")
Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 14
Identification of Potential Key Genes and Regulatory Markers in Essential Thrombocythemia Through Integrated Bioinformatics Analysis and Clinical Validation
Authors Wang J , Wu Y, Uddin MN, Chen R, Hao JP
Received 4 March 2021
Accepted for publication 16 June 2021
Published 5 July 2021 Volume 2021:14 Pages 767—784
DOI https://doi.org/10.2147/PGPM.S309166
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Jie Wang,1,2 Yun Wu,3 Md Nazim Uddin,2,4 Rong Chen,5 Jian-Ping Hao5
1Department of Pharmacy, First Affiliated Hospital of Xinjiang Medical University, Urumqi, 830011, People’s Republic of China; 2School of Basic Medicine and Clinical Pharmacy, China Pharmaceutical University, Nanjing, 211198, People’s Republic of China; 3Department of General Medicine, First Affiliated Hospital of Xinjiang Medical University, Urumqi, 830011, People’s Republic of China; 4Bangladesh Council of Scientific and Industrial Research (BCSIR), Dhaka, 1205, Bangladesh; 5Department of Hematology, First Affiliated Hospital of Xinjiang Medical University, Urumqi, 830011, People’s Republic of China
Correspondence: Jian-Ping Hao No. 137 Liyushan S. Road, Urumqi, Xinjiang, People’s Republic of China
Tel +86 13579876416
Email [email protected]
Introduction: Essential thrombocytosis (ET) is a group of myeloproliferative neoplasms characterized by abnormal proliferation of platelet and megakaryocytes. Research on potential key genes and novel regulatory markers in essential thrombocythemia (ET) is still limited.
Methods: Downloading array profiles from the Gene Expression Omnibus database, we identified the differentially expressed genes (DEGs) through comprehensive bioinformatic analysis. GO, and REACTOME pathway enrichment analysis was used to predict the potential functions of DEGs. Besides, constructing a protein–protein interaction (PPI) network through the STRING database, we validated the expression level of hub genes in an independent cohort of ET, and the transcription factors (TFs) were detected in the regulatory networks of TFs and DEGs. And the candidate drugs that are targeting hub genes were identified using the DGIdb database.
Results: We identified 63 overlap DEGs that included 21 common up-regulated and 42 common down-regulated genes from two datasets. Functional enrichment analysis shows that the DEGs are mainly enriched in the immune system and inflammatory processes. Through PPI network analysis, ACTB, PTPRC, ACTR2, FYB, STAT1, ETS1, IL7R, IKZF1, FGL2, and CTSS were selected as hub genes. Interestingly, we found that the dysregulated hub genes are also aberrantly expressed in a bone marrow cohort of ET. Moreover, we found that the expression of CTSS, FGL2, IKZF1, STAT1, FYB, ACTR2, PTPRC, and ACTB genes were significantly under-expressed in ET (P< 0.05), which is consistent with our bioinformatics analysis. The ROC curve analysis also shows that these hub genes have good diagnostic value. Besides, we identified 4 TFs (SPI1, IRF4, SRF, and AR) as master transcriptional regulators that were associated with regulating the DEGs in ET. Cyclophosphamide, prednisone, fluorouracil, ruxolitinib, and lenalidomide were predicted as potential candidate drugs for the treatment of ET.
Discussion: These dysregulated genes and predicted key regulators had a significant relationship with the occurrence of ET with affecting the immune system and inflammation of the processes. Some of the immunomodulatory drugs have potential value by targeting ACTB, PTPRC, IL7R, and IKZF1 genes in the treatment of ET.
Keywords: essential thrombocythemia, hub genes, regulatory markers, candidate drugs, bioinformatics analysis
Introduction
Essential thrombocythemia (ET) belongs to a class of BCR/ABL negative myeloproliferative neoplasm (MPN) that is characterized by unexplained proliferation of megakaryocytes in the bone marrow.1 Generally speaking, the increased platelet is natural to be ignored by patients due to the lack of typical clinical characters and could result in persistent high platelet counts, even cause an embolism. However, the pathogenesis of the increased number of megakaryocytes is still unclear.2 When thrombotic complications, bleeding, and leukemic transformation occur, which could severely affect the patient’s quality of life.3 In recent years, targeted therapeutic drugs are being explored as a mono-or combination therapy in this field, such as ruxolitinib, pacritinib, fedratinib.4 Although it seems that some existing researches focus on ET pathogenesis, there still limited treatment is available on ET; also, the mechanism of ET is complicated, and more genetic mutations involved.5,6 Thus, identifying molecular biomarkers associated with ET is crucially essential for personalized therapy.
Gene chip is a new nucleic acid analysis and detection technology, and the relevant analysis results have been stored in public databases.7 Therefore, gene expression profiling is an appropriate application field of gene chip technology, and integrating and re-analyzing these genomic data to identifying specific biomarkers that are closely related to the disease provides new avenues.8 Recently, with the massive generation of tumor sequencing data, bioinformatics is developing rapidly in hematological malignancies.9 Gene Expression Omnibus (GEO) database serves as a public gene expression profile repository for high-throughput experimental data, and this allows us to comprehensively clarify the pathogenesis of ET from extensive data analysis at the multi-gene level.10 In this study, we will thoroughly analyze genomic data in more detail to explore potential biological therapeutic targets.
Materials and Methods
Microarray Data Analysis and DEG Identification
The gene expression profiles were obtained from The National Center for Biotechnology Information-Gene Expression Omnibus Data Sets (NCBI-GEO Datasets, https://www.ncbi.nlm.nih.gov/gds/). The GSE61629 expression data was based on the GPL570 platform, which uses HG-U133_Plus_2 Affymetrix Human Genome U133 Plus 2.0 arrays and includes 54 total RNA samples from whole blood [21control subjects, 8 ET patients, 21 polycythemia vera (PV) patients, 4 primary myelofibrosis (PMF) patients].11 The GSE124281 expression data was based on the GPL10558 platform, which profiling by uses Illumina HumanHT-12 V4.0 expression bead chip and includes 22 RNA samples from whole blood (8 control subjects, 5 ET patients, 9 PMF patients). Another dataset, GSE123732, was used as an independent cohort for validating the results. The expression data of GSE123732 was based on the GPL10558 Illumina HumanHT-12 V4.0 expression bead chip platform and contains 6 RNA samples from bone marrow mononuclear cells (3 ET patients and 3 healthy individuals). In this study, the raw data from Affymetrix were processed using the robust multiarray (RMA) method for background correction, normalization quality control, and gene expression index summarization in the “Affy” package.12 The raw data from Illumina were processed using the “Lumi,”13 “org.Hs.eg.db”,14 and “Limma”15 packages from Bioconductor in the R program (Version 3.6.2) were used to annotate the microarray data and identify DEGs between patients with ET and control subjects. Genes with the absolute value of |log2FC|>0.5 and P < 0.05 were used as the cut-off criteria of DEG analysis. The DEGs data were processed by “pheatmap”16 and “ggrepel”17 packages in the R program to draw a heatmap and volcano plot of the significantly changed genes. Venn diagrams were used to show up-regulated common genes, down-regulated common genes, and up-regulated versus down-regulated common genes.
Functional Enrichment Analysis of DEGs
The Gene Ontology (GO) and the pathway enrichment analysis of DEGs were carried out using the ClusterProfiler package of the R program (Version 3.6.2).18 GO terms enrichment analysis include biological process (BP), molecular function (MF), cellular component (CC). The pathway analysis was carried out using REACTOME (http://www.reactome.org) online database, and P-value <0.05 was considered the cut-off criterion.
Prediction of the Master Transcription Factors (TFs) Significantly Regulating the DEGs
To predict master TFs that are remarkably associated with the regulating of DEGs, we have utilized the iRegulon plugin of Cytoscape software (version 3.8.0) to detect regulons from all DEGs.19 For this purpose, we used an extensive collection of TF motifs and a large collection of ChIP-seq tracks. The iRegulon method depends on a ranking-and-recovery system where all genes of the human genome are scored by a motif discovery step integrating the clustering of binding sites within cis-regulatory modules (CRMs) and the potential distal location of CRMs upstream or downstream of the transcription start site (TSS ±10 kb). The recovery step calculates the normalized enrichment score (NES) of TFs for each set of genes, input for each of the individual analyses, leading to the prediction of the TFs based on NES and their putative direct target genes which exist in the input lists. This method optimizes the association of TFs to motifs using both explicit annotations and predictions of TF orthologs and motif similarity. A transcription factor normalized enrichment score was computed for each group where a normalized enrichment score > 4.0 corresponds, and the maximum false discovery rate (FDR) on motif similarity set as 0.001.
Construction and Analysis of Protein–Protein Interaction (PPI) Network
The network of predicted associations for DEGs was searched through the STRING online database (STRING, http://string‐db.org, version 11.0).20 DEGs with a Combined Score ≥0.4 was set as the cut-off criterion to construct the PPI network, and then the PPI network was visualized Cytoscape software (version 3.8.0).21 The Molecular Complex Detection (MCODE) plugin of Cytoscape software was used to detect the functional clusters of the PPI network,22 cluster finding parameters sets as Node Score Cutoff=0.2; K-Core= 2; and Max.Depth= 100. Then the top 10 Hub genes were filtered from the results of the CytoHubba plugin of Cytoscape software,23 in this work, and we ranked hub nodes by the Maximal Centrality Clique (MCC) method.
