")
Back to Journals » Journal of Inflammation Research » Volume 15
Identification of Significant Modules and Targets of Xian-Lian-Jie-Du Decoction Based on the Analysis of Transcriptomics, Proteomics and Single-Cell Transcriptomics in Colorectal Tumor
Authors Yu CT, Chen T, Lu S, Hu W, Zhang Q , Tan J, Sun D, Li L, Sun X , Xu C, Lai Y, Fan M, Shen Z, Shen W, Cheng H
Received 22 October 2021
Accepted for publication 31 December 2021
Published 1 March 2022 Volume 2022:15 Pages 1483—1499
DOI https://doi.org/10.2147/JIR.S344861
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Cheng-Tao Yu,1,* Tongqing Chen,1,* Sicheng Lu,1,* Wenlong Hu,1 Qinchang Zhang,1 Jiani Tan,1 Dongdong Sun,1 Liu Li,1 Xin Sun,1 Changliang Xu,1 Yueyang Lai,1 Minmin Fan,1 Zhengjie Shen,2 Weixing Shen,1 Haibo Cheng1
1The First Clinical Medical College, Jiangsu Collaborative Innovation Center of Traditional Chinese Medicine Prevention and Treatment of Tumor, Nanjing University of Chinese Medicine, Nanjing, People’s Republic of China; 2Medical Oncology Department, The Affiliated Zhangjiagang Hospital of Soochow University, Suzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Weixing Shen; Haibo Cheng, The First Clinical Medical College, Jiangsu Collaborative Innovation Center of Traditional Chinese Medicine Prevention and Treatment of Tumor, Nanjing University of Chinese Medicine, Nanjing, People’s Republic of China, Tel +86 13815857118, Fax +86 2585811006, Email [email protected]; [email protected]
Purpose: Colorectal cancer (CRC) remains the third most common tumor worldwide. Ulcerative colitis (UC) could cause chronic inflammation and ulcers in the colon and rectum. UC is a risk factor for a high incidence of CRC, and the incidence of UC-associated CRC (UC-CRC) is still increasing. Chinese medicine prescription, Xian-Lian-Jie-Du decoction (XLJDD), has been proven its efficacy in some UC-CRC patients. However, the mechanism of XLJDD in treating UC-CRC remains unknown. This study aimed to investigate the mechanism of XLJDD in treating UC-CRC.
Methods: We constructed an AOM/DSS mouse model that could simulate the various stages of UC-CRC in humans. XLJDD and its 5 main components are used to treat the AOM/DSS model, respectively. With the power of high-throughput sequencing technology, we described the mechanism of XLJDD from transcriptomics, proteomics, and single-cell transcriptomics.
Results: Our results showed that XLJDD could effectively suppress the occurrence and development of colorectal tumors. Using the weighted correlation network analysis (WGCNA), several mRNA and protein modules that respond to XLJDD have been identified. Moreover, two essential genes, Mfsd2a and Ccdc85c, were caught our attention. They were prognostic markers in CRC patients, and their expression could be significantly modulated by XLJDD, showing their potential as effective targets of XLJDD. In addition, we also discovered that XLJDD could affect the cell composition of the colorectal tumor environment, especially in the infiltration of B cells.
Conclusion: We demonstrated that XLJDD could prevent the initiation and development of colorectal tumors by modulating the expression of Mfsd2a and Ccdc85c and reducing the infiltration of B cells in the tumor microenvironment of colorectal tumor.
Keywords: colorectal tumor, Mfsd2a, Ccdc85c, inflammation pathway, B cell infiltration
Introduction
According to the International Agency for Research on Cancer (IARC) investigation, colorectal cancer (CRC) is the third most common and the second most deadly type among 36 kinds of cancers. It is estimated that there will be more than 1.9 million new cases and 935,000 deaths in 2020, accounting for approximately 1/10 of total cancer deaths.1 Ulcerative colitis (UC) is a long-term disease that causes chronic inflammation and ulcers in the colon and rectum, a risk factor for a high incidence of CRC.2,3 Although the exact mechanism by which UC-associated CRC (UC-CRC) is still unclear, there is growing evidence that the abnormal alteration of transcriptomes, proteomics, and tumor environment may contribute to their occurrence and progression.2,4 Currently, the drugs for UC-CRC are minimal. Therefore, it is significant to develop new drugs for the clinical treatment of UC-CRC.
The drug development for tumors is firstly carried out on animal models. So far, a large number of animal models have been applied for the study of drugs, including tumor subcutaneous tumor model, orthotopic tumor model, and Azoxymethane (AOM)/Dextran sodium sulfate (DSS) model.5–7 Among these, the AOM/DSS mouse model is wildly used to nominate UC-CRC.8,9 The combination of AOM and the inflammatory agent DSS has reduced the latency. The regular use of DSS in drinking water could make colon tumors grow rapidly within ten weeks, while the incubation period of other models is 30 weeks at least.10 In addition, increasing evidence suggests that the process of AOM/DSS-induced colorectal tumors is similar to that of UC-CRC in humans, making it possible for us to study the effects and mechanisms of drugs on UC-CRC.6
The early studies of the AOM/DSS model mainly focus on a single molecular mechanism. Nowadays, we can explore the pathogenesis of the AOM/DSS model comprehensively by high-throughput sequencing technology, such as transcriptomics sequencing, proteomics sequencing, and single-cell transcriptomics sequencing.11–17 The application of transcriptomics and proteomics could display thousands of RNA and protein expression profiles in one picture. Through single-cell RNA sequencing technology, the map of thousands of cells could also be described simultaneously. The demand for big data analysis has prompted the rapid development of bioinformatics in recent years.16,17 For example, Subramanian et al proposed the gene set enrichment analysis (GSEA) software, which could help us get significant pathways from a large amount of gene expression profiles data.18 Langfelder et al opened up the R package of weighted correlation network analysis (WGCNA), a systems biology method describing gene association patterns between different samples. WGCNA method could identify highly coordinated gene sets and candidate biomarkers based on the interconnectivity of genes.19
Chinese medicine prescriptions are based on the guidance of traditional Chinese medicine theory, which is composed of a variety of medicines according to a certain quality ratio for specific diseases.20 Many Chinese medicine prescriptions have been studied by Asian countries and have proven to be effective for certain chronic diseases.21 Our laboratory has been committed to evaluating the clinical efficacy and researching potential molecular mechanisms about traditional Chinese medicine prescriptions and has assessed the classic recipe named Xiao-Ai-Jie-Du decoction (XAJDD), established by Pro. Zhou Zhongying, a master of Chinese medicine.22,23 Recently, we focused on another Chinese medicine prescription, Xian-Lian-Jie-Du decoction (XLJDD), developed by Pro. Cheng Haibo, due to its excellent effect of CRC progression control based on our clinical observation. However, the mechanism of XLJDD on UC-CRC has not been revealed so far.
Materials and Methods
AOM/DSS-Induced Mouse Model
C57BL/6N male mice (6 weeks old, bodyweight 18–22 g, animal certificate number: SCXK2016-0006) were purchased from Beijing Wei Tong Li Hua Laboratory Animal Technology Co., Ltd. (Beijing, China) and were housed under controlled conditions of temperature (21±2°C) and humidity (40–60%) with 12 h light/dark cycle. Food and water were supplied as required, and cages were changed every week.
The mice were i.p. injected with azoxymethane (AOM, 10 mg/kg body weight). After one week, mice were treated with 2% dextran sodium sulfate (DSS) in drinking water for seven days, allowing 5 mL DSS solution per mouse per day. Then, DSS was replaced by drinking autoclaved water for four weeks. In the second cycle and third cycle, the mice were treated with 2% dextran sodium sulfate (DSS) in drinking water for seven days and then replaced by drinking autoclaved water for two weeks. The drug was administered once a day starting the day after DSS drinking and was administered six consecutive days in one week. The body weights of mice were recorded per week, and the survival and general health of the mice were recorded daily. The XLJDD concentration is calculated by mass conversion based on the effective clinical. Previous research by our team set up three concentrations of high (26 g/Kg), middle (12.9 g/Kg), and low (6.5 g/Kg) for pre-experiment. The experimental results determined that the concentration of 12.9 g/Kg in the XLJDD animal experiment was appropriate (Figure S1A–C). In detail, the concentrations of P1-P5 were 2.55 g/Kg, 2.25 g/Kg, 2.7 g/Kg, 1.35 g/Kg, 4.05 g/Kg. The drugs were administered through oral gavage and the control group received an equal volume of saline.
