")
Back to Journals » OncoTargets and Therapy » Volume 14
Identification of Ten Mitosis Genes Associated with Tamoxifen Resistance in Breast Cancer
Authors Sun X, Ding S, Lu S, Wang Z, Chen X , Shen K
Received 24 November 2020
Accepted for publication 10 May 2021
Published 4 June 2021 Volume 2021:14 Pages 3611—3624
DOI https://doi.org/10.2147/OTT.S290426
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjay Singh
Xi Sun, Shuning Ding, Shuangshuang Lu, Zheng Wang, Xiaosong Chen, Kunwei Shen
Department of General Surgery, Comprehensive Breast Health Center, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, 200025, People’s Republic of China
Correspondence: Kunwei Shen; Xiaosong Chen
Shanghai Jiao Tong University School of Medicine, No. 197, Rui Jin Er Road, Shanghai, 200025, People’s Republic of China
Tel +86-21-64370045-602102
Email [email protected]; [email protected]
Background: Endocrine therapy is the backbone therapy in estrogen receptor α (ER)-positive breast cancer, and tamoxifen resistance is a great challenge for endocrine therapy. Tamoxifen-resistant and sensitive samples from the international public repository, the Gene Expression Omnibus (GEO) database, were used to identify therapeutic biomarkers associated with tamoxifen resistance.
Materials and Methods: In this study, integrated analysis was used to identify tamoxifen resistance-associated genes. Differentially expressed genes (DEGs) were identified. Gene ontology and pathway analysis were then analyzed. Weighted correlation network analysis (WGCNA) was performed to find modules correlated with tamoxifen resistance. Protein–protein interaction (PPI) network was used to find hub genes. Genes of prognostic significance were further validated in another GEO dataset and cohort from Shanghai Ruijin Hospital using RT-PCR.
Results: A total of 441 genes were down-regulated and 123 genes were up-regulated in tamoxifen-resistant samples. Those up-regulated genes were mostly enriched in the cell cycle pathway. Then, WGCNA was performed, and the brown module was correlated with tamoxifen resistance. An overlap of 81 genes was identified between differentially expressed genes (DEGs) and genes in the brown module. These genes were also enriched in the cell cycle. Twelve hub genes were identified using PPI network, which were involved in the mitosis phase of the cell cycle. Finally, 10 of these 12 genes were validated to be up-regulated in tamoxifen-resistant patients and were associated with poor prognosis in ER-positive patients.
Conclusion: Our study suggested mitosis-related genes are mainly involved in tamoxifen resistance, and high expression of these genes could predict poor prognosis of patients receiving tamoxifen. These genes may be potential targets to improve efficacy of endocrine therapy in breast cancer, and inhibitors targeted these genes could be used in endocrine-resistant patients.
Keywords: breast cancer, tamoxifen resistance, WGCNA, PPI, mitosis
Introduction
As the most common cancer in the female, breast cancer is a great threat to world health.1 It is the most common cause of cancer death in developing countries and second to lung cancer in more developed countries.2 Breast cancer is a heterogeneous disease. Approximately 70% breast cancers are estrogen receptor α (ER)-positive.3 Endocrine therapy is used as the backbone therapy in ER-positive patients by blocking the ER pathway. Tamoxifen, a selective ER modulator, has dual agonistic/antagonistic effects on ER transcription, depending on its effect and location.4 Tamoxifen can cause cell cycle arrest in the G1 phase, inhibiting the proliferation and leading to apoptosis of breast cancer cells.5 In ER-positive patients, recurrence rates were reduced by almost 50% throughout the first 10 years, and the death rate was reduced by 25–30% after administration of 5-year tamoxifen.6,7 However, 30% patients who have taken tamoxifen for 5 years will have suffered from recurrence within 15 years. Therefore, finding new therapeutic biomarkers associated with tamoxifen resistance is important to overcome tamoxifen resistance.
Nowadays, gene sequencing has been widely used to identify biomarkers related to tumor biology.8,9 Large-scale sequencing made people have a better understanding of the heterogeneity, pathobiology and mechanism of cancers.3 As tamoxifen-resistant patient samples are difficult to obtain, no large-scale sequencing data about tamoxifen resistance have been systematically analyzed due to the limitation of sample size. However, high-throughput microarray and next-generation sequence datasets have been submitted by research groups to the international public repository, the Gene Expression Omnibus (GEO) database, and data in this database are freely available for integrated analysis.10 In our study, tamoxifen-resistant patients were derived from the GEO database in datasets GSE26971, GSE17705 and GSE45255, and integrated analysis was employed to have a better understanding of the mechanism of tamoxifen resistance.
Analysis of differentially expressed genes has mainly focused on the up-regulation and down-regulation of different genes, ignoring the interaction of different genes. Weighted gene co-expression network analysis (WGCNA) is a systems biology method that can be used to construct correlation networks and find modules of genes highly correlated to clinical traits.11,12 Candidate biomarkers or hub genes related to disease can be identified on the basis of correlation network.
In our study, 44 tamoxifen-resistant and 44 tamoxifen-sensitive patients who were matched with clinicopathological parameters were included. Differentially expressed genes (DEGs) were analyzed after normalization and gene ontology (GO) term enrichment analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis were performed.13,14 Modules correlated to tamoxifen resistance were identified using WGCNA. An interaction of genes in module and DEGs were selected as candidate genes. The protein–protein interaction (PPI) network was constructed using candidate genes, and hub genes were identified according to the degree in the network.15 Genes with prognostic significance were considered as important genes involved in tamoxifen resistance, and these genes were all involved in mitosis. This study sheds new light on the biological mechanisms of tamoxifen resistance and identifies new targets for tamoxifen-resistant patients.
Materials and Methods
Dataset
Microarray datasets GSE26971, GSE17705 and GSE45255 were downloaded from the National Center for Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/). These microarrays were all generated using an Affymetrix Human Genome U133A microarray (HG-U133A). Dataset GSE6532 generated using the Affymetrix Human Genome U133 Plus 2.0 microarray (HG-U133_Plus_2) was used as a validation dataset. Patients who had taken tamoxifen after resection and suffered from distant metastasis within 2 years were defined as tamoxifen-resistant patients according to the International Consensus Guidelines for Advanced Breast Cancer (ABC). Patients who had no recurrence were defined as tamoxifen-sensitive patients in our study. All paired tamoxifen-sensitive and resistant patients had been administered tamoxifen in our analysis. Then tamoxifen-sensitive patients were 1:1 matched with tamoxifen-resistant patients according to their age, chemotherapy and tumor stage. A total of 17 patients from GSE26971, 19 patients from GSE17705 and 8 patients from GSE45255 were included in the tamoxifen-resistant group.
Processing of Microarray Data
The “getGEOSuppFiles” function in the “GEOquery” package of R was used to identify the raw data, and “cel” style files were downloaded from the GEO database using the “GEOquery” package of R. Raw data were converted to expression data using the affy package. Background adjustment and normalization were done using the gcrma package. Batch effect was removed using the combat function in the sva package. DEGs were calculated using the limma package, and statistical significance was defined as adjusted P<0.05 and foldchange ≥1.5.
GO and KEGG Analyses
Gene ontology (GO) was used to annotate biological processes, molecular functions and cellular components of genes, and Kyoto Encyclopedia of Genes and Genomes (KEGG) was used to annotate the gene pathways. GO functional analysis and KEGG pathway analysis were both performed separately in up-regulated and down-regulated DEGs. The cluster profiler was used to analyze the functional annotation. We also performed enrichment analysis using the DAVID website (https://david.ncifcrf.gov/home.jsp). Adjusted P-value <0.05 was considered as a significant enrichment.