Expression Level Differentially Expressed Hub Genes in a Bone Marrow Cohort of ET
The expression levels of top hub genes were further investigated using an independent cohort (GSE123732) associated with ET. We downloaded this dataset from NCBI that contains the ET patients with healthy controls (n=6). We used ET patients with matched healthy controls for validating our findings. We employed the “limma” package in the R program from the Bioconductor project (http://www.bioconductor.org/) for identifying differential expression of significant hub genes in ET. Hub genes with |log2FC| >0.50 and P < 0.05 were considered as statistically significant.
qRT-PCR Verify the Expression Level of Hub Genes in the Clinical Samples
To clarify whether the expression profile of key genes also had consistent results in our clinical samples, we further validated the expression levels of hub genes in peripheral blood from 10 ET patients (diagnosed according to the WHO 2016 diagnostic criteria, including five patients with JAK2 V617F mutation) using qPCR. Total RNA was isolated from 1 mL of whole blood using Invitrogen Trizol (Carlsbad, CA, USA), followed by standard phenol-chloroform extraction. Complementary DNA (cDNA) was quantified using a reverse transcription kit (K1622, Thermo Fisher Scientific, USA), and the expression levels of hub genes were performed using SYBR Green Master Mix (SYBR GREEN, Beijing, China). The housekeeping gene GAPDH was used as an internal control. The primers were synthesized by Shanghai Biotechnology Co., Ltd (Table 1). qRT-PCR was performed on ViiATM 7 system software (Thermo Fisher Scientific, ABI7500, USA). The results were normalized to the expression of GAPDH and expressed as fold change (2−ΔΔCT). The qRT-PCR experiment on each clinical sample with three biological replicates. Peripheral whole blood from 10 anonymous healthy volunteers was used as control samples.
 |
Table 1 The Primers for qRT-PCR |
ROC Curve Analysis of Hub Genes
The receiver operating characteristic (ROC) curve was applied to examine the diagnostic value of differential hub genes in mRNA expression for distinguishing control subjects from ET patients. The statistical analysis and visualization were performed using GraphPad Prism 8.0 software (version 8.3.0). The hub genes with an area under the ROC curve (AUC)≥0.5 consider significant sensitivity and specificity, and 0.8≤AUC≤1.0 indicates that hub genes may have remarkable diagnostic value.
Identification of Food and Drug Administration (FDA)-Approved Drug-Hub Gene Interaction
We identified the potential candidate drugs that target the differentially dysregulated hub genes by using the DGIdb.24 DGIdb collects drug-gene interaction data from 30 disparate sources, including ChEMBL, DrugBank, Ensembl, NCBI Entrez, PharmGKB, PubChem, Clinical Trial Databases, and literature in NCBI PubMed. The drug-gene interactions supported by at least one database and PubMed reference were identified. We selected the only drugs that have been approved by the FDA.
Results
Identification of Differentially Expressed Genes in ET
For identifying DEGs in MPNs, we obtained gene expression data from three datasets of patients with Essential Thrombocythemia and healthy controls. A total of 864 DEGs were identified from GSE124281, and 717 DEGs were identified from GSE61629, respectively. It should be noted that the datasets GSE124281 and GSE61629 were both include ET, PV, and PMF samples, so we have further selected expression profiling before treatment in ET patients and healthy controls from the raw data. Expectedly, we got very few genes between up and down groups. Figure 1A and B show the heatmap and volcano plot of DEGs; these DEGs were well clustered between ET patients and healthy controls. After that, the Venn diagram in R language was used to identify the 63 overlapping DEGs between two gene expression profiles. Among 63 DEGs, we identified 21 common up-regulated and 42 common down-regulated genes, as well as up-regulated versus down-regulated common genes between two gene expression profiles (Figure 1C).
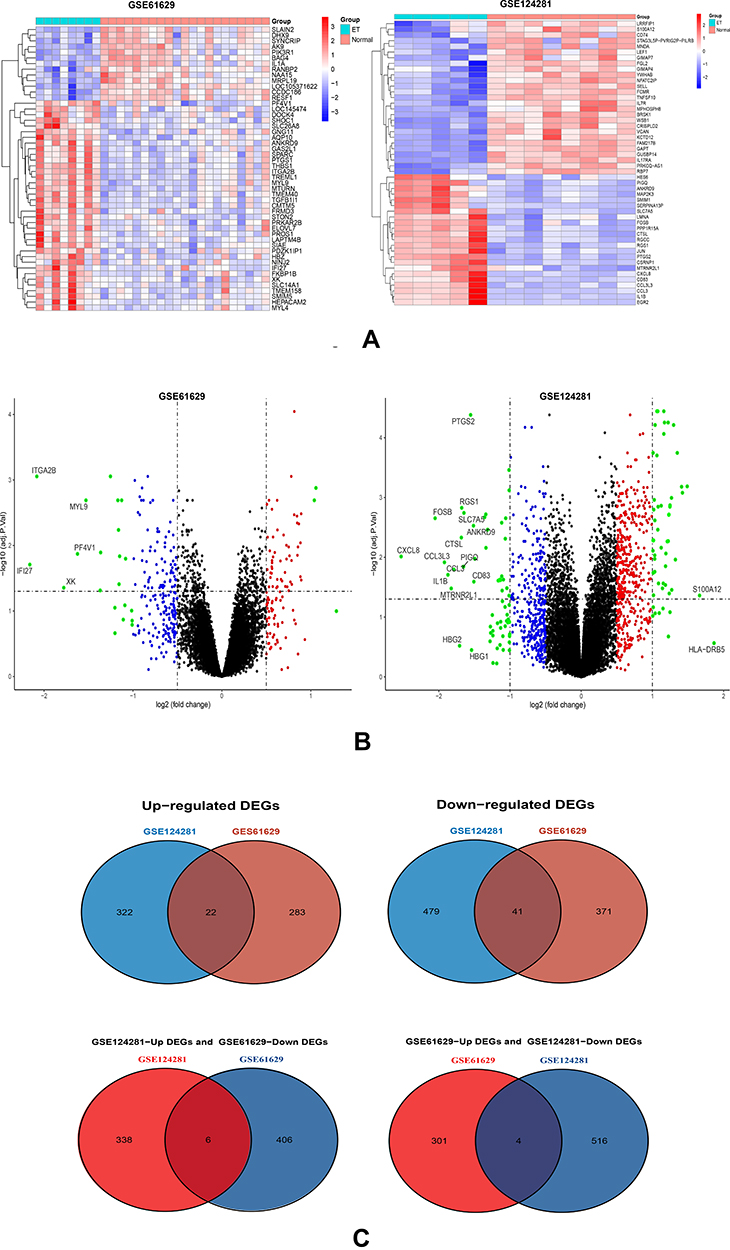 |
Figure 1 Identification of DEGs in two datasets (GSE61629 and GSE124281). (A) Heatmap of the top 50 DEGs in GSE621629 and GSE124281 datasets, respectively. (B) Volcano plot of DEGs in GSE61629 and GSE124281 datasets. Red and blue plots represent genes with [logFC]>0.5 and P<0.05, and black plots represent genes with no significant difference. Furthermore, the green plots represent up-regulated and down-regulated genes with [logFC]>1 and P<0.05, and the labeled genes represent these genes with [logFC]>1.5 and P<0.05. (C) Venn diagram of commonly changed DEGs in the two datasets. (including 22 common up-regulated genes, 41 common-down-regulated genes, six genes have interacted between GSE124281 up-regulated DEGs and GSE61629 downregulated DEGs, four genes have interacted between GSE61629 up-regulated DEGs GS124281 downregulated DEGs). DEGs, differentially expressed genes. DEGs, differentially expressed genes. |
GO Analysis and Function Enrichment of DEGs
The results of GO analysis demonstrated that biological processes (BP) of DEGs are mainly enriched in the neutrophil degranulation, neutrophil activation, and mediated involved in neutrophil-mediated immune response, regulation of hemopoiesis, receptor signaling pathway via STAT, and interleukin-6-mediated signaling pathway (Table 2, Figure 2A). For cellular component (CC), GO analysis results showed that the DEGs were primarily enriched in the focal adhesion and cell-substrate junction (Table 2, Figure 2B). Regarding molecular function (MF), the DEGs are mainly enriched in RNA polymerase II-specific DNA-binding transcription factor binding and DNA-binding transcription factor binding (Table 2, Figure 2C). Besides, the most significant pathways identified through REACTOME pathway analysis were present in Table 3, and the results have shown that the common DEGs were mainly related to the immune system, such as innate immune system, neutrophil degranulation, and interleukin-6 signaling.
