")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 13
Identification of the First PAX4-MODY Family Reported in Brazil
Authors Abreu GM , Soares CAPD , Tarantino RM , da Fonseca ACP , de Souza RB, Pereira MFC , Cabello PH , Rodacki M, Zajdenverg L , Zembrzuski VM , Campos Junior M
Received 4 April 2020
Accepted for publication 27 May 2020
Published 24 July 2020 Volume 2020:13 Pages 2623—2631
DOI https://doi.org/10.2147/DMSO.S256858
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Juei-Tang Cheng
Gabriella de Medeiros Abreu,1,* Camila de Almeida Pereira Dias Soares,1,* Roberta Magalhães Tarantino,2,3 Ana Carolina Proença da Fonseca,1 Ritiele Bastos de Souza,1 Maria de Fátima Carvalho Pereira,4 Pedro Hernan Cabello,1,5 Melanie Rodacki,2 Lenita Zajdenverg,2 Verônica Marques Zembrzuski,1 Mário Campos Junior1
1Human Genetics Laboratory, Oswaldo Cruz Institute, Oswaldo Cruz Foundation, Rio de Janeiro, Brazil; 2Diabetes and Nutrology Section, Internal Medicine Department, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil; 3Ambulatory of Diabetes, State Institute for Diabetes and Endocrinology Luiz Capriglione, Rio de Janeiro, Brazil; 4Clinical Pathology Department, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil; 5Laboratory of Genetics, School of Health Science, University of Grande Rio, Rio de Janeiro, Brazil
*These authors contributed equally to this work
Correspondence: Mário Campos Junior Junior
Human Genetics Laboratory,Oswaldo Cruz Institute, Oswaldo Cruz Foundation, Pavilhão Leônidas Deane, Sala 615, Avenida Brasil 4365, Rio de Janeiro 21040-360, RJ, Brazil
Tel +55 21 3865 8192
Email [email protected]
Purpose: The aim of this study was to sequence the coding region of the PAX4 gene in a Brazilian cohort with clinical manifestations of monogenic diabetes.
Patients and Methods: This study included 31 patients with autosomal dominant history of diabetes, age at diagnosis ≤ 40 years, BMI < 30 kg/m2, and no mutations in GCK or HNF1A, HNF4A, and HNF1B. Screening of the PAX4 coding region was performed by Sanger sequencing. In silico algorithms were used to assess the potential impact of amino acid substitutions on protein structure and function. Additionally, PAX4-MODY family members and 158 control subjects without diabetes were analyzed for the identified mutation.
Results: The molecular analysis of PAX4 has detected one missense mutation, p.Arg164Gln (c.491G>A), segregating with diabetes in a large Brazilian family. The mutation was absent among the control group. The index case is a woman diagnosed at 32 years of age with polyneuropathy and treated with insulin. She did not present diabetic renal disease or retinopathy. Family members with the PAX4 p.Arg164Gln mutation have a heterogeneous clinical manifestation and treatment response, with age at diagnosis ranging from 24 years to 50 years.
Conclusion: To the best of our knowledge, this is the first study to report a PAX4-MODY family in Brazil. The age of PAX4-MODY diagnosis in the Brazilian family seems to be higher than the classical criteria for MODY. Our results reinforce the importance of screening large monogenic diabetes families for the understanding of the clinical manifestations of rare forms of diabetes for the specific and personalized treatment.
Keywords: diabetes mellitus, monogenic diabetes, MODY, PAX4, mutation
Introduction
In the past years, mutations in genes that disrupt the secretion and signaling of insulin have been recognized as causative factors for monogenic forms of diabetes mellitus (DM). Among these genes, there are critical transcription factors, such as HNF4A,1 HNF1A,2 HNF1B,3 PDX1,4 NEUROD1,5 KLF11,6 and PAX4.7 The Paired Box Gene 4 (PAX4; OMIM*167413), also known as MODY9 gene, encodes a transcription factor that plays an important role in the development of β-cells and δ-cells. PAX4 acts in the differentiation of β-cells and δ-cells precursors in the early pancreas and latter maintaining β-cells in differentiated state.8 In vivo experiments demonstrated that newborn mice that are knockout for both Pax4 alleles exhibit growth retardation and dehydration, dying 3 days after birth.8 To date, several variants in the PAX4 gene have been associated with a number of DM types, including type 1 DM (T1D),9 type 2 DM (T2D),10 Ketosis-Prone Diabetes (KPD),11 as well as monogenic diabetes.7 Mutations associated with monogenic diabetes were first identified in two patients of Thai origin, who did not present mutations in the other known MODY genes.7 More than one decade after this initial report, the number of studies supporting the involvement of PAX4 mutations in monogenic diabetes remains limited to a few cases, and restricted to Asian populations.7,12,15 Due to its rarity, the clinical characteristics of PAX4-MODY remain unclear, compromising its diagnosis. In this context, the identification of new cases will be helpful to better understand the PAX4-MODY phenotype. This study aimed to screen the coding region of PAX4 gene in a sample of Brazilian patients with a clinical suspicion of monogenic diabetes. To the best of our knowledge, this is the first study to describe a PAX4 mutation in a large Brazilian family with autosomal dominant diabetes.