One hundred and ten male mice were divided into eleven groups randomly, including control group (Normal saline), NF-MOD model group (AOM-DSS model with normal diet), HDF-MOD model group (AOM-DSS model with high-fat diet), NF-XLJDD group (normal diet AOM-DSS model with XLJDD), HDF-XLJDD group (high-fat diet AOM-DSS model with XLJDD), P1 model group (XLJDD ingredients 1), P2 model group (XLJDD ingredients 2), P3 model group (XLJDD ingredients 3), P4 model group (XLJDD ingredients 4), P5 model group (XLJDD ingredients 5), and positive control group (Folic Acid). All animal experiments were approved by our Animal Ethics Committee of Nanjing University of Chinese Medicine (201911A027, 201905A014, 202105A042). The animal experiments followed the guidelines for the ethical review of laboratory animal welfare in Nanjing University of Chinese Medicine.
Preparation of XLJDD
All herbs were identified by Professor Si-li Zou (Nanjing University of Chinese Medicine). Agrimoniae Herba (Xian-He-Cao), Coptidis Rhizoma (Huang-Lian), Astragali Radix Praeparata Cum Melle (Zhi-Huang-Qi), Coicis Semen (Yi-Yi-Ren) were immersed in water (1:10, w/v) for 0.5 h and then heated to boiling and maintained for 2 h. The aqueous extract was filtered, and the residue was decocted again with water (1:8, w/v) under the same conditions. The filtrate was combined and 60% alcohol precipitation for 1 day. The solution was removed via evaporation in a rotary evaporator and concentrated to 2 g drug per milliliters. The extract was stored at −20°C.
UPLC-LTQ-MS Analysis for XLJDD
The significant constituents in XLJJD were determined using a UPLC-LTQ-Orbitrap-MS approach. The qualitative analysis was performed on a series Ultimate 3000 UPLC system (Dionex, USA) coupled with the LTQ-Orbitrap Velos Pro mass spectrometer (Thermo, USA). The separation was carried out on an ACQUITY UPLC BEH C18 1.7 µm (2.1 mm×100 mm, Waters) at 30°C with 0.4 mL/min flow rate. (A) 0.1% formic acid in water and (B) acetonitrile were used as the mobile phase. The gradient program of the mobile phase was as follows: 0–4.5 min, 5% B; 4.5 min-6.0 min, 5–15% B; 6.0–7.5 min, 15% B; 7.5–9 min, 15–18% B; 9–11 min, 18–27% B; 11–16 min, 27–35% B; 16–18 min, 35–60% B; 18–20 min, 60–5% B. The samples were maintained at 4°C during the whole analysis, and the injection volume was 2 μL. Sample introduction and ionization were detected by electrospray ionization (ESI) in the positive ion mode. MS data acquisition was performed in a range of m/z = 100–1000. The MS parameters were as follows: the capillary temperature was 275 °C, the sheath gas (N2) flow was 40 kPa, the auxiliary gas (He) flow was 15 kPa. The second-order mass spectrometry was acquired in data-dependent analysis mode with dynamic exclusion enabled and selected the top peak of the upper level for collision-induced dissociation (CID). All operations were guided and helped by the staff of the public instrument platform of Nanjing University of Chinese Medicine. The compounds detailed results have been shown in Figure 1A and Supplementary Materials 1. These works were performed by Pro. Shulan Su from Nanjing University of Chinese Medicine.
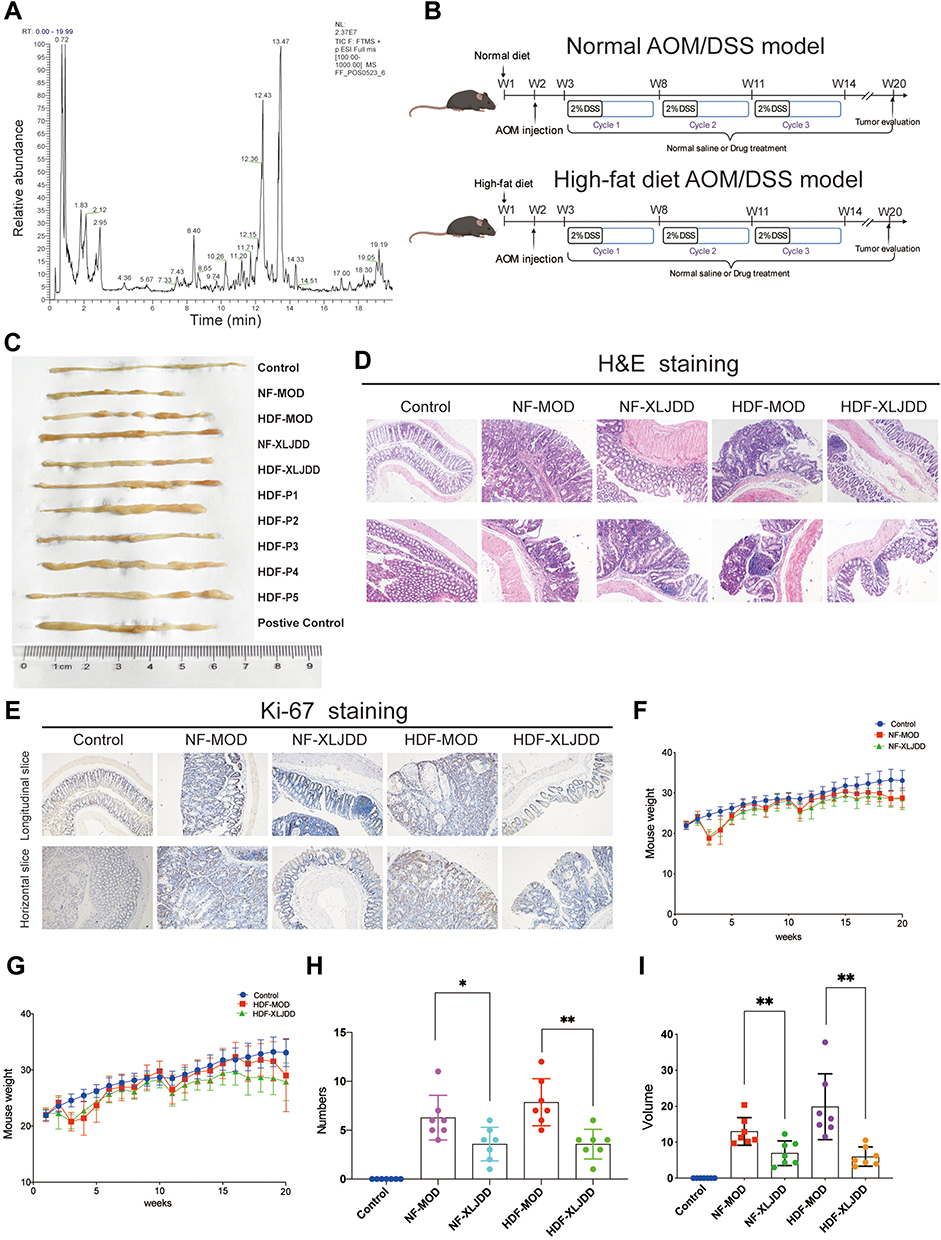 |
Figure 1 Alleviating effects of XLJDD on AOM/DSS induced tumor progression. (A) Total chromatograms of typical extracts from Xian-Lian-Jie-Du decoction using UPLC-LTQ-Orbitrap-MS. (B) A schematic overview of the AOM/DSS mice model construction. (C) The morphological display of the colorectal tissues in each group. (D) H&E staining of colorectal paraffin sections (longitudinal and horizontal slice of paraffin sections). (E) Immunohistochemical staining of Ki-67 expression in colorectal tissues. (F and G) Body weight of mice was monitored weekly. (H and I) The number and volume of nodules were measured. Results are represented as mean ± SD, n=7; *P<0.05, **P<0.01. |
Transcriptomics Sequencing
RNA sequencing was performed at Novogene Bioinformatics Technology Co. Ltd. (Beijing, China). According to the manufacturer’s protocol, the total RNA was extracted using Trizol (Invitrogen, USA). RNA degradation and contamination were monitored on 1% agarose gels. RNA purity was checked using the NanoPhotometer® spectrophotometer (IMPLEN, CA, USA). The wavelengths ratio of A260/A280 and A260/A230 were used to control the quality of RNA. RNA concentration was measured using Qubit® RNA Assay Kit in Qubit® 2.0 Fluorometer (Life Technologies, CA, USA). RNA integrity was assessed using the RNA Nano 6000 Assay Kit of the Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA). Library quality was assessed on the Agilent Bioanalyzer 2100. The clustering of the index-coded samples was performed on a cBot Cluster Generation System using TruSeq SR Cluster Kit v3-cBot-HS (Illumia) according to the manufacturer’s instructions. After cluster generation, the library preparations were sequenced on an Illumina HiSeq 2500/2000 platform, and 50 bp single-end reads were generated. The expression profiles were analyzed by standard procedure.