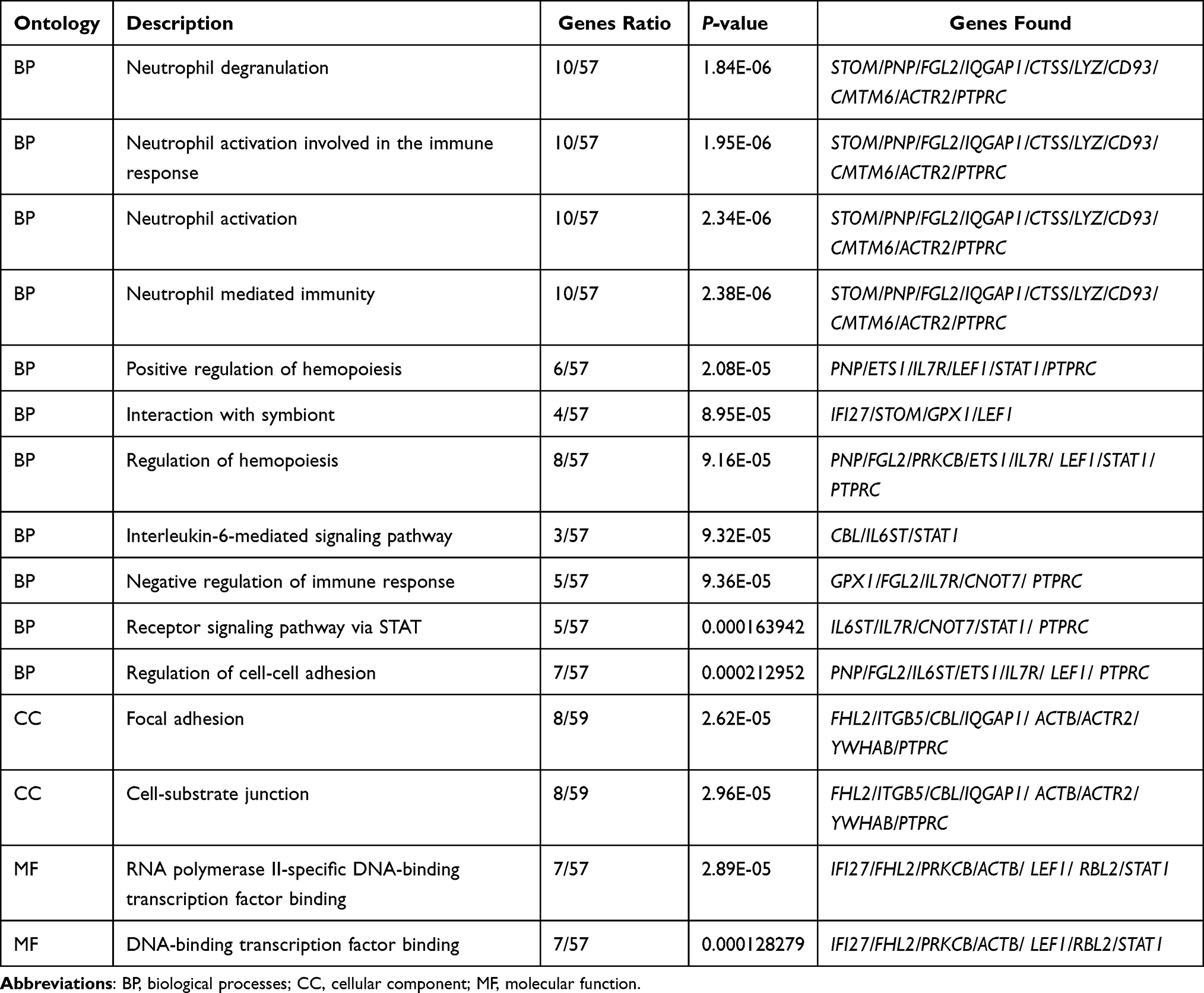 |
Table 2 Gene Ontology Term Enrichment Analysis of DEGs in Essential Thrombocythemia |
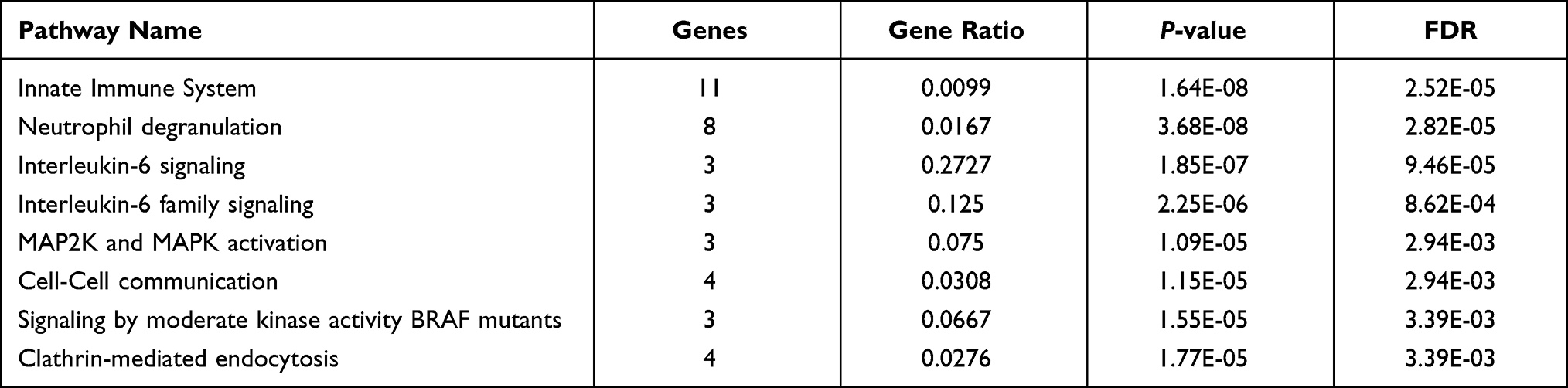 |
Table 3 Signaling Pathway Enrichment Analysis of DEGs in REACTOME |
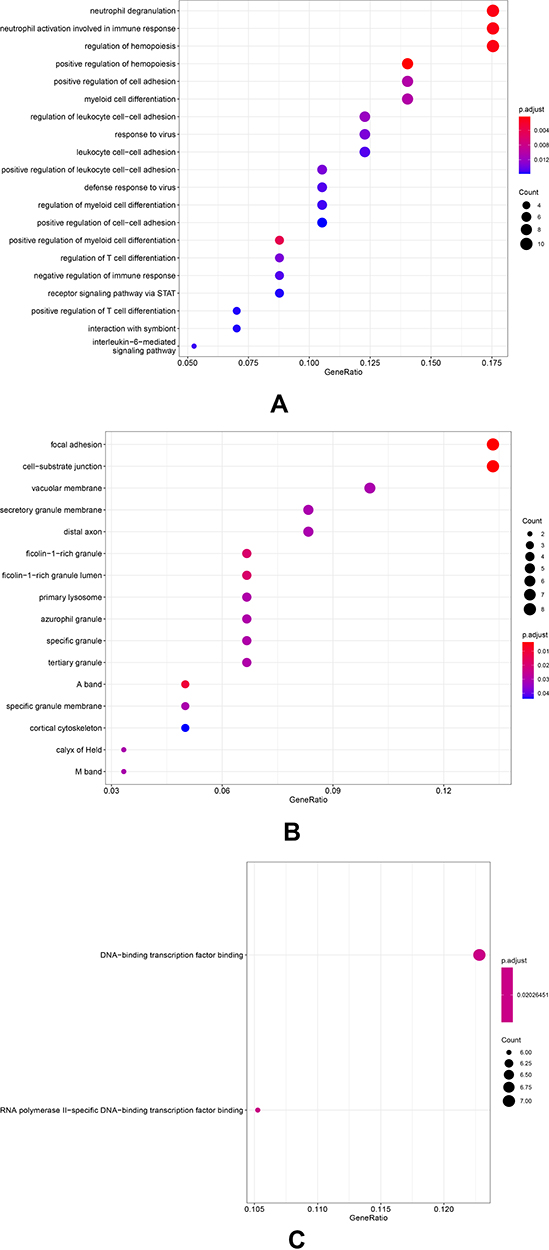 |
Figure 2 GO term enrichment analysis of DEGs. (A) Biological process. (B) Cellular component. (C) Molecular function. DEGs, differentially expressed genes. GO, Gene Ontology. |
Identification of TFs is Significantly Associated with the Overlap DEGs
We identified four TFs: SPI1, IRF4, SRF, and AR as master transcriptional regulators that are associated with regulating the DEGs in ET. Furthermore, we identified 8 (GTF2A1, TEAD4, E2F3, MYB, SP9, GATA2, CEBPA, IKZF2) and 6 (SPIB, FLI1, SPI1, ELK1, LEF1, CREB3) TFs for the up-regulated and the down-regulated genes in ET, respectively. The master TFs associated with the DEGs were displayed in the regulatory networks. In the networks, TEAD4 (TEA Domain Transcription Factor 4) and MYB (MYB Proto-Oncogene, Transcription Factor) both target 11 up-regulated genes (Figure 3A). SPI1(Spi-1 Proto-Oncogene, Hematopoietic Transcription Factor) and SPIB (Spi-B Transcription Factor) target 26 and 23 down-regulated genes separately (Figure 3B). Besides, IRF4 (Interferon Regulatory Factor 4, Transcription Factor) targets 35 up and down-regulated genes (Figure 3C). The results of TFs prediction will provide insights into the development and pathogenesis of ET.
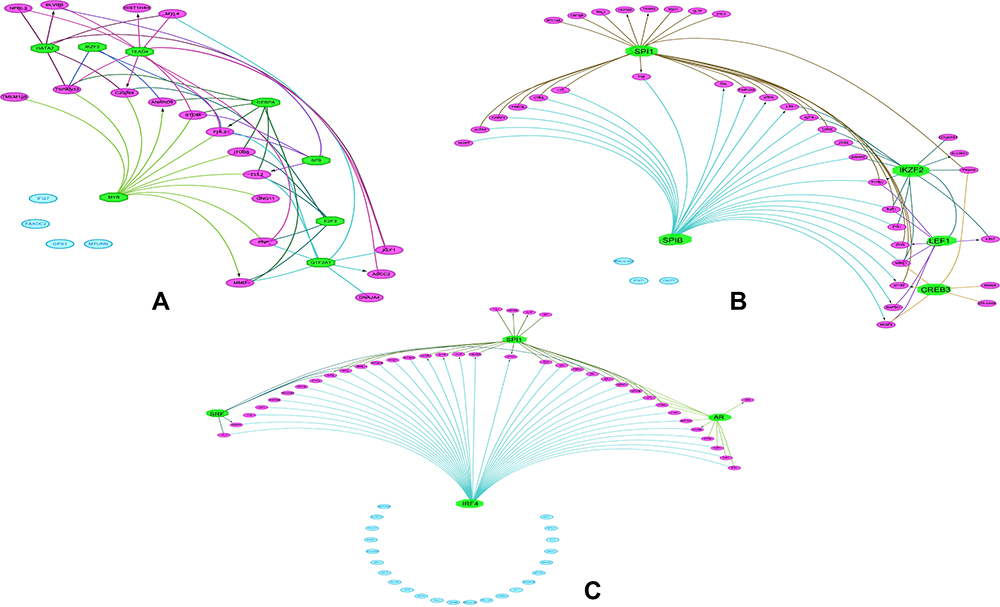 |
Figure 3 Regulatory networks of the TFs and their targeted DEGs identified by iRegulon. (A) Regulatory network of the TFs and their targeted up-regulated genes. (B) Regulatory network of the TFs and their targeted down-regulated genes. (C) Regulatory network of the TFs and their targeted up and down-regulated genes. The green color octagon indicates TFs, the purple color oval indicates the DEGs regulated by TFs, and the blue color oval indicates that TFs do not restrict them. TFs, transcriptional regulators; DEGs, differentially expressed genes. |
PPI Network Analysis and Hub Gene Identification
Based on the STRING database (version 11.0), we constructed a PPI network reflecting the functional association between DEGs and visualized the network analysis results in Cytoscape software (version 3.8.0) with 33 nodes and 54 edges, as shown in Figure 4. Then, we identified two clusters in the PPI network by utilizing the MCODE application in Cytoscape. Cluster 1 consists of four nodes and six edges, including FYB, FGL2, PTPRC, CTSS (Figure 5A). Cluster 2 contained three nodes and three edges, including CD93, STOM, CMTM6 (Figure 5B). The top 10 hub genes inside the network were identified with the CytoHubba plugin, including ACTB, PTPRC, ACTR2, FYB, STAT1, ETS1, IL7R, IKZF1, FGL2, and CTSS (Table 4, Figure 5C).