Patients and Methods
Subjects
In this cross-sectional observational study, 31 unrelated patients with DM (13 males and 18 females; average age at diagnosis: 19.7±10.9 years) were recruited from the Clementino Fraga Filho University Hospital and from the State Institute for Diabetes and Endocrinology Luiz Capriglione, Rio de Janeiro, Brazil. In this study, the inclusion criteria were as follows: 1) age at onset equal to or less than 40 years old; 2) a positive family history of diabetes in at least two generations; and 3) negative β-cells anti-GAD (Glutamic Acid Decarboxylase) and anti-IA-2 (Islet Antigen-2) autoantibodies. We excluded patients with T1D, obesity (Body Mass Index [BMI] ≥30 kg/m2 or ≥95th percentile for age at diagnosis), history of diabetic ketoacidosis at diabetes onset, clinical signs of insulin resistance, and the presence of secondary causes of diabetes. Clinical information was obtained through a review of the medical chart. All patients were previously screened for GCK or HNF1A (based on the clinical phenotype),16 HNF4A and HNF1B mutations and did not show mutations. Additionally, family members were screened for the novel variant, as well as 158 healthy controls (59 males and 99 females; average age: 32.03±8.41 years; BMI average: 22.48±1.40 kg/m2). The control group inclusion criteria were as follows: 1) fasting plasma glucose (FPG) <100 mg/dL and glycated hemoglobin (HbA1c) <5.7%; 2) BMI ≤24.9 kg/m2; and 3) Individuals without a family history of diabetes. The Ethics and Research Committee of the Clementino Fraga Filho University Hospital (CAAE n° 04232512.4.0000.5257) and of the State Institute for Diabetes and Endocrinology Luiz Capriglione (CAAE n° 04232512) approved this study protocol. All participants were informed about the aim of this study and provided verbal and written consent.
Molecular Genetics
Genomic DNA from the probands and nondiabetic controls were isolated from peripheral blood leukocytes using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany). The proband’s family had their genomic DNA extracted from buccal epithelial cells.17 Screening of the entire coding region and exon-intron boundaries of the PAX4 gene was done (Supplemental Table S1). PCR products were purified by ExoSAP-IT Reagent (Applied Biosystems, Vilnius, Lithuania). Sanger sequencing was performed using the Big Dye Terminator Kit v3.1 (Applied Biosystems, Austin, TX, USA), conducted on an ABI 3130 Automatic Genetic Analyzer (Applied Biosystems).
Bioinformatic Analysis
The PAX4 variants identified were checked against PubMed, Clinvar, dbSNP (https://www.ncbi.nlm.nih.gov/), Human Genome Mutation Database (HGMD) (http://www.hgmd.cf.ac.uk/ac/), ExAC Browser (http://exac.broadinstitute.org), 1000 Genomes project database (http://www.internationalgenome.org, and Online Archive of Brazilian Mutations (ABraOM; http://abraom.ib.usp.br/index.php),18 in order to investigate their previous occurrence in these public databases. To assess the potential impact of the missense mutations identified, in silico pathogenicity prediction algorithms were used, including SIFT,19 PolyPhen-2,20 PROVEAN,21 Revel,22 WS-SNPs&GO,23 MutPred,24 SNAP,25 Fathmm,26 M-CAP,27 CADD,28 Mutation assessor,29 Align-GVGD,30 PANTHER-PSEP,31 and Mutation Taster.32 The Ensembl reference transcript ENST00000341640.2 of PAX4 gene (Genome release GRCh37.p13) was used as reference (https://www.ensembl.org/index.html).
Results
In this study, we screened the entire coding region of the PAX4 gene in 31 unrelated probands from Brazil. The participants have clinical characteristics of monogenic diabetes. Our results showed a missense mutation p.Arg164Gln (c.491G>A) segregating with DM in a large Brazilian family. This variant was absent among the 158 normoglycemic controls analyzed and was not found in the ABraOM database. We also found the variant p.Arg133Trp (c.397C>T) in heterozygous state in three patients (9.67%) and in one homozygous patient (3.22%), and the common missense p.His321Pro (c.962A>C) variant in 28 probands (C allele frequency: 0.677). The synonymous p.Gln173Gln (c.519A>G) and p.Gly150Gly (c.450C>T) variants were found in five patients (16.12%) and in one patient (3.22%), respectively.
The arginine residue in position 164 of the PAX4 homeodomain is evolutionary conserved among several species (Figure 1). The change of the arginine amino acid to glutamine in the 164 position was predicted to be harmful by all 15 algorithms (Table 1). The arginine is an amino acid charged positively while glutamine belongs to uncharged polar side groups. This mutation was registered in dbSNP under the access number rs587780414; it was found with allele frequency of 0.00004119 in ExAC. However, we did not find any previously association of this mutation to DM (Table 2).