Proteomics Sequencing
DIA quantitative proteomics analysis was performed by Novogene Bioinformatics Technology Co. Ltd. (Beijing, China). DIA quantitative proteomics analysis was used to identify differentially expressed proteins between the various group. The protein of colorectal tissues was extracted using a protein-extraction reagent. Protein purity was confirmed by SDS-PAGE, and protein concentration was determined by the Protein Quantitative Kit (Bradford). After Trypsin treatment, the filtrate was collected and lyophilized. After DDA spectrum library construction, the LC-MS/MS Analysis-DIA mode was performed. Analyses mainly included raw data filtering and quality control, protein function annotation.
Single-Cell RNA Sequencing
Single-cell RNA sequencing analysis was performed by Genergy Bio-Technology Co. Ltd. (Shanghai, China). Single-cell suspension was loaded onto the Chromium controller (10X Genomics) at a concentration of 1000 cells per μL (Single Cell 3′ library and Gel Bead Kit v.3) as described in the manufacturer’s protocol. Generation of gel beads in emulsion (GEMs), barcoding, GEM-RT clean-up, complementary DNA amplification, and library construction were all performed as per the manufacturer’s protocol. Qubit was used for library quantification before pooling. The final library pool was sequenced on the Illumina HiSeq 2500 platform using 150-base-pair paired-end reads.
Bioinformatic Analysis
Gene set enrichment analysis (GSEA) software (version 4.0, www.broadinstitute.org/gsea/) was employed to identify gene sets that were significantly overrepresented between two groups, such as the NF-MOD group and NF-XLJDD group. GSEA v4.0 software was used to explore the distribution of members of the gene sets from the MSigDB database.18 In this bioinformatics analysis, if the most members in a gene set were positively or negatively correlated with the XLJDD treatment, the set was termed associated with XLJDD.
According to the WGCNA operation process, we input the expression data detected by each group into the R studio software. The interesting gene modules can be identified by the WGCNA algorithm, and the intramodular connectivity and gene significance based on the correlation of a gene expression profile with a sample trait was used to identify key genes further validation. WGCNA was a freely accessible R package for the construction of weighted gene co-expression networks.19
Immunohistochemistry Assay
The tissues were fixed in 4% paraformaldehyde, dehydrated, and embedded in paraffin. After producing paraffin sections, sections were cut at 4 μm and baked at 60°C overnight. The slices were treated by heat-mediated antigen retrieval in citrate buffer. Pieces were washed with PBS, followed by permeabilization with 0.2% Triton X-100. The portions were then treated with 2% H2O2 to inactivate endogenous peroxidase activity. The slices were washed with PBS 3 times and incubated for 2 h with 3% normal goat serum in PBS. Primary antibody was incubated overnight at 4°C, and secondary antibodies were incubated for 1 h at room temperature. The slices were subsequently treated with DAB substrate–chromogen solution for 10 min and counterstained with hematoxylin, then dehydrated. The photograph was taken, and the positive cell percentage was calculated.
Western Blot Assay
Antibodies: Bax (ab32503, Abcam,1:5000), Bcl-2 (ab196495, Abcam, 1:1000), Ccdc85c (D263389, BBI Life Science, 1:400), Mfsd2a (D261903, BBI Life Science, 1:1000), β-actin (ab227387, Abcam, 1:5000). Secondary antibody was IgG-HRP secondary antibody (7074P2, Cell Signaling Technology, 1:5000).
Briefly, Proteins were extracted from colorectal tissue, and protein concentrations of tissues were quantified by a BCA protein assay kit (Thermo Scientific, Waltham, MA, USA). Protein were separated by 10% sodium dodecyl sulfate-polyacrylamide gel and transferred to PVDF membrane, then blocked by 5% non-fat milk in PBST buffer (PBS buffer containing 0.05% Tween-20) for 1 h at room temperature. The membranes were incubated overnight with primary antibodies at 4°C. After 3 times washing them in PBST buffer, the membranes were incubated with the secondary antibodies for 1 h at room temperature. Signals were detected by using an ECL substrate (Thermo Scientific) and exposure with the Tanon 5500 imaging system. The intensity of the bands was quantified by densitometry.
Clinical Sample
The colorectal tissues of UC-associated colorectal tumor patients with and without XLJDD were obtained from the Jiangsu Provincial Hospital of Chinese Medicine, Nanjing University of Traditional Chinese Medicine (Nanjing, China). These samples were identified by two pathologists independently. Written consent approving the usage of tissues in our study was obtained from each patient. This study was approved by the Ethics Committees of the Jiangsu Provincial Hospital of Traditional Chinese Medicine, Nanjing University of Chinese Medicine (2020NL-09404). The collection of clinical samples was conducted in accordance with the Declaration of Helsinki.
Mouse Colorectal Orthotopic Tumor Model
2×106 CT26 cells were injected as the subcutaneous tumor in BALB/c mice. After 14 days, the mice were sacrificed, the tumor tissue was peeled off, and the scissors were cut into 1 mm3 size and soaked in normal saline for later use. Make tumor tissue in situ inoculated. After the mouse was anesthetized with isoflurane, the left lower abdominal incision was disinfected, the cecum was pulled out, and the serosal membrane was slightly scraped with a needle at the junction of the blind knot to cause slight blood oozing. Subsequently, the prepared tumor mass adhered to the scraping site with Histoacryl adhesive, then carefully returned to the abdomen, and the peritoneum was sutured. The tissues of colorectal orthotopic tumor model in this study was provided by Pro.Tan Jiani.
Statistical Analysis
Data were expressed as the mean ± standard deviation from at least three independent experiments. Survival analysis was performed with the GEPIA, and the Log rank test were used for comparisons.24 Statistical results were analyzed using Prism software (GraphPad Software). Student’s t-test was applied to compare two experimental groups. A probability of 0.05 or less was considered as statistical significance.
Results
Alleviating Effects of XLJDD on AOM/DSS Induced Tumor Progression
To explore the efficacy of XLJDD in preventing UC-CRC, we constructed AOM/DSS mouse model. We built two kinds of AOM/DSS mouse models, one for normal diet (NF-MOD) and the other for high-fat diet (HDF-MOD). The XLJDD was applied to treat in both of models, and the five main ingredients of the drug (P1-P5) were used only in the HDF AOM/DSS model (Figure 1B). As the results showed, the colorectal length of the control group was relatively normal, and the intestinal mucosa was smooth. There was no congestion, edema, or apparent hyperplasia. In the NF-MOD group and HDF-MOD group, the colorectal length was shortened, the surface of the intestinal mucosa was rough, the hyperemia, edema, and local blood spots appeared. Compared with the model group, the symptoms mentioned above could be relieved after XLJDD treatment. Although each medicine component (P1-P5) had an effect on reducing symptoms at different levels, its effect seemed to be weaker than the XLJDD group (Figure 1C).
Next, we evaluated the area of the tumor lesions by H&E and Ki-67 staining. The results of H&E staining displayed that the cells in the control group were arranged regularly, the cell nucleus was located at the base, flat or cubic. In the NF-MOD and HDF-MOD groups, the epithelial cells disappeared in polarity, merged, presented a nested, the nucleus to cytoplasm ratio increased, the nucleus was stained deeply, suggesting the process of tumors generation. In the XLJDD treatment group, the arrangement of epithelial cells was relatively regular, and the above symptoms have been relieved to some extent (Figure 1D). The results of Ki-67 staining showed that epithelial cells in NF-MOD and HDF-MOD group were significantly more proliferating than the control group, but the numbers of these proliferating cells in the XLJDD group were relatively small (Figure 1E). This finding suggested that XLJDD may limit the abnormal proliferation of epithelial cells. Subsequently, we appraised the body weight, the number, and the volume of nodules in these groups. The results showed that no significant difference in the body weight of each group, but the treatment of XLJDD could decrease the number and volume of nodules (Figure 1F–I).
Weighted Gene Co-Expression Network Analysis of Transcriptome Profiles
To elucidate the underlying mechanism of XLJDD, we detected the mRNA expression profiles of each group by transcriptomics technology. A total of 21,898 mRNAs were detected, and multiple differentially expressed genes were screened out (Tables S1–S7). Considering that the method of screening for differential genes could not clarify the mechanism of XLJDD from a global perspective, we decided to adopt WGCNA, a systematic data-analyzed method, to explore the critical genes in each group.