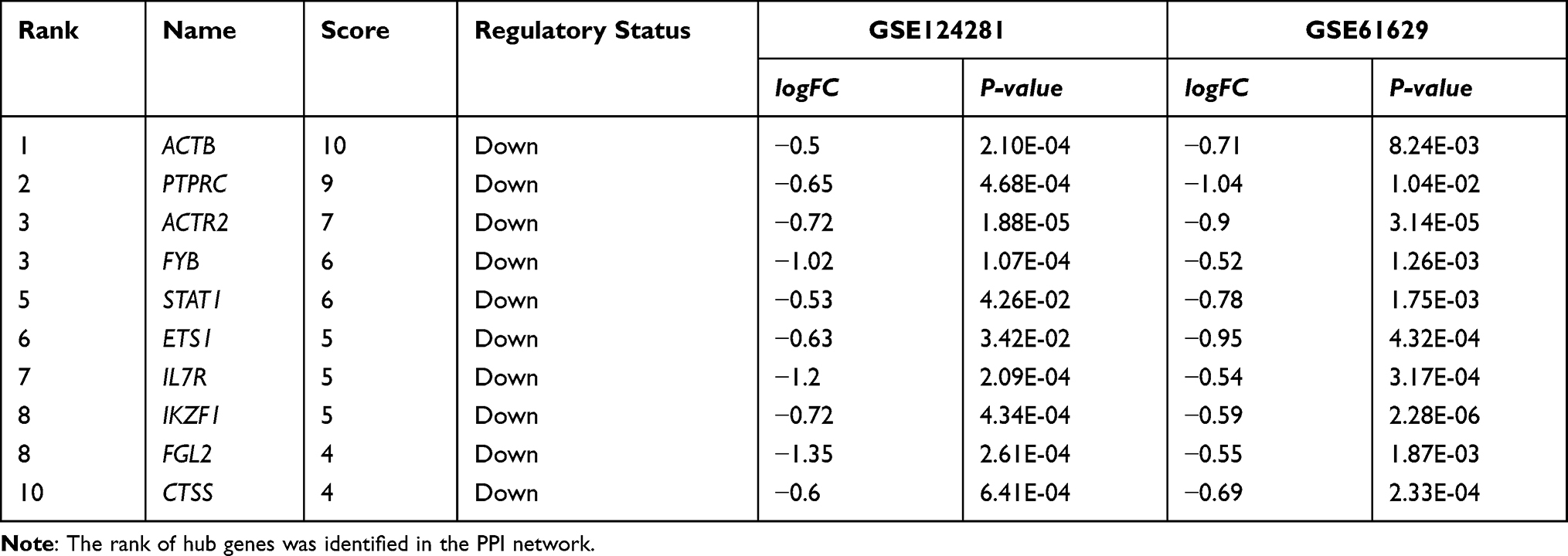 |
Table 4 The Rank of Hub Genes in the PPI Network of DEGs |
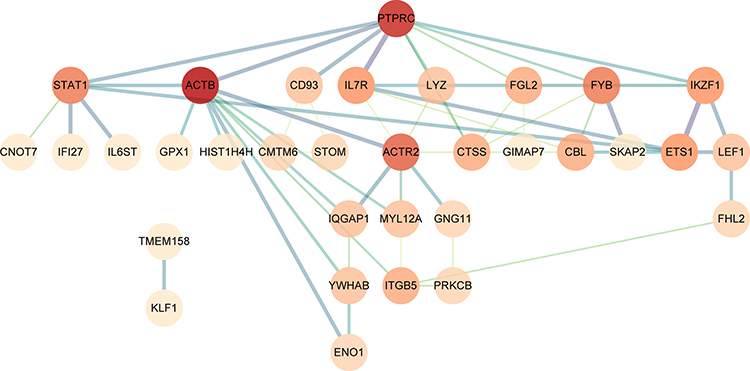 |
Figure 4 PPI network of DEGs in the STRING database. Based on the STRING online database, the DEGs PPI network was constructed containing 63 DEGs. The different colors in the figure indicate the connectivity of genes, and the deep color shows the genes with the highest connectivity in the PPI network. PPI, protein–protein interaction; DEGs, differentially expressed genes. |
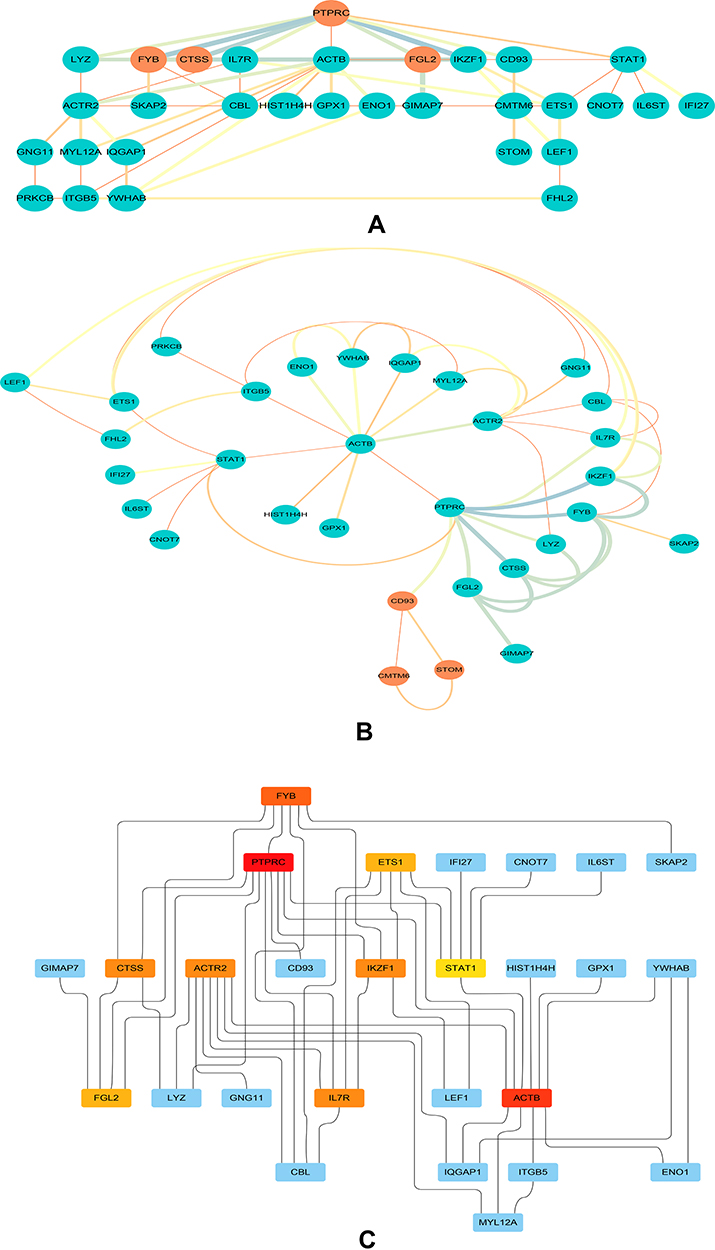 |
Figure 5 Clusters and hub genes identified in the PPI network. (A) Cluster 1 in the PPI network. The orange nodes represent the screened genes in cluster 1. (B) Cluster 2 in the PPI network. The orange nodes represent the screened genes in cluster 2. (C) Ten hub gene identification in a PPI network based on the MCC method. The dark (red) nodes show the genes with higher MCC scores in the PPI network. PPI, protein–protein interaction; MCC, maximal centrality clique. |
Differentially Expressed Hub Genes are Dysregulated in a Bone Marrow Cohort of ET
To investigate the hub genes expression in bone marrow, we re-analyzed the significant hub genes in an independent cohort. Interestingly, we found that the top ten down-regulated hub genes (ACTB, PTPRC, ACTR2, FYB, STAT1, ETS1, IL7R, IKZF1, FGL2, and CTSS) were also down-regulated in a bone marrow cohort containing ET and healthy samples (Figure 6). It indicates that these aberrantly expressed hub genes may have a vital contribution to the ET. ACTB, PTPRC, ACTR2, FYB, STAT1, IL7R, and FGL2 dysregulated hub genes are also downregulated in a bone marrow cohort of ET (P< 0.05). It indicates the consistency of our findings in this study. However, three of the down-regulated hub genes (ETS1, IKZF1, and CTSS) showed similar expression differences between ET and healthy samples but not significant (P> 0.05). This result Indicating that the hub genes are also deregulated in the bone marrow of ET patients.
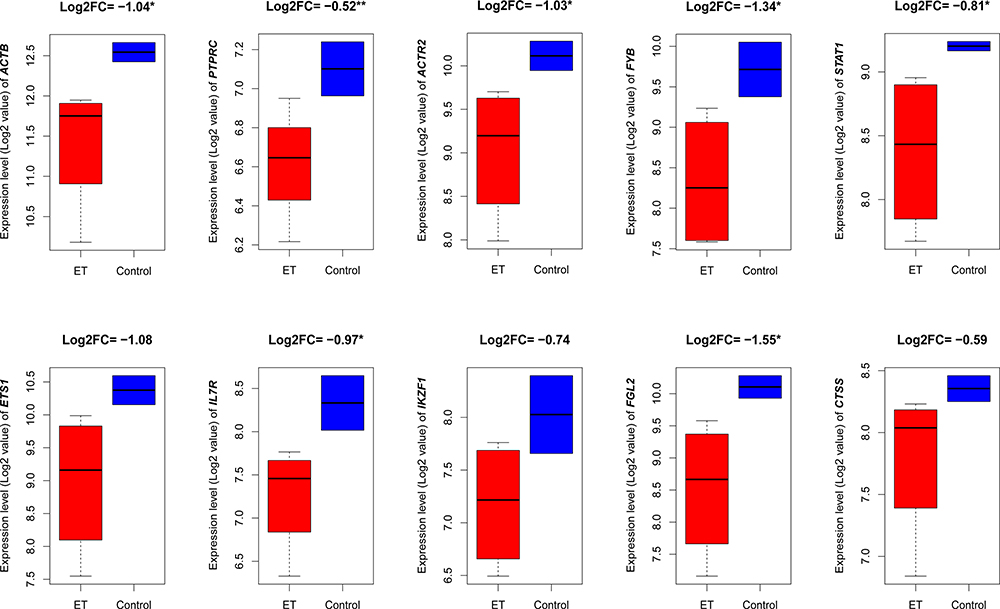 |
Figure 6 Validated the expression of the identified hub genes in an independent cohort group. The top 10 hub genes were both lower expressed in ET samples from the GSE123732 database, and the seven genes as ACTB, PTPRC, ACTR2, FYB, STAT1, IL7R, and FGL2 with a significant difference between ET and control. *P<0.05, **P<0.01. ET, essential thrombocythemia. |
Results of Expression Validation of Hub Genes in Clinical Samples
By further validating the hub genes in 20 clinical samples (10 ET vs 10 healthy individuals), we found that the expression of 8 of hub genes (CTSS, FGL2, IKZF1, STAT1, FYB, ACTR2, PTPRC, and ACTB) was significantly down-regulated in the ET samples (P<0.05), which was consistent with our analysis. However, two of the hub genes (IL7R and ETS1) were significantly up-regulated (P<0.05) in both ET samples (Figure 7). This result further indicates that these hub genes, which we identified in our previous analysis, might be recognized as potential key genes in ET.