 |
Table 1 In silico Prediction of Missense Mutations Identified in PAX4 Gene |
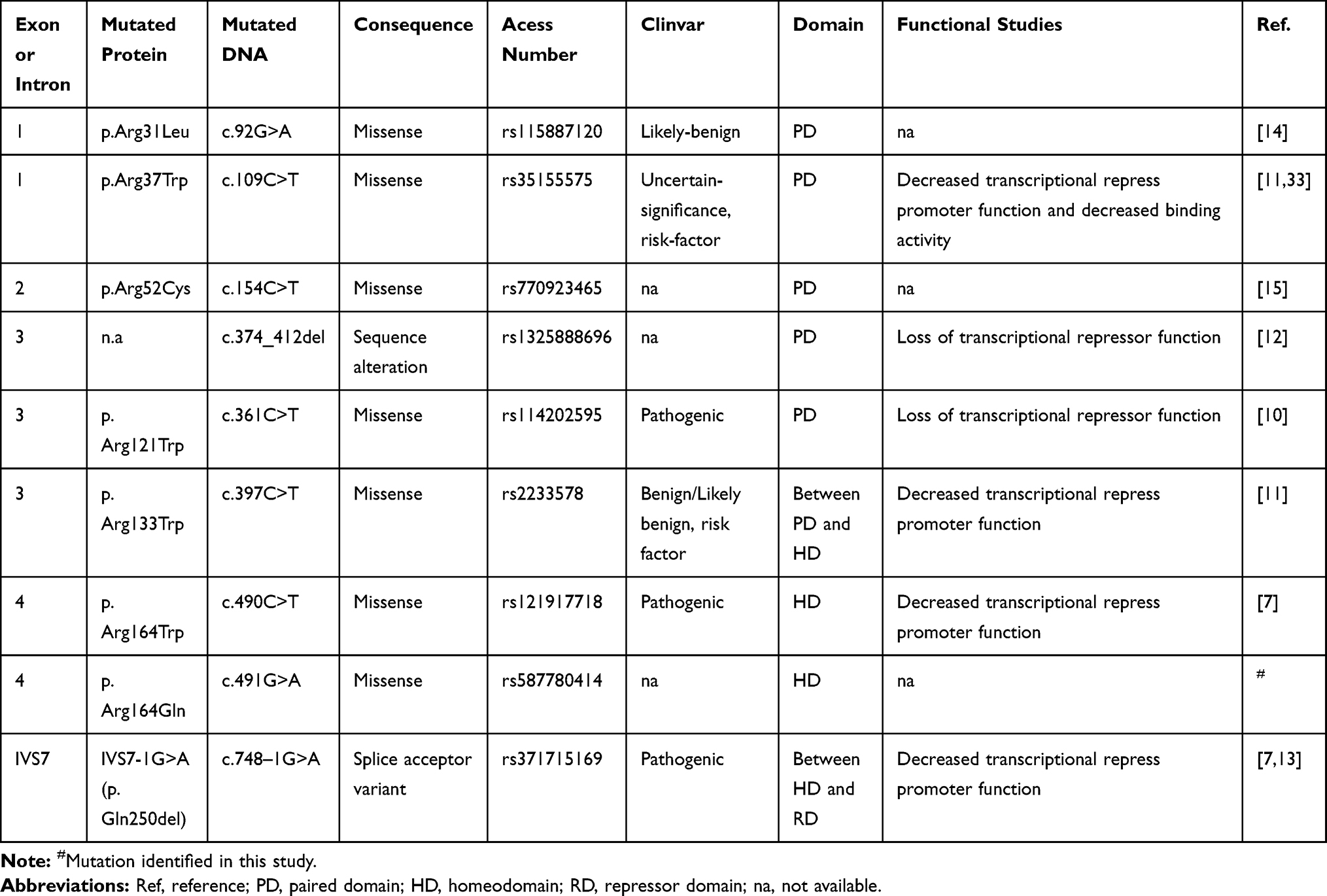 |
Table 2 Characterization of Mutations in PAX4 Gene Associated to Diabetes Mellitus |
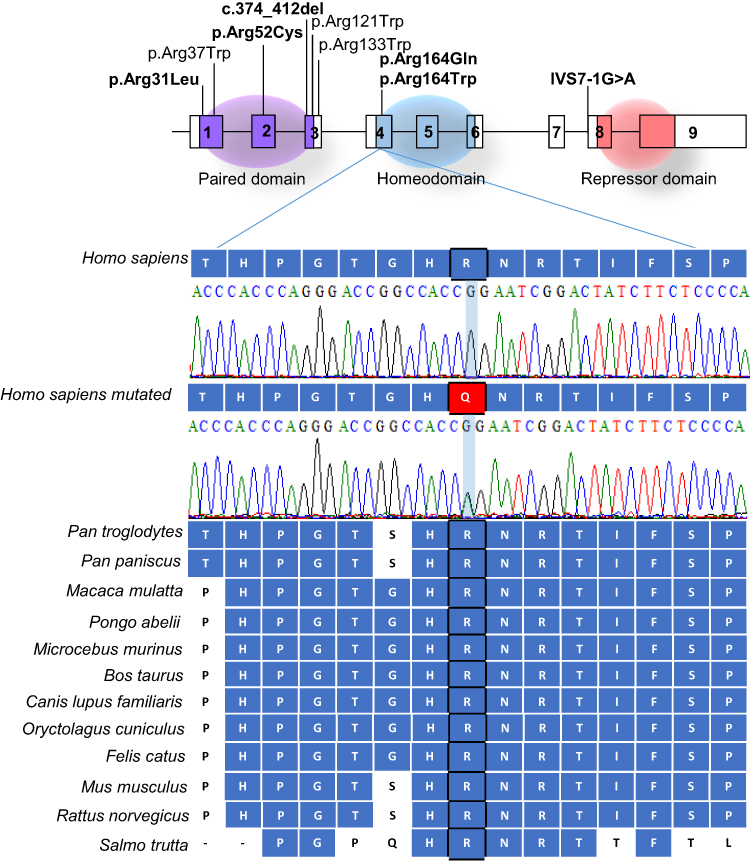 |
Figure 1 Diagrammatic representation of PAX4 gene and protein domains. Pathogenic mutations described associated to diabetes mellitus are pointed in the figure and PAX4-MODY are show in bold. Electropherograms of PAX4 exon 4 wild type and p.Arg164Gln (c.491G>A) in the patient DM35. Alignment by Clustal W (1.81) of PAX4 gene across species are presented (below). |
We identified the PAX4 p.Arg164Gln in the heterozygous state in a normal weight woman (BMI: 24.8 kg/m2; Current age: 45 years). She was diagnosed with diabetes during her second pregnancy at the age of 32 years (BMI at diagnosis: 21.68 kg/m2). She reported polyneuropathy and did not present diabetic renal disease or retinopathy until that moment. The patient was treated with insulin therapy since the diagnosis of DM. The family pedigree is shown in Figure 2 and clinical features are summarized in Table 3.
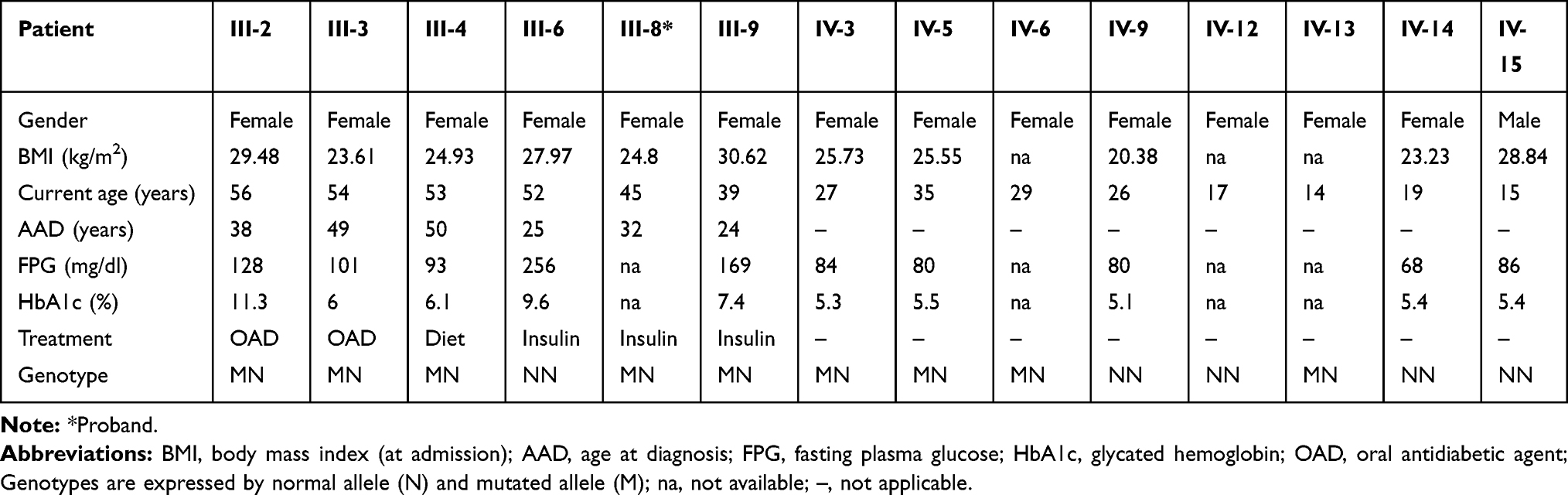 |
Table 3 Clinical Features and Laboratory Parameters of the Family 35 Members |
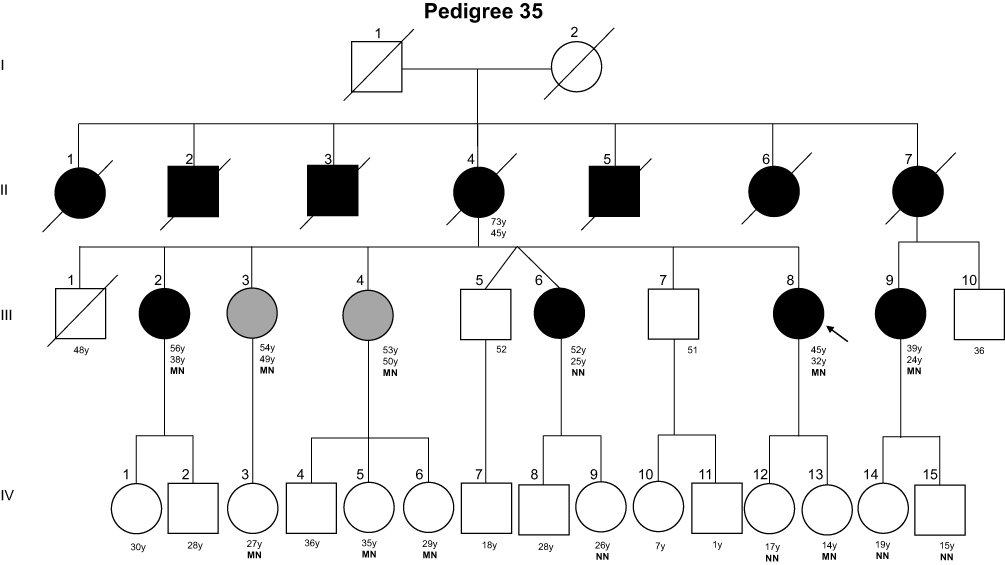 |