According to the WGCNA construction guidelines, we constructed a co-expression network based on the expression profiles of each group. We identified 12 gene modules, and each represented by a color (Figure 2A). Through conservative analysis, it could be known that all modules have passed the conventional test, and these 12 modules are all credible (Figure 2B). Then, functional enrichment analyses were performed for the genes of each module to find the corresponding pathway. For example, the gene of the purple module was mainly involved in the response of inflammatory factors, and the black module was engaged primarily in the focal adhesion pathway (Figure 2C). Finally, we identified the most related modules to the phenotype by constructing an association network between gene modules and phenotypes. As the results showed, only the purple module was most associated with the NF-XLJDD group, suggesting that the genes of the purple model may be activated by XLJDD (Figure 2D and E). In addition, we found that the purple module contains 14 differential genes between NF-XLJDD and NF-MOD groups (Figure 2F). Based on the enrichment analysis, we realized that these genes were closely related to cell death-related pathways, indicating that they may be essential for the function of XLJDD to induce tumor cell death (Figure 2G). A GSEA analysis between the NF-XLJDD and the NF-MOD groups displayed that the interferon-related pathways were significantly enriched, suggesting that XLJDD inducing cell death may be involved in the function of interferon (Figure 2H). Considering that interferon can induce the death of tumor cells by activating apoptosis, we then detected the marker gene of apoptosis, and experimental evidence confirmed that apoptosis occurred (Figure S1D). Above all, these results suggested that the purple module, responsible for inflammatory factor response, was closely associated with XLJDD treatment, and 14 differential genes of this module may mediate the tumor cell-killing effect of XLJDD.
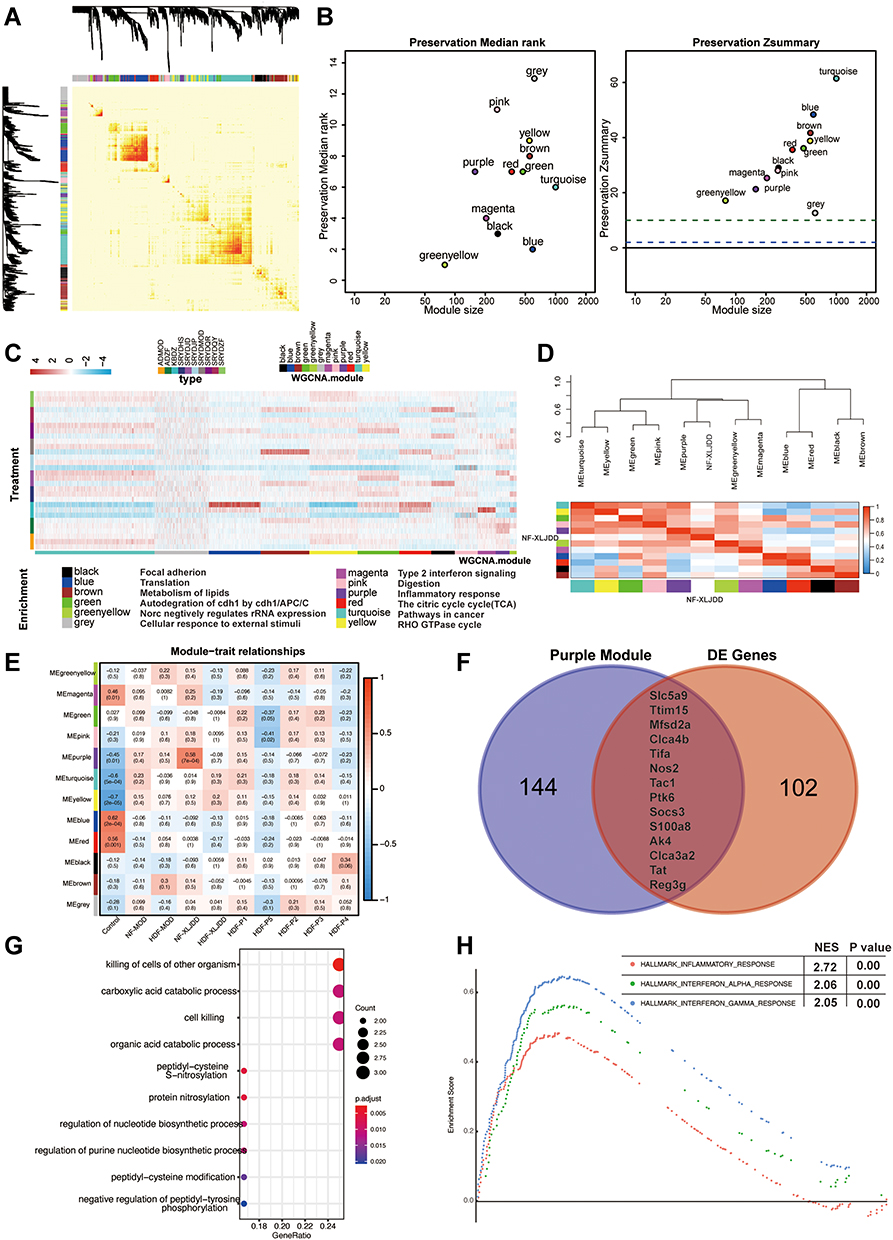 |
Figure 2 Weighted gene co-expression network analysis of transcriptome profiles. (A) Topological overlap matrix (TOM) plot showing the correlations of each module. (B) Module preservations median rank and Zsummary score of all modules were presented. (C) Functional enrichment analyses for different modules corresponding to the colors. (D) The correlations between the modules and the NF-XLJDD group. (E) Heatmap of correlations between modules and each group. Colors correspond to correlations, while red means positively correlated and blue means negatively correlated. Correlations values and P values are also labeled. (F) Venn diagrams of comparing purple module and differentially expressed genes (DE genes) between NF-XLJDD and NF-MOD groups. (G) The enrichment analysis of 14 differential genes. (H) Gene set enrichment analysis showed enrichment pathways between the NF-XLJDD and the NF-MOD groups. |
Mfsd2a May Be a Tumor Suppressor Activated by XLJDD
As mentioned above, we found that the differential genes may be critical for XLJDD. Next, we analyzed their homologous gene expression trend in CRC patients and their clinical correlation. In the human TCGA-COAD dataset, we found that Mfsd2a, Nos2, Trim15, Tifa, and Ptk6 were significantly upregulated in CRC tissues, while Slc5a9, Tac1, Socs3, S100a8, Tat were significantly downregulated in CRC tissues, and the expression levels of Ak4 and Reg3g were not significant (Figure 3A). Interestingly, although the TCGA dataset showed that Mfsd2a was highly expressed in CRC tumor tissues, the prognostic data have shown that CRC patients with high Mfsd2a expression lived longer (Figure 3B). This seemingly contradictory evidence prompted us to search the literature on Mfsd2a. Then, we learned that Mfsd2a was expressed in CRC cells and CD8+T cells, vascular cells, and other cells in the tumor microenvironment. Mfsd2a expressed in different cells may have completely distinct functions.25 Ungaro et al reported that endothelial progenitor cells that overexpress Mfsd2a could reduce colitis in mice, declaring that Mfsd2a had the potential to resolve intestinal inflammation in patients with colitis.26 In our results, evidence suggested that XLJDD activated the expression of Mfsd2a (Figure 3C), suggesting that XLJDD may prevent the progression of UC-CRC dependent on Mfsd2a. To confirm the relationship between Mfsd2a and XLJDD, we examined them in the colon tissues of mouse orthotopic colorectal tumor models and the UC-CRC patient. IHC assays showed that expression of Mfsd2a was increased when XLJDD was treated (Figure 3E). The results of qPCR revealed that the mRNA levels of Mfsd2a could be upregulated, which suggested that the changes in Mfsd2a could be produced at the transcription level (Figure 3D). These results indicated that the regulatory relationship between Mfsd2a and XLJDD could be conservative.
 |
Figure 3 Mfsd2a may be a tumor suppressor activated by XLJDD. (A) Candidate genes expression in cancer tissues (Red) is compared with normal tissues (Black) in the TCGA-COAD dataset. (B) Kaplan-Meier survival analyses of Mfsd2a expression (low vs high). (C)The expression of Mfsd2a in our results of transcriptomic sequencing. (D) The expression results of Mfsd2a measured by qPCR. (E) Representative images for IHC staining of Mfsd2a. *P<0.05, **P<0.01. |
Weighted Gene Co-Expression Network Analysis of Proteomics Expression Profiling
Then, we tried to explore the mechanism of XLJDD from the protein level. A total of 6856 proteins were detected, and eight protein modules with various expression patterns were identified through the WGCNA method (Figure 4A–C). We found the functional pathways corresponding to each module by the enrichment analysis. For example, we found that the black module corresponds to the cell apoptosis process, the brown module corresponds to the focal adhesion pathway, and the green module corresponds to the citrate cycle process (Figure 4D). After that, we constructed the phenotype-module correlation matrix and observed that the NF-XLJDD group was significantly positively correlated with the blue module, and the HDF-XLJDD group was significantly positively associated with the black module and the green module. Moreover, we also discovered that the primary component P2 was positively correlated with the red module, and P4 was positively correlated with the green and turquoise module (Figure 4E–G). Comparing the differentially expressed proteins in the corresponding modules, we found that some proteins may be important for the function of XLJDD. For instance, Rpl6, Ccdc85c, Ctrb1, Mgp, Naaladl1, and Rpl36a may play a vital role in the NF-XLJDD group. Meak7, Seroina1b, Mpz, and Seroina1a may be valid in the HDF-XLJDD group. Some proteins, such as Daglb and Haxb, may be essential for P4 (Figure 4H, Tables S8–S14).