WGCNA Analysis
Weighted correlation network analysis (WGCNA) was performed to find modules of highly correlated genes using the WGCNA package. The “goodSamplesGenes” function in WGCNA package was used to check for missing values, and hierarchical cluster analysis was used to detect outliers, with cut height of 140. After checking data for missing values and identification of outlier microarray samples, an appropriate soft power was selected to meet the requirements of a scale-free network. Genes with a threshold of variation coefficient of expression >0.25 was used to perform WGCNA analysis. One-step network construction was used to construct networks, and modules were identified. Eigengenes were correlated with external traits to identify modules that were significantly associated with the measured clinical traits. A scatterplot of gene significance (GS) vs module membership (MM) in the brown module was plotted to show the correlation of GS and MM.
PPI Network Construction and Hub Gene Identification
STRING (http://string-db.org) is a database of known and predicted protein–protein interactions, and it was used to construct a PPI network. The Cytoscape software and cytoHubba app were then employed to analyze the interactive relationship of the candidate proteins. The cytoHubba app in Cytoscape was used to identify hub genes ranked by degree method to rank the top 20 hub genes.
Candidate Genes Validation and RT-PCR Detection
Ten pairs of matched tamoxifen-resistant and sensitive breast cancer tissues were obtained between January 2009 and December 2011 at the Comprehensive Breast Health Center, Shanghai Ruijin Hospital. As these involved human participants they were reviewed and approved by the independent Ethical Committees of Ruijin Hospital, Shanghai Jiaotong University School of Medicine.
TRIZOL reagent (TaKaRa, Kusatsu, Japan) was used for the isolation of total RNA, and 1000 ng RNA was reverse-transcribed into cDNA using Primescript RT Reagent (TaKaRa, Japan). RT-PCR was performed using FastStart Universal SYBR Green Master (Roche, Switzerland) in a real-time PCR instrument (Applied Biosystems, USA), and β-actin was used as endogenous control. The primers are listed in Supplementary Table 1.
Cell Culture
The MCF-7 cell line was obtained from the American Type Culture Collection (ATCC, USA) and was cultured in Roswell Park Memorial Institute 1640 (RPMI-1640, Thermo Fisher, USA), supplemented with 10% fetal bovine serum and 100 μg/mL penicillin-streptomycin (Hyclone, USA).
Lentivirus Preparation and Infection
We used pLKO.1 (Addgene Plasmid, 10878) to generate lentiviral shRNA plasmids; shRNA plasmids targeting MELK, RACGAP1 and MAD2L1 were constructed. The sequence of shRNAs used were: MELK, 5ʹ-GACAUCCUAUCUAGCUGCA-3ʹ; MAD2L1, 5ʹ-CUACUGAUCUUGAGCUCAU-3ʹ; RACGAP1, 5ʹ-CAACUAAGCGAGGAGCAAATT-3ʹ. Lentivirus was generated by transfection of HEK293T cells with packaging vectors (pMD2.G and psPAX) and transducing vector and concentrated with PEG6000 (Sigma, USA). Forty-eight hours post-infection, puromycin (1 μg/mL; 60210ES25, YEASEN) was used to select positively infected cells.