Figure 2 Pedigree of family 35. Filled black symbols, grey symbols, and empty symbols represent diabetic patients, impaired glucose tolerance individuals, and healthy individuals, respectively. The present age of the individuals are shown below the symbols in years (y), followed by age of diagnosis in years, and genotype. Genotypes are expressed by normal allele (N) and mutated allele (M); An arrow indicates the index case. |
The index-case’s mother (II-4) was diagnosed with DM at 45 years of age and died at 73 years with chronic kidney disease. The patient also reported three deceased uncles (individuals II-2, II-3, and II-5), three deceased aunts (individuals II-1, II-6, and II-7), and a cousin (individual III-9) with diabetes and four sisters with hyperglycemia (individuals III-2, III-3, III-4, and III-6). Thirteen family members were available for genetic testing and eight of them were found to be carrying the p.Arg164Gln, of which four exhibited hyperglycemia.
The proband’s older sister (individual III-2) is an overweight woman of 56 years old (BMI= 29.48 kg/m2; FPG= 128 mg/dl; HbA1c= 11.3%) diagnosed with DM at 38 years. She carried the mutation p.Arg164Gln in a heterozygous state. She has been on oral antidiabetic agents (OAD) treatment for 8 years (Metformin 1500 mg/day; Gliclazide 60 mg/day) and has hypertension. The carrier proband’s sister (individual III-3) is 49 years, non-obese (BMI= 23.61 kg/m2), and was diagnosed at 49 years with FPG of 104 mg/dL, glucose 2 hours post dextrose of 142 mg/dL, and HbA1c 6%. She is on Metformin 1000 mg/day. Like her, the carrier sister (individual III-4) was diagnosed with impaired glucose tolerance (IGT) at the age of 50 years and