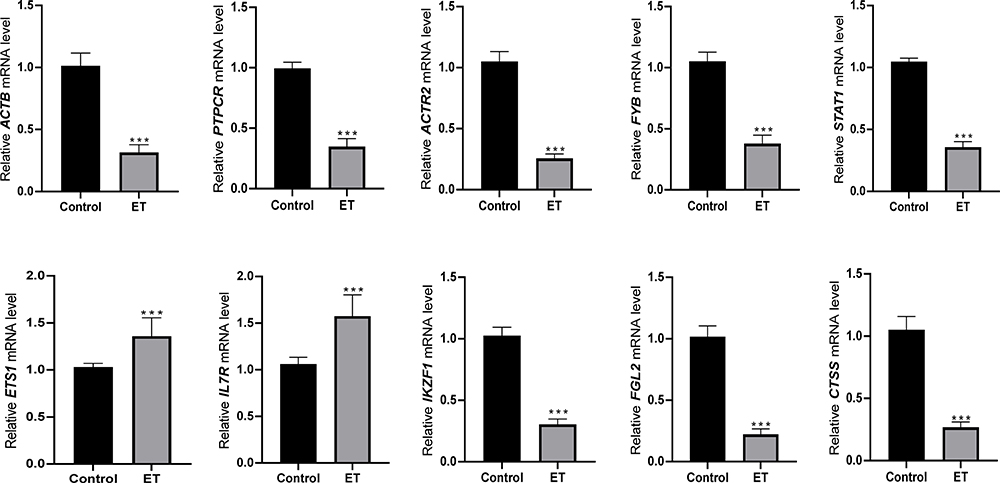 |
Figure 7 Validated the expression of the identified hub genes in clinical samples. Eight of the hub genes (CTSS, FGL2, IKZF1, STAT1, FYB, ACTR2, PTPRC, and ACTB) were both lower expressed in ET samples, and two hub genes (IL7R and ETS1) were higher expressed in ET samples. ***P<0.001. ET, essential thrombocythemia. |
The Expression Levels of Hub Genes are Associated with the Diagnostic Value in ET
To evaluate the diagnostic value of the dysregulated hub genes, we analyzed the correlation of hub gene expression levels. The performance of these ten hub genes in the ET group and healthy individuals was analyzed by the receiver operating characteristic (ROC) curve. The results show that the expression of these hub genes showed excellent diagnostic value between ET patients and healthy. All the hub genes showed a significant sensitivity and specificity between ET patients and healthy [area under the ROC curve (AUC)≥0.5] (Figure 8). ROC curves showed their high diagnostic value as biomarkers for ET (such as ACTB AUC: 0.975 in GSE 124281 and 0.768 in GSE61629; PTPRC AUC: 1.0 in GSE 124281 and 0.768 in GSE61629; STAT1 AUC: 0.8 in GSE 124281 and 0.5 in GSE61629; EST1 AUC: 0.8 in GSE 124281 and 0.893 in GSE61629; CTSS AUC: 0.975 in GSE 124281 and 0.774 in GSE61629). This result indicating the diagnostic significance of these aberrantly expressed hub genes in ET patients, and these genes may be promising targets for developing diagnostic markers to manage patients with ET.
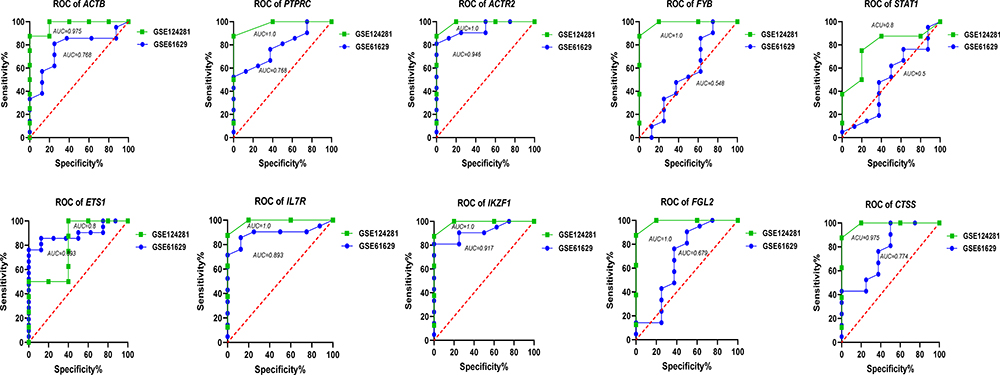 |
Figure 8 ROC curves employed to assess the diagnostic value of the hub genes. The ROC curve of the top 10 hub genes both showed a significant sensitivity and specificity in two databases (all the genes with AUC≥0.5). This result indicating these genes may have good diagnostic value for patients with ET. ROC, receiver operating characteristic. |
Identification of Candidate Drugs Targeting Hub Genes
We screened all ten hub genes for drug-gene interactions by using DGIdb, and we identified FDA-approved drugs that potentially target the protein products of four hub genes (PTPRC, ACTB, IL7R, IKZF1). Still, the interaction types between target genes and drugs are uncertain (Table 5). The results showed that cyclophosphamide, prednisone, fluorouracil, ruxolitinib, and lenalidomide both had been identified as potential medicine for ET treatment. However, as far as we know, the inhibitory effects of ACTB, PTPRC, IL7R, and IKZF1 have not been tested for the treatment of ET. Our data suggest that these genes may be promising targets for developing anticancer drugs to treat patients with ET.
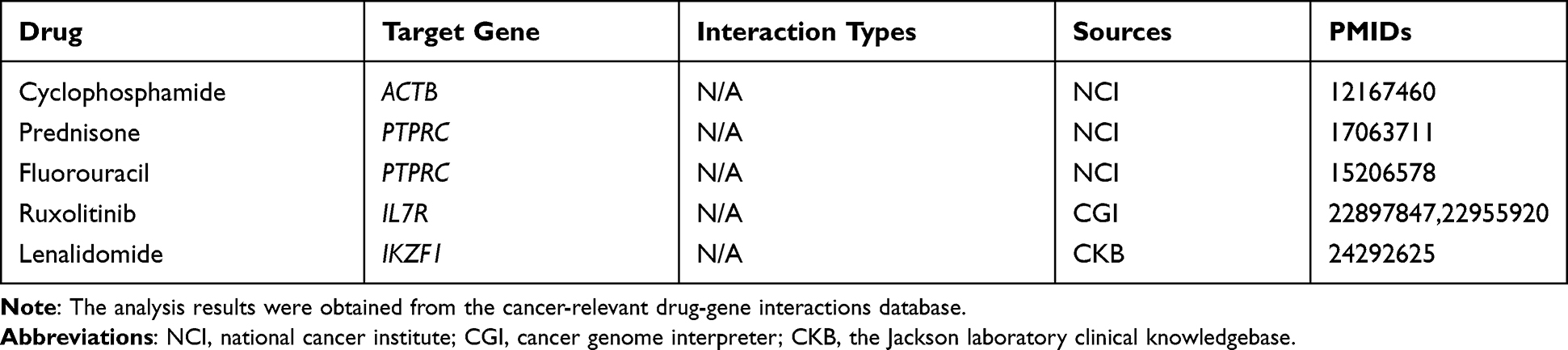 |
Table 5 FDA-Approved Drugs Potentially Targeting Five Hub Genes |
Discussion
The Philadelphia chromosome-negative myeloproliferative neoplasms (MPNs) are a group of clonal hematopoietic stem cell disorders. As far as we know, there are signaling pathways that are highly sensitive to cytokines and aberrantly activated in MPNs. However, the molecular mechanisms leading to the alteration of these signaling pathways are still unknown.25 Since 2005, the discovery of mutations in JAK2, CALR, and MPL genes has changed the classification and diagnosis of BCR/ABL-negative MPN. The JAK2V617F positive variations are in 50–60% of ET patients,26,27 MPL gene mutations are in 3%-5%, and CALR mutations are in 60–88% of JAK2 V617F mutation-negative ET patients.28,29 It is noteworthy that there are still 10–15% of patients with triple-negative gene mutations, and their genetic abnormalities are unknown. Recently, with the development of second-generation sequencing (NGS) and other whole-genome analysis techniques, many different gene mutations have been found in MPNs, including TET2, DNMT3A, IDH1/2, TP53, and EZH2.30
Nevertheless, these gene mutations also exist in other diseases, which are not specific to MPNs, and have a low frequency of mutations (< 10%).31 Some of these genes are directly involved in the pathogenesis of ET, and some are directly involved in epigenetic regulation. They can induce JAK2 mutations, and often earlier than JAK2 variation, suggesting that the pathogenesis of ET is the result of multiple gene disorders, not a single gene mutation.32 Recently, the rapid development of bioinformatics technology provides us with an advanced method to study the pathogenesis of disease more comprehensively and identify possible core targets, which can also offer the potential for the diagnosis and treatment of ET.
In this study, we selected two microarrays (accession GSE57793 and GSE124281) that met the inclusion criteria from the GEO databases and screened a total of 63 overlap DEGs. GO analysis results showed that the DEGs significantly enriched in the immune system and regulation of hemopoiesis, like neutrophil mediated immune response, Interleukin-6-mediated signaling pathway, and receptor signaling pathway via STAT. Neutrophils are an essential part of the human immune system,33 they could make an active immune response, and the formation of the neutrophil extracellular trap (NET) has been known as a component of innate immunity.34 It is currently recognized that NET could be triggered by a variety of proinflammatory cytokines, such as increased levels of IL-6, IL-1, IL-8, and TNFα.35 Although NETs are mainly involved in host defense, they also provide a suitable scaffold for the formation of blood clots and induce a robust procoagulant response by binding red blood cells, platelets, and von Willebrand factor (VWF).36 Through the contact phase, activation NETs will link the processes between innate immunity and thrombosis, and it explains how NET promotes the function of immune thrombosis. Recent research found that neutrophils isolated from patients with JAK2 V617F mutations in MPNs showed up-regulation of NADPH oxidase activity, which is closely related to the increase in ROS production.37 Furthermore, the other study showed that inhibition of JAK2 could lead to a reduction in neutrophils induced superoxide production.38 The results have further confirmed that the abnormal activation of JAK2 V617F is related to the excessive activation of neutrophil NADPH oxidase, and the neutrophils could play a vital role in the activation of oxidative stress and the occurrence of myeloproliferative neoplasms. These conclusions also provide a new explanation for the occurrence of embolism in ET patients.