Colony Formation Assay
Cells transfected with scramble or shRNA were cultured into 6-well plates with 5000 cells and cultured for 15 days. After seeding cells for 24 hours, tamoxifen with concentration of 2 μm/mL was added into every well for 15 days.
The colonies were fixed in 75% absolute ethanol for 10 minutes, washed twice with PBS and stained with Giemsa (Sigma, USA) for 15 min, then dried at room temperature. The colonies containing 50 or more cells in each well were counted.
Cell Viability Analysis
Cells were plated into 96-well in triplicates and then treated with tamoxifen of different concentrations. Cell viability was measured using Cell Titer-Glo 2.0 Assay (G9243, Promega), and data were collected on Synergy H4 Hybrid Reader (BioTek).
Survival Analysis
The key genes were identified as the intersecting genes of the brown module and DEGs. ER-positive patients treated with tamoxifen in three datasets with disease metastasis survival within five years were divided into two groups according to the medium expression of key genes. Kaplan–Meier (K-M) plot was plotted using survival package.
Results
Identification of DEGs in Tamoxifen-Resistant Patients
The DEGs of tamoxifen-resistant patients were analyzed, and a total of 564 DEGs were identified, including 441 down-regulated genes and 123 up-regulated genes compared to tamoxifen-sensitive patients (Figure 1A). DEGs are also listed in a volcano plot (Figure 1B). In KEGG pathway analysis (Figure 1C), the up-regulated genes are significantly enriched in pathways including cell cycle, cellular senescence and human T-cell leukemia virus 1 infection. The down-regulated genes are mainly enriched in complement and coagulation cascades, PPAR signaling pathway and arachidonic acid metabolism. In GO functional analysis (Figure 1D), these up-regulated genes are mainly enriched in terms including tubulin binding, microtubule binding and ATPase activity. The down-regulated DEGs are enriched in enzyme inhibitor activity and glycosaminoglycan binding.
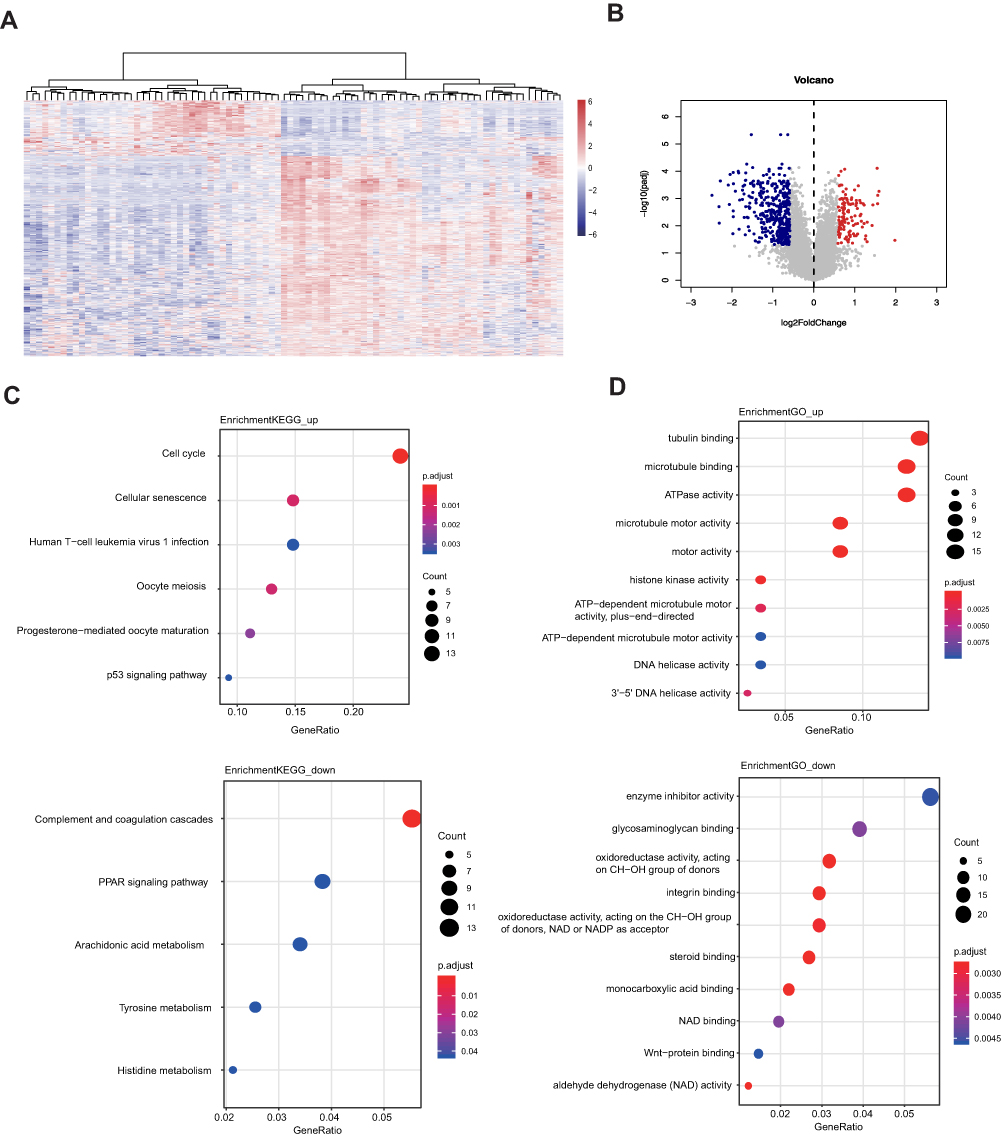 |
Figure 1 Differentially expressed genes identified in tamoxifen-resistant and sensitive patients. (A) Heatmap and (B) volcano plot of differentially expressed genes in primary tamoxifen-resistant and sensitive patients. (C) KEGG pathway analysis of up-regulated and down-regulated DEGs. (D) GO functional analysis of up-regulated and down-regulated DEGs. |
Identification of Key Modules Using WGCNA Analysis
After excluding outliers, WGCNA analysis was performed to identify key modules correlated with tamoxifen resistance (Supplementary Figure 1). Soft threshold power was set to 6 to ensure a scale-free network (Figure 2). A total of 37 clusters were identified based on the criteria of a cut height = 0.25 and a minimum of 30 genes (Figure 3). To identify modules correlated to tamoxifen resistance, the module–trait relationship was analyzed, and the brown module with 576 genes was significantly related to tamoxifen resistance (Figure 4A). A scatterplot of module membership and gene significance indicates significant correlation between the brown module and tamoxifen resistance (Figure 4B).