has been managed with nutritional therapy (FPG= 93 mg/dL; HbA1c= 6.1%). The sister with DM (individual III-6) did not present the mutation. She received the diagnosis in her second gestation at the age of 25 years old. She has been treated with fast-acting insulin analog. The family reported that the proband’s older brother (individual III-1) had schizophrenia and died at 48 years old due to a heart attack, and DM was not reported.
The proband’s cousin with DM (individual III-9) also presented the mutation tested (FPG=169 mg/dL; HbA1c= 7.4%) and received the diagnosis in her second gestation at 24 years. She has diabetic retinopathy. Her mother with DM (individual II-7) was diagnosed at 36 years in her second gestation and had been on dialysis before dying.
In the younger examined generation, all eight individuals do not have DM (individuals IV-3, IV-5, IV-6, IV-9, IV-12, IV-13, IV-14, and IV-15). Four of them presented the genetic variant, including three proband’s nieces (individuals IV-3, IV-5, and IV-6), of 27 years, 35 years, and 29 years old, respectively, and the proband’s younger daughter (individual IV-13), of 14 years old.
Discussion
Variants in the PAX4 gene have been associated to the risk of non-monogenic types of DM in the past years. However, a few mutations in this gene have also been described as the cause of monogenic diabetes (Table 4), and, to the best of our knowledge, this is the first monogenic diabetes case (PAX4-MODY) reported in a Brazilian family.