Regulation and misregulation of TFs are associated with human diseases since TFs crucially controlled the genes and gene expression programs.39 In human T cell acute lymphoblastic leukemia, the core transcriptional machinery regulates molecular pathogenesis.40 It was demonstrated that a crucial transcription factor, GATA1, is associated with the pathogenesis of ET.41 Therefore, identifying more master TFs that regulating gene expressions in human diseases, including ET, is one of the substantial targets. To identify regulatory networks of master transcriptional regulators (MMTRs), we utilized iRegulon, a plugin of Cytoscape, to identify MMTRs. We found that the TFs such as IRF4, SRF, AR, SPI1, GTF2A1, TEAD4, E2F3, MYB, SP9, GATA2, CEBPA, IKZF2, SPIB, FLI1, ELK1, LEF1, and CREB3 were significantly associated with the DEGs. Based on our research, we have confirmed IRF4, TEAD4, MYB, SPI1, and SPIB with the most regulatory relationships.
IRF4 belongs to the IRF (Interferon Regulatory Factor) family of transcription factors and is significant in the regulation of interferon in the regulation of interferon-induced genes.42,43 It has been confirmed with lymphocyte-specific and negatively regulate Toll-like receptor (TLR) signaling and is essential for the activation of the innate and adaptive immune system.44,45 Recently, IRF4 also has been reported in the literature that it can bind to the interferon-stimulated response element (ISRE) of MHC class I promoters and is specifically involved in the pathogenesis of lymphatic system cancer.46 Furthermore, research has found that IRF4 is highly expressed in B cells and plasma cells, as well as controls the differentiation of B cells to plasma cells and the conversion of immunoglobulin classes.47 There was also a study that hypothesizes that direct targeting of IRF4 may be a potential therapeutic strategy for the treatment of multiple myeloma.48
TEAD4 (also known as Transcription Enhancement Factor 3, TEF-3) is a crucial member of the TEAD family.49 Studies have reported TEAD4 plays a vital role in the Hippo signaling pathway, thereby regulating cell proliferation, migration, and epithelial-mesenchymal transition (EMT) induction.50 In the study of the expression pattern and biological function of TEAD4, it was observed that the expression level of TEAD4 was increased in a variety of tumor tissues, such as lung adenocarcinoma, colorectal cancer, breast cancer, etc., and patients with higher TEAD4 expression tend to have higher poor overall survival.51–53 More and more convincing evidence also shows that TEAD4 does play a role in cancer development, and it is also a new prognostic marker for various cancers.54
MYB has been confirmed to play an essential role in the regulation of hematopoiesis.55 As far as we know, MYB has abnormal expression or rearrangement or undergoes translocation in leukemia and lymphoma and is considered to be an oncogene.56 In recent years, the implementation of advanced molecular biology and genetic manipulation techniques in vitro and in vivo has further revealed the critical role of MYB in many types of tumors, including leukemia, colon cancer, breast cancer, and adenoid cystic carcinoma.57–59 In general, the high level of MYB expression is related to the blockade of cell differentiation and continued proliferation, which leads to carcinogenicity.60 Clinically, MYB has been found to have abnormal expression, rearrangement, or translocation in many types of leukemia and lymphoma, so it is considered to be an oncogene, and its strange expression or rearrangement or translocation is closely related to poor prognosis.61 Still, there a study has found that lower levels of MYB expression have been shown to promote an ET-like disease in mice and megakaryocytic development in humans.62
SPI1 (also named hematopoietic transcription factor PU.1) encodes an ETS (Erythroblast Transformation Specific) domain transcription factor, which activates gene expression during the development of myeloid cells and B lymphocytes, and can also specifically participate in the differentiation or activation of macrophages or B cells.63 In recent years, research has found that SPI1 as a critical regulator of many steps in the hematopoietic process, not only can limit the self-renewal of blood stem cells but also the imbalance of its expression or activity leads to leukemia.64,65
Moreover, SPIB (SPI1 Related Transcription Factor) also can act as a lymphoid-specific enhancer and promote the development of plasmacytoid dendritic cells (pDCs), which can produce large amounts of interferon.66 SPIB has been shown to regulate many genes essential for BCR-mediated signals, including Igβ heavy chain, Ig light chain (α and α), mb-1 (Igα), and tyrosine kinase BTK.67 Recently, a study has found that SPIB with the diffuse large B-cell lymphoma (DLBCL).68 Its expression is essential for the survival of patients with DLBCL and contributes to cell apoptosis through the PI3K-AKT pathway.69 Besides, gene expression profiling, and IHC staining have both detected SPIB in some solid malignant tumors, indicating that SPI1 and SPIB may be abnormally expressed in In many types of tumors.70 Also, the other TFs such as GATA2, CEBPA, FLI1, SPI1, CREB3 may also have been implicated in the pathogenesis of leukemic transformation of myeloproliferative neoplasm.71–75 These results of TFs identification revised us to an in-depth understanding of the regulatory network of MMTRs in ET.
In the procession of REACTOME pathway analysis, we also found that the DEGs were mainly enriched in the innate immune system, neutrophil degranulation, interleukin-6 signaling. As far as we know, the abnormal expression of cytokines related to inflammation and immune regulation was ubiquitous in MPN patients, especially those with ET.76 Thus, driving clonal evolution by inducing mutations in additional inflammatory and immunomodulatory genes can further enhance cytokine and cytokine release, thereby promoting MPNs development.77 Also, recent clinical trials have proved that IL-6 can mediate the occurrence and development of MPN through the IL-6/JAK2/STAT3 axis, and current clinical trials about new drugs target the IL- 6/JAK2/STAT3 signal axis have achieved encouraging results.78 Besides, the REACTOME database used to conduct pathway enrichment analysis also presented additional enrichment in both MAP2K and MAPK activation, signaling by moderate kinase activity BRAF mutants, and the role of these classic signal pathway has been confirmed related with MPNs through an effect on the immune system and inflammation processes.79–81
Subsequently, we further analyzed the protein interaction network in the STRING database and the top 10 hub genes screened out, such as ACTB, PTPRC, ACTR2, FYB, STAT1, ETS1, IL7R, IKZF1, FGL2, and CTSS, were both down-regulated in two profiles. Interestingly, we found that the hub genes we identified were both down-regulated in a bone marrow cohort, and we also confirmed that the expression level of 8 identified hub genes was down-regulated in our clinical samples. However, it is worth mentioning that two of the hub genes (IL7R and ETS1) are contrary to the results of previous findings, and the result indicating that the deregulation of hub genes might be started from patients’ bone marrow and exist in peripheral blood. Besides, we used multiple databases to identify and analyzed the therapeutic drugs targeting these genes, and five target drugs were finally determined. Studies have found that ACTB appeared to be strongly associated with the JAK2 V617F mutation in ET patients, and the drug interaction analysis showed that ACTB was target by cyclophosphamide.82 PTPRC (also named CD45) is protein tyrosine phosphatase (PTP) as a natural counter-product of PTK activity. A single-Cell RNA-Seq research has determined that the PTPRC gene can be stably expressed in MPN stem and progenitor cells.83 Then, the target drug identification shows that prednisone and fluorouracil have been detected as the potential drug for targeting PTPRC.84 The defect of the IL7R gene may be related to severe combined immunodeficiency (SCID), and IL7R could lead to the activation of downstream JAK/STAT and PI3K/Akt/mTOR signaling cascades, so loss-of-function mutations in IL7R could play an essential role in the oncogenesis of MPNs.85 IL7R signals through the JAK/STAT (specifically, JAK1) and PI3K/mTOR pathways and gain-of-function mutations result in their constitutive activation; there is research that finds that Ruxolitinib would be expected to abrogate this signaling.86 IKZF1 gene encodes a transcription factor that is associated with MPN transformation, and lenalidomide could significantly reduce the abundance of IKZF1 in the multiple myeloma cell line.87,88
Although we have verified the relationship between the above four genes and MPN through the literature, as well as potential therapeutic drugs that target these genes, the relationship between STAT1 and MPN is still worthy of our attention. STAT1 is one of the members of the STAT family, which is involved in cell proliferation, differentiation, immune response, and other essential biological processes.89 Studies have shown that under the stimulation of IFN, the phosphorylation of STAT1 will activate downstream gene expression, and the megakaryocytes of mice lacking STAT1 showed a decrease in polyploidy. This evidence indicates that STAT1 signal transduction plays an essential function in the differentiation of normal megakaryocytes. Under the background of JAK2V617F, the deletion of the STAT1 gene can lead to the occurrence and development of ET and aggravate the disease burden.90 Altogether, these dysregulated hub genes and key regulators are associated with complications and pathogenesis of ET. It should be emphasized that this study still has some limitations. The GSE124281 and GSE61629 were originated from different experimental platforms (GPL570 vs GPL1058), which may lead to internal heterogeneity of the study. Another limitation of this study is that the sample size (n=7) of the bone marrow cohort is small, and our findings need to be further validated with a larger clinical sample.
Conclusion
Above all, the hub genes and key regulators were screened out as the critical markers in ET, and these markers could mediate the occurrence, development, and diagnosis of MPN by affecting the immune system and inflammatory processes. The hub genes we screened in this present study both have essential diagnostic value, and we found that the deregulation of hub genes might be started from bone marrow and exist in peripheral blood. Besides, we find that cyclophosphamide, prednisone, fluorouracil, ruxolitinib, and lenalidomide could be potential treatments through targeting on CTB, PTPRC, IL7R, IKZF1. Due to the complex pathogenesis of ET and many influencing factors, it is necessary to explore and sort out the genes and interaction clusters with logical relationships and practical significance, then to find out the entry point of the follow-up research work.
Data Sharing Statement
All data utilized, produce, or analyzed during this study are included in this article.
Ethics Approval and Consent to Participate
All clinical studies were conducted in integrity, in compliance with accepted ethical principles, and were approved by the Research Ethics Committee of the First Affiliated Hospital of Xinjiang Medical University (reference number: 220200320). The study was performed in accordance with the principles of the Declaration of Helsinki. All the participants provided written informed consent to be involved in this study. The ethics committee mentioned approval of this consent procedure. All individual information related to the samples included in the study was anonymous.