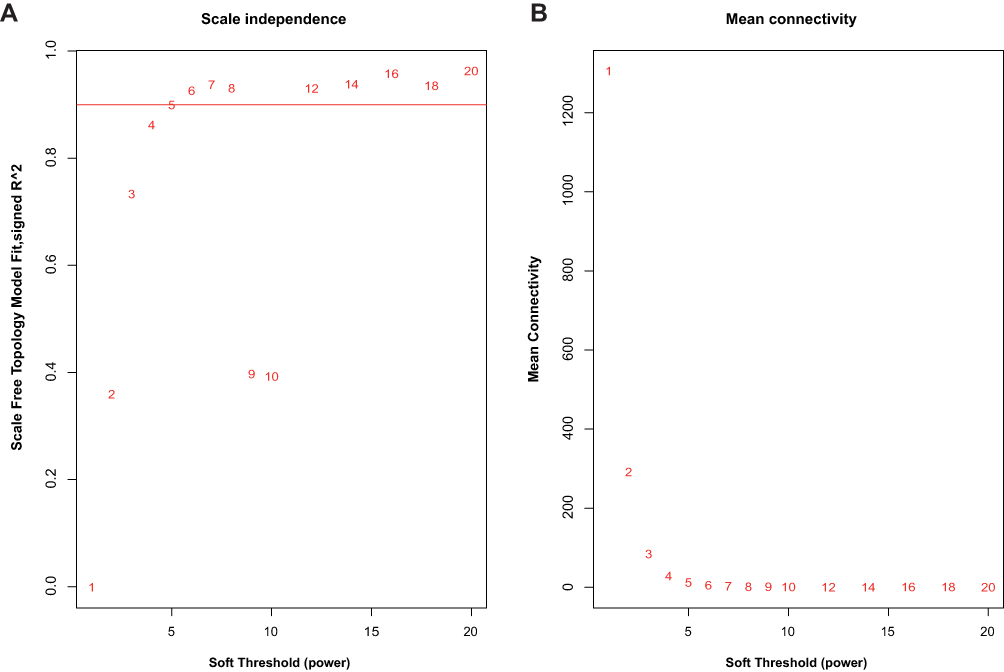 |
Figure 2 Soft threshold power identified in the weighted gene co-expression network analysis (WGCNA). (A) Analysis of scale-free fit index for various soft threshold powers. (B) Analysis of mean connectivity for various soft threshold powers. |
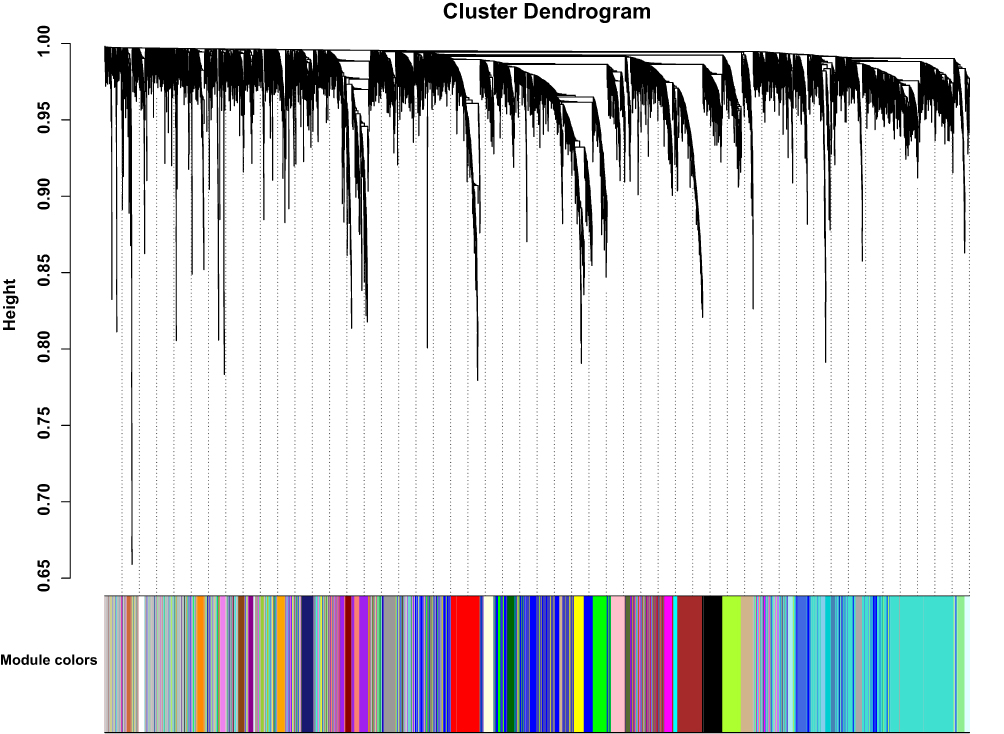 |
Figure 3 Co-expression network modules for mRNA. |
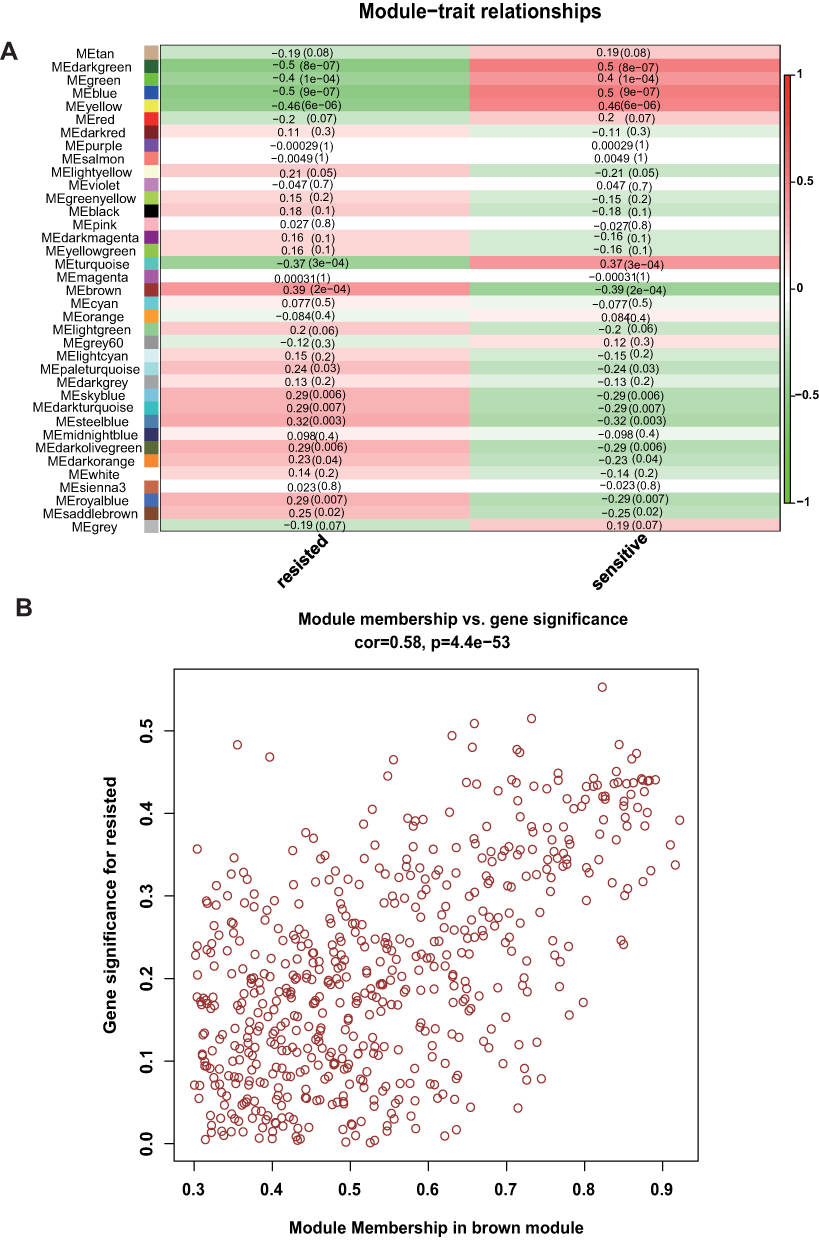 |
Figure 4 The brown module was identified with tamoxifen resistance. (A) Heatmap of trait correlation with tamoxifen resistance. The brown module was highly correlated with tamoxifen resistance. (B) A scatter plot of module membership in the brown module and gene significance. |
Key Genes Identified with Tamoxifen Resistance
To identify genes mostly related to tamoxifen resistance, genes in the brown module and DEGs were overlapped and a total of 81 genes were identified (Figure 5A). To explore the main function of these genes, KEGG pathway analysis was used, and pathways including cell cycle, oocyte meiosis and cellular senescence were enriched in these genes (Figure 5B). KEGG using DAVID was also calculated, and cell cycle was still the top pathway (Supplementary Figure 2). The enrichment analysis was similar to that in up-regulated genes of Figure 1C, indicating that those up-regulated genes in these pathways play an important role in tamoxifen resistance.
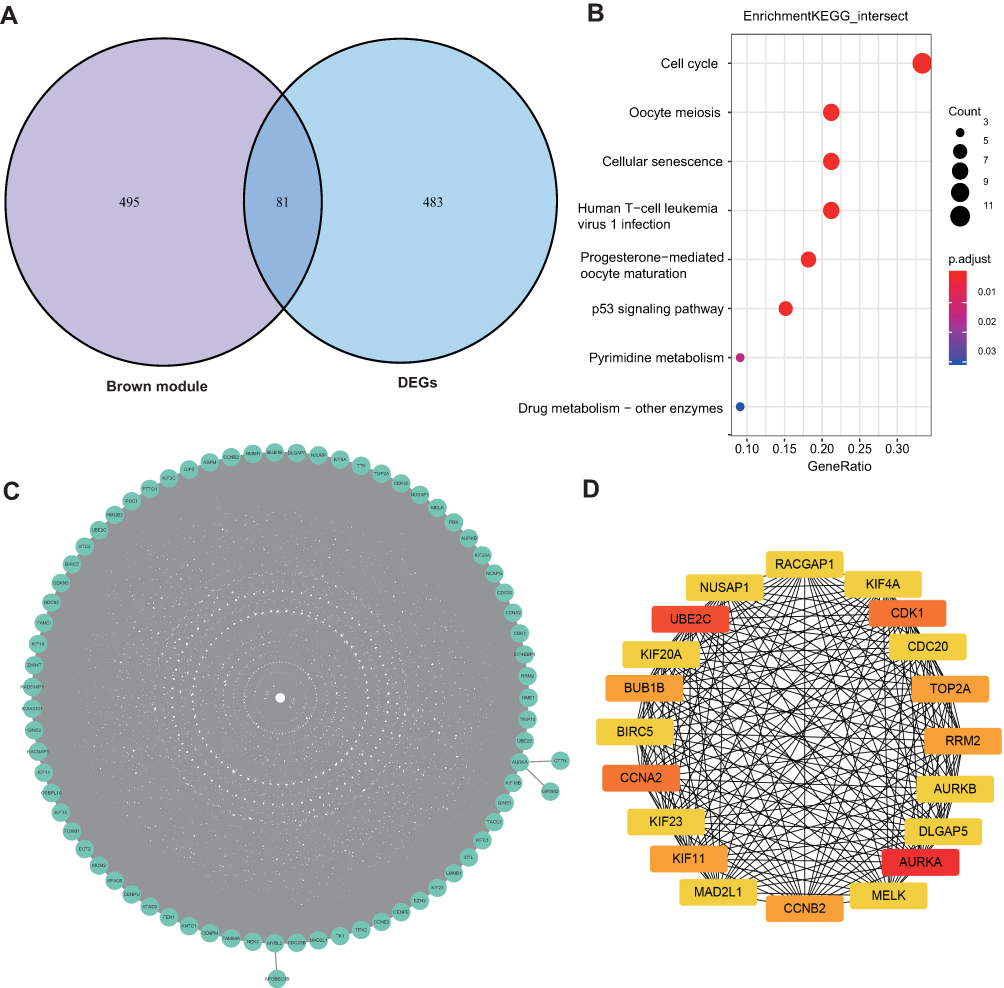 |
Figure 5 Hub genes identified with tamoxifen resistance. (A) Venn plot of DEGs and genes in the brown module. A total of 81 genes overlapped. (B) KEGG pathway analysis of the 81 overlapped genes. (C) Protein–protein interaction network of the 81 overlapped genes. (D) Protein–protein interaction network of 21 hub genes. The intensity of red indicates the scores of degrees. |
A PPI network was constructed to investigate the interaction between these 81 genes, which consists of 81 nodes and 1941 edges (Figure 5C). To explore hub genes in this network, cytoHubba was used and the top 20 genes were identified according to the degree in the network. AURKA, UBE2C, CCNA2, CDK1, KIF11, RRM2, TOP2A, BUB1B, CCNB2, MELK, BIRC5, NUSAP1, CDC20, KIF20A, KIF4A, MAD2L1, AURKB, DLGAP5, RACGAP1 and KIF23 were the top 20 hub genes (Figure 5D), which were all up-regulated in tamoxifen-resistant patients (Table 1). Survival analysis found that high expression of 12 genes (AURKA, BIRC5, CCNA2, CCNB2, DLGAP5, KIF4A, KIF20A, KIF23, MELK, MAD2L1, RACGAP1, UBE2C) was associated with worse survival when patients were treated with tamoxifen (Figure 6). All these 12 mitotic genes have a prognostic role for ER-positive breast cancer using Kaplan–Meier plotter website (Supplementary Figures 3 and 4).