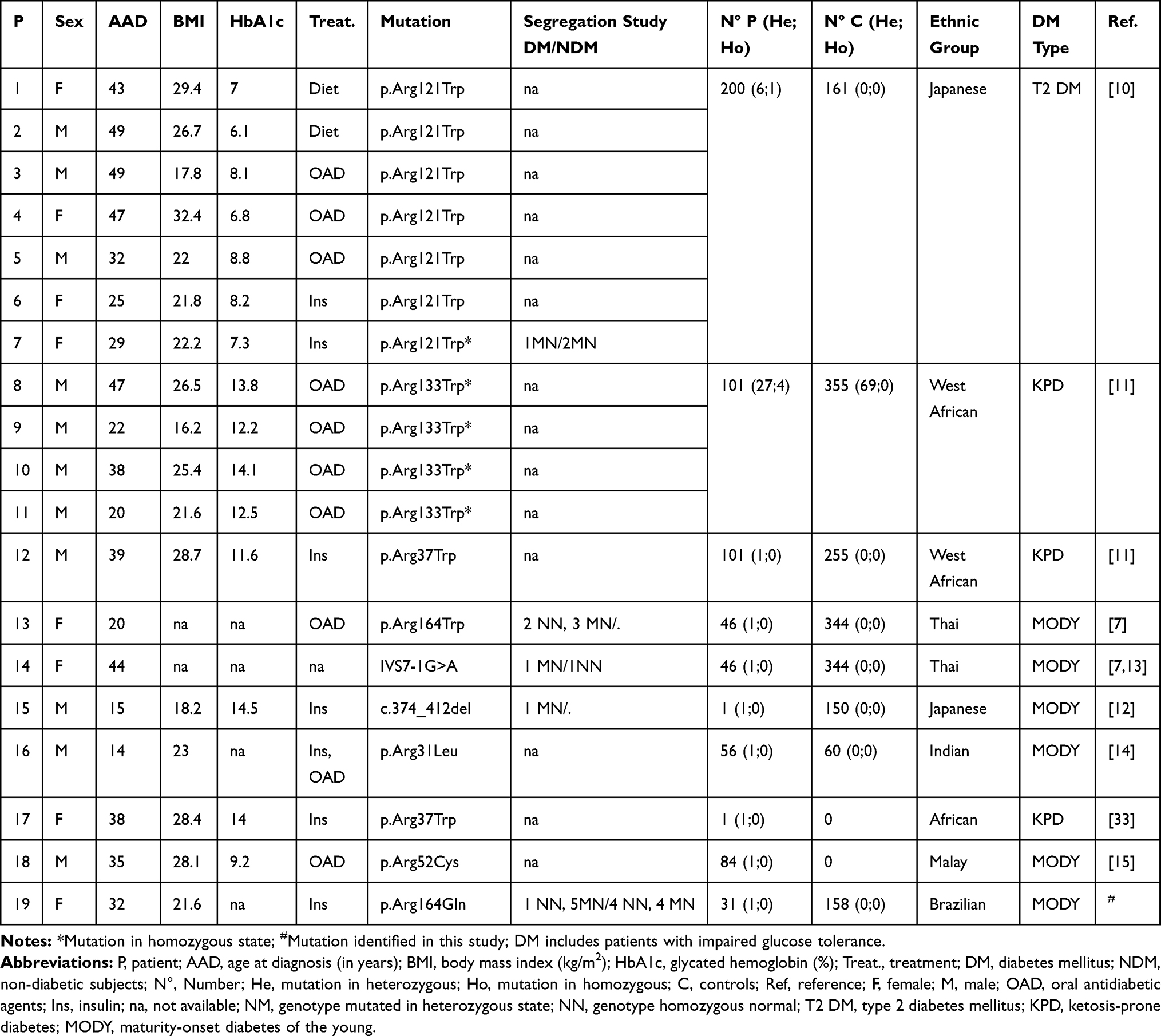 |
Table 4 Clinical Characteristics of Patients with DM with Mutations in PAX4 Gene |
Shimajiri et al10 described the p.Arg121Trp mutation in seven Japanese patients with T2D and absent among 161 controls (Table 4). One of these patients, a woman diagnosed at the age of 29 years, carried this variant in the homozygous state. The variant p.Arg121Trp segregated from her heterozygous parents, who were cousins, to the patient and to her heterozygous sister. Severe diabetes was presented only in the homozygous proband. In our sample, we identified the p.Arg133Trp in three patients in heterozygosis and in one patient in homozygosis. This variant was described as benign/risk factor by ClinVar and it was predicted to be benign by the majority of the in silico tools analyzed (Table 1). Mauvais-Jarvis et al11 previously reported an association of the p.Arg133Trp in homozygous state to ketosis-prone diabetes (KPD), a rare form of T2D. In vivo and in vitro studies showed that this variant alters the protein function (Table 2). They also observed the p.Arg37Trp mutation in a patient from Cameroon. This variant was later described co-segregating in a heterozygous form with BLK p.Phe112Ser (c.335C>T) in a Nigerian woman with KPD.33 Further case-control studies should be carried out to evaluate the association of these variants with different forms of diabetes.
Plengvidhya et al7 described in Thai families the first association of mutations in PAX4 to MODY diabetes. They observed one missense mutation, p.Arg164Trp, in the PAX4 homeodomain in a female patient diagnosed at the age of 20 years and treated with OAD. In vitro analysis showed that p.Arg164Trp decreased PAX4 repression activity. They also found an intronic variant (IVS7-1G>A) in one women with DM diagnosed at 44 years of age7 and after, in her daughter who was diagnosed at 30 years of age with gestational DM and required insulin treatment.13 Another four non-tested members from this family showed several complications, such as diabetic renal disease and retinopathy, and three of them died of end-stage renal failure.7,13 Similarly, two non-tested members from the Brazilian family reported in our study had diabetic end-stage renal disease (Figure 2; individuals II-4 and II-7); and one mutated patient (Figure 2; individual III-9) had diabetic retinopathy 15 years after disease onset. The guanine to adenine change in the last nucleotide of intron 7 (IVS7-1G>A) disrupts mRNA splicing and results in an in-frame deletion p.Gln250del (exon 8). Similar to p.Arg164Trp, the PAX4 p.Gln250del have its repressor activity of glucagon and insulin promoter impaired. Studies in vitro showed that this mutation increased susceptibility to apoptosis within high glucose condition.13
Jo et al12 found a frameshift deletion (c.374_412del) in a 15-year-old Japanese proband on insulin treatment. His father was diagnosed at 30 years old with T2D and had his glucose controlled only by nutritional therapy. This deletion leads to the loss of PAX4 homeodomain, decreasing its repression activity. Another two missense mutations, p.Arg31Leu14 and p.Arg52Cys,15 were found in an Indian and in a Malay patient, respectively. Both exhibited clinical hallmarks of monogenic diabetes.