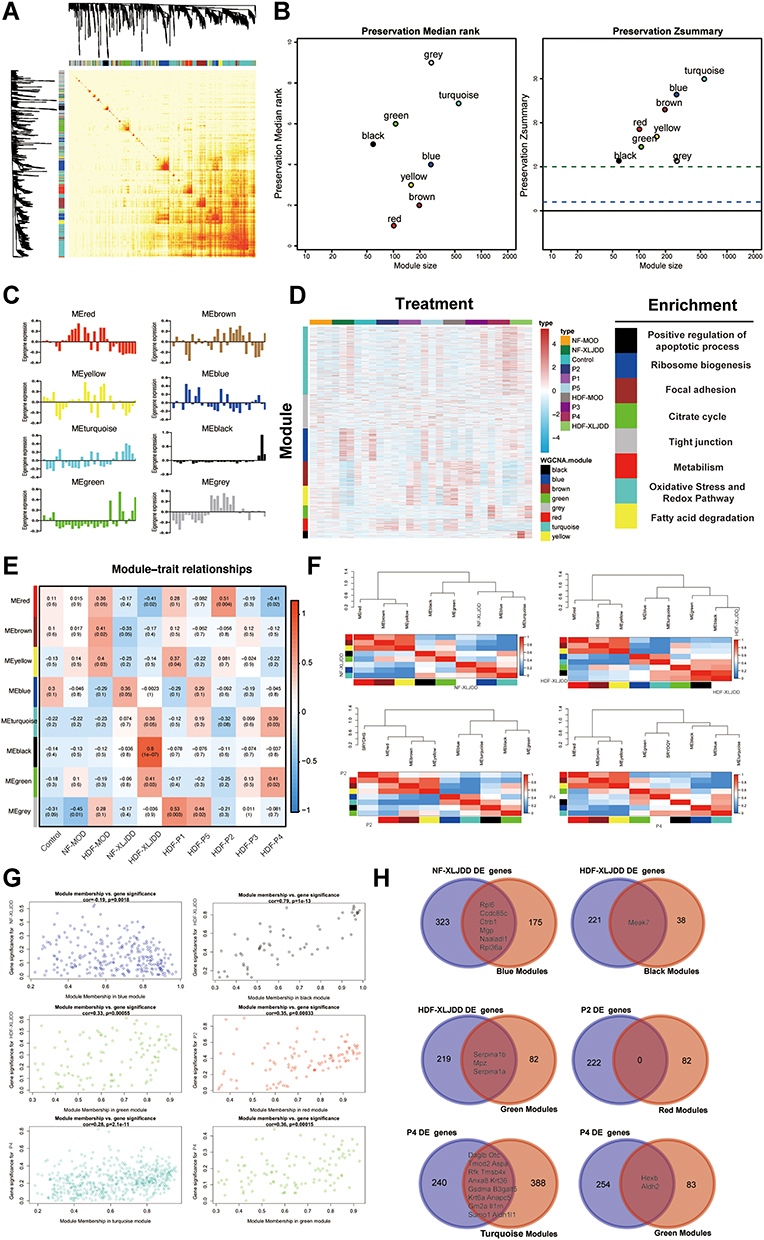 |
Figure 4 Weighted gene co-expression network analysis of proteomics profiles. (A) Topological overlap matrix (TOM) plot showing the correlations of each module. (B) Module preservations median rank and Zsummary score of all modules were presented. (C) The Eigengenes expression pattern in each group. (D) The correlations between various modules and their corresponding pathways. (E) Heatmap of correlations between modules and each group. Colors correspond to correlations, while red means positively correlated and blue means negatively correlated. Correlations values and P values are also labeled. (F) The correlations between the functional modules and NF-XLJDD, HDF-XLJDD, P2, and P4 groups, respectively. (G) The maps of module membership with gene significance were displayed. (H) The Venn diagram of differential genes in NF-XLJDD, HDF-XLJDD, P2, and P4 and their corresponding modules. |
Ccdc85c May Be an Effective Target for XLJDD
GSEA analysis was first applied to seek protein pathways altered by XLJDD. We found that adipogenesis, fatty_acid_metabolism, and oxidative_phosphorylation pathways were significantly enriched in the NF-XLJDD group and HDF-XLJDD group compared to NF-MOD and HDF-MOD groups, respectively. The hypoxia, myogenesis, and oxidative_phosphorylation pathways were enriched in the P2 group. In the P4 group, E2F_targets, G2M_checkpoint, and p53_pathway was enriched (Figure 5A). Then, we tried to identify the essential protein that supported the function of XLJDD. Joint analysis with the results of Figure 4H and TCGA dataset, we noted that Ccdc85c was modulated by XLJDD and downregulated in the tumor tissue of CRC patients. The survival time of patients with high expression of Ccdc85c was significantly longer than that of patients with low expression (Figure 5B and C). The qPCR results revealed that the mRNA levels of Ccdc85c could not be altered, which suggested that the changes in Ccdc85c protein levels are not produced at the transcription level. (Figure 5D).
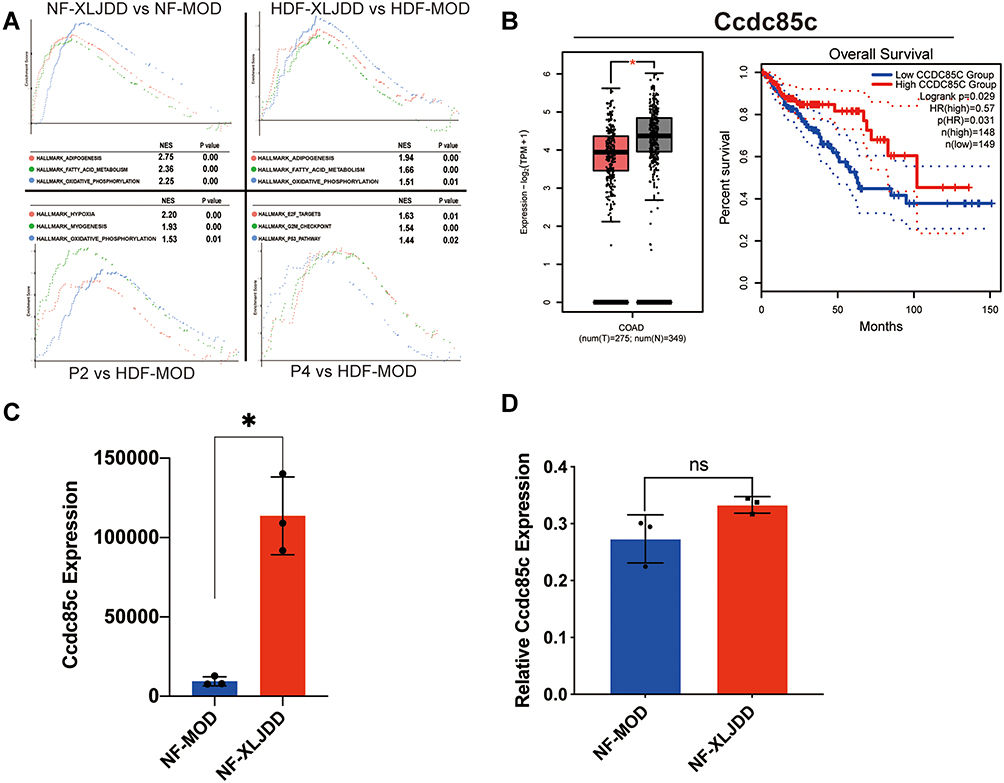 |
Figure 5 Ccdc85c may be an effective target for XLJDD. (A) Gene set enrichment analysis showed enrichment pathways between the two groups. (B) The expression levels of Ccdc85c and Kaplan-Meier survival analysis from TCGA data. (C) The expression of Ccdc85c in our results of proteomics sequencing. (D) The expression results of Ccdc85c measured by qPCR. *P<0.05, ns, not significant. |
The Alteration of Tumor Microenvironment Caused by XLJDD
Based on the above results, we have discovered critical genes in the NF-XLJDD group through transcriptome and proteome profiles. However, we still have not explored the mechanism of the HDF-XLJDD group. Currently, increasing studies have shown that the tumor microenvironment was the key to the occurrence and development of tumor cells, and many drugs have been identified to target the tumor microenvironment for tumor therapy.27 Thus, we next decided to explore the mechanism of XLJDD in the HDF-XLJDD group from the perspective of the tumor microenvironment. We selected the control, HDF-MOD, and HDF-XLJDD groups for single-cell RNA sequencing. Our results showed that a total of 45,388 cells were captured for analysis. The captured cells were subjected to dimensionality reduction analysis, and a total of 29 clusters were obtained (Figure 6A–E). We found that the ratio of cells was significantly changed in the HDF-MOD group compared to the control group but were rescued in the HDF-XLJDD group. For example, the ratio of cluster 2 and cluster 17 increased significantly in the HDF-MOD group, while their percentage in the HDF-XLJDD group decreased and was close to that of the control group (Figure 6F). The above results suggested that XLJDD may play as a tumor suppressor by affecting the composition of tumor microenvironment cells in the HDF-XLJDD group.