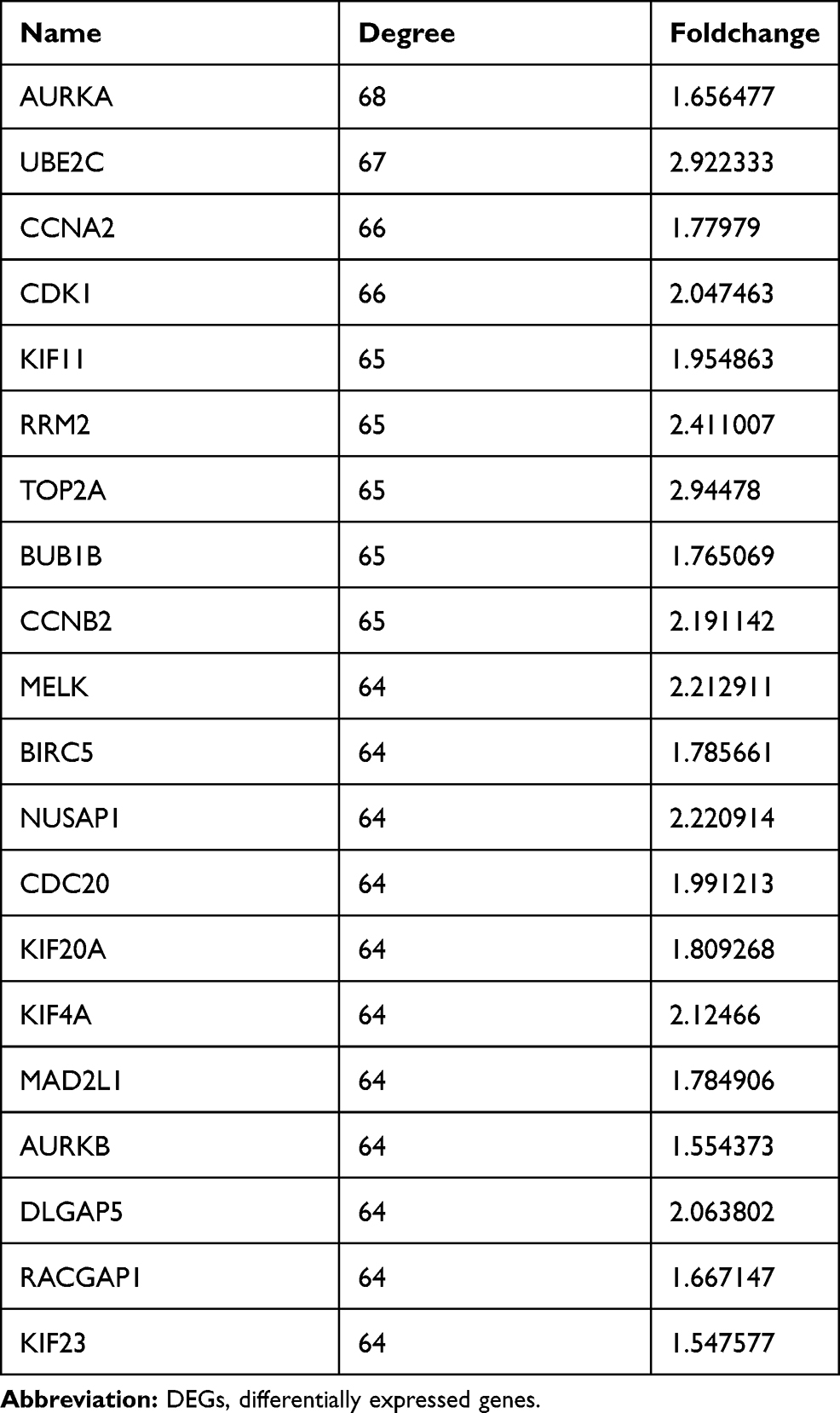 |
Table 1 Top 20 Genes in PPI Network Ranked by Degree Method and Foldchange in DEGs |
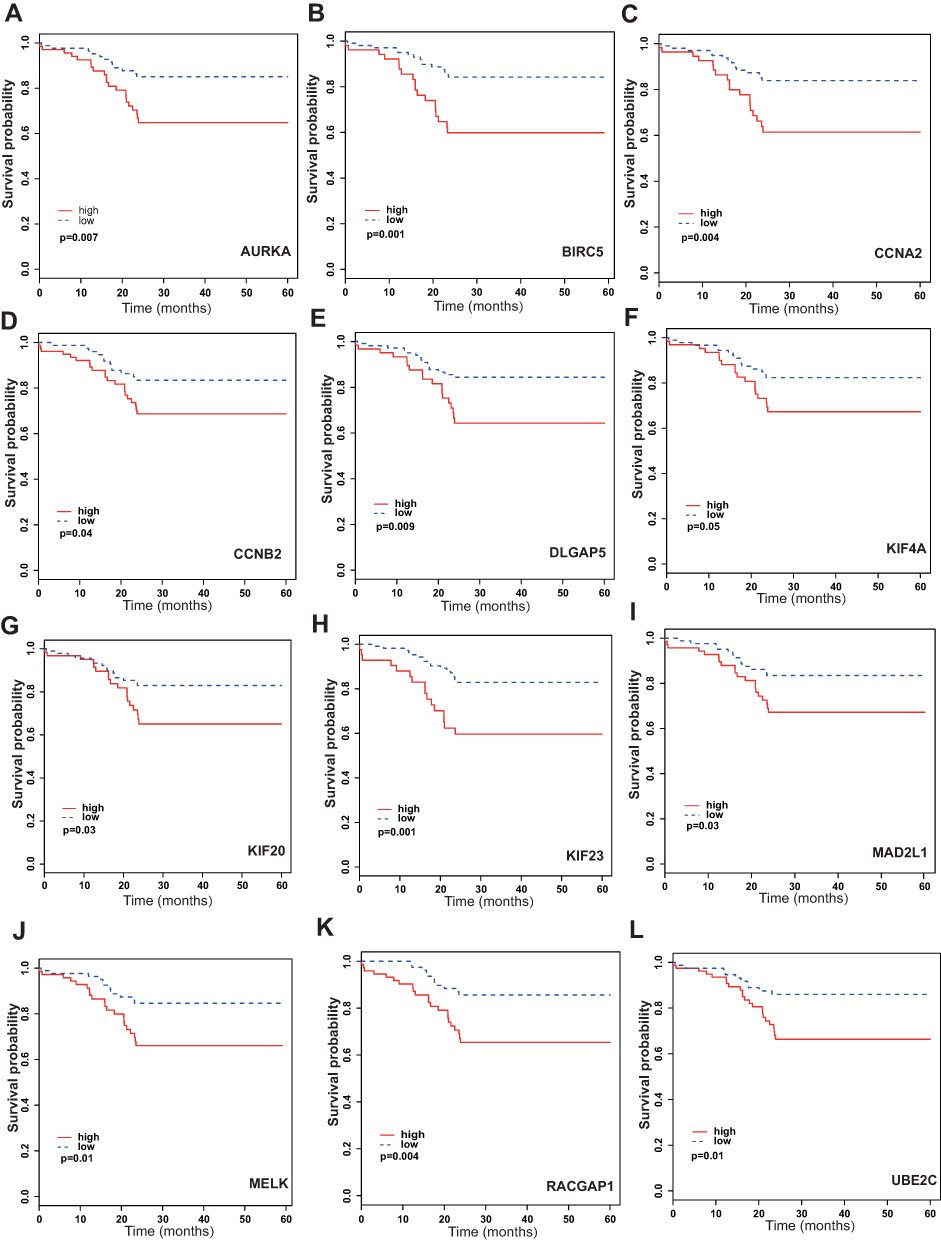 |
Figure 6 Survival analysis of 12 significant genes for disease-free survival. (A) K-M plot of AURKA for patients treated with tamoxifen. (B) K-M plot of BIRC5 for patients treated with tamoxifen. (C) K-M plot of CCNA2 for patients treated with tamoxifen. (D) K-M plot of CCNB2 for patients treated with tamoxifen. (E) K-M plot of DLGAP5 for patients treated with tamoxifen. (F) K-M plot of KIF4A for patients treated with tamoxifen. (G) K-M plot of KIF20 for patients treated with tamoxifen. (H) K-M plot of KIF23 for patients treated with tamoxifen. (I) K-M plot of MAD2L1 for patients treated with tamoxifen. (J) K-M plot of MELK for patients treated with tamoxifen. (K) K-M plot of RACGAP1 for patients treated with tamoxifen. (L) K-M plot of UBE2C for patients treated with tamoxifen. |
Furthermore, these 12 genes were validated in the GSE6532, and a total of 10 genes were up-regulated in tamoxifen-resistant patients (Figure 7A).
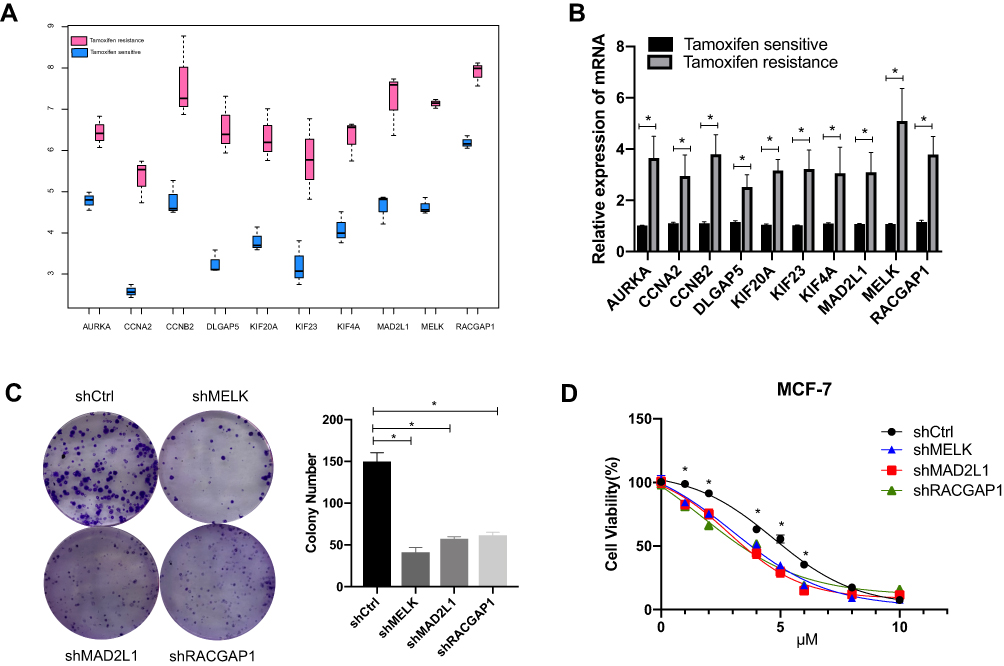 |
Figure 7 Validation of ten genes in public database and samples from our own database. (A) Validation of significant genes in GSE6532. All genes are statistically significant. (B) Validation of significant genes in 10 pairs of patient samples in Ruijin cohort. (C) Colony formation assay for cells with knockdown of MELK, MAD2L1 and RACGAP1 treated with tamoxifen. (D) Cell viability assay for cells with knockdown of MELK, MAD2L1 and RACGAP1 treated with tamoxifen. *P<0.05. |
Validation of Candidate Genes for Tamoxifen Resistance