Acknowledgments
We thank Drs Jian-hua Wang, Jian-hua Yang, and Xuan-sheng Ding for critically reviewing the entire manuscript and providing valuable comments on the completeness of the work.
Author Contributions
All listed authors made important contributions to the study concept and study design as well as to the interpretation of the data. Jie Wang, Yun Wu, and Md. Nazim Uddin were responsible for the analysis of the study data, also the production of all the images and tables. Jie Wang, Yun Wu, Md. Nazim Uddin, and Jian-ping Hao drafted the manuscript and substantially revised its content. Jie Wang, Rong Chen, and Jian-ping Hao were responsible for diagnostics, sample collection, and PCR validation experiments in the clinical validation. All listed authors reviewed all versions of the article prior to submission, during revision, in the final version accepted for publication, and agree to take responsibility and be accountable for the contents of the article.
Funding
The present study was supported by the research funds from the Natural Science Foundation of Xinjiang Uygur autonomous region (grant no. 2019D01C309).
Disclosure
The authors declare that they have no competing interests.
References
1. Tefferi A, Pardanani A, Solomon CG. Essential Thrombocythemia. N Engl J Med. 2019;381(22):2135–2144. doi:10.1056/NEJMcp1816082
2. Sankar K, Stein BL, Rampal RK. Thrombosis in the Philadelphia Chromosome-Negative Myeloproliferative Neoplasms. Cancer Treat Res. 2019;179:159–178.
3. Ayres-Silva JP, Bonamino MH, Gouveia ME, et al. Genetic Alterations in Essential Thrombocythemia Progression to Acute Myeloid Leukemia: a Case Series and Review of the Literature. Front Oncol. 2018;8:32. doi:10.3389/fonc.2018.00032
4. O’Sullivan JM, Hamblin A, Yap C, et al. The poor outcome in high molecular risk, hydroxycarbamide-resistant/intolerant ET is not ameliorated by ruxolitinib. Blood. 2019;134(23):2107–2111. doi:10.1182/blood.2019001861
5. Lau WW, Hannah R, Green AR, Göttgens B. The JAK-STAT signaling pathway is differentially activated in CALR-positive compared with JAK2V617F-positive ET patients. Blood. 2015;125(10):1679–1681. doi:10.1182/blood-2014-12-618074
6. Milosevic Feenstra JD, Nivarthi H, Gisslinger H, et al. Whole-exome sequencing identifies novel MPL and JAK2 mutations in triple-negative myeloproliferative neoplasms. Blood. 2016;127(3):325–332. doi:10.1182/blood-2015-07-661835
7. Barrett T, Wilhite SE, Ledoux P, et al. NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Res. 2013;41(Database issue):D991–995. doi:10.1093/nar/gks1193
8. Clough E, Barrett T. The Gene Expression Omnibus Database. Methods Mol Biol. 2016;1418:93–110.
9. Ghaedi H, Bastami M, Zare-Abdollahi D, et al. Bioinformatics prioritization of SNPs perturbing microRNA regulation of hematological malignancy-implicated genes. Genomics. 2015;106(6):360–366. doi:10.1016/j.ygeno.2015.10.004
10. Guo C, Li Z. Bioinformatics Analysis of Key Genes and Pathways Associated with Thrombosis in Essential Thrombocythemia. Med Sci Monit. 2019;25:9262–9271. doi:10.12659/MSM.918719
11. Hasselbalch HC, Thomassen M, Riley CH, et al. Whole blood transcriptional profiling reveals deregulation of oxidative and antioxidative defence genes in myelofibrosis and related neoplasms. Potential implications of downregulation of Nrf2 for genomic instability and disease progression. PLoS One. 2014;9(11):e112786. doi:10.1371/journal.pone.0112786
12. Gautier L, Cope L, Bolstad BM, Irizarry RA. affy--analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20(3):307–315. doi:10.1093/bioinformatics/btg405
13. Du P, Kibbe WA, Lin SM. lumi: a pipeline for processing Illumina microarray. Bioinformatics. 2008;24(13):1547–1548. doi:10.1093/bioinformatics/btn224
14. Carlson M. org.Hs.eg.db: genome wide annotation for Human: bioconductor; 2019.
15. Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi:10.1093/nar/gkv007
16. Kolde R. pheatmap: pretty Heatmaps. Available from: https://CRAN.R-project.org/package=pheatmap.
17. Slowikowski K. ggrepel: automatically Position Non-Overlapping Text Labels with ‘ggplot2ʹ. Available from: https://CRAN.R-project.org/package=ggrepel.
18. Bindea G, Mlecnik B, Hackl H, et al. ClueGO: a Cytoscape plugin to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25(8):1091–1093. doi:10.1093/bioinformatics/btp101
19. Janky R, Verfaillie A, Imrichová H, et al. iRegulon: from a gene list to a gene regulatory network using large motif and track collections. PLoS Comput Biol. 2014;10(7):e1003731. doi:10.1371/journal.pcbi.1003731
20. Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607–607D613. doi:10.1093/nar/gky1131
21. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi:10.1101/gr.1239303
22. Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003;4:2. doi:10.1186/1471-2105-4-2
23. Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, Lin CY. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;8(Suppl 4):S11. doi:10.1186/1752-0509-8-S4-S11
24. Cotto KC, Wagner AH, Feng YY, et al. DGIdb 3.0: a redesign and expansion of the drug-gene interaction database. Nucleic Acids Res. 2018;46(D1):D1068–1068D1073. doi:10.1093/nar/gkx1143
25. Tefferi A. Myeloproliferative neoplasms: a decade of discoveries and treatment advances. Am J Hematol. 2016;91(1):50–58. doi:10.1002/ajh.24221
26. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. doi:10.1182/blood-2016-03-643544
27. Saeidi K. Myeloproliferative neoplasms: current molecular biology and genetics. Crit Rev Oncol Hematol. 2016;98:375–389.
28. Shirane S, Araki M, Morishita S, et al. JAK2, CALR, and MPL mutation spectrum in Japanese patients with myeloproliferative neoplasms. Haematologica. 2015;100(2):e46–48. doi:10.3324/haematol.2014.115113
29. Elala YC, Lasho TL, Gangat N, et al. Calreticulin variant stratified driver mutational status and prognosis in essential thrombocythemia. Am J Hematol. 2016;91(5):503–506. doi:10.1002/ajh.24338
30. Nangalia J, Green TR. The evolving genomic landscape of myeloproliferative neoplasms. Hematology Am Soc Hematol Educ Program. 2014;2014(1):287–296.
31. Tefferi A. Novel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1. Leukemia. 2010;24(6):1128–1138. doi:10.1038/leu.2010.69
32. Tefferi A, Pardanani A. Myeloproliferative Neoplasms: a Contemporary Review. JAMA Oncol. 2015;1(1):97–105. doi:10.1001/jamaoncol.2015.89
33. Lewis CM, Pegrum GD. Immune complexes in myeloproliferative disorders. Lancet. 1977;2(8049):1151–1153. doi:10.1016/s0140-6736(77)91539-2
34. Marin Oyarzún CP, Carestia A, Lev PR, et al. Neutrophil extracellular trap formation and circulating nucleosomes in patients with chronic myeloproliferative neoplasms. Sci Rep. 2016;6:38738.
35. Marin Oyarzún CP, Heller PG. Platelets as Mediators of Thromboinflammation in Chronic Myeloproliferative Neoplasms. Front Immunol. 2019;10:1373. doi:10.3389/fimmu.2019.01373
36. Wolach O, Sellar RS, Martinod K, et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci Transl Med. 2018;10(436):436. doi:10.1126/scitranslmed.aan8292
37. Hurtado-Nedelec M, Csillag-Grange MJ, Boussetta T, et al. Increased reactive oxygen species production and p47phox phosphorylation in neutrophils from myeloproliferative disorders patients with JAK2 (V617F) mutation. Haematologica. 2013;98(10):1517–1524. doi:10.3324/haematol.2012.082560
38. Dutzmann J, Daniel JM, Bauersachs J, Hilfiker-Kleiner D, Sedding DG. Emerging translational approaches to target STAT3 signalling and its impact on vascular disease. Cardiovasc Res. 2015;106(3):365–374. doi:10.1093/cvr/cvv103
39. Lee TI, Young RA. Transcriptional regulation and its misregulation in disease. Cell. 2013;152(6):1237–1251. doi:10.1016/j.cell.2013.02.014
40. Sanda T, Lawton LN, Barrasa MI, et al. Core transcriptional regulatory circuit controlled by the TAL1 complex in human T cell acute lymphoblastic leukemia. Cancer Cell. 2012;22(2):209–221. doi:10.1016/j.ccr.2012.06.007
41. Rinaldi CR, Martinelli V, Rinaldi P, Ciancia R, Del Vecchio L. GATA1 is overexpressed in patients with essential thrombocythemia and polycythemia vera but not in patients with primary myelofibrosis or chronic myelogenous leukemia. Leuk Lymphoma. 2008;49(7):1416–1419. doi:10.1080/10428190802087462
42. Nam S, Lim JS. Essential role of interferon regulatory factor 4 (IRF4) in immune cell development. Arch Pharm Res. 2016;39(11):1548–1555. doi:10.1007/s12272-016-0854-1
43. Man K, Gabriel SS, Liao Y, et al. Transcription Factor IRF4 Promotes CD8(+) T Cell Exhaustion and Limits the Development of Memory-like T Cells during Chronic Infection. Immunity. 2017;47(6):1129–1141.e5. doi:10.1016/j.immuni.2017.11.021
44. Negishi H, Ohba Y, Yanai H, et al. Negative regulation of Toll-like-receptor signaling by IRF-4. Proc Natl Acad Sci U S A. 2005;102(44):15989–15994. doi:10.1073/pnas.0508327102
45. Li Q, Yan Y, Liu J, et al. Toll-Like Receptor 7 Activation Enhances CD8+ T Cell Effector Functions by Promoting Cellular Glycolysis. Front Immunol. 2019;10:2191. doi:10.3389/fimmu.2019.02191
46. Wang C, Zhang L, Ke L, et al. Primary effusion lymphoma enhancer connectome links super-enhancers to dependency factors. Nat Commun. 2020;11(1):6318. doi:10.1038/s41467-020-20136-w
47. Pignarre A, Chatonnet F, Caron G, Haas M, Desmots-Loyer F, Fest T. Plasmablasts derive from CD23-negative activated B cells after the extinction of IL-4/STAT6 signaling and IRF4 induction. Blood. 2020.