Here, we report a rare missense mutation in the PAX4 gene, p.Arg164Gln, in a large Brazilian family. Interestingly, this mutation is located in the same residue of the first mutation described associated to PAX4-MODY in a Thai family by Plengvidhya et al.7 The age at diagnosis of the hyperglycemic members from the family described here ranged from 24 years to 50 years. Whereas in the Thai family described, members were treated with OAD or diet, in the Brazilian family the treatment was variable (Diet: 1; OAD: 2; Insulin: 3). In addition, two proband’s sisters presented impaired glucose tolerance; the same was observed in the proband’s brother from a Thai family. It seems that phenotypes can vary between affected members from the same family, from severe to mild clinical presentations, as also observed by other studies of PAX4-MODY families,7,12 imposing a challenge for establishing a clinical pattern for PAX4-MODY. The age at diagnosis observed in the patients with the p.Arg164Gln mutation from the Brazilian family was remarkably high. Among the five mutated patients from the third generation, three presented diabetes symptoms after 35 years of age; the age at diagnosis was higher than that expected for MODY most common forms. This late development could explain the absence of DM in the younger carrier individuals of this family. Unexpectedly, one sister with DM (Figure 2; individual III-6) did not show the mutation p.Arg164Gln. She reported weight gain at the time of diagnosis, which could represent a phenocopy of diabetes. This is similar to the two sisters described in the Thai family, who presented impaired glucose tolerance and did not carry the mutation.7
Our study has some limitations; the proband’s biochemical exams were not available and she abandoned treatment and medical care. We did not have access to the two brothers (Figure 2; individuals III-5 and III-7) and the cousin (Figure 2; individual III-10) without DM, which could reinforce the role of PAX4 p.Arg164Gln as the cause of DM in this family.
Until now, PAX4-MODY had been described only in families with Asian origins. To our knowledge, this is the first study to report a PAX4-MODY in a family in South America. Functional studies are needed to better understand the role of PAX4 p.Arg164Gln mutation in the cause of monogenic diabetes and its contribution to the clinical profile of PAX4-MODY patients.
Acknowledgments
The authors are grateful to the patients and their families and to the DNA Sequencing Platform from Oswaldo Cruz Institute (RPT01A). This work was supported by the National Council for Scientific and Technological Development (CNPq), Coordination for the Improvement of Higher Education Personnel (CAPES), Carlos Chagas Filho Rio de Janeiro State Research Foundation (FAPERJ), and Oswaldo Cruz Institute (IOC).
Disclosure
The authors declare no conflict of interest.
References
1. Yamagata K, Furuta H, Oda N, et al. Mutations in the hepatocyte nuclear factor-4α gene in maturity-onset diabetes of the young (MODY1). Nature. 1996;384(6608):458–460. doi:10.1038/384458a0
2. Yamagata K, Oda N, Kaisaki PJ, et al. Mutations in the hepatocyte nuclear factor-1α gene in maturity-onset diabetes of the young (MODY3). Nature. 1996;384(6608):455–458. doi:10.1038/384455a0
3. Horikawa Y, Iwasaki N, Hara M, et al. Mutation in hepatocyte nuclear factor–1β gene (TCF2) associated with MODY. Nat Genet. 1997;17(4):384–385. doi:10.1038/ng1297-384
4. Stoffers DA, Ferrer J, Clarke WL, Haneber JF. Early-onset type-II diabetes mellitus (MODY4) linked to IPF1. Nat Genet. 1997;17:138–139. doi:10.1038/ng0797-270
5. Malecki MT, Jhala US, Antonellis A, et al. Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nat Genet. 1999;23(3):323–328. doi:10.1038/15500
6. Neve B, Fernandez-Zapico ME, Ashkenazi-Katalan V, et al. From the cover: role of transcription factor KLF11 and its diabetes-associated gene variants in pancreatic beta cell function. Proc Natl Acad Sci. 2005;102(13):4807–4812. doi:10.1073/pnas.0409177102
7. Plengvidhya N, Kooptiwut S, Songtawee N, et al. PAX4 mutations in thais with maturity onset diabetes of the young. J Clin Endocrinol Metab. 2007;92(7):2821–2826. doi:10.1210/jc.2006-1927
8. Sosa-Pineda B, Chowdhury K, Torres M, Oliver G, Gruss P. The Pax4 gene is essential for differentiation of insulin-producing β cells in the mammalian pancreas. Nature. 1997;386(6623):399–402. doi:10.1038/386399a0