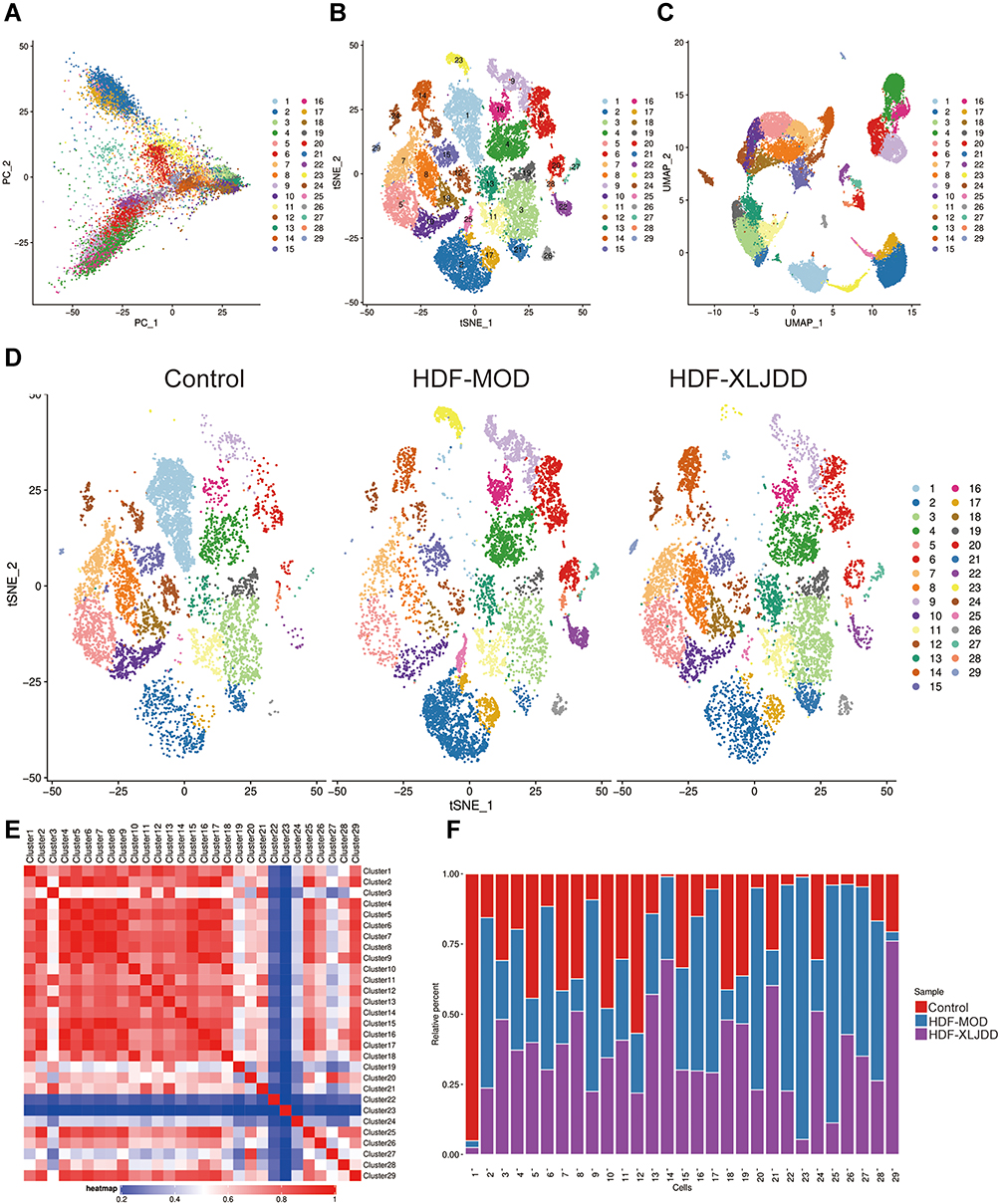 |
Figure 6 The alteration of tumor microenvironment caused by XLJDD. (A) The PCA result of single-cell sequencing profiles. (B) t-distributed stochastic neighbor embedding (t-SNE) plot of all cells from control, HDF-MOD, and HDF-XLJDD group. (C) Uniform manifold approximation and projection (UMAP) plot of all cells from control, HDF-MOD, and HDF-XLJDD group. (D) The t-SNE plot of control, HDF-MOD, and HDF-XLJDD group. (E) The correlations between the various clusters. (F) Histogram of the ratio of clusters in control, HDF-MOD and HDF-XLJDD group. |
Interestingly, through cell type prediction, we found that cell clusters rescued by XLJDD, such as cluster 2, cluster 17, cluster 23, and cluster 25, were predicted to be B cells (Figure 7A). Further analysis results showed that B cells can be divided into different sub-categories based on its marker molecules, such as (CD74+ EBF1+ CD79A+ H2Aa+) B cells and (IgIc1+ Igha+ Igkc+ Jchain+) B cells (Figure 7B). Moreover, we found that the Oxidative phosphorylation pathway was enriched, regardless of the treatment of XLJDD. However, only the Ferroptosis pathway was enriched after the treatment of XLJDD in these four clusters. These results hinted that the regulation of B cells by XLJDD may be mediated by the ferroptosis pathway (Figure 7C–J). Above all, XLJDD could suppress tumorigenesis and the development in the HDF-XLJDD groups by altering the tumor microenvironment, especially the composition ratio of B cells.