48. Agnarelli A, Chevassut T, Mancini EJ. IRF4 in multiple myeloma-Biology, disease and therapeutic target. Leuk Res. 2018;72:52–58. doi:10.1016/j.leukres.2018.07.025
49. Chen M, Huang B, Zhu L, Chen K, Liu M, Zhong C. Structural and Functional Overview of TEAD4 in Cancer Biology. Onco Targets Ther. 2020;13:9865–9874. doi:10.2147/OTT.S266649
50. Lin KC, Moroishi T, Meng Z, et al. Regulation of Hippo pathway transcription factor TEAD by p38 MAPK-induced cytoplasmic translocation. Nat Cell Biol. 2017;19(8):996–1002. doi:10.1038/ncb3581
51. Wang C, Nie Z, Zhou Z, et al. The interplay between TEAD4 and KLF5 promotes breast cancer partially through inhibiting the transcription of p27Kip1. Oncotarget. 2015;6(19):17685–17697. doi:10.18632/oncotarget.3779
52. Liu Y, Wang G, Yang Y, et al. Increased TEAD4 expression and nuclear localization in colorectal cancer promote epithelial-mesenchymal transition and metastasis in a YAP-independent manner. Oncogene. 2016;35(21):2789–2800. doi:10.1038/onc.2015.342
53. Gu C, Huang Z, Chen X, et al. TEAD4 promotes tumor development in patients with lung adenocarcinoma via ERK signaling pathway. Biochim Biophys Acta Mol Basis Dis. 2020;1866(12):165921. doi:10.1016/j.bbadis.2020.165921
54. Zhou Y, Huang T, Cheng AS, Yu J, Kang W, To KF. The TEAD Family and Its Oncogenic Role in Promoting Tumorigenesis. Int J Mol Sci. 2016;17(1).
55. Mitra P. Transcription regulation of MYB: a potential and novel therapeutic target in cancer. Ann Transl Med. 2018;6(22):443. doi:10.21037/atm.2018.09.62
56. Liu X, Xu Y, Han L, Yi Y. Reassessing the Potential of Myb-targeted Anti-cancer Therapy. J Cancer. 2018;9(7):1259–1266. doi:10.7150/jca.23992
57. Inoue K, Fry EA. Novel Molecular Markers for Breast Cancer. Biomark Cancer. 2016;8:25–42. doi:10.4137/BIC.S38394
58. Malaterre J, Pereira L, Putoczki T, et al. Intestinal-specific activatable Myb initiates colon tumorigenesis in mice. Oncogene. 2016;35(19):2475–2484. doi:10.1038/onc.2015.305
59. Cao L, Mitra P, Gonda TJ. The mechanism of MYB transcriptional regulation by MLL-AF9 oncoprotein. Sci Rep. 2019;9(1):20084. doi:10.1038/s41598-019-56426-7
60. Wang X, Angelis N, Thein SL. MYB - A regulatory factor in hematopoiesis. Gene. 2018;665:6–17. doi:10.1016/j.gene.2018.04.065
61. Zuber J, Rappaport AR, Luo W, et al. An integrated approach to dissecting oncogene addiction implicates a Myb-coordinated self-renewal program as essential for leukemia maintenance. Genes Dev. 2011;25(15):1628–1640. doi:10.1101/gad.17269211
62. Tapper W, Jones AV, Kralovics R, et al. Genetic variation at MECOM, TERT, JAK2 and HBS1L-MYB predisposes to myeloproliferative neoplasms. Nat Commun. 2015;6:6691. doi:10.1038/ncomms7691
63. Kastner P, Chan S. PU.1: a crucial and versatile player in hematopoiesis and leukemia. Int J Biochem Cell Biol. 2008;40(1):22–27. doi:10.1016/j.biocel.2007.01.026
64. Delestré L, Cui H, Esposito M, et al. Senescence is a Spi1-induced anti-proliferative mechanism in primary hematopoietic cells. Haematologica. 2017;102(11):1850–1860. doi:10.3324/haematol.2016.157636
65. Seki M, Kimura S, Isobe T, et al. Recurrent SPI1 (PU.1) fusions in high-risk pediatric T cell acute lymphoblastic leukemia. Nat Genet. 2017;49(8):1274–1281. doi:10.1038/ng.3900
66. Sasaki I, Kaisho T. Transcriptional control of dendritic cell differentiation. Curr Top Microbiol Immunol. 2014;381:257–278. doi:10.1007/82_2014_378
67. Du W, Xu X, Niu Q, et al. Spi-B-Mediated Silencing of Claudin-2 Promotes Early Dissemination of Lung Cancer Cells from Primary Tumors. Cancer Res. 2017;77(18):4809–4822. doi:10.1158/0008-5472.CAN-17-0020
68. Takagi Y, Shimada K, Shimada S, et al. SPIB is a novel prognostic factor in diffuse large B-cell lymphoma that mediates apoptosis via the PI3K-AKT pathway. Cancer Sci. 2016;107(9):1270–1280. doi:10.1111/cas.13001
69. Montes-Moreno S, Ramos-Medina R, Martínez-López A, et al. SPIB, a novel immunohistochemical marker for human blastic plasmacytoid dendritic cell neoplasms: characterization of its expression in major hematolymphoid neoplasms. Blood. 2013;121(4):643–647. doi:10.1182/blood-2012-08-447599
70. Ho YJ, Lin YM, Huang YC, Yeh KT, Lin LI, Lu JW. Tissue microarray-based study of hepatocellular carcinoma validating SPIB as potential clinical prognostic marker. Acta Histochem. 2016;118(1):38–45. doi:10.1016/j.acthis.2015.11.005
71. Forrester AM, Grabher C, McBride ER, et al. NUP98-HOXA9-transgenic zebrafish develop a myeloproliferative neoplasm and provide new insight into mechanisms of myeloid leukaemogenesis. Br J Haematol. 2011;155(2):167–181. doi:10.1111/j.1365-2141.2011.08810.x
72. Fluhr S, Boerries M, Busch H, et al. CREBBP is a target of epigenetic, but not genetic, modification in juvenile myelomonocytic leukemia. Clin Epigenetics. 2016;8:50. doi:10.1186/s13148-016-0216-3
73. Crispino JD, Horwitz MS. GATA factor mutations in hematologic disease. Blood. 2017;129(15):2103–2110. doi:10.1182/blood-2016-09-687889
74. Vainchenker W, Raslova H. Megakaryocyte polyploidization: role in platelet production. Platelets. 2020;31(6):707–716. doi:10.1080/09537104.2019.1667497
75. Zhang L, Li J, Xu H, et al. Myc-Miz1 signaling promotes self-renewal of leukemia stem cells by repressing Cebpα and Cebpδ. Blood. 2020;135(14):1133–1145.
76. Lussana F, Rambaldi A. Inflammation and myeloproliferative neoplasms. J Autoimmun. 2017;85:58–63. doi:10.1016/j.jaut.2017.06.010
77. Čokić VP, Mitrović-Ajtić O, Beleslin-čokić BB, et al. Proinflammatory Cytokine IL-6 and JAK-STAT Signaling Pathway in Myeloproliferative Neoplasms. Mediators Inflamm. 2015;2015:453020. doi:10.1155/2015/453020
78. Johnson DE, O’Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol. 2018;15(4):234–248.
79. Braun LM, Zeiser R. Immunotherapy in Myeloproliferative Diseases. Cells. 2020;9(6):1559. doi:10.3390/cells9061559
80. Kamata T, Dankort D, Kang J, et al. Hematopoietic expression of oncogenic BRAF promotes aberrant growth of monocyte-lineage cells resistant to PLX4720. Mol Cancer Res. 2013;11(12):1530–1541.
81. Williams DA. Pairing JAK with MEK for improved therapeutic efficiency in myeloproliferative disorders. J Clin Invest. 2019;129(4):1519–1521. doi:10.1172/JCI127582
82. Xie HJ, Lundgren S, Broberg U, Finnström N, Rane A, Hassan M. Effect of cyclophosphamide on gene expression of cytochromes p450 and beta-actin in the HL-60 cell line. Eur J Pharmacol. 2002;449(3):197–205. doi:10.1016/S0014-2999(02)01995-7
83. Craver B, Nguyen Q, Ramanathan G, Fleischman AG. Single-Cell RNA-Seq to Assess Differential Responses to Tnfα in Human Hematopoietic Stem and Progenitor Cells in Myeloproliferative Neoplasm. Blood. 2019;134(Supplement_1):2518. doi:10.1182/blood-2019-123480
84. Kakiuchi S, Ohara S, Ogata S, Miura D, Kasahara Y, Izawa Y. Flow cytometric analyses on lineage-specific cell surface antigens of rat bone marrow to seek potential myelotoxic biomarkers: status after repeated dose of 5-fluorouracil. J Toxicol Sci. 2004;29(2):101–111. doi:10.2131/jts.29.101
85. Dhanyamraju PK, Iyer S, Smink G, et al. Transcriptional Regulation of Genes by Ikaros Tumor Suppressor in Acute Lymphoblastic Leukemia. Int J Mol Sci. 2020;21(4):1377. doi:10.3390/ijms21041377
86. Maude SL, Tasian SK, Vincent T, et al. Targeting JAK1/2 and mTOR in murine xenograft models of Ph-like acute lymphoblastic leukemia. Blood. 2012;120(17):3510–3518. doi:10.1182/blood-2012-03-415448
87. Nguyen HM, Gotlib J. Insights into the molecular genetics of myeloproliferative neoplasms. Am Soc Clin Oncol Educ Book. 2012;411–418. doi:10.14694/EdBook_AM.2012.32.85
88. Krönke J, Udeshi ND, Narla A, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014;343(6168):301–305. doi:10.1126/science.1244851
89. Arulogun SO, Choong HL, Taylor D, et al. JAK1 somatic mutation in a myeloproliferative neoplasm. Haematologica. 2017;102(8):e324–324e327. doi:10.3324/haematol.2017.170266
90. Czech J, Cordua S, Weinbergerova B, et al. JAK2V617F but not CALR mutations confer increased molecular responses to interferon-α via JAK1/STAT1 activation. Leukemia. 2019;33(4):995–1010. doi:10.1038/s41375-018-0295-6
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.