cancer Med. 2020;9(12):4290–4298. doi:10.1002/cam4.3047
6. Zhang Q, Wang J, Liu M, et al. Weighted correlation gene network analysis reveals a new stemness index-related survival model for prognostic prediction in hepatocellular carcinoma. Aging (Albany NY). 2020;12(13):13502–13517. doi:10.18632/aging.103454
7. Wei Z, Liu Y, Qiao S, et al. Identification of the potential therapeutic target gene UBE2C in human hepatocellular carcinoma: an investigation based on GEO and TCGA databases. Oncol Lett. 2019;17(6):5409–5418. doi:10.3892/ol.2019.10232
8. Ritchie M, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi:10.1093/nar/gkv007
9. Reiner-Benaim AJ. FDR control by the BH procedure for two-sided correlated tests with implications to gene expression data analysis. Biom J. 2007;49(1):107–126. doi:10.1002/bimj.200510313
10. Horvath S, Dong JJ, Miyano S. Geometric interpretation of gene coexpression network analysis. PLoS Comput Biol. 2008;4(8):e1000117. doi:10.1371/journal.pcbi.1000117
11. Xu N, Chen S, Lin T, et al. Development and validation of hub genes for lymph node metastasis in patients with prostate cancer. J Cell Mol Med. 2020;24(8):4402–4414. doi:10.1111/jcmm.15098
12. Tang D, Zhao X, Zhang L, Wang Z, Wang CJ. Identification of hub genes to regulate breast cancer metastasis to brain by bioinformatics analyses. J Cell Biochem. 2019;120(6):9522–9531. doi:10.1002/jcb.28228
13. Wang Y, Chen L, Wang G, et al. Fifteen hub genes associated with progression and prognosis of clear cell renal cell carcinoma identified by coexpression analysis. J Cell Physiol. 2019;234(7):10225–10237. doi:10.1002/jcp.27692
14. Jiang C, Wu S, Jiang L, et al. Network-based approach to identify biomarkers predicting response and prognosis for HER2-negative breast cancer treatment with taxane-anthracycline neoadjuvant chemotherapy. PeerJ. 2019;7:e7515. doi:10.7717/peerj.7515
15. Liu X, Huang Q, Yin X, et al. Strong correlation between the expression of CHEK1 and clinicopathological features of patients with multiple myeloma. Crit Rev Eukaryot Gene Expr. 2020;30(4):349–357. doi:10.1615/CritRevEukaryotGeneExpr.2020027084
16. Di Leo G, Sardanelli FJ. Statistical significance: p value, 0.05 threshold, and applications to radiomics-reasons for a conservative approach. European Radiology Experimental. 2020;4(1):18. doi:10.1186/s41747-020-0145-y
17. Chen L, Zhang Y, Wang S, Zhang Y, Huang T, Cai YJ. Prediction and analysis of essential genes using the enrichments of gene ontology and KEGG pathways. PLoS One. 2017;12(9):e0184129. doi:10.1371/journal.pone.0184129
18. Ma X, Zhou L, Zheng S. Transcriptome analysis revealed key prognostic genes and microRNAs in hepatocellular carcinoma. PeerJ. 2020;8:e8930. doi:10.7717/peerj.8930
19. Cui H, Shan H, Miao M, et al. Identification of the key genes and pathways involved in the tumorigenesis and prognosis of kidney renal clear cell carcinoma. Sci Rep. 2020;10(1):4271. doi:10.1038/s41598-020-61162-4
20. Gilberto S, Peter MJ. Dynamic ubiquitin signaling in cell cycle regulation. J Cell Biol. 2017;216(8):2259–2271. doi:10.1083/jcb.201703170
21. Zheng K, He Z, Kitazato K, Wang YJT. Selective autophagy regulates cell cycle in cancer therapy. Theranostics. 2019;9(1):104–125. doi:10.7150/thno.30308
22. Liu X, Li Y, Meng L, et al. Reducing protein regulator of cytokinesis 1 as a prospective therapy for hepatocellular carcinoma. Cell Death Dis. 2018;9(5):534. doi:10.1038/s41419-018-0555-4
23. Kang S, Chun TJE. Structural heterogeneity of the mammalian polycomb repressor complex in immune regulation. Exp Mol Med. 2020;52(7):1004–1015. doi:10.1038/s12276-020-0462-5
24. Ge Z, Leighton J, Wang Y, et al. Integrated genomic analysis of the ubiquitin pathway across cancer types. Cell Rep. 2018;23(1):213–226.e3. doi:10.1016/j.celrep.2018.03.047
25. Zhang W, Zhang J, Xu C, et al. Ubiquitin-specific protease 7 is a drug-able target that promotes hepatocellular carcinoma and chemoresistance. Cancer Cell Int. 2020;20:28. doi:10.1186/s12935-020-1109-2
26. Bufalieri F, Lospinoso Severini L, Caimano M, Infante P, Di Marcotullio LJC. DUBs activating the hedgehog signaling pathway: a promising therapeutic target in cancer. Cancers (Basel). 2020;12(6):1518. doi:10.3390/cancers12061518
27. Swan R, Poh L, Cowell I, Austin CJ. Small molecule inhibitors confirm ubiquitin-dependent removal of TOP2-DNA covalent complexes. Mol Pharmacol. 2020;98(3):222–233. doi:10.1124/mol.119.118893
28. Lyu W, Shu Y, Liu Y, Dong PJ. Topoisomerase II alpha promotes gallbladder cancer proliferation and metastasis through activating phosphatidylinositol 3-kinase/protein kinase B/mammalian target of rapamycin signaling pathway. Chin Med J (Engl). 2020;133:2321–2329. doi:10.1097/cm9.0000000000001075
29. Xiong G, Li L, Chen X, et al. Up-regulation of CKAP2L expression promotes lung adenocarcinoma invasion and is associated with poor prognosis. Onco Targets Ther. 2019;12:1171–1180. doi:10.2147/ott.S182242
30. Sui X, Kong N, Ye L, et al. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 2014;344(2):174–179. doi:10.1016/j.canlet.2013.11.019
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.