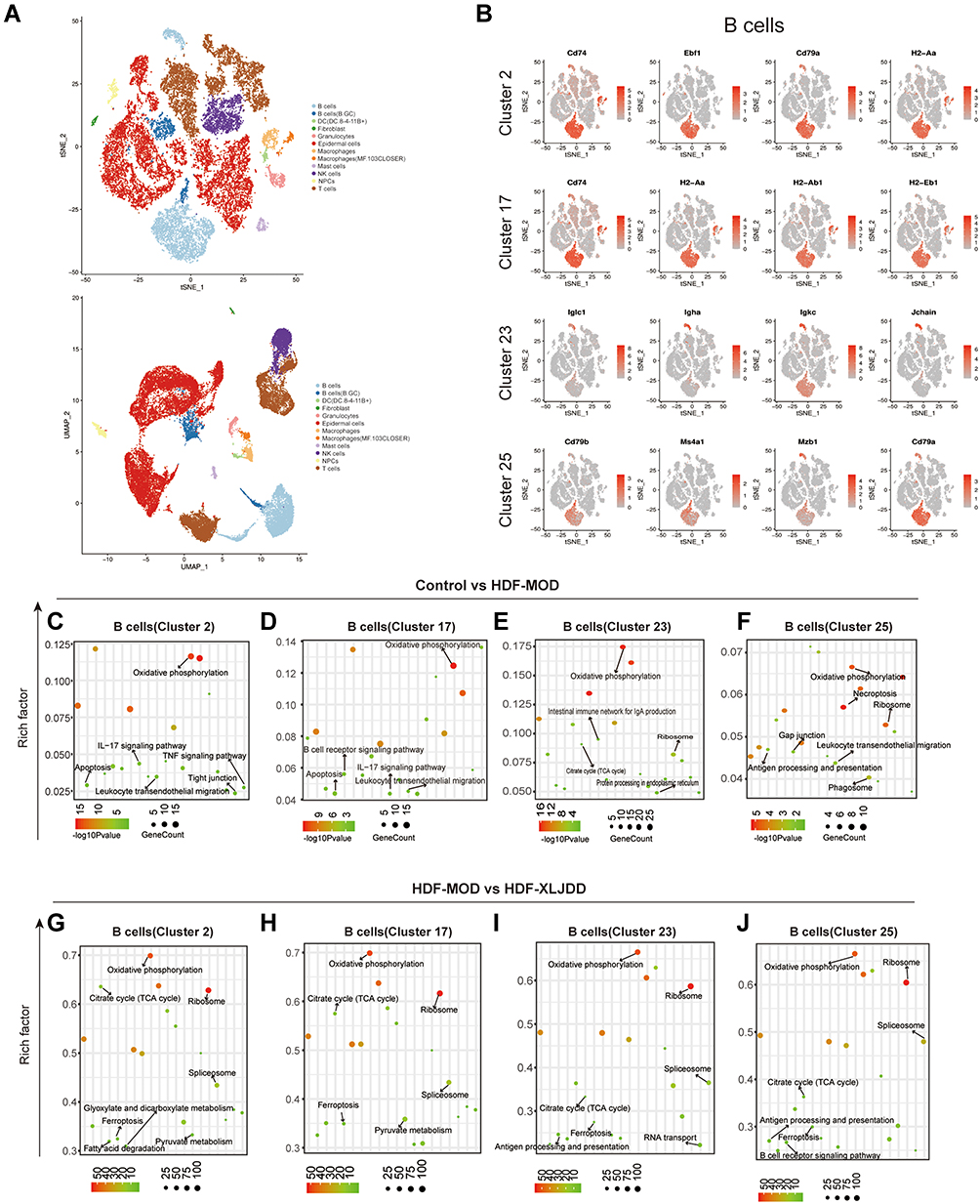 |
Figure 7 Identification of B cells clusters by Single-cell Atlas. (A) The t-SNE plot and UMAP plot showed the predicted type of cells. (B) The t-SNE plot of cluster with marker genes expression, the red dots represent the cells with high expression of the marker gene in the total detected cells. (C–F) The enrichment analysis of differential genes in B cells (cluster 2, cluster 17, cluster 23, cluster 25) between control and HDF-MOD groups. (G–J) The enrichment analysis of differential genes in B cells (cluster 2, cluster 17, cluster 23, cluster 25) between HDF-MOD and HDF-XLJDD groups. |
Discussion
Our team has been committed to studying the mechanism of classic Chinese medicine prescriptions and clinically effective prescriptions for several years, such as the famous Xiao-Ai-Jie-Du prescription. Recently, our attention has been attracted by another Chinese medicine prescription, XLJDD. It was found that some UC-CRC patients taking XLJDD could effectively relieve tumor progression. In order to illustrate the mechanism of XLJDD, our team has planned to carry out clinical trials, animals, and molecular experiments to explain its potential mechanism of action. At present, animal experiments, including AOM/DSS models, orthotopic models of colorectal tumors, and subcutaneous tumor models, have been completed.
In this study, we mainly used the AOM/DSS model to simulate the progress of UC-CRC to study the mechanism of XLJDD on the treatment of UC-CRC. We used orthotopic models and clinical sample tissues to clarify high-throughput sequencing results. It is worth mentioning that multiple high-throughput sequencing technologies were adopted in our study. As a result, thousands of genes and proteins were detected, and the immune cell infiltration map of the tumor microenvironment was also displayed. As mentioned above, multiple important modules have been identified, and the corresponding pathway, such as the cell-killing pathway, was enriched. In the single-cell sequencing results, we also found that XLJDD could regulate the infiltration capacity of multiple immune cells in the tumor microenvironment. The proportion of B cells in the XLJDD group was significantly lower than that in the model group, which was almost similar to the control group, suggesting that the XLJDD’s modulation of immune cell infiltration in the tumor microenvironment was indispensable for its therapeutic effect. Previously, other researchers have also identified multiple key tumor molecules through high-throughput technology.28,29 For example, Manoochehri et al found that CDC25A is a key gene for the prognosis of colorectal cancer.30 Khoei et al found that miR-299-5p is an important prognostic factor for a variety of tumors.31
Although high-throughput technology can provide an objective perspective for us to understand the data of XLJDD and enrich our understanding of the mechanism of action, it also brings us some confusion. As shown in Figure S1E, even for the same sample, the consistency of the RNA expression data and the protein expression data was still quite different, suggesting that the expression of the RNA may not necessarily lead to the expression of the protein. There are many reasons for this, which may be caused by multiple steps such as post-transcriptional modification,32 alternative splicings,33 protein translation,34 and post-translational modification.35 It brings difficulties for us to analyze mRNA-protein data jointly. However, analyzing these two types of data separately can also allow us to explain the mechanism of XLJDD from different perspectives.
As the results showed, we found two crucial targets of XLJDD, Mfsd2a, and Ccdc85c from transcriptomics sequencing and proteomics sequencing, respectively. Research on these two genes in tumors was relatively rare, especially in colorectal tumors. In lung cancer, Spinola et al reported that Mfsd2a was a typical tumor suppressor gene, which could play a role by blocking the progression of the G1 phase and impairing the invasion ability of cells.36 Shi et al found the lower expression of Mfsd2a in gastric cancer tissues. The expression of Mfsd2a in moderate/well-differentiated gastric cancer was significantly higher than that in poorly differentiated gastric cancer, and the expression in early gastric cancer was significantly higher than that in advanced gastric cancer. The prognosis of Mfsd2a-positive patients was significantly better than that of Mfsd2a-negative patients.37 In this study, we found that XLJDD can significantly activate the expression of Mfsd2a. Experimental evidence showed that this regulatory relationship could be repeated in orthotopic colorectal tumors and UC-CRC patients, suggesting this was a conservative regulatory relationship. Interestingly, the high expression of Mfsd2a in vascular endothelial progenitor cells can effectively prevent UC, and Mfsd2a activation was essential for the function of CD8+ T cells. The higher the proportion of CD8+ T cells in the tumor microenvironment, the better the patient’s prognosis.38 Besides, our results also showed that the function of XLJDD could not depend on TNF-α (Figure S1E), suggesting that XLJDD may prevent UC-CRC through a non-classical pathway. We speculated that the XLJDD-Mfsd2a regulatory axis enables many kinds of cells to work together to suppress tumors in the tumor microenvironment. Compared with Mfsd2a, the reports of Ccdc85 were even rarer. Guo et al reported that Ccdc85 could activate beta-catenin and promote liver cancer progression.39 We found that XLJDD could alter the expression of Ccdc85, but the specific mechanism remains to be further studied.
B cells are classically known to positively modulate immune responses and inflammation through antibody production and promote T-cell activation and proliferation through antigen presentation.40 Recently, an emerging role for B cells with immunosuppressive or regulatory functions in modulating anti-tumor immune responses has been described.41 Conversely, some studies described that B cells showed to possess a protective rather than an immunosuppressive function. In this study, our results showed that B cells were significantly upregulated in the colorectal model group but downregulated in the XLJDD-treated group, returning to a similar ratio to the control group. This evidence showed that B cells might play a role in promoting tumorigenesis and development in our system.
Conclusion
In conclusion, we demonstrated that XLJDD could prevent the initiation and development of colorectal tumors by activating the expression of Mfsd2a and the expression of Ccdc85c and reducing the infiltration of B cells in the colorectal tumor environment. In future research, we will study the functions of Mfsd2a and Ccdc85c in tumor cells and tumor microenvironment cells to explore their potential molecular mechanisms. In addition, we will also actively explore the possibility of XLJDD combined with modulators of target genes, hoping for clinical efficacy.
Acknowledgments
We would like to thank Dr. Zhengjie Shen from The Affiliated Zhangjiagang Hospital of Soochow University for his experimental advice, and Pro. Shulan Su from Nanjing University of Chinese Medicine for her work to perform UPLC-TQ-MS analysis for XLJDD.
Funding
This work was supported by Grants from the National Natural Science Foundation of China (81804073, 82004310, 81930117).
Disclosure
The authors declare that there are no competing financial interests and conflicts of interest in this work.