Ten of these genes (AURKA, CCNA2, CCNB2, DLGAP5, KIF4A, KIF20A, KIF23, MELK, MAD2L1, RACGAP1) were then further validated in 10 paired tamoxifen-resistant and sensitive breast cancer patients from the Ruijin cohort. Tamoxifen-resistant patients were all resisted, and their clinical characteristics were matched. Detailed characteristics for these patients are listed in Supplementary Table 2. RT-PCR results demonstrated that these ten genes were all over-expressed in tamoxifen-resistant patients, and expression of these ten genes in tamoxifen-resistant patients ranges from 2.2- to 4.7-fold that of tamoxifen-sensitive patients (P<0.05, Figure 7B).
To validate the function of these genes in tamoxifen resistance, MELK, RACGAP1 and MAD2L1 were knocked down in MCF-7 cells because function of these genes in tamoxifen resistance was not reported while other genes were reported. As shown in colony formation assay and cell viability assay, knocking down these genes sensitized cells to tamoxifen compared with control (Figure 7C and D). This validated the robustness of our analysis.
Discussion
In our study, we collected data from the GEO database, and an integrated analysis was used to identify genes associated with tamoxifen resistance in ER-positive breast cancer patients. Analysis of differentially expressed genes ignores correlation between different genes. In our study, we combined WGCNA with DEGs to identify differentially expressed genes with high correlation. The interaction of different proteins is an important mechanism to regulate cell biology. So, PPI network identified hub genes which were mostly engaged in tamoxifen resistance.
A total of 564 DEGs were identified, including 441 down-regulated genes and 123 up-regulated genes. The up-regulated genes were enriched in the cell cycle pathway, which is the most common pathway studied in tamoxifen resistance. The brown module with 576 genes was identified using WGCNA. An overlap of genes between DEGs and the brown module was also enriched in the cell cycle. A total of 10 mitosis genes with prognostic significance were identified as hub genes associated with tamoxifen resistance and were validated in the public database and in our cohort samples.