9. Biason-Lauber A, Boehm B, Lang-Muritano M. et al. Association of childhood type 1 diabetes mellitus with a variant of PAX4: possible link to beta cell regenerative capacity. Diabetologia. 2005;48(5):900–905. doi:10.1007/s00125-005-1723-5
10. Shimajiri Y, Sanke T, Furuta H, et al. A missense mutation of Pax4 gene (R121W) is associated with type 2 diabetes in Japanese. Diabetes. 2001;50(12):2864–2869. doi:10.2337/diabetes.50.12.2864
11. Mauvais-Jarvis F, Smith SB, Le May C, et al. PAX4 gene variations predispose to ketosis-prone diabetes. Hum Mol Genet. 2004;13(24):3151–3159. doi:10.1093/hmg/ddh341
12. Jo W, Endo M, Ishizu K, Nakamura A, Tajima T. A novel PAX4 mutation in a Japanese patient with maturity-onset diabetes of the young. Tohoku J Exp Med. 2011;223(2):113–118. doi:10.1620/tjem.223.113
13. Sujjitjoon J, Kooptiwut S, Chongjaroen N, Tangjittipokin W, Plengvidhya N, Yenchitsomanus PT. Aberrant mRNA splicing of paired box 4 (PAX4) IVS7-1G>A mutation causing maturity-onset diabetes of the young, type 9. Acta Diabetol. 2016;53(2):205–216. doi:10.1007/s00592-015-0760-x
14. Chapla A, Mruthyunjaya MD, Asha HS, et al. Maturity onset diabetes of the young in India - a distinctive mutation pattern identified through targeted next-generation sequencing. Clin Endocrinol (Oxf). 2015;82(4):533–542. doi:10.1111/cen.12541
15. Ang SF, Lim SC, Tan CS, et al. A preliminary study to evaluate the strategy of combining clinical criteria and next generation sequencing (NGS) for the identification of monogenic diabetes among multi-ethnic Asians. Diabetes Res Clin Pract. 2016;119:13–22. doi:10.1016/j.diabres.2016.06.008
16. Tarantino RM, Abreu GDM, Fonseca A, et al. MODY probability calculator for GCK and HNF1A screening in a multiethnic background population. Arch Endocrinol Metab. 2019;1(8):1–7. doi:10.20945/2359-3997000000173
17. Aidar M, Line SRP. A simple and cost-effective protocol for DNA isolation from buccal epithelial cells. Braz Dent J. 2007;18(2):148–152. doi:10.1590/S0103-64402007000200012
18. Naslavsky MS, Yamamoto GL, de Almeida TF, et al. Exomic variants of an elderly cohort of Brazilians in the ABraOM database. Hum Mutat. 2017;38(7):751–763. doi:10.1002/humu.23220
19. Sim N-L, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012;40(W1):W452–W457. doi:10.1093/nar/gks539
20. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi:10.1038/nmeth0410-248
21. Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31(16):2745–2747. doi:10.1093/bioinformatics/btv195
22. Ioannidis NM, Rothstein JH, Pejaver V, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. 2016;99(4):877–885. doi:10.1016/j.ajhg.2016.08.016
23. Capriotti E, Calabrese R, Fariselli P, Martelli P, Altman RB, Casadio R. WS-SNPs&GO: a web server for predicting the deleterious effect of human protein variants using functional annotation. BMC Genomics. 2013;14(Suppl 3):S6. doi:10.1186/1471-2164-14-S3-S6
24. Pejaver V, Urresti J, Lugo-Martinez J, et al. MutPred2: inferring the molecular and phenotypic impact of amino acid variants. bioRxiv. 2017:134981. doi:10.1101/134981.
25. Bromberg Y, Rost B. SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res. 2007;35(11):3823–3835. doi:10.1093/nar/gkm238
26. Shihab HA, Gough J, Cooper DN, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden markov models. Hum Mutat. 2013;34(1):57–65. doi:10.1002/humu.22225
27. Jagadeesh KA, Wenger AM, Berger MJ, et al. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet. 2016;48(12):1581–1586. doi:10.1038/ng.3703
28. Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47(D1):D886–D894. doi:10.1093/nar/gky1016
29. Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39(17):e118. doi:10.1093/nar/gkr407
30. Tavtigian SV, Byrnes GB, Goldgar DE, Thomas A. Classification of rare missense substitutions, using risk surfaces, with genetic- and molecular-epidemiology applications. Hum Mutat. 2008;29(11):1342–1354. doi:10.1002/humu.20896
31. Tang H, Thomas PD. PANTHER-PSEP: predicting disease-causing genetic variants using position-specific evolutionary preservation. Bioinformatics. 2016;32(14):2230–2232. doi:10.1093/bioinformatics/btw222
32. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361–362. doi:10.1038/nmeth.2890
33. Schmidt W, Lankers H. Co-inheritance of PAX4 and BLK mutations (MODY 7 and 9) in a 38 year old African Patient with Ketosis-Prone Diabetes. Presented at European Congress of Endocrinology ECE Munick, DEU. Endocrine abstract. 2016;41:EP401. doi:10.1530/endoabs.41.EP401
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.