References
1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
2. Rogler G. Chronic ulcerative colitis and colorectal cancer. Cancer Lett. 2014;345(2):235–241. doi:10.1016/j.canlet.2013.07.032
3. Yashiro M. Ulcerative colitis-associated colorectal cancer. World J Gastroenterol. 2014;20(44):16389–16397. doi:10.3748/wjg.v20.i44.16389
4. Kobayashi T, Siegmund B, Le Berre C, et al. Ulcerative colitis. Nat Rev Dis Primers. 2020;6(1):74. doi:10.1038/s41572-020-0205-x
5. Yassin M, Sadowska Z, Djurhuus D, et al. Upregulation of PD-1 follows tumour development in the AOM/DSS model of inflammation-induced colorectal cancer in mice. Immunology. 2019;158(1):35–46. doi:10.1111/imm.13093
6. De Robertis M, Massi E, Poeta ML, et al. The AOM/DSS murine model for the study of colon carcinogenesis: from pathways to diagnosis and therapy studies. J Carcinog. 2011;10:9. doi:10.4103/1477-3163.78279
7. Parang B, Barrett CW, Williams CS. AOM/DSS model of colitis-associated cancer. Methods Mol Biol. 2016;1422:297–307.
8. Wang G, Ma N, He F, et al. Taurine attenuates carcinogenicity in ulcerative colitis-colorectal cancer mouse model. Oxid Med Cell Longev. 2020;2020:7935917. doi:10.1155/2020/7935917
9. Wang W, Li X, Zheng D, et al. Dynamic changes and functions of macrophages and M1/M2 subpopulations during ulcerative colitis-associated carcinogenesis in an AOM/DSS mouse model. Mol Med Rep. 2015;11(4):2397–2406. doi:10.3892/mmr.2014.3018
10. Tanaka T, Kohno H, Suzuki R, Yamada Y, Sugie S, Mori H. A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci. 2003;94(11):965–973. doi:10.1111/j.1349-7006.2003.tb01386.x
11. Hong M, Tao S, Zhang L, et al. RNA sequencing: new technologies and applications in cancer research. J Hematol Oncol. 2020;13(1):166. doi:10.1186/s13045-020-01005-x
12. Reuter JA, Spacek DV, Snyder MP. High-throughput sequencing technologies. Mol Cell. 2015;58(4):586–597. doi:10.1016/j.molcel.2015.05.004
13. Baslan T, Hicks J. Unravelling biology and shifting paradigms in cancer with single-cell sequencing. Nat Rev Cancer. 2017;17(9):557–569. doi:10.1038/nrc.2017.58
14. Tan HT, Lee YH, Chung MC. Cancer proteomics. Mass Spectrom Rev. 2012;31(5):583–605. doi:10.1002/mas.20356
15. Li X, Wang W, Chen J. Recent progress in mass spectrometry proteomics for biomedical research. Sci China Life Sci. 2017;60(10):1093–1113. doi:10.1007/s11427-017-9175-2
16. Oliver GR, Hart SN, Klee EW. Bioinformatics for clinical next generation sequencing. Clin Chem. 2015;61(1):124–135. doi:10.1373/clinchem.2014.224360
17. Hwang B, Lee JH, Bang D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp Mol Med. 2018;50(8):1–14. doi:10.1038/s12276-018-0071-8
18. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. doi:10.1073/pnas.0506580102
19. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 2008;9:559. doi:10.1186/1471-2105-9-559
20. Normile D. Asian medicine. The new face of traditional Chinese medicine. Science. 2003;299(5604):188–190. doi:10.1126/science.299.5604.188
21. Xu HY, Zhang YQ, Liu ZM, et al. ETCM: an encyclopaedia of traditional Chinese medicine. Nucleic Acids Res. 2019;47(D1):D976–D982. doi:10.1093/nar/gky987
22. Fan H, Liu S, Shen W, et al. Identification of the absorbed components and metabolites of Xiao-Ai-Jie-Du decoction and their distribution in rats using ultra high-performance liquid chromatography/quadrupole time-of-flight mass spectrometry. J Pharm Biomed Anal. 2020;179:112984. doi:10.1016/j.jpba.2019.112984
23. Qiu WI, Chen HB, Jiang ZQ, Zhou HG. [Effect of Xiaoai Jiedu Recipe on mIRNA Expression Profiles in H(2)(2) Tumor-bearing Mice]. Zhongguo Zhong Xi Yi Jie He Za Zhi. 2016;36(9):1112–1118. Chinese.
24. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):W98–W102. doi:10.1093/nar/gkx247
25. Eser Ocak P, Ocak U, Sherchan P, Zhang JH, Tang J. Insights into major facilitator superfamily domain-containing protein-2a (Mfsd2a) in physiology and pathophysiology. What do we know so far? J Neurosci Res. 2020;98(1):29–41. doi:10.1002/jnr.24327
26. Ungaro F, Tacconi C, Massimino L, et al. MFSD2A promotes endothelial generation of inflammation-resolving lipid mediators and reduces colitis in mice. Gastroenterology. 2017;153(5):1363–1377. doi:10.1053/j.gastro.2017.07.048
27. Roma-Rodrigues C, Mendes R, Baptista PV, Fernandes AR. Targeting tumor microenvironment for cancer therapy. Int J Mol Sci. 2019;20(4):840. doi:10.3390/ijms20040840
28. Manoochehri H, Sheykhhasan M, Samadi P, Pourjafar M, Saidijam M. System biological and experimental validation of miRNAs target genes involved in colorectal cancer radiation response. Gene Rep. 2019;17:100540. doi:10.1016/j.genrep.2019.100540
29. Manoochehri H, Jalali A, Tanzadehpanah H, Taherkhani A, Saidijam M. Identification of key gene targets for sensitizing colorectal cancer to chemoradiation: an integrative network analysis on multiple transcriptomics data. J Gastrointest Cancer. 2021. doi:10.1007/s12029-021-00690-2
30. Manoochehri H, Asadi S, Tanzadehpanah H, Sheykhhasan M, Ghorbani M. CDC25A is strongly associated with colorectal cancer stem cells and poor clinical outcome of patients. Gene Rep. 2021;25:101415. doi:10.1016/j.genrep.2021.101415
31. Gholamzadeh Khoei S, Manoochehri H, Saidijam M. Systemic biological study for identification of miR-299-5p target genes in cancer. Meta Gene. 2020;24:100655. doi:10.1016/j.mgene.2020.100655
32. Zhao BS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol. 2017;18(1):31–42. doi:10.1038/nrm.2016.132
33. Bonnal SC, Lopez-Oreja I, Valcarcel J. Roles and mechanisms of alternative splicing in cancer - implications for care. Nat Rev Clin Oncol. 2020;17(8):457–474. doi:10.1038/s41571-020-0350-x
34. Merrick WC, Pavitt GD. Protein synthesis initiation in eukaryotic cells. Cold Spring Harb Perspect Biol. 2018;10(12):a033092. doi:10.1101/cshperspect.a033092
35. Han ZJ, Feng YH, Gu BH, Li YM, Chen H. The post-translational modification, SUMOylation, and cancer (Review). Int J Oncol. 2018;52(4):1081–1094. doi:10.3892/ijo.2018.4280
36. Spinola M, Falvella FS, Colombo F, et al. MFSD2A is a novel lung tumor suppressor gene modulating cell cycle and matrix attachment. Mol Cancer. 2010;9:62. doi:10.1186/1476-4598-9-62
37. Shi X, Huang Y, Wang H, Zheng W, Chen S. MFSD2A expression predicts better prognosis in gastric cancer. Biochem Biophys Res Commun. 2018;505(3):699–704. doi:10.1016/j.bbrc.2018.09.156
38. Maimela NR, Liu S, Zhang Y. Fates of CD8+ T cells in tumor microenvironment. Comput Struct Biotechnol J. 2019;17:1–13. doi:10.1016/j.csbj.2018.11.004
39. Guo Y, Chai B, Jia J, et al. KLF7/VPS35 axis contributes to hepatocellular carcinoma progression through CCDC85C-activated β-catenin pathway. Cell Biosci. 2021;11(1):73. doi:10.1186/s13578-021-00585-6
40. LeBien TW, Tedder TF. B lymphocytes: how they develop and function. Blood. 2008;112(5):1570–1580. doi:10.1182/blood-2008-02-078071
41. Lu Y, Zhao Q, Liao JY, et al. Complement Signals Determine Opposite Effects of B Cells in Chemotherapy-Induced Immunity. Cell. 2020;180(6):1081–1097, e1024. doi:10.1016/j.cell.2020.02.015
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.