Studies of tamoxifen resistance mainly focus on acquired resistance due to limitation of sample size for tamoxifen resistance.16,17 According to the ESMO International Consensus Guidelines for Advanced Breast Cancer (ABC), acquired resistance was defined as patients who relapse while on adjuvant endocrine therapy (ET) but after the first 2 years, or relapse within 12 months of completing adjuvant ET, or have progression disease (PD) ≥6 months after initiating ET for ABC, while on ET. In our study, primary tamoxifen-resistant patients were studied using data from the public repository, and the mitosis phase of the cell cycle was identified as the main pathway. The most well-known mechanism of tamoxifen resistance is the mutation of ESR1.18 And recently, CDK4/6 inhibitors were combined with endocrine therapy to overcome endocrine resistance.19,20 In MONALEESA-7, 26.3% pre- and perimenopausal patients received tamoxifen. Patients receiving tamoxifen and ribociclib had longer median progression-free survival of 22.1 months (95% CI 16.6–24.7) than patients in the placebo group who had progression-free survival of 11.0 months (9.1–16.4) (HR 0.59, 95% CI 0.39–0.88). In our analysis, we illustrated the role of cell cycle-related genes in tamoxifen resistance, and these genes may serve as targets to overcome tamoxifen resistance. Except genes regulating G1/S transition like CCNE2 (Figure 5C), hub genes were all involved in mitosis. CCNE2 is a regulatory subunit of cyclin-dependent kinase 2. It interacts with CDK2 and forms a catalytically active kinase complex.21 This complex phosphorylates histone H1 and Rb and promotes the transition of G1/S. It has been reported that cell cycle and mitosis genes were involved in tamoxifen insensitivity and suppression of these genes could re-inhibit growth.22,23 Our study further indicated that therapy targeting mitosis is a potential strategy in overcoming tamoxifen resistance. As endocrine therapy is a standard adjuvant therapy of ER-positive breast cancer patients and there was no dataset with patients both receiving and not receiving tamoxifen, we cannot evaluate the interaction of genes regarding prognosis and efficacy to tamoxifen. When we assessed the prognostic role of these mitotic genes in ER breast cancers, high expression of these genes indicated poor survival for ER breast cancers though the treatment information was unknown.
AURKA, Aurora kinase A, controls many processes of the G2/M transition.24–26 AURKA was identified as a marker for endocrine resistance in early estrogen receptor-positive breast cancer, and knockdown of AURKA made tamoxifen-resistant cells re-sensitized to tamoxifen treatment.27 CCNA2 is highly expressed from S phase to early mitosis and binds to CDK1 during the transition from G2 to M phase.28 It was overexpressed in tamoxifen-resistant cells. CCNB2 levels gradually increase during S and G2 phase and peak at mitosis.29,30 It was reported that ER-positive patients who have high expression of CCNA2 or CCNB2 have inferior survival.31,32 KIF4A, KIF20A and KIF23 are all mitotic kinesins and have a highly conserved motor domain involving ATP-binding and microtubule-binding sequences.33 They all promote the proliferation of breast cancers, and treatment with tamoxifen reduced the expression of these three proteins. Knockout of KIF4A re-sensitized cancer cells to tamoxifen.34 Maternal embryonic leucine zipper kinase (MELK) is a member of both the sucrose-non-fermenting (snf)1 and the AMP-activated protein kinase (AMPK) families.35 It has been reported that MELK is overexpressed in many cancers, and high expression of this gene correlated with a poor survival.35,36 MAD2L1 is a key protein in mediating spindle checkpoint activation.37,38 The disks large-associated protein 5 (DLGAP5), a microtubule-associated protein, is responsible for stabilizing and correct formation of microtubules, and the bipolar arrays of dynamic microtubules are critical in forming the mitotic spindle. It is overexpressed in many cancers and indicates a poor prognosis in these cancers.39–41 Rac GTPase-activating protein 1 (RACGAP1) links the mitotic spindle to the plasma membrane to secure the final cut during cytokinesis in animal cells. RACGAP1 was identified as an oncogene in many cancers.42,43 The role of MAD2L1, DLGAP5 and RACGAP1 in tamoxifen resistance was validated in our study, and knockdown of these genes sensitizes breast cancer to tamoxifen.
Conclusion
In conclusion, this present study provides evidence that mitosis genes contributed to tamoxifen resistance. We concluded that ten mitosis genes showed high expression in tamoxifen-resistant patients and lead to poor analysis in patients receiving tamoxifen and in ER-positive breast cancer patients. Knockdown of three genes (MAD2L1, DLGAP5, RACGAP1) makes cells more sensitive to tamoxifen. Our results indicates that mitosis is an important biological process of tamoxifen resistance, and mitotic genes may serve as potential targets to overcome tamoxifen resistance.
Abbreviations
ER, estrogen receptor α; GEO, Gene Expression Omnibus; DEGs, differentially expressed genes; WGCNA, weighted correlation network analysis; PPI, protein–protein interaction; GO, gene ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; HG-U133A, human genome U133A microarray; GS, gene significance; MM, module membership; K-M, Kaplan–Meier; AURKA, Aurora kinase A; MELK, maternal embryonic leucine zipper kinase; AMPK, AMP-activated protein kinase; DLGAP5, disks large-associated protein 5; RACGAP1, Rac GTPase-activating protein 1.
Ethics Approval and Informed Consent
The investigations involving human participants were reviewed and approved by the independent Ethical Committees of Ruijin Hospital, Shanghai Jiaotong University School of Medicine and all subjects gave written informed consent. The ethics number for our analysis is 2020-309. This study was conducted in accordance with the Declaration of Helsinki.
Author Contributions
XS participated in all experimental work and drafted the paper. XC and KS designed the article. SD, ZW and SL collected the data. All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
The authors appreciate the financial supported by the National Natural Science Foundation of China (Grant Number: 81772797), Shanghai Municipal Education Commission-Gaofeng Clinical Medicine Grant Support (20172007); Ruijin Hospital, Shanghai Jiao Tong University School of Medicine-“Guangci Excellent Youth Training Program” (GCQN-2017-A18). All these financial sponsors had no role in the study design, collection, analysis or interpretation of data.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. doi:10.3322/caac.21387
2. Harbeck N, Gnant M. Breast cancer. Lancet. 2017;389(10074):1134–1150. doi:10.1016/S0140-6736(16)31891-8
3. Sorlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98(19):10869–10874. doi:10.1073/pnas.191367098
4. Yang G, Nowsheen S, Aziz K, Georgakilas AG. Toxicity and adverse effects of Tamoxifen and other anti-estrogen drugs. Pharmacol Ther. 2013;139(3):392–404. doi:10.1016/j.pharmthera.2013.05.005
5. Riggins RB, Schrecengost RS, Guerrero MS, Bouton AH. Pathways to tamoxifen resistance. Cancer Lett. 2007;256(1):1–24. doi:10.1016/j.canlet.2007.03.016
6. Davies C, Godwin J, Gray R, et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet. 2011;378(9793):771–784.
7. Early Breast Cancer Trialists’ Collaborative Group. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;365(9472):1687–1717. doi:10.1016/S0140-6736(05)66544-0
8. Liu J, Lichtenberg T, Hoadley KA, et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell. 2018;173(2):400–416.e411.
9. Zhou ZR, Wang XY, Yu XL, et al. Building radiation-resisted model in triple-negative breast cancer to screen radioresistance-related molecular markers. Ann Transl Med. 2020;8(4):108. doi:10.21037/atm.2019.12.114
10. Barrett T, Wilhite SE, Ledoux P, et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 2013;41(Database issue):D991–995. doi:10.1093/nar/gks1193
11. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 2008;9(1):559. doi:10.1186/1471-2105-9-559
12. Jia R, Zhao H, Jia M. Identification of co-expression modules and potential biomarkers of breast cancer by WGCNA. Gene. 2020;750:144757. doi:10.1016/j.gene.2020.144757
13. Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45(D1):D353–d361. doi:10.1093/nar/gkw1092
14. Gene Ontology Consortium. Gene ontology consortium: going forward. Nucleic Acids Res. 2015;43(Databaseissue):D1049–1056. doi:10.1093/nar/gku1179
15. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi:10.1101/gr.1239303
16. Nayar U, Cohen O, Kapstad C, et al. Acquired HER2 mutations in ER(+) metastatic breast cancer confer resistance to estrogen receptor-directed therapies. Nat Genet. 2019;51(2):207–216. doi:10.1038/s41588-018-0287-5
17. Fanning SW, Jeselsohn R, Dharmarajan V, et al. The SERM/SERD bazedoxifene disrupts ESR1 helix 12 to overcome acquired hormone resistance in breast cancer cells. eLife. 2018;7. doi:10.7554/eLife.37161
18. Jeselsohn R, Buchwalter G, De Angelis C, Brown M, Schiff R. ESR1 mutations—a mechanism for acquired endocrine resistance in breast cancer. Nat Rev Clin Oncol. 2015;12(10):573–583. doi:10.1038/nrclinonc.2015.117
19. Tripathy D, Im SA, Colleoni M, et al. Ribociclib plus endocrine therapy for premenopausal women with hormone-receptor-positive, advanced breast cancer (MONALEESA-7): a randomised Phase 3 trial. Lancet Oncol. 2018;19(7):904–915. doi:10.1016/S1470-2045(18)30292-4
20. Turner NC, Slamon DJ, Ro J, et al. Overall survival with palbociclib and fulvestrant in advanced breast cancer. N Engl J Med. 2018;379(20):1926–1936. doi:10.1056/NEJMoa1810527
21. Payton M, Coats S. Cyclin E2, the cycle continues. Int J Biochem Cell Biol. 2002;34(4):315–320. doi:10.1016/S1357-2725(01)00137-6
22. Salazar MD, Ratnam M, Patki M, et al. During hormone depletion or tamoxifen treatment of breast cancer cells the estrogen receptor apoprotein supports cell cycling through the retinoic acid receptor alpha1 apoprotein. Breast Cancer Res. 2011;13(1):R18. doi:10.1186/bcr2827
23. Bergamaschi A, Christensen BL, Katzenellenbogen BS. Reversal of endocrine resistance in breast cancer: interrelationships among 14-3-3zeta, FOXM1, and a gene signature associated with mitosis. Breast Cancer Res. 2011;13(3):R70. doi:10.1186/bcr2913
24. Berdnik D, Knoblich JA. Drosophila aurora-A is required for centrosome maturation and actin-dependent asymmetric protein localization during mitosis. Curr Biol. 2002;12(8):640–647. doi:10.1016/S0960-9822(02)00766-2
25. Hirota T, Kunitoku N, Sasayama T, et al. Aurora-A and an interacting activator, the LIM protein Ajuba, are required for mitotic commitment in human cells. Cell. 2003;114(5):585–598. doi:10.1016/S0092-8674(03)00642-1
26. Marumoto T, Honda S, Hara T, et al. Aurora-A kinase maintains the fidelity of early and late mitotic events in HeLa cells. J Biol Chem. 2003;278(51):51786–51795. doi:10.1074/jbc.M306275200
27. Thrane S, Pedersen AM, Thomsen MB, et al. A kinase inhibitor screen identifies Mcl-1 and aurora kinase A as novel treatment targets in antiestrogen-resisted breast cancer cells. Oncogene. 2015;34(32):4199–4210. doi:10.1038/onc.2014.351
28. Baumann K. Genome stability: cyclin’ on mRNA. Nat Rev Mol Cell Biol. 2016;17(11):676–677. doi:10.1038/nrm.2016.142
29. Bailly E, Pines J, Hunter T, Bornens M. Cytoplasmic accumulation of cyclin B1 in human cells: association with a detergent-resisted compartment and with the centrosome. J Cell Sci. 1992;101(Pt 3):529–545. doi:10.1242/jcs.101.3.529
30. Pines J. Cyclins: wheels within wheels. Cell Growth Differ. 1991;2(6):305–310.
31. Liu R, Guo CX, Zhou HH. Network-based approach to identify prognostic biomarkers for estrogen receptor-positive breast cancer treatment with tamoxifen. Cancer Biol Ther. 2015;16(2):317–324. doi:10.1080/15384047.2014.1002360
32. Zhou H, Lv Q, Guo Z. Transcriptomic signature predicts the distant relapse in patients with ER+ breast cancer treated with tamoxifen for five years. Mol Med Rep. 2018;17(2):3152–3157. doi:10.3892/mmr.2017.8234
33. Diefenbach RJ, Mackay JP, Armati PJ, Cunningham AL. The C-terminal region of the stalk domain of ubiquitous human kinesin heavy chain contains the binding site for kinesin light chain. Biochemistry. 1998;37(47):16663–16670. doi:10.1021/bi981163r
34. Zou JX, Duan Z, Wang J, et al. Kinesin family deregulation coordinated by bromodomain protein ANCCA and histone methyltransferase MLL for breast cancer cell growth, survival, and tamoxifen resistance. Mol Cancer Res. 2014;12(4):539–549. doi:10.1158/1541-7786.MCR-13-0459
35. Hiwatashi K, Ueno S, Sakoda M, et al. Expression of maternal embryonic leucine zipper kinase (MELK) correlates to malignant potentials in hepatocellular carcinoma. Anticancer Res. 2016;36(10):5183–5188. doi:10.21873/anticanres.11088
36. Wang Y, Lee YM, Baitsch L, et al. MELK is an oncogenic kinase essential for mitotic progression in basal-like breast cancer cells. eLife. 2014;3:e01763. doi:10.7554/eLife.01763
37. Chen X, Cheung ST, So S, et al. Gene expression patterns in human liver cancers. Mol Biol Cell. 2002;13(6):1929–1939. doi:10.1091/mbc.02-02-0023
38. Garber ME, Troyanskaya OG, Schluens K, et al. Diversity of gene expression in adenocarcinoma of the lung. Proc Natl Acad Sci U S A. 2001;98(24):13784–13789. doi:10.1073/pnas.241500798
39. Fragoso MC, Almeida MQ, Mazzuco TL, et al. Combined expression of BUB1B, DLGAP5, and PINK1 as predictors of poor outcome in adrenocortical tumors: validation in a Brazilian cohort of adult and pediatric patients. Eur J Endocrinol. 2012;166(1):61–67. doi:10.1530/EJE-11-0806
40. Schneider MA, Christopoulos P, Muley T, et al. AURKA, DLGAP5, TPX2, KIF11 and CKAP5: five specific mitosis-associated genes correlate with poor prognosis for non-small cell lung cancer patients. Int J Oncol. 2017;50(2):365–372. doi:10.3892/ijo.2017.3834
41. Weinberger P, Ponny SR, Xu H, et al. Cell cycle M-phase genes are highly upregulated in anaplastic thyroid carcinoma. Thyroid. 2017;27(2):236–252. doi:10.1089/thy.2016.0285
42. Lawson CD, Der CJ. Filling GAPs in our knowledge: ARHGAP11A and RACGAP1 act as oncogenes in basal-like breast cancers. Small GTPases. 2018;9(4):290–296. doi:10.1080/21541248.2016.1220350
43. Saigusa S, Tanaka K, Mohri Y, et al. Clinical significance of RacGAP1 expression at the invasive front of gastric cancer. Gastric Cancer. 2015;18(1):84–92. doi:10.1007/s10120-014-0355-